")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Lisdexamfetamine prodrug activation by peptidase-mediated hydrolysis in the cytosol of red blood cells
Authors Sharman J, Pennick M, Murkett K
Received 1 July 2014
Accepted for publication 23 September 2014
Published 28 November 2014 Volume 2014:10 Pages 2275—2280
DOI https://doi.org/10.2147/NDT.S70382
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Johannah Sharman, Michael Pennick
Shire, Basingstoke, UK
Abstract: Lisdexamfetamine dimesylate (LDX) is approved as a once-daily treatment for attention-deficit/hyperactivity disorder in children, adolescents, and adults in some countries. LDX is a prodrug comprising d-amphetamine covalently linked to l-lysine via a peptide bond. Following oral administration, LDX is rapidly taken up from the small intestine by active carrier-mediated transport, probably via peptide transporter 1. Enzymatic hydrolysis of the peptide bond to release d-amphetamine has previously been shown to occur in human red blood cells but not in several other tissues. Here, we report that LDX hydrolytic activity resides in human red blood cell lysate and cytosolic extract but not in the membrane fraction. Among several inhibitors tested, a protease inhibitor cocktail, bestatin, and ethylenediaminetetra-acetic acid each potently inhibited d-amphetamine production from LDX in cytosolic extract. These results suggest that an aminopeptidase is responsible for hydrolytic cleavage of the LDX peptide bond, although purified recombinant aminopeptidase B was not able to release d-amphetamine from LDX in vitro. The demonstration that aminopeptidase-like activity in red blood cell cytosol is responsible for the hydrolysis of LDX extends our understanding of the smooth and consistent systemic delivery of d-amphetamine by LDX and the long daily duration of efficacy of the drug in relieving the symptoms of attention-deficit/hyperactivity disorder.
Keywords: lisdexamfetamine, LDX, prodrug, peptidase, hydrolysis, attention-deficit/hyperactivity disorder
Introduction
Lisdexamfetamine dimesylate (LDX) is an effective treatment for the symptoms of attention-deficit/hyperactivity disorder (ADHD) in children, adolescents, and adults.1 LDX is a prodrug in which a peptide bond covalently links d-amphetamine with the naturally occurring amino acid l-lysine, rendering the parent molecule pharmacologically inactive. The absorption and subsequent hydrolysis of LDX to release the active d-amphetamine moiety are clinically significant because these processes underlie the low interpatient and intrapatient variability in active drug exposure2 and the long duration of efficacy necessary in a once-daily ADHD medication.3,4
Following oral administration, LDX is efficiently absorbed from the small intestine by carrier-mediated active transport, most likely via peptide transporter-1.5,6 LDX is neither a substrate nor an inhibitor of the cytochrome P450 enzymes and, therefore, has low potential for pharmacokinetic drug–drug interactions.7 We have previously shown that the hydrolysis of LDX takes place in human blood but not in human intestine, kidney, liver, or pancreas.5 Within the blood, the bioconversion of LDX was associated with red blood cells but not with platelets, peripheral blood mononuclear cells (PBMCs), or polymorphonuclear leukocytes (PMNs).5 In these studies, the rate of LDX hydrolysis was hematocrit-dependent, but substantial conversion to d-amphetamine remained when hematocrit values were only 10% or 25% of normal.5 Lysis of red blood cells did not affect the rate of LDX hydrolysis.5 Furthermore, the release of d-amphetamine from LDX was unimpaired in blood from donors with abnormal red blood cells resulting from sickle-cell disease.8
To build on the identification of human red blood cells as the site of enzymatic conversion of LDX prodrug to pharmacologically active d-amphetamine, we now describe in vitro investigations into the identity and subcellular localization of the human red blood cell enzyme activity responsible for LDX hydrolysis.
