")
Back to Journals » International Journal of Nanomedicine » Volume 12
Liposomal formulation of a methotrexate lipophilic prodrug: assessment in tumor cells and mouse T-cell leukemic lymphoma
Authors Alekseeva AA, Moiseeva EV , Onishchenko NR , Boldyrev IA, Singin AS, Budko AP , Shprakh ZS, Molotkovsky JG, Vodovozova EL
Received 23 January 2017
Accepted for publication 6 April 2017
Published 15 May 2017 Volume 2017:12 Pages 3735—3749
DOI https://doi.org/10.2147/IJN.S133034
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Israel (Rudi) Rubinstein
Anna A Alekseeva,1 Ekaterina V Moiseeva,1 Natalia R Onishchenko,1 Ivan A Boldyrev,1 Alexander S Singin,2,† Andrey P Budko,2 Zoya S Shprakh,2 Julian G Molotkovsky,1 Elena L Vodovozova1
1M.M. Shemyakin and Yu.A. Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, 2N.N. Blokhin Russian Cancer Research Center, the Ministry of Health of the Russian Federation, Moscow, Russian Federation
†Alexander S Singin passed away on October 4, 2011
Abstract: In a previous study, a formulation of methotrexate (MTX) incorporated in the lipid bilayer of 100-nm liposomes in the form of diglyceride ester (MTX-DG, lipophilic prodrug) was developed. In this study, first, the interactions of MTX-DG liposomes with various human and mouse tumor cell lines were studied using fluorescence techniques. The liposomes composed of egg phosphatidylcholine (PC)/yeast phosphatidylinositol/MTX-DG, 8:1:1 by mol, were labeled with fluorescent analogs of PC and MTX-DG. Carcinoma cells accumulated 5 times more MTX-DG liposomes than the empty liposomes. Studies on inhibitors of liposome uptake and processing by cells demonstrated that the formulation used multiple mechanisms to deliver the prodrug inside the cell. According to the data from the present study, undamaged liposomes fuse with the cell membrane only 1.5–2 hours after binding to the cell surface, and then, the components of liposomal bilayer enter the cell separately. The study on the time course of plasma concentration in mice showed that the area under the curve of MTX generated upon intravenous injection of MTX-DG liposomes exceeded that of intact MTX 2.5-fold. These data suggested the advantage of using liposomal formulation to treat systemic manifestation of hematological malignancies. Indeed, the administration of MTX-DG liposomes to recipient mice bearing T-cell leukemic lymphoma using a dose-sparing regimen resulted in lower toxicity and retarded lymphoma growth rate as compared with MTX.
Keywords: liposomes, methotrexate, lipophilic prodrug, endocytosis, hematological malignancies, leukemia/lymphoma
Corrigendum for this paper has been published
Introduction
Methotrexate (MTX) – a folic acid antimetabolite – is a cytostatic agent widely used in the treatment of cancer and autoimmune diseases; it is in the World Health Organization Model List of Essential Medicines.1 The MTX competitively inhibits dihydrofolate reductase, thus depriving the cell of tetrahydrofolate, an essential cofactor for the biosynthesis of purine nucleotides and thymidylate. The efficiency of treatment with MTX is limited by systemic toxicity and frequent development of drug resistance; the latter is mainly associated with mutations that lead to a decrease in the activity of reduced folate carrier (RFC) controlling the entrance of folate and the analogs into the cell.2 MTX encapsulation in a nanoparticulate carrier is a promising means to improve its pharmacological properties. Such a carrier would protect the drug from premature nontarget interactions with biomolecules in circulation. Being able to enter a cell via pinocytosis, a carrier also would allow overcoming the resistance of tumor cells caused by impaired transmembrane transport. Indeed, in the last decade, a burst of research has been conducted on MTX-bearing drug delivery systems,3,4 including polyamide dendrimers,5 polymer nanogels,6 nanostructured carriers composed of triglycerides and surfactants,7 quantum dots,8 polyethylene glycol (PEG)-phospholipid micelles,9 and liposomes.10,11 Most of these carriers have insufficient drug-loading capacity. Recently, peptide-targeted PEGylated liposomes encapsulating MTX caused a significant improvement in clinical scores in the experimental autoimmune encephalomyelitis in mice.12 Meanwhile, the antitumor therapy with this antimetabolite agent – even in the case of low-dose regimens – requires several times higher dosages (and thus liposome-loading capacity) than the anti-inflammatory therapy.
An acknowledged method to encapsulate water-soluble drugs in nanosized liposomes is the remote loading technique. Unfortunately, it can only be applied to weak amphipathic acids or bases, which MTX molecule does not belong to.13 As for the passive encapsulation, the calculated loading capacity of a 100-nm liposome for a highly water-soluble drug does not exceed 2–3 mol% to total lipids. In a previous study, a liposomal formulation of MTX incorporated in the fluid lipid bilayer in the form of a tailored lipophilic conjugate, that is, dioleoylglyceride ester of α-COOH in the glutamate moiety of MTX (MTX-DG), was developed.14 In the cell, MTX-DG should easily be hydrolyzed with the release of MTX owing to the low substrate specificity of esterases distributed in most tissues in abundance.15 A short hydrophilic spacer (N-methylenecarbonyl-β-alanine) between the bulky MTX moiety and diglyceride membrane anchor minimizes the disruption of the bilayer packing to load as much as 10 mol% prodrug (to total lipids) into 100-nm liposomes (Figure 1). The liposomes are stable in buffer for at least 3 weeks; even after freezing, thawing, and ultrasonic treatment, they retain the prodrug in the lipid bilayer.14,16 In a panel of in vitro tests, overall, the formulation showed good hemotolerance.16
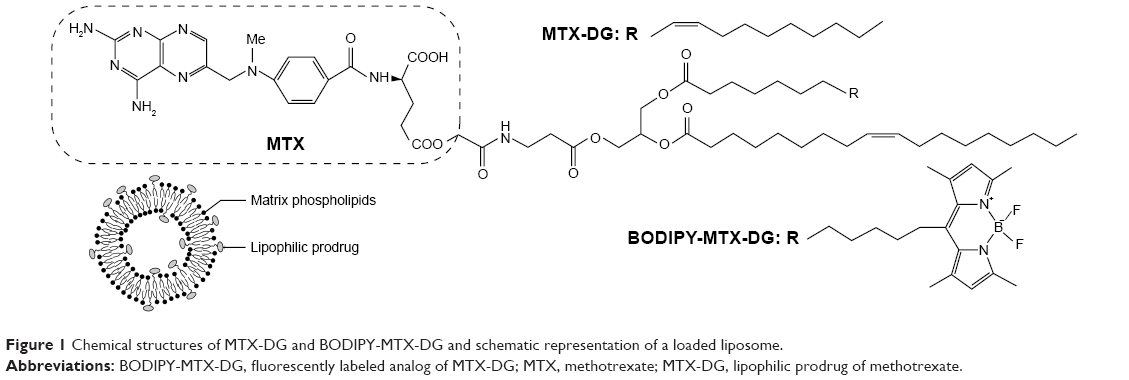 | Figure 1 Chemical structures of MTX-DG and BODIPY-MTX-DG and schematic representation of a loaded liposome. |
The liposomal formulation of MTX-DG (MTX-DG liposomes) successfully surmounted the resistance of cultured RFC-deficient human T-lymphoblastic cells toward MTX.14 Most likely, MTX-DG enters the cell incorporated in the liposomes or, more precisely, into liposomal bilayer. Whether any specific interactions of MTX moiety with folate-binding molecules on cell surface are involved in the process remains a question.
In this work, the interactions of the MTX-DG formulation with a number of human tumor cell lines were studied using fluorescence techniques to elucidate the mechanism of MTX-DG liposome internalization by cells. The in vitro data evidence that the whole liposomes bind to the cell surface using multiple mechanisms to deliver the prodrug inside the cell (clathrin-independent endocytosis is involved). Then, the plasma retention of MTX-DG and MTX generated upon intravenous (IV) injection of MTX-DG liposomes in mice compared with intact MTX administered IV were evaluated. The data obtained highlighted the therapeutic potency of the delivery system to treat systemic hematological diseases. Taking into account both the latter results and continuing prevalent use of MTX in the treatment of leukemia and lymphoma in clinic, the antitumor effect of these preparations was tested in a mouse T-cell leukemic lymphoma. The advantages of the liposomal formulation were as follows: toxicity was decreased and lymphoma growth was significantly delayed in tumor-bearing mice treated with MTX-DG liposomes as compared with free MTX.
Materials and methods
Materials
Phosphatidylcholine (PC) from egg yolk and phosphatidylinositol (PI) from Saccharomyces cerevisiae were obtained from Reakhim (Moscow, Russian Federation). rac-1,2-Dioleoylglyceride ester conjugate of MTX (MTX-DG),17 1-palmitoyl-2-[7-(Me4-BODIPY-8)heptanoyl]-sn-glycero-3-phosphocholine (BODIPY-PC),18 and BODIPY-labeled analog of MTX-DG (BODIPY-MTX-DG)19 (Figure 1) were synthesized as previously reported.