Methods
Blood collection and fractionation
Fresh human blood was collected from three healthy male donors using ethylenediaminetetra-acetic acid (EDTA) as an anticoagulant. Experiments were usually started within 2 hours of blood collection. Samples were pooled if they lacked obvious hemolysis (assessed visually after microcentrifugation of a small aliquot for 10 minutes). A portion of pooled whole blood was removed; then, the remainder was centrifuged at approximately 2,000× g for 10 minutes at 4°C, and the plasma supernatant was collected. Red blood cells were not separated from other blood cells during preparation of lysates, cytosolic extracts, or membrane fractions. Nevertheless, we refer to these as deriving from red blood cells in this paper because other blood cells have been previously shown not to hydrolyze LDX.5 To prepare red blood cell lysates, the pellet was resuspended in Hanks’ balanced salt solution (HBSS), pH 7.4, and washed once in HBSS by repeat centrifugation and suspension. It was then snap frozen for 30 minutes on dry ice, thawed at room temperature, homogenized with a handheld device, and diluted with five volumes of HBSS. Red blood cell cytosolic extracts were prepared by pelleting and washing as above, followed by the addition of three volumes of HBSS, freeze-thawing, and collection of the supernatant following centrifugation at 80,000× g for 30 minutes at 4°C. The resulting pellet was reconstituted in four volumes of HBSS to form the membrane fraction.
Incubation of LDX
A stock solution of LDX (Albany Molecular Research, Inc., Albany, NY, USA) was prepared at a concentration of 100 μg/mL free-base equivalent in water and used fresh or stored at -20°C. Duplicate 1.98 mL samples of blood or fractionated blood were equilibrated to 37°C for 5 minutes before addition of 20 μL of stock LDX to produce a final LDX concentration of 1 μg/mL. At 0, 15, 30, 60, 120, and 240 minutes after addition of LDX, 50 μL aliquots were removed, and the reaction was quenched in 0.5 mL of acetonitrile with vortex mixing for 30 seconds. Negative control incubations (excluding red blood cell-derived tissue) were conducted for up to 4 hours.
Inhibitors
Bis(4-nitrophenyl) phosphate (BNPP) (Sigma-Aldrich, St Louis, MO, USA) was prepared in HBSS and used at a 1 mM final concentration. Protease inhibitor cocktail (Sigma-Aldrich) was prepared in HBSS to produce final incubation concentrations of 2 mM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), 14 μM E-64, 130 μM bestatin, 1.0 μM leupeptin, 0.3 μM aprotinin, and 1 mM EDTA. In some experiments, pellets were resuspended in protease inhibitor cocktail or BNPP in HBSS. Each inhibitor in the cocktail was also obtained separately from Sigma-Aldrich and was prepared in HBSS for use at the same final concentration as in the cocktail. Blood tissue samples were incubated with inhibitors 30 minutes before the addition of LDX. All protease inhibitor stock solutions were freshly made on the day of the experiment and stored on wet ice.
Aminopeptidase B assay
Purified human recombinant aminopeptidase B (OriGene, Rockville, MD, USA) was used at a concentration of 10.9 μg/mL total protein in HBSS. The formation of β-naphthylamine from l-lysine-2-naphthylamide (2 μM) was monitored in 200 μL volumes in 96-well plates by fluorescence at 389 nm with excitation at 250 nm. Incubations were conducted in the absence or presence of LDX (1 μg/mL) or bestatin (130 μM) at 37°C and monitored at 10-minute intervals for up to 100 minutes.
Quantification of LDX and d-amphetamine
To each acetonitrile-quenched sample, 50 μL of 1% formic acid in water, 10 μL of methanol, and 50 μL of internal mass spectrometry standards (300 ng/mL of deuterium-labeled amphetamine-d6 in water or 100 ng/mL each of warfarin, carbutamide, reserpine, and ritonavir in acetonitrile) were added. The samples were vortex mixed for 5 minutes and centrifuged at approximately 2,000× g for 15 minutes at 10°C, then a 400 μL aliquot of the supernatant was transferred to a 96-well plate, dried under nitrogen, and dissolved in 5% (volume/volume) acetonitrile in water containing 0.1% (volume/volume) formic acid. Mass spectrometry calibration standards of 10, 20, 100, 200, 1,000, and 2,000 ng/mL were prepared in whole blood or blood fractions as appropriate. A 5 μL aliquot of each sample was analyzed by high-performance liquid chromatography and tandem electrospray mass spectrometry. No statistical analyses were performed to compare concentrations of LDX or d-amphetamine in different samples.
Results
LDX hydrolysis in whole and fractionated human blood
Across all experiments using incubations of LDX with whole blood (Figures 1 and 2), the intact prodrug was lost with a mean half-life (t½) of 1.6 hours (standard deviation [SD] 0.5). Across all such experiments (Figures 1 and 2), 82% (SD 8) of intact LDX was lost after 4 hours, resulting in mean d-amphetamine concentrations of 446 ng/mL (SD 20). In contrast, LDX was completely stable in plasma for 4 hours, and d-amphetamine levels remained below the limit of detection (10 ng/mL) (Figure 1).
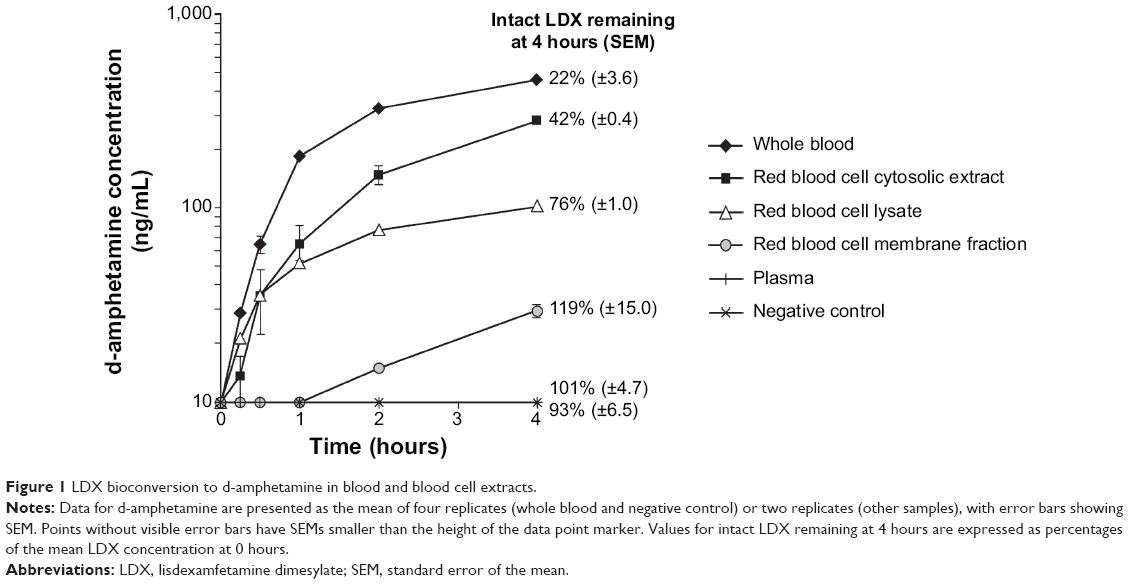 | Figure 1 LDX bioconversion to d-amphetamine in blood and blood cell extracts. |
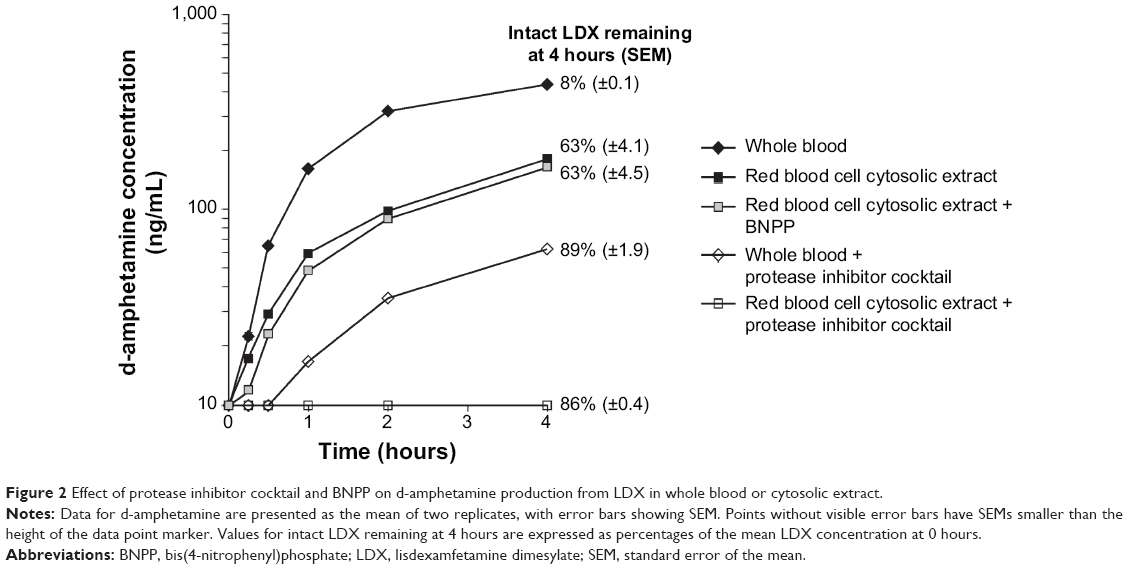 | Figure 2 Effect of protease inhibitor cocktail and BNPP on d-amphetamine production from LDX in whole blood or cytosolic extract. |
After 4 hours’ incubation with red blood cell lysate, 24% of intact LDX was lost and d-amphetamine levels reached 102 ng/mL (Figure 1). In the membrane fraction, there