Fluorescent probes and tracers used in the present study were the following: BODIPY-PC, BODIPY-MTX-DG, and L-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (Rhod-PE) obtained from Avanti Polar Lipids (Alabaster, AL, USA); ER-Tracker™ Red (E34250, ~1 μM working solution), Transferrin Alexa Fluor® 555 conjugate (T35352, 20 μg/mL working solution), and wheat germ agglutinin (WGA) Alexa Fluor® 555 conjugate (W32464, 10 μg/mL working solution) obtained from Invitrogen (Carlsbad, CA, USA); calcein (bis[N,N-di(carboxymethyl)aminomethyl]fluorescein tetrasodium salt) and Hoechst 33342 obtained from Sigma Aldrich (St Louis, MO, USA). Rabbit polyclonal antibody raised against amino acids 1–257 representing full-length folate receptor alpha (FRα) of human origin (FL-257, 1:50 dilution) and bovine anti-rabbit IgG-PE secondary antibody (sc-3750, 1:400 dilution) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Inorganic salts, ethylenediaminetetraacetic acid (EDTA), chlorpromazine, cytochalasin B, colchicine, and nocodazole were purchased from Sigma Aldrich. Purified deionized water was prepared by Milli-Q plus System (Millipore Co., Billerica, MA, USA). The other solvents were of analytical or chromatographic grade. The evaporation processes were performed in vacuo at temperatures <40°C.
Liposome preparation and characterization
Prodrug-loaded liposomes (PC–PI–MTX-DG, 8:1:1; L-MTX-DG) were prepared as described earlier.16 Empty liposomes were composed of PC–PI, 9:1. Briefly, dry lipid films were hydrated in phosphate-buffered saline (PBS: 1.5 mM KH2PO4, 1.1 mM NaH2PO4 ⋅ 2H2O, 6.3 mM Na2HPO4, 2.7 mM KCl, and 136.8 mM NaCl) containing 1 mM EDTA, pH 7.0, and subjected to 6–10 cycles of freezing/thawing (liquid nitrogen/+40°C). Then, the suspension was extruded at an ambient temperature through two-stacked polycarbonate membrane filters with pore sizes of 100 nm (Nucleopore, obtained from Avanti Polar Lipids) 10 times on a mini-extruder (Avanti Polar Lipids). The size of the liposomes was controlled by dynamic light scattering by using Zetasizer Nano ZS equipment (Malvern Instruments Ltd., Malvern, UK) in at least 3 runs per sample. Zeta potential values were obtained for diluted (0.9–1.0 mg/mL) liposome suspensions in 10 mM KCl solution buffered with 1 mM potassium phosphate buffer, pH 7.4, by using the Zetasizer equipment. The samples were equilibrated 3 minutes in the prerinsed disposable cuvettes before 5 measurements per sample were performed at 25°C under the voltage of 150 V and current intensity of 4 mA. In order to visualize liposomes, 0.5 mol% of BODIPY-PC or 1 mol% of Rhod-PE and/or 1 mol% of BODIPY-MTX-DG were added at the stage of lipid film formation. Prodrug concentration was controlled via ultraviolet (UV) spectrophotometry after liposome disruption with 20 volumes of ethanol (MTX-DG: λmax =302 nm, ε =25,000 M−1 cm−1) on an SF-256-UVI two-beam spectrophotometer (LOMO Fotonika, St Petersburg, Russian Federation). The formulations were stored at 4°C and used for in vitro experiments within 10 days. For in vivo experiments, liposomal dispersions typically contained 2.6 mM MTX-DG and were used within 3 days.
Accumulation of liposomes by cultured cells studied by flow cytometry
In order to assess liposome accumulation by cells in vitro, liposome bilayers were labeled with a fluorescent PC conjugate, BODIPY-PC, and empty liposomes (without the prodrug) were used as control. Cells were cultured at 37°C in 5% CO2 atmosphere in Dulbecco’s Modified Eagle’s Medium or RPMI-1640 medium (Gibco BRL, Gaithersburg, MD, USA) supplemented with 300 μg/mL L-glutamine, 50,000 IU penicillin, 50 μg/mL streptomycin, and 10% fetal calf serum (PAA, Wien, Austria). In the case of adherent cultures (human lung carcinoma cells A549, human pancreatic cancer cells CoLo-357, human kidney embryonic cells HEK 293T, and murine fibroblast cells 3T3 were obtained from Bank of Cell Cultures of the Institute of Cytology, Russian Academy of Sciences, St Petersburg, Russian Federation), the cells were seeded on 24-well plates (Corning Inc., New York, NY, USA) to allow the formation of a confluent monolayer. On the next day, culture medium was replaced by liposome suspension (100 μM total lipid in a serum-free medium) and incubated for various time periods at 37°C. Then, the cells were rinsed with PBS, detached with 0.02% EDTA solution (10 minutes, 37°C), and analyzed by flow cytometry. For suspended human T-lymphocyte Jurkat cells (Bank of Cell Cultures of the Institute of Cytology), the growth medium was replaced with a serum-free medium; then, the cells were concentrated by centrifugation, diluted with liposome suspension (1×106 cell/mL, 100 μM final lipid concentration in the serum-free medium), and incubated for various time periods at 37°C under gentle stirring. Then, they were rinsed with PBS by centrifugation and analyzed by flow cytometry.
For flow cytometry measurements, cell suspensions were diluted with equal volumes of 1% bovine serum albumin solution in PBS and 0.3 μg/mL propidium iodide solution in PBS (the latter being used to assess cell viability) and analyzed using FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA) equipped with a 488-nm argon ion laser. Duplicate measurements with 10,000 events were recorded for each sample. Side/forward scatter and propidium iodide fluorescence signals were used to gate the cell subsets of interest and to eliminate debris, dead cells, and cell aggregates. The data were analyzed by using CELL Quest software.
The inhibition of MTX-DG liposome binding to A549 and CoLo-357 cells was assessed by the treatment of cells with excess of free MTX (100- and 1,000-fold excess over MTX-DG concentration) or anti-FRα antibody (10 μg/mL). Cell monolayers on 24-well plates were incubated with free MTX or anti-FRα antibody for 1 hour at 37°C. Then, with or without washing with PBS, MTX-DG liposomes were added (100 μM total lipids, 10 μM MTX-DG). After a 1-hour incubation, the cells were rinsed with PBS, detached with 0.02% EDTA solution, and analyzed by flow cytometry. FRα expression by A549 and CoLo357 cells was verified by indirect immunostaining of fixed permeabilized cells with anti-FRα antibody.
The different mechanisms of liposome uptake and cellular processing were studied by the use of a number of inhibitors, such as chlorpromazine (blocks reversibly clathrin-mediated pathway), cytochalasin B (inhibits actin polymerization and actin-dependent movement), colchicine (inhibits tubulin polymerization), and nocodazole (depolymerizes microtubules). A549 or CoLo-357 cell monolayers on 24-well plates were washed with PBS, incubated with the inhibitors at concentrations causing <5% cell death in serum-free medium for 1 hour (except for colchicine, where incubation was 5 minutes), and then coincubated with liposomes for 1 hour at 37°C (100 μM total lipids). In the case of chlorpromazine, the cells were washed with PBS prior to the incubation with liposomes (as this inhibitor is known to disturb liposome membrane20); in addition, the incubation with liposomes was limited to 30 minutes, as the effect of chlorpromazine is reversible. The untreated control cells were incubated with liposomes for 1 hour or 30 minutes in the serum-free medium, and then they were rinsed with PBS, detached with 0.02% EDTA solution (10 minutes, 37°C), and analyzed by flow cytometry.
Binding and intracellular trafficking of MTX-DG liposomes studied by confocal microscopy
The experiments were conducted on A549 cells seeded on cover slides (~0.5×106 cells/mL) and incubated for 24 hours in a humidified atmosphere of 5% CO2 at 37°C.
In order to examine the early stages of liposome–cell interactions, the cells with BODIPY-PC-labeled liposomes and trackers of intracellular organelles were coincubated. The cells were incubated with Hoechst 33342 for 30 minutes to stain nuclei, washed with PBS, and coincubated with the trackers and liposomes, added in the serum-free medium to a final lipid concentration of 100 μM, for 30 minutes at 37°C. Before being fixed with 1% paraformaldehyde solution, the cells were washed with PBS. For tracking of glycocalyx and Golgi apparatus (WGA conjugate) and endoplasmic reticulum (ER-Tracker), coincubations were performed for 30 minutes. In the case of early endosome tracker (transferrin [Trf] conjugate), coincubation was shortened to 15 minutes.
In order to study the intracellular delivery of MTX-DG liposomes to ER after longer time periods, first, the cells were cooled to 4°C and incubated with liposomes (100 μM in the serum-free medium) for 60 minutes on ice. At 4°C, the liposomes can only be adsorbed on the cell surface without internalization. Then, the cells were washed with PBS. By washing off the unbound liposomes, it was ensured that further processing of all the liposomes proceeds roughly from the same location, that is, the cell membrane. Finally, the cells were incubated at 37°C in a complete medium for various time periods. Then, 30 minutes before the end of the incubation, ER-Tracker and Hoechst 33342 were added.