was no detectable loss of intact LDX, and the mean d-amphetamine concentration was 29 ng/mL after 4 hours (Figure 1). Across all experiments using incubations of LDX with cytosolic extract (Figures 1 through 3), about 50% of the intact prodrug was lost after 4 hours (mean t½, 4.1 hours; SD 0.99), and mean d-amphetamine levels reached 223 ng/mL (SD 53).
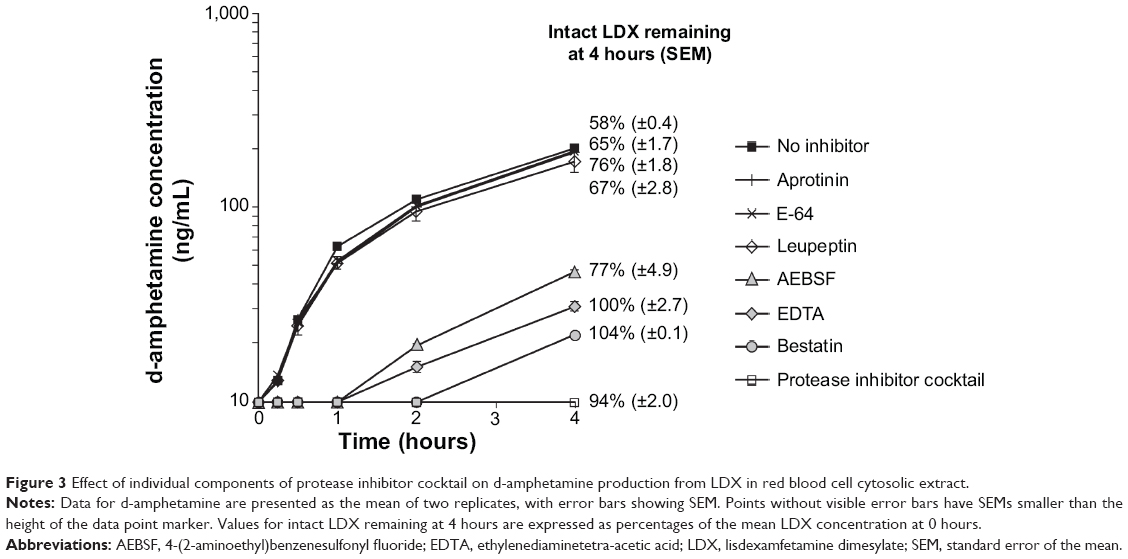 | Figure 3 Effect of individual components of protease inhibitor cocktail on d-amphetamine production from LDX in red blood cell cytosolic extract. |
Inhibition of LDX hydrolysis in blood cell cytosolic extract
When whole blood or red blood cell cytosolic extract was incubated with the protease inhibitor cocktail, more than 85% of LDX remained after 4 hours and d-amphetamine concentrations were 63 ng/mL or below the limit of detection, respectively (Figure 2). In contrast, the carboxylesterase/amidase inhibitor BNPP had no effect on LDX turnover or d-amphetamine production in cytosolic extract (Figure 2).