In order to assess the disassembling of MTX-DG liposome components, the cells were cooled to 4°C and incubated with the liposomes (100 μM in the serum-free medium) labeled with BODIPY-MXT-DG and Rhod-PE for 30 minutes on ice; then, the cells were washed with PBS and incubated for various time periods at 37°C in a complete medium.
Prior to examination on a Nikon TE 2000 Eclipse confocal microscope (Nikon Corp., Tokyo, Japan), slides were mounted using Mowiol 4.88 (Calbiochem, San Diego, CA, USA) solution as a polymerizing agent. Images were analyzed by using ImageJ 1.48 software.
Plasma retention of liposomal MTX-DG versus intact MTX in mice
Hybrid F1(CBA × C57BL/6) female mice, 20–22 g each, 6–8 weeks old, supplied by nursery for laboratory animals, Russian Academy of Medical Sciences (Stolbovaya, Moscow Region, Russian Federation), were injected IV (intra vena caudalis) with free MTX (10 mg/kg) or equivalent dose of MTX-DG in liposomes (0.2 mL per mouse). Then, at selected time points, the mice (n=3) were decapitated, blood samples were collected, and serum was isolated by centrifugation at 600× g for 10 minutes.
For MTX determination with RP-HPLC, an aliquot of serum (0.5 mL) was denatured by a 10-minute incubation with 0.05 mL 63% HClO4 followed by centrifugation at 2,000× g for 20 minutes. The supernatant was analyzed by using Gilson HPLC System (Viffiers-le-Bel, France) with 305/306 Pump, 199 UV/Vis Detector, and Rheodyne 7725i Injector, on a Zorbax C8, 4.6×250 mm column (Agilent Technologies, Santa Clara, CA, USA); the mobile phase consisted of water–acetonitrile (9:1, v/v); and UV detection was at 313 nm. The injection volume, flow rate, and run time were 50–100 μL, 1 mL/min, and 12 minutes per sample, respectively. MTX was clearly separated with the retention time of 10.1 minutes and the sensitivity of 10–50 ng/mL. The extraction of known amounts of MTX from control serum of noninjected mice was used to plot the calibration curve.
For MTX-DG determination with HPLC, an aliquot of serum (0.02 mL) was extracted with 0.48 mL of chloroform–methanol (65:25, v/v), vortexed, and centrifuged at 5,000× g for 10 minutes. The supernatant was analyzed on a LiChrosorb® Si-60-5, 4.6×250 mm, Chrompack column (Merck, Darmstadt, Germany); the mobile phase consisted of chloroform–ethanol–water (74:25:1, v/v/v); and UV detection was at 313 nm. The injection volume, flow rate, and run time were 20 μL, 1 mL/min, and 15 minutes per sample, respectively. MTX-DG was separated with the retention time of 14.1–14.5 minutes and the sensitivity of 50 ng/mL. MTX-DG solutions of known concentrations in chloroform–methanol (65:25, v/v) were added to the control serum samples of noninjected mice to construct the calibration curve.
Serum concentrations at each time point were averaged, and the data were analyzed in order to calculate the pharmacokinetic variables of free MTX, MTX-DG, and MTX derived from MTX-DG (MTXmetab) by using WinNonLin computer software (Pharsight 2.1, Mountain View, CA, USA). Biexponential model of clearance (concentration = Ae−a × time + Be−b × time, where A, a, B, and b are the variables determined in the model) was used.
Transplanted mouse T-cell leukemic lymphoma
Breeding nucleus of the A/WySnJCitMoise (A/Sn) mouse strain was maintained in a thoroughly controlled conventional environment at M.M. Shemyakin and Yu.A. Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences; each mouse was labeled individually and was followed up as a patient through the entire life span.21 Both A/Sn male and female mice had a low incidence (<5%) of spontaneous multiple T-cell lymphomas with leukemic manifestations. The ASF-LL tumor for transplantation was originally obtained from a spontaneous T-cell leukemic lymphoma derived in an old A/Sn female mouse.22
The tumor transplantation was performed by subcutaneous (SC) inoculation of ASF-LL cells (106 per mouse, n=6) into A/Sn male mice near right fore limb (day 0). The mice were euthanatized on days 13 (n=3) and 20 (n=3); tumors, blood, and other organs were collected. Blood smears and tumor imprints were routinely stained by true Gimsa for cytological analysis; tumors and organs were fixed in buffered formalin (pH =7.2) and cut for histopathological analysis; the slides were routinely stained with hematoxylin-eosin staining or periodic acid staining.21 Peripheral lymphocytes from the blood samples were separated by Ficoll gradient centrifugation, washed, and labeled with anti-CD25 (phycoerythrin-conjugated) and anti-CD62L (fluorescein isothiocyanate-conjugated; Pharmingen, San Jose, CA, USA) for immunophenotyping by flow cytometry (FACScan, Becton Dickinson, San Jose, CA, USA).
All the animal experiments were conducted in accordance with the “Guide for the Care and Use of Laboratory Animals” (US Department of Health and Human Services, National Institute of Health Publication No 93–23, revised 1985) and were approved by the Institutional Animal Care and Use Committee (http://www.ibch.ru/downloads/documents/553/Institutional_Policy_on_the_Use_of_Laboratory_Animals.pdf).
Antitumor effect of MTX-DG liposomes versus intact MTX in vivo
Two experiments with female mice of different ages (6 and 12 months) were carried out. At day 0, A/Sn female mice (n=18) were inoculated with ASF-LL cells as described earlier. The mice were treated with 4 IV injections of MTX (10 mg/kg; n=6) or equivalent dose of L-MTX-DG (n=6) in 0.2 mL of PBS with 1 mM EDTA, pH 7.0, on days 3, 5, 10, and 12. Control mice (n=6) received 0.2 mL of PBS with 1 mM EDTA via the same route.
The mice were monitored each third day for palpable tumor appearance (1–4 mm of size). For larger tumor nodules, individual mean tumor diameter (TD, mm) as an average of 3 perpendicular TDs was used as a measure of tumor size.23 The mice were monitored daily for health control and survival: animals at the end point of leukemic lymphoma (weight loss >20% and/or food rejection independently of tumor size) were immediately euthanatized.21
The significance of differences in averages was determined by using the Student’s t-test, and the results were presented as mean ± standard deviation.
Results
Liposome characterization
Table 1 lists the physicochemical characteristics of liposomes used in this study. The formulations met the nanomedicine size criterion.24 They were negatively charged due to the presence of the relatively high PI content (10 mol%) in the bilayer. In MTX-DG liposomes, the carboxyl groups of MTX moiety were exposed on the surface and caused further increase in the negative charge as compared with empty liposomes. Both the liposome diameters and zeta potential values were well reproduced among different batches (eg, Kuznetsova et al16 for the previously established characteristics).
 | Table 1 Physicochemical characteristics of the liposomes under studya |
Accumulation of liposomes by cultured cells studied by flow cytometry
At least 5 times more MTX-DG liposomes accumulated in cultured tumor cells of epithelial origin after a 1-hour incubation as compared with MTX-DG-free liposomes (Figure 2A presents the data for A549 and CoLo357 cells). Malignant cells accumulated MTX-DG liposomes more intensively than conditionally normal 3T3 (murine fibroblasts) and HEK293T (human kidney embryonic cells) cells (Figure 2A). As for the acute T-cell lymphocyte leukemia cell line (Jurkat), the presence of MTX-DG in the bilayer resulted in a 30%–40% increase in liposome accumulation in cells (Figure 2A).