When the individual components of the protease inhibitor cocktail were tested in blood cell cytosolic extract, the time courses of d-amphetamine production in the presence of aprotinin, E-64, or leupeptin were almost superimposable upon that observed in the absence of inhibitor; in all cases, d-amphetamine concentrations at 4 hours exceeded 170 ng/mL (Figure 3). In contrast, addition of either EDTA or bestatin prevented detectable loss of intact LDX and reduced the d-amphetamine yield to 22–31 ng/mL at 4 hours. In the presence of AEBSF, some loss of intact LDX was evident, and the concentration of d-amphetamine after 4 hours was 47 ng/mL.
Assay for LDX hydrolysis by aminopeptidase B
Treatment of LDX with a commercial preparation of aminopeptidase B did not result in detectable d-amphetamine release from LDX, despite linear, bestatin-sensitive production of β-naphthylamide from a lysine-2-naphthylamine reporter substrate (data not shown).
Discussion
LDX has previously been shown to be stable when incubated with isolated platelets, PMNs, or PBMCs, with negligible production of d-amphetamine.5 In contrast, in the presence of isolated red blood cells, LDX was lost with a t½ of 1.0 hours, and d-amphetamine was produced.5 The present results build on these earlier observations by demonstrating, for the first time, that peptidase activity present in the cytosolic extract, but not the membrane fraction, is responsible for the hydrolysis of LDX by red blood cells.
The biotransformation of LDX to d-amphetamine when incubated with intact red blood cells and red blood cell cytosol was not inhibited by BNPP, but it was inhibited by a broad-spectrum inhibitor cocktail, implicating protease activity rather than carboxylesterase or amidase activity in the hydrolysis of LDX. When the components of the protease inhibitor cocktail were tested individually, the LDX-cleaving activity of the red blood cell cytosol was not inhibited by aprotinin, E-64, or leupeptin, suggesting that serine, cysteine, or thiol proteases were unlikely to be involved. In contrast, hydrolysis of LDX was inhibited by bestatin, AEBSF, and EDTA. The dipeptide analog bestatin is a selective inhibitor of aminopeptidases (eg, aminopeptidase B, and leukotriene A4 hydrolase). AEBSF is a broad-spectrum serine protease inhibitor,9 but has also been reported to inhibit eukaryotic aminopeptidases.10 Finally, inhibition by the chelating agent EDTA implicates a divalent cation-requiring protease. Together, the activities of these inhibitors are strongly suggestive of the involvement of metallo-aminopeptidase activity in the hydrolysis of LDX in red blood cell cytosol.
Previous studies have ruled out several classes of enzyme as being responsible for the hydrolysis of LDX. Incubation of LDX with dipeptidyl peptidase IV, cathepsin G, elastase, and trypsin, or in simulated gastric or intestinal fluid, all failed to elicit significant loss of LDX or generation of d-amphetamine.5 Peptidase activity of red blood cell cytosol has been described,11 and proteomic studies have identified peptidases in the cytosol.12 Aminopeptidase B is a red blood cell cytosolic enzyme with a preference for lysine13 and was, therefore, considered a likely candidate to mediate LDX hydrolysis. However, failure of recombinant human aminopeptidase B to convert LDX to d-amphetamine in vitro suggests that it is not the enzyme responsible. Other candidate peptidases (eg, leukotriene A4 hydrolase) remain to be investigated.