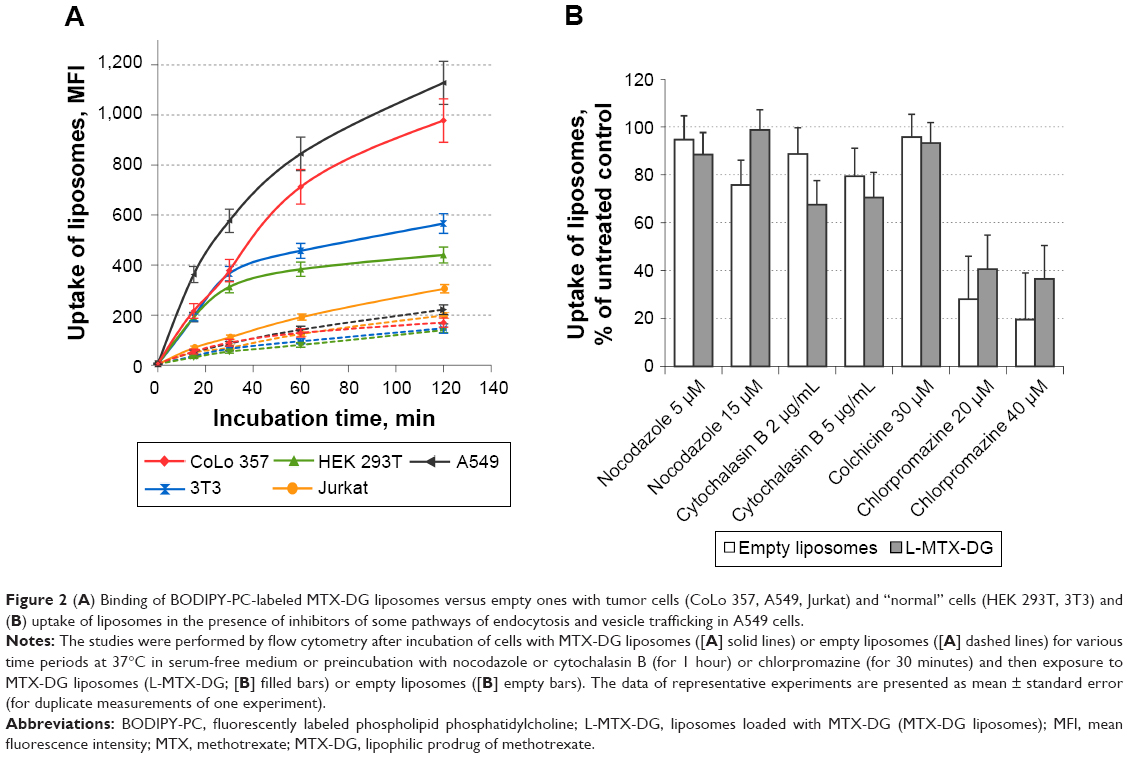 | Figure 2 (A) Binding of BODIPY-PC-labeled MTX-DG liposomes versus empty ones with tumor cells (CoLo 357, A549, Jurkat) and “normal” cells (HEK 293T, 3T3) and (B) uptake of liposomes in the presence of inhibitors of some pathways of endocytosis and vesicle trafficking in A549 cells. |
Then, the specific interactions between MTX-DG liposomes and tumor cells that could be mediated by RFC or FRα by competitive inhibition of binding with excess of free MTX and anti-FRα antibody, respectively, were probed. The A549 and CoLo357 cells were positive for FRα – the level of specific immunofluorescence observed with flow cytometry was 2–2.5 times higher than in the case of binding with nonspecific secondary antibodies (Figure S1A). The excess amounts of MTX did not affect liposome binding to cells, neither did anti-FRα antibody (Figure S1B).
In order to reveal the possible mechanisms of liposome entrance to the cell (and intracellular trafficking), different endocytosis pathways were blocked with chlorpromazine, cytochalasin B, colchicine, and nocodazole. In all the cell lines tested, the inhibitors caused elusive effects, except for chlorpromazine. The latter caused a concentration-dependent decrease in liposome accumulation in all the cell lines (shown by the example of A549 cells in Figure 2B; in CoLo-357, similar results were obtained, data not shown). Noteworthy, the inhibition was more pronounced in the case of MTX-DG-free liposomes. Methyl-β-cyclodextrin (inhibits caveolin-mediated and, to a lesser extent, clathrin-mediated endocytosis due to cholesterol extraction from cytoplasmic membrane) caused a >50% cell death in the range of concentrations required for the inhibitory studies. Survived cells accumulated several times more liposomes than control (untreated) cells apparently due to the increased cell membrane permeability (data not shown).
Binding and intracellular trafficking of MTX-DG liposomes studied by confocal microscopy
In order to study binding and internalization of MTX-DG liposomes by tumor cells with confocal microscopy, A549 cells were chosen, which spread the most on the well surface allowing for better monitoring of the location of liposomes labeled with a fluorescent analog of the MTX prodrug, BODIPY-MTX-DG (Figure 1).
In the experiment, tumor A549 cells were first preincubated with MTX-DG liposomes at 4°C to prevent endocytosis; then, unbound liposomes were withdrawn by washing, and the cells were put in physiological conditions. This preliminary treatment of samples synchronized liposome internalization by the cells to ensure that the duration of further incubation at 37°C corresponded to the period of liposome processing by cells (and excludes binding and processing of new portions of liposomes). Confocal microscopy data analysis showed that, upon internalization by cells, MTX-DG liposomes moved slowly inward (Figure 3). In the scans, ER red staining covered the entire cytoplasm diffusively, whereas MTX-DG liposomes (green fluorescence of BODIPY-MTX-DG label) presented a punctuated vesicular pattern. Thus, the liposomes seemed not to be incorporated in the ER structures. The vesicles did not coincide with the tracker of early endosomes either (Trf; data not shown).
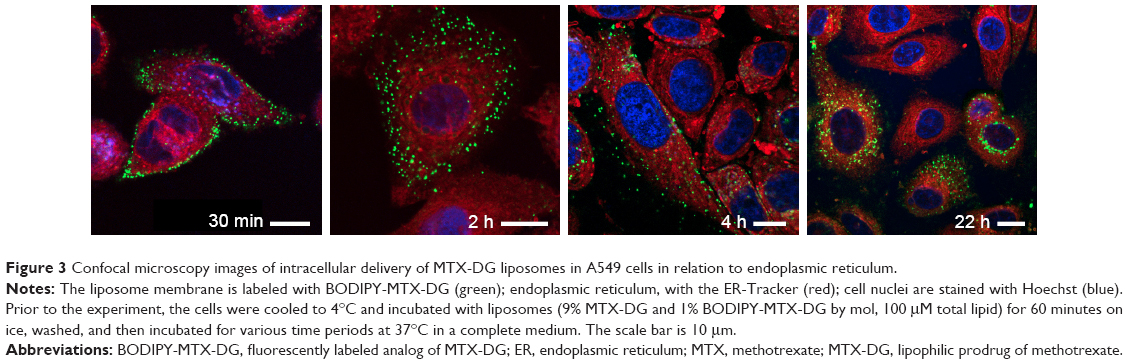 | Figure 3 Confocal microscopy images of intracellular delivery of MTX-DG liposomes in A549 cells in relation to endoplasmic reticulum. |
In order to study the early stages of liposome–cell interactions, the cells with liposomes were incubated immediately at 37°C without preliminary binding at 4°C and washing off the unbound liposomes. Over the first 15–30 minutes, the liposomes, undoubtedly, localized only in the glycocalyx (as evidenced by colocalization with the WGA, the tracker of Golgi/glycocalyx). Early endosomes, which look like red spots of different size (Figure 4), apparently did not contain any of the liposome components. In the following 1.5 hours, the liposomes were already observed in the cytoplasm, but also as vesicles independent of Trf, and the same image was observed after 3 hours of incubation (Figure S2).
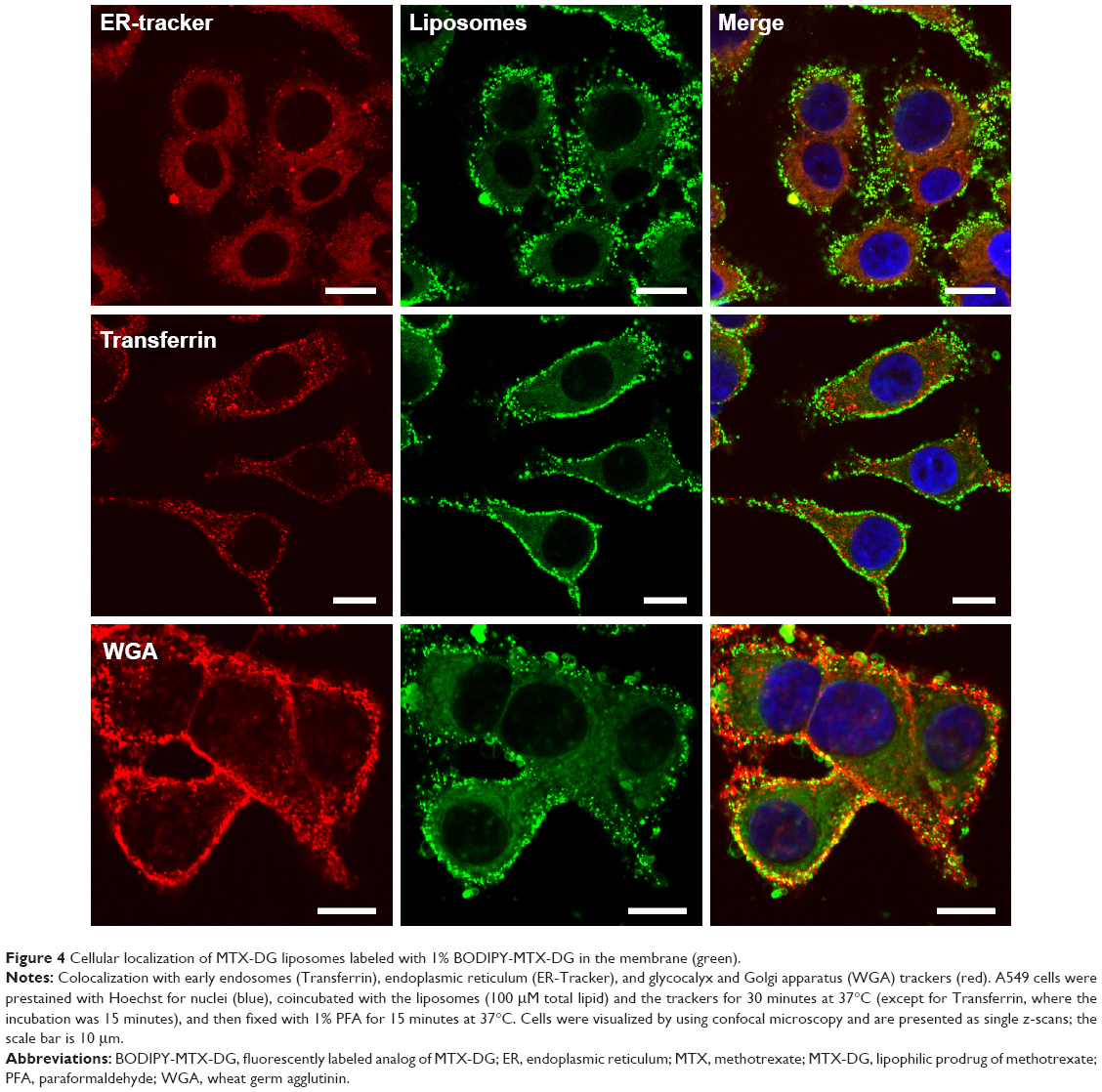 | Figure 4 Cellular localization of MTX-DG liposomes labeled with 1% BODIPY-MTX-DG in the membrane (green). |
Intracellular transport of the individual components of MTX-DG liposomes – matrix lipids and the prodrug – was monitored by using both the prodrug analog BODIPY-MTX-DG and rhodamine-labeled phospholipid Rhod-PE (red fluorescent probe, arbitrarily represents matrix lipids), in order to label the lipid bilayer. After incubations with the liposomes at 4°C, the cells were washed and left at physiological conditions for various time periods before fixation. The results of confocal microscopy (Figure 5) confirmed the slow entry of MTX-DG into the cell.
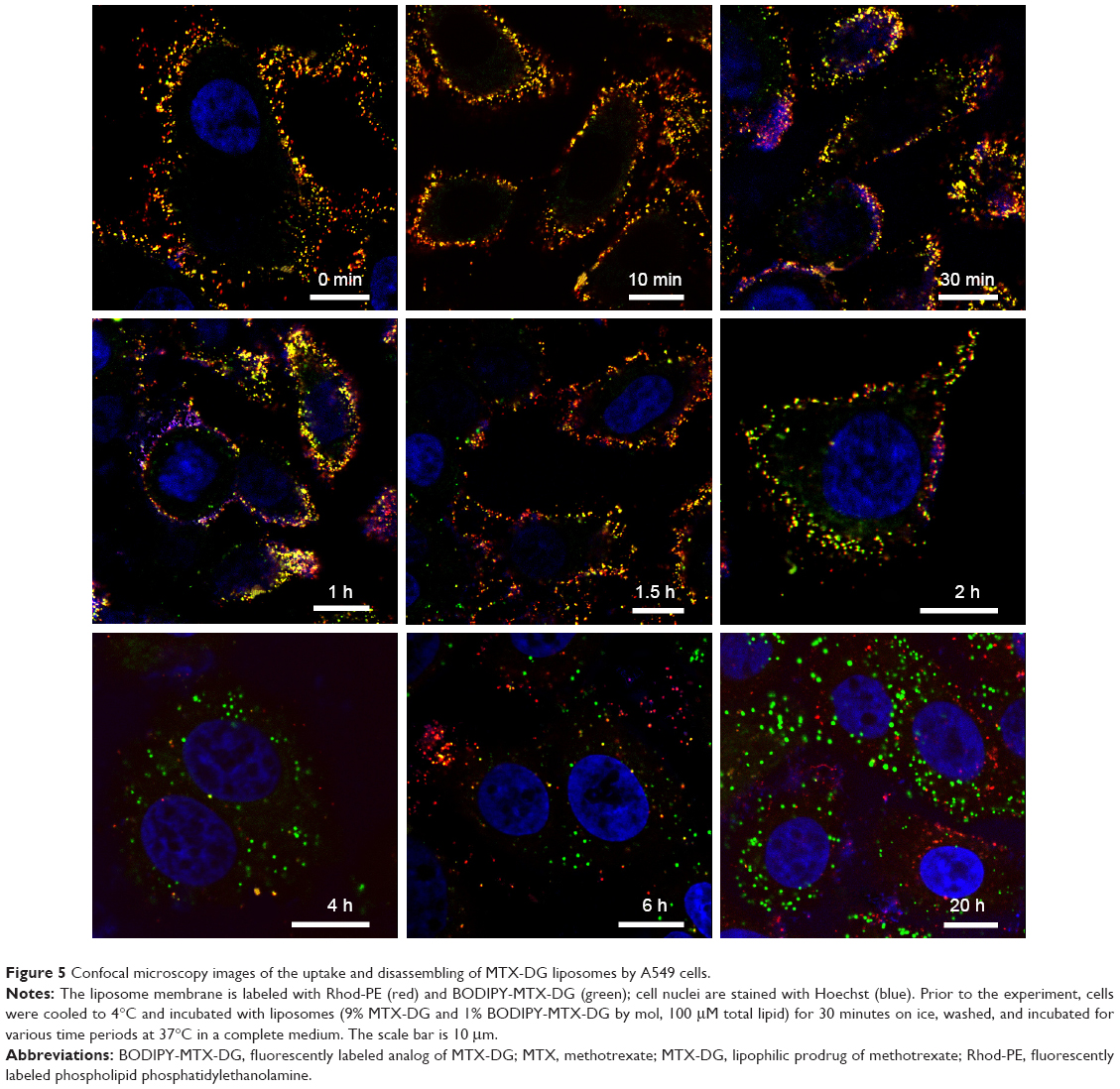 | Figure 5 Confocal microscopy images of the uptake and disassembling of MTX-DG liposomes by A549 cells. |
In the first minutes and even after 1 hour of incubation, liposomes were seen as yellow and yellow-red bright spots (overlap of green and red fluorescence) contouring plasma membrane. The patterns started to change only after 1.5–2 hours, when green spots appeared beside the yellow and yellow-red ones. The separation of red and green fluorophores could be seen most clearly after 4 hours and later on. (In addition, the essential decrease of the red fluorescence observed during the prolonged incubations may be due to oxidative metabolism of the rhodamine fluorophore.25) It was concluded that intact MTX-DG liposomes bind to the cell surface and then enter the cell only 1.5–2 hours later, most likely not as the whole units but rather as the fragments of liposomal bilayer or even separate molecules fused with the cell membrane. The delay of cell internalization of MTX-DG liposomes (Figures 3 and 5) can be explained by the liposome negative charge. In the cytoplasm, they form vesicular structures trafficking intact for >20 hours (Figures 3 and 5). It is difficult to attribute them to any specific cell organelle. Unlikely, these vesicles originate from clathrin-coated pits where Trf receptor is located, because no colocalization of the liposomes with Trf was observed at any time of incubation (Figure 4). Whatever the following intracellular traffic of the prodrug could be, the data from the present study evidenced that MTX-DG liposomes provide the entrance of MTX-DG into the cell.
Plasma retention of liposomal MTX-DG versus intact MTX in mice
The plasma levels of MTX-DG and MTX after IV injections of MTX-DG liposomes and free MTX, respectively, were determined in the equivalent molar concentration in mice. Figure 6 shows the data plotted against time. Pharmacokinetic analysis (Table 2) evidenced that MTX-DG quickly distributed in organs and tissues (t1/2α =3.6 minutes) and was rapidly eliminated from plasma (t1/2β ~40 minutes). The same parameters for MTX were approximately 4 times lower. Mean residence time (MRT) of the MTX-DG prodrug considerably exceeded the values obtained for MTX. However, the values of area under the curve (AUC) were very close.