The demonstration that d-amphetamine is generated from LDX in red blood cell cytosol raises the question of the nature of the transport of LDX across the red blood cell plasma membrane. While investigating LDX transport was not an objective of the present work, the dipeptide-like properties of the drug, combined with the capacity of red blood cells to take up cationic dipeptides14 and the presence of high-affinity amino acid transporters in the red blood cell plasma membrane,15 are suggestive of a mechanism for future investigation. Furthermore, it is noteworthy that LDX is actively taken up from the gastrointestinal tract via peptide transporter-1.5,6 The rapid production of d-amphetamine from LDX in whole blood and intact red blood cells suggests that the transport of LDX across the red blood cell membrane is not rate limiting. The likely explanation for the reported failure of EDTA to inhibit LDX hydrolysis in whole blood or intact red blood cells is that EDTA is membrane impermeable.5
The prodrug nature of LDX, its rapid uptake from the gut, and its rate-limiting hydrolysis in the blood to produce active d-amphetamine have several important clinical implications. The clinical properties of LDX depend upon red blood cell cytosolic aminopeptidase activity, which is here identified as responsible for activation of the prodrug. First, oral administration of LDX results in systemic exposure to d-amphetamine that is proportional to the LDX dose and that exhibits low intraindividual and interindividual variability.2 The profile of d-amphetamine delivery by LDX underlies the long daily duration of action of LDX in relieving the hyperactive, inattentive, and impulsive symptoms experienced by patients with ADHD.3,4
Secondly, a direct comparison of d-amphetamine pharmacokinetics following intravenous administration of LDX or an equivalent dose of immediate-release d-amphetamine in adult stimulant abusers indicated that the mean maximum plasma drug concentration (Cmax) was lower (LDX, 20.7 ng/mL [SD 1.5]; d-amphetamine, 74.2 ng/mL [SD 58.7]) and the mean Tmax (time to reach Cmax) was longer (LDX, 2.5 hours [SD 1.3]; d-amphetamine, 0.2 hours [SD 0.2]) for the prodrug compared with immediate-release d-amphetamine. This may have contributed to the reduced subjective “drug-liking” effects of LDX compared with immediate-release d-amphetamine in these individuals.16,17
Thirdly, the demonstrations that substantial hydrolysis of LDX remained if the prodrug was incubated with red blood cells when hematocrit levels were 10% or 25% of normal, and that concentration–time curves for both LDX and d-amphetamine were virtually superimposable when LDX was incubated with red blood cells from healthy donors or individuals with sickle-cell disease, suggest that the high-capacity hydrolysis of LDX by red blood cells is tolerant of anemia and impaired red blood cell function.
Finally, aminopeptidase inhibitors are being evaluated as anti-inflammatory, immunomodulatory, analgesic, and anticancer agents,18,19 and bestatin is used as a cancer treatment in some countries.20 Thus, although the entry of bestatin into red blood cells is reportedly inefficient,21 the demonstration that it inhibits the hydrolysis of LDX suggests that potential interactions between LDX and therapeutic aminopeptidase inhibitors are possible. Previous work showed that none of the seven major human cytochrome P450 isoforms was inhibited by LDX, suggesting that interactions of LDX, suggesting that other drug–drug interactions are unlikely to be common.7
Limitations of this study include the following. First, red blood cells were not separated from other blood cells because platelets, PMNs, and PBMCs have been previously shown not to mediate LDX hydrolysis. Second, the purity of the cytosolic and membrane fractions was not verified (for example by assay of known markers) and the possibility therefore exists that the low level of LDX hydrolysis observed in membrane fraction was due to contamination with residual cytosolic protein. Third, blood from donors was pooled for analysis, preventing assessment of whether rates of LDX bioconversion vary between individuals.
In conclusion, the biotransformation of the prodrug stimulant LDX to produce active d-amphetamine is an important determinant of the pharmacodynamic profile of the drug in patients with ADHD. The present in vitro investigations demonstrate, for the first time, that the hydrolysis of LDX occurs in the cytosolic extract, but not in the membrane fraction, of human red blood cells. Although the inhibitor profile of the biotransformation is suggestive of aminopeptidase activity, the enzyme responsible remains to be identified.
Acknowledgments
Funding for this research was provided by Shire Development LLC. Oxford PharmaGenesis™ Ltd provided writing and editorial support for this publication with funding from Shire International GmbH. Matt Cottingham, DPhil, and Eric Southam, PhD, of Oxford PharmaGenesis™ Ltd provided writing assistance under the direction of the authors. Editorial assistance in editing, fact checking, formatting, proofreading, and submission was also provided by Oxford PharmaGenesis™ Ltd. Shire develops and markets drugs to treat psychiatric disorders, including ADHD.
Disclosure
Michael Pennick and Johannah Sharman are former employees of Shire and may have owned stocks or stock options. The authors do not have other conflicts of interest in this work.