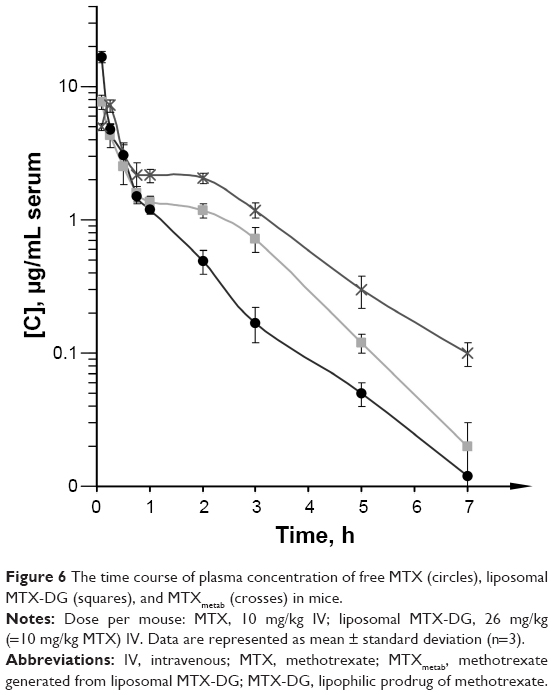 | Figure 6 The time course of plasma concentration of free MTX (circles), liposomal MTX-DG (squares), and MTXmetab (crosses) in mice. |
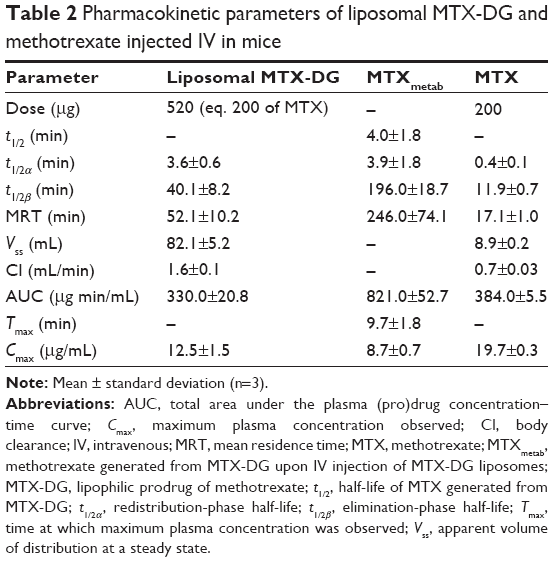 | Table 2 Pharmacokinetic parameters of liposomal MTX-DG and methotrexate injected IV in mice |
Then, the plasma retention of MTX formed upon IV injection of MTX-DG liposomes (MTXmetab) was assessed. The data presented in Table 2 (Column 2) evidenced that MTX-DG loses the lipophilic part of the molecule rather quickly, generating MTX itself. The values of Tmax and t1/2β for MTXmetab suggested prolonged blood circulation of MTX upon IV injection of MTX-DG liposomes. The concentration of MTXmetab in plasma persistently exceeded the concentration obtained for intact MTX starting from 15 minutes after IV injection; by 3 hours, the ratio of the concentrations in plasma reached an order of magnitude (Figure 6).
Quite moderate superiority of MTXmetab AUC over intact MTX (by a factor of ~2.5) let us suppose that the liposomal delivery system would better suit the treatment of a systemic hematological disease, not a solid tumor, where much longer circulation time is highly desirable for the efficient accumulation of the drug in tumor site.
Antitumor effect of MTX-DG liposomes versus intact MTX in the mouse T-cell leukemic lymphoma
In a previous study, a transplantable mouse model of T-cell leukemic lymphoma, ASF-LL, was developed.22 The ASF-LL is characterized by CD4+/CD25+ phenotype of the tumor cells and a pronounced leukemic constituent with extensive dissemination to many organs. In the present study, the blood samples of lymphoma-bearing control mice were examined to estimate the time of leukemic manifestation. CD25 marker was used for this end as the number of CD25+ cells in peripheral vessels in an intact mouse does not exceed 2%–5%. CD25+ ASF-LL cells, which have been transplanted SC at day 0, were found in various proportions in blood samples of the recipient mice at day 13 after transplantation (mean TD was 9.3±2.2 mm; Figure S3 presents the representative fluorescence-activated cell sorting analysis dot plots obtained from individual mouse blood lymphocytes and lymphoma cells). Cytology analysis revealed the presence of characteristic “flower” cells in murine blood and lymphoma samples (Figure S4). On day 20 (mean TD was 18.9±3.1 mm), histopathology analysis revealed tumor cells disseminated to all lymphoid organs tested (ie, lymph nodes, thymus, bone marrow, and spleen), liver, kidney, lung, pancreas, brain, and urogenital tract (Figure S5).
The antitumor effect of MTX formulations in the ASF-LL model was studied by using a dose-sparing regimen of treatment (10 mg/kg MTX on days 3, 5, 10, and 12 after tumor transplantation). In the preliminary short-term experiments, this regimen has been sufficient for a significant lymphoma growth inhibition by treatment with intact MTX (data not shown). Based on the data from the present study on prolonged blood circulation of MTX generated upon IV injection of MTX-DG liposomes, it was expected that the effect of liposomal formulation should clearly surpass the effect of intact MTX. In both the experiments (recipient mice aged 6 and 12 months), treatment with free MTX resulted in a significant delay in palpable lymphoma appearance (P<0.001) and tumor growth kinetics (P<0.02) as compared with these parameter values in the control group. Treatment with the L-MTX-DG led to the most pronounced delay of lymphoma appearance and tumor growth rate compared with controls (Figure 7A and C). Treatment with the liposomes significantly retarded tumor appearance and growth as compared with MTX starting from day 14 after transplantation (P<0.05).
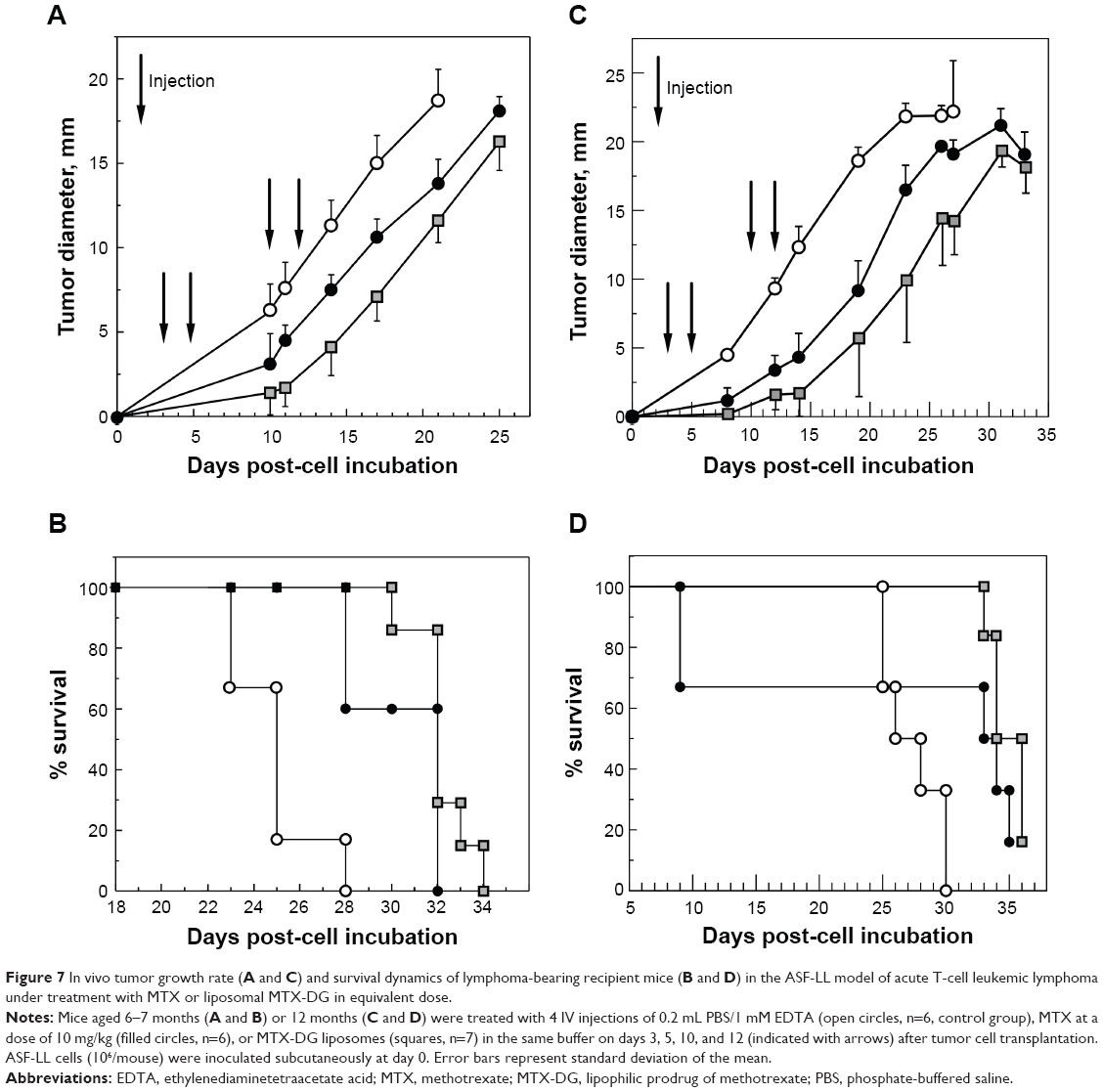 | Figure 7 In vivo tumor growth rate (A and C) and survival dynamics of lymphoma-bearing recipient mice (B and D) in the ASF-LL model of acute T-cell leukemic lymphoma under treatment with MTX or liposomal MTX-DG in equivalent dose. |
Both the experiments showed clearly that tumor growth delay in the groups treated with the MTX-DG liposomes resulted in a significant survival improvement compared with controls (Figure 7B and D). In the first experiment, the mean survival time of the mice treated with MTX differed from that in the control group (30±2 and 25±2 days, respectively; P<0.01) yielding 23% of increase in the mean life span (Figure 7B). The mean survival time of mice that received L-MTX-DG (32±1 days) exceeded that in the control group (P<0.001), resulting in a 29% improvement, although there were no significant differences between the MTX-DG-treated and MTX-treated groups. In the second experiment (Figure 7D), however, free MTX caused pronounced toxicity in the old recipients; therefore, mean life span for the mice treated with MTX did not exceed this parameter value in the control group (26±3 and 28±2 days, respectively), whereas the mean survival time of mice that received L-MTX-DG (35±1 days) exceeded the parameter value in the control group (P<0.001) resulting in a 25% improvement. Meanwhile, 4 injections of MTX-DG liposomes were well tolerated by all of the old mice without any signs of toxicity; the tumor growth rate was significantly delayed as compared with the free MTX-treated group (P<0.05 from day 14 till day 31 posttransplantation, Figure 7D).
Discussion
The study concerned both the mechanisms of interactions between liposomes loaded with the MTX-DG lipophilic prodrug of MTX in the bilayer and tumor cells and primary evaluation of the antitumor potency of this formulation in vivo.
First, how MTX-DG conjugate present in the bilayer influences liposome binding with tumor cells was the main interest of this study; particularly, whether MTX moieties located at the surface of liposomes can be provided for targeting of the formulation. If MTX fragment should be used as a targeting ligand, it should be accommodated on a long flexible hydrophilic linker exposing the folate analog residue at a sufficient distance from the carrier surface. Even though MTX-DG molecule lacks such a linker, all the tested cells of epithelial origin, especially tumor cells, persistently showed a significant increase in binding with the prodrug-loaded liposomes as compared with the empty ones (Figure 2A). The difference in the binding potential between the epithelial and lymphoid cells could be explained by the differences in experimental setup (adherent versus suspended cells allow for different presentation of the cell surface for interactions), smaller size of lymphocytes (as compared with epithelial cells), or specific biological properties of the latter. The increase of binding could be due to the interactions of MTX moieties with the RFC molecules, which play the key role in MTX cellular uptake.2 However, even highly excessive amounts of free MTX did not inhibit binding of the formulation to the tumor cells (Figure S1B). Then, whether FRα could be involved in liposome binding with cells was tested. Although FRα-mediated transport of MTX is unlikely to be of clinical importance,26 it may be involved in the effects produced by MTX nanocarriers.5 However, neither anti-FRα antibody (Figure S1B) nor folic acid inhibited L-MTX-DG binding to cells. Notably, the tested carcinoma cells do not belong to lineages highly expressing FR, and an increase in the receptor expression by the privation of vitamin B9 in cultural medium was not endeavored. Thus, it was assumed that there were specific interactions (beyond the aforesaid ones) between the tumor cells and MTX-DG liposomes. Otherwise, it was unclear why the latter, while having higher negative charge than empty liposomes (zeta potential values were ~-53 and −42 mV, respectively; Table 1), bind tumor cells significantly more effectively, as most of the cells express negatively charged (mainly sulfated) proteoglycans on their surface.27
The sole nanoparticulate drug delivery system exploiting the FR-targeting potential of MTX that was found is the MTX-conjugated pluronic-based micelles reported by Chen et al.28 Interestingly, the A549 cells did not show any increase in binding with the micelles as compared with MTX-free micelles after 60 minutes of incubation.28 Presumably, in contrast to MTX-DG liposomes, MTX moieties in the micelles are shielded by polyethylene glycol chains, which could decrease the efficacy of targeting.
In further studies on the mechanisms of endocytosis of MTX-DG liposomes, the impressive effect of chlorpromazine let us first imply the involvement of the classical cell entry pathway through clathrin-coated pits (Figure 2B). The effect of chlorpromazine was even greater in the case of MTX-DG-free liposomes; that is, empty liposomes relied more on this pathway than did MTX-DG liposomes. Surprisingly, tracing colocalization of the BODIPY-MTX-DG probe with Trf in early endosomes characteristic for clathrin-mediated endocytosis was not successful (Figures 4 and S2). Perhaps, the reason for the apparent contradiction is that chlorpromazine is able not only to deplete cell membrane from clathrin-associated machinery (and thus block clathrin-mediated endocytosis) but also to inhibit some clathrin-independent mechanisms.29 The pronounced inhibition of MTX-DG liposome binding by cytochalasin B also argued in favor of a clathrin-independent mechanism due to the key role of actin in clathrin-independent pathways.30 On the other hand, clathrin also mediates the formation of endosomes that cannot be recycled, and such endosomes do not contain Trf. As for caveolae-mediated uptake, its inhibition using methyl-β-cyclodextrin was also not successful for the reasons stated above (see the “Results” section). Yet, it was assumed that caveolae unlikely participate in the endocytosis of the formulation, as indirectly evidenced by the absence of binding of MTX-DG liposomes with FR, which predominantly localized in caveolae.31 Using the BODIPY-MTX-DG probe to label MTX-DG liposomes, some well-defined vesicular structures formed in the cytoplasm upon several hours of incubation (Figure 3) were observed. It was concluded that MTX-DG liposomes enter the cell using multiple mechanisms, including clathrin-independent endocytosis (and not involving FR).
In a previous study, it was reported that the cytotoxicity of MTX-DG liposomes in culture of human T-lymphoblastic leukemia cells was only 10-fold lower than that of MTX per se;14 typically, the cytotoxicity of an intact drug is even much higher than that of the encapsulated formulation of the same drug assayed in vitro. This may implicate facilitated unloading/disassembling of liposomes inside cell followed by the hydrolysis of the prodrug by intracellular esterases, the gradual decomposition of liposomes during incubation in the culture medium with subsequent passive diffusion of the prodrug into the cell, or both. In the present study, the spots of colocalized fluorescence of MTX-DG analog and Rhod-PE contoured cell membranes even after the whole cell membrane should have been internalized (taken that the time required for nonspecialized cells to uptake their entire surface is ~1 hour32). Thus, the spots could not be due to liposomes (or fragments thereof) fused with the plasma membrane and disassembling of the formulation occurred only after fusion with plasma membrane or receptor-mediated entrance in the cell (Figure 5).
Taken together, the aforesaid results evidenced that the formulation allowed for the transportation of the lipophilic prodrug of MTX into tumor cells and has a potentiality for selective delivery to tumor tissue.
In a previous in vitro study of the formulation, MTX-DG remained stable for up to 24 hours of incubation in human plasma, which evidenced shielding of the conjugate ester bonds from the unfavorable premature hydrolysis by plasma esterases.14 The data from the present study on the retention in plasma showed rather rapid clearance of MTX-DG accompanied by the generation of MTX (MTXmetab, t1/2=4 minutes; Table 2). In addition to the difference in plasma enzyme activities in vivo in circulation and in the in vitro model, species-specific differences in the esterase activities should be considered as another possible reason for such contradictory/conflicting results. Both esterase expression and hydrolyzing patterns significantly differ between humans and rodents (eg, Bahar et al33). Another explanation could be the uptake of MTX-DG liposomes by the reticuloendothelial cells followed by intracellular cleavage of the MTX prodrug and its recycling into bloodstream. In order to prolong liposome circulation in bloodstream, PEG-conjugated phospholipids are commonly used as steric stabilizers.34 Presumably, PEGylation of the present formulation – instead of incorporation of PI as a potential liposome stabilizer35 – could provide for substantially improved stability of L-MTX-DG in circulation and thus increased AUC and t1/2, yet it could decrease the targeting potential of MTX moiety due to its shielding.
The antitumor effect of MTX-DG liposomes was studied in the mouse T-cell leukemic lymphoma ASF-LL.22 T-cell lymphomas are weakly immunogenic, highly aggressive tumors that are frequently resistant to therapy.36 Some pathological characteristics of the ASF-LL model resemble the acute form of adult T-cell leukemia/lymphoma of retroviral nature in human patients.37 Indeed, in both species, retroviruses are involved, tumor cells demonstrate flower appearance, and extensive growth in central nervous system is observed (Figures S3 and S4). MTX is a basic component of many therapeutic interventions against acute leukemia.38 It was hypothesized that L-MTX-DG could suppress or prevent the leukemic manifestation of the lymphoma, which might be present at the time of treatment (day 13 after transplantation), as it has been demonstrated on untreated mice.
The results from the present study demonstrated that MTX-DG liposomes inhibit lymphoma growth more efficiently than free MTX does (Figure 7A and C). The present study used dose-sparing regimen of therapy. For comparison, a frequently used regimen in the treatment of acute leukemic lymphoma caused by xenograft transplantation of tumor in mice includes intraperitoneal injections with 5 mg/kg of MTX daily for 5 days, repeated every 14 days for 8 weeks with the total number of injections up to 20.39 Usage of aged recipients (10 and 12 months) in the experiments highlighted the lower toxicity of MTX-DG liposomes compared with MTX, promising a safe treatment with prolonged regimen(s).
Conclusion
To the authors’ knowledge, the present work is the first attempt to treat leukemic lymphoma with a liposomal formulation of MTX. Moreover, only two early reports concerning antitumor effect of similar formulations were found. First, MTX encapsulated in negatively charged liposomes showed contradictory results in the Ehrlich ascites-bearing mice.40 Second, MTX-bearing immunoliposomes targeted to human renal carcinoma cells significantly prolonged the survival of mice in an ascites tumor model.41 The current study filled in the gaps concerning various mechanisms for the intracellular delivery of the MTX-DG liposome cargo and showed the parameters of the prodrug retention in plasma. The successive investigation into the formulation behavior and mechanisms of action in in vitro and in vivo models allowed moving on to an in vivo antitumor activity assessment, which proved it promising to treat hematological malignancies. Further explicit studies on the pharmacokinetics and biodistribution of liposomal MTX-DG, as well as adjustment of treatment regimen, are required.