References
Steer C, Froelich J, Soutullo CA, Johnson M, Shaw M. Lisdexamfetamine dimesylate: a new therapeutic option for attention-deficit hyperactivity disorder. CNS Drugs. 2012;26(8):691-705. | ||
Ermer J, Homolka R, Martin P, Buckwalter M, Purkayastha J, Roesch B. Lisdexamfetamine dimesylate: linear dose-proportionality, low intersubject and intrasubject variability, and safety in an open-label single-dose pharmacokinetic study in healthy adult volunteers. J Clin Pharmacol. 2010;50(9):1001-1010. | ||
Wigal SB, Kollins SH, Childress AC, Squires L; 311 Study Group. A 13-hour laboratory school study of lisdexamfetamine dimesylate in school-aged children with attention-deficit/hyperactivity disorder. Child Adolesc Psychiatry Ment Health. 2009;3(1):17. | ||
Wigal T, Brams M, Gasior M, Gao J, Squires L, Giblin J; 316 Study Group. Randomized, double-blind, placebo-controlled, crossover study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder: novel findings using a simulated adult workplace environment design. Behav Brain Funct. 2010;6:34. | ||
Pennick M. Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr Dis Treat. 2010;6: 317-327. | ||
Ermer JC, Haffey MB, Doll WJ, et al. Pharmacokinetics of lisdexamfetamine dimesylate after targeted gastrointestinal release or oral administration in healthy adults. Drug Metab Dispos. 2012;40(2):290-297. | ||
Krishnan S, Moncrief S. An evaluation of the cytochrome p450 inhibition potential of lisdexamfetamine in human liver microsomes. Drug Metab Dispos. 2007;35(1):180-184. | ||
Pennick M. Metabolism of the prodrug lisdexamfetamine dimesylate in human red blood cells from normal and sickle cell disease donors. J Drug Assess. 2013;2:17-20. | ||
Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102(12): 4639-4750. | ||
Rhoads ML, Fetterer RH, Urban JF Jr. Secretion of an aminopeptidase during transition of third- to fourth-stage larvae of Ascaris suum. J Parasitol. 1997;83(5):780-784. | ||
Grdisa M, Vitale L. Types and localization of aminopeptidases in different human blood cells. Int J Biochem. 1991;23(3):339-345. | ||
Goodman SR, Daescu O, Kakhniashvili DG, Zivanic M. The proteomics and interactomics of human erythrocytes. Exp Biol Med (Maywood). 2013;238(5):509-518. | ||
Abramic M, Vitale L. Basic amino acids preferring broad specificity aminopeptidase from human erythrocytes. Biol Chem Hoppe Seyler. 1992;373(7):375-380. | ||
Lochs H, Morse EL, Adibi SA. Uptake and metabolism of dipeptides by human red blood cells. Biochem J. 1990;271(1):133-137. | ||
Ellory JC, Swietach P, Gibson JS. Amino acid transport. In: Bernhardt I, Ellory JC, editors. Red Cell Membrane Transport in Health and Disease. Berlin: Springer Verlag; 2003:303–319. | ||
Jasinski DR, Krishnan S. Human pharmacology of intravenous lisdexamfetamine dimesylate: abuse liability in adult stimulant abusers. J Psychopharmacol. 2009;23(4):410-418. | ||
Jasinski DR, Krishnan S. Abuse liability and safety of oral lisdexamfetamine dimesylate in individuals with a history of stimulant abuse. J Psychopharmacol. 2009;23(4):419-427. | ||
Chen L, Teng Y, Xu W. Progress in the development of bestatin analogues as aminopeptidases inhibitors. Curr Med Chem. 2011; 18(7):964-976. | ||
Jia MR, Wei T, Xu WF. The analgesic activity of bestatin as a potent APN inhibitor. Front Neurosci. 2010;4:50. | ||
Hitzerd SM, Verbrugge SE, Ossenkoppele G, Jansen G, Peters GJ. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids. 2014;46(4):793-808. | ||
Scornik OA, Botbol V. Cellular uptake of 3H-bestatin in tissues of mice after its intravenous injection. Drug Metab Dispos. 1997;25(7): 798-804. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.