Acknowledgments
The work received financial support from the Russian Foundation for Basic Research (projects nos 13-04-00069, 15-04-07415, 16-04-01585, and 16-34-01237). The authors are grateful to Dr Elena Svirshchevskaya (M.M. Shemyakin and Yu.A. Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences) for her help with facilities and equipment for the working with cell cultures. The late Dr Alexander S Singin was a leading specialist in clinical pharmacokinetics of MTX in Russia. In this work, he contributed to the development of a method to determine the lipophilic prodrug of MTX in blood plasma of the mice.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. WHO Model List of Essential Medicines. Adults – 19th List (April 2015); 2015 [Revised November 2015]. Available from: http://www.who.int/medicines/publications/essentialmedicines/en/. Accessed January 18, 2017. | ||
McGuire J. Anticancer antifolates: current status and future directions. Curr Pharm Des. 2003;9(31):2593–2613. | ||
Abolmaali SS, Tamaddon AM, Dinarvand R. A review of therapeutic challenges and achievements of methotrexate delivery systems for treatment of cancer and rheumatoid arthritis. Cancer Chemother Pharmacol. 2013;71(5):1115–1130. | ||
Khan ZA, Tripathi R, Mishra B. Methotrexate: a detailed review on drug delivery and clinical aspects. Expert Opin Drug Deliv. 2012;9(2):151–169. | ||
Thomas TP, Joice M, Sumit M, et al. Design and in vitro validation of multivalent dendrimer methotrexates as a folate-targeting anticancer therapeutic. Curr Pharm Des. 2013;19(37):6594–6605. | ||
Abolmaali S, Tamaddon A, Kamali-Sarvestani E, Ashraf M, Dinarvand R. Stealth nanogels of histinylated poly ethyleneimine for sustained delivery of methotrexate in collagen-induced arthritis model. Pharm Res. 2015;32(10):3309–3323. | ||
Ferreira M, Chaves LL, Lima SA, Reis S. Optimization of nanostructured lipid carriers loaded with methotrexate: a tool for inflammatory and cancer therapy. Int J Pharm. 2015;492(1–2):65–72. | ||
Johari-Ahar M, Barar J, Alizadeh AM, Davaran S, Omidi Y, Rashidi MR. Methotrexate-conjugated quantum dots: synthesis, characterisation and cytotoxicity in drug resistant cancer cells. J Drug Target. 2016;24(2):120–133. | ||
Li Y, Lin J, Wu H, et al. Orthogonally functionalized nanoscale micelles for active targeted codelivery of methotrexate and mitomycin C with synergistic anticancer effect. Mol Pharm. 2015;12(3):769–782. | ||
Srisuk P, Thongnopnua P, Raktanonchai U, Kanokpanont S. Physico-chemical characteristics of methotrexate-entrapped oleic acid-containing deformable liposomes for in vitro transepidermal delivery targeting psoriasis treatment. Int J Pharm. 2012;427(2):426–434. | ||
Prabhu P, Shetty R, Koland M, et al. Investigation of nano lipid vesicles of methotrexate for anti-rheumatoid activity. Int J Nanomedicine. 2012;7:177–186. | ||
Ding Q, Si X, Liu D, et al. Targeting and liposomal drug delivery to CD40L expressing T cells for treatment of autoimmune diseases. J Control Release. 2015;207:86–92. | ||
Zucker D, Marcus D, Barenholz Y, Goldblum A. Liposome drugs’ loading efficiency: a working model based on loading conditions and drug’s physicochemical properties. J Control Release. 2009;139(1):73–80. | ||
Kuznetsova N, Kandyba A, Vostrov I, et al. Liposomes loaded with lipophilic prodrugs of methotrexate and melphalan as convenient drug delivery vehicles. J Drug Deliv Sci Technol. 2009;19(1):51–59. | ||
Liederer BM, Borchardt RT. Enzymes involved in the bioconversion of ester-based prodrugs. J Pharm Sci. 2006;95(6):1177–1195. | ||
Kuznetsova NR, Sevrin C, Lespineux D, et al. Hemocompatibility of liposomes loaded with lipophilic prodrugs of methotrexate and melphalan in the lipid bilayer. J Control Release. 2012;160(2):394–400. | ||
Vodovozova EL, Evdokimov DV, Molotkovsky JG. Synthesis of a lipid derivative of the antitumor agent methotrexate. Russ J Bioorg Chem. 2004;30(6):599–601. | ||
Boldyrev IA, Zhai X, Momsen MM, Brockman HL, Brown RE, Molotkovsky JG. New BODIPY lipid probes for fluorescence studies of membranes. J Lipid Res. 2007;48(7):1518–1532. | ||
Vlasenko YV, Alekseeva AS, Vodovozova EL. Synthesis of a fluorescent analogue of methotrexate lipophilic prodrug. Russ J Bioorg Chem. 2014;40(1):114–117. | ||
Huth US, Schubert R, Peschka-Süss R. Investigating the uptake and intracellular fate of pH-sensitive liposomes by flow cytometry and spectral bio-imaging. J Control Release. 2006;110(3):490–504. | ||
Moiseeva E. Anti-breast cancer drug testing. Original approaches. Novel set of mouse models. Lambert Academic Publishing; 2009. Available from: https://www.lap-publishing.com/catalog/details//store/tr/book/978-3-8383-1691-8/anti-breast-cancer-drug-testing. Accessed January 18, 2017. | ||
Chaadaeva AV, Tenkeeva II, Moiseeva EV, Svirshchevskaia EV, Demushkin VP. Antitumor activity of the plant remedy peptide extract PE-PM in a new mouse T-lymphoma/eukemia model. Biomed Khim. 2009;55(1):81–88. Russian. | ||
Vodovozova E, Moiseeva E, Grechko G, et al. Antitumour activity of cytotoxic liposomes equipped with selectin ligand SiaLeX, in a mouse mammary adenocarcinoma model. Eur J Cancer. 2000;36(7):942–949. | ||
Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011;8(6):2101–2141. | ||
Sarvazyan N, Swift L, Martinez-Zaguilan R. Effects of oxidants on properties of fluorescent calcium indicators. Arch Biochem Biophys. 1998;350(1):132–136. | ||
Jackman AL, Theti DS, Gibbs DD. Antifolates targeted specifically to the folate receptor. Adv Drug Deliv Rev. 2004;56(8):1111–1125. | ||
Mislick KA, Baldeschwieler JD. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc Natl Acad Sci U S A. 1996;93(22):12349–12354. | ||
Chen Y, Zhang W, Sha X, et al. The therapeutic effect of methotrexate-conjugated Pluronic-based polymeric micelles on the folate receptor-rich tumors treatment. Int J Nanomedicine. 2015;10(1):4043–4057. | ||
Dutta D, Donaldson JG. Search for inhibitors of endocytosis: intended specificity and unintended consequences. Cell Logist. 2012;2(4):203–208. | ||
Howes MT, Mayor S, Parton RG. Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr Opin Cell Biol. 2010;22(4):519–527. | ||
Smart E, Ying Y, Mineo C, Anderson R. A detergent-free method for purifying caveolae membrane from tissue culture cells. Proc Natl Acad Sci U S A. 1994;92(22):10104–10108. | ||
Amyere M, Mettlen M, Van Der Smissen P, et al. Origin, originality, functions, subversions and molecular signalling of macropinocytosis. Int J Med Microbiol. 2002;291(6–7):487–494. | ||
Bahar FG, Ohura K, Ogihara T, Imai T. Species difference of esterase expression and hydrolase activity in plasma. J Pharm Sci. 2012;101(10):3979–3988. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85(18):6949–6953. | ||
Erter J, Alinari L, Darabi K, et al. New targets of therapy in T-cell lymphomas. Curr Drug Targets. 2010;11(4):482–493. | ||
Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27(3):453–459. | ||
Kako S, Akahoshi Y, Harada N, et al. Meta-analysis and meta-regression analysis to compare the outcomes of chemotherapy for T- and B-lineage acute lymphoblastic leukemia (ALL): the use of dexamethasone, L-asparaginase, and/or methotrexate may improve the outcome of T-lineage ALL. Ann Hematol. 2016;95(1):87–92. | ||
Kang MH, Harutyunyan N, Hall CP, Papa RA, Lock RB. Methotrexate and aminopterin exhibit similar in vitro and in vivo preclinical activity against acute lymphoblastic leukaemia and lymphoma. Br J Haematol. 2009;145(3):389–393. | ||
Freise J, Schmidt FW, Magerstedt P. Effect of liposome-entrapped methotrexate on Ehrlich ascites tumor cells and uptake in primary liver cell tumor. J Cancer Res Clin Oncol. 1979;94(1):21–27. | ||
Singh UV, Udupa N. Methotrexate loaded chitosan and chitin microspheres – in vitro characterization and pharmacokinetics in mice bearing Ehrlich ascites carcinoma. J Microencapsul. 1998;15(5):581–594. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.