")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 15
Lipoprotein Glomerulopathy, First Case Report from Canada
Authors Ting JA , McRae SA, Schwartz D, Barbour SJ, Riazy M
Received 4 March 2022
Accepted for publication 3 June 2022
Published 21 June 2022 Volume 2022:15 Pages 207—214
DOI https://doi.org/10.2147/IJNRD.S364890
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Julie Anne Ting,1 Susanna A McRae,1,2 Daniel Schwartz,1 Sean J Barbour,1 Maziar Riazy1,2
1Division of Nephrology, University of British Columbia, Vancouver, BC, V5Z 1M9, Canada; 2Department of Pathology, University of British Columbia, Vancouver, BC, V6Z 1Y6, Canada
Correspondence: Maziar Riazy, Department of Pathology, St. Paul’s Hospital, 1081 Burrard Street, Vancouver, BC, V6Z 1Y6, Canada, Tel +1 604-682-2344, extension 62228 or 62225, Fax +1 604-806-8701, Email [email protected]
Abstract: Lipoprotein glomerulopathy (LPG) is caused by a mutation in the apolipoprotein E gene (APOE) gene and is characterized by lipoprotein thrombi in glomerular capillaries. Here, we describe a case of LPG, the first to be reported from Canada and the first case of LPG in North America to be associated with the APOE Tokyo/Maebashi mutation (p.Leu162_Lys164del, traditional nomenclature 142_144del). A 49-year-old man of Chinese descent with a previous diagnosis of dyslipidemia and a new diagnosis of hypertension was found to have proteinuria on routine urinalysis. Renal biopsy showed markedly dilated glomerular capillaries filled with pale staining mesh-like material that stained positive for Oil-Red-O, consistent with lipoprotein thrombi. APOE gene sequencing confirmed the diagnosis of LPG. The patient was treated with fenofibrate and perindopril. His lipid profile normalized and proteinuria dropped to minimal levels. Repeat renal biopsy 2 years after the first showed resolution of lipoprotein thrombi but with rare residual granular densities by electron microscopy consistent with lipoprotein in the subendothelial space, supporting the hypothesis that this subendothelial material contains precursors to lipoprotein thrombi.
Keywords: apolipoprotein E, apoE Tokyo/Maebashi, proteinuria, dyslipidemia, fibrate, lipoprotein glomerulopathy
Introduction
Lipoprotein glomerulopathy (LPG) is a condition first described in Japan in 1989.1 Since then, approximately 200 cases have been reported, mostly from China and Japan, with only a handful of cases reported outside of Asia.2 LPG usually presents as progressively worsening proteinuria in a patient of East Asian descent, with males more commonly affected than females.3 Patients often, but not always, have co-existent hypertriglyceridemia. It is characterized on renal biopsy by lipoprotein thrombi in glomerular capillaries. We report a case of a 49-year-old man presenting with proteinuria and renal biopsy that showed LPG. Repeat renal biopsy two years later showed disease resolution in response to therapy.
Case Report
A 49-year-old male of Chinese descent was found to have proteinuria with urine albumin-to-creatinine ratio (ACR) of 153.8 mg/mmol on routine laboratory testing. His creatinine at that time was 109 µmol/L with an associated estimated GFR of 68 mL/min/1.73.2 He was asymptomatic to his renal disease, apart from newly diagnosed hypertension for which he had been started on perindopril 4 mg daily a week prior to his first nephrology assessment. Another previous medical history was significant for dyslipidemia diagnosed six years ago, although he had only started taking rosuvastatin 10 mg daily 3–6 months prior. He was also a current cigarette smoker with a 30-year smoking history.
Family history was negative for any kidney disease. His father died of pancreatic cancer. His mother died of liver cancer. All other immediate family members were healthy.
On examination, BP was 136/80 mmHg. Physical examination was otherwise normal with no stigmata of connective tissue disease or manifestations of lipoidosis. Serum creatinine was 109 µmol/L and serum albumin was 39 g/L. The patient had subnephrotic range proteinuria with ACR 153.8 and 24 h urine protein of 2.66 g/day. Urinalysis was positive for 0.3 mg/L hemoglobin, but urine microscopy showed only 1–3 erythrocytes/hpf. Antinuclear antibody was weakly positive, but the autoimmune review of systems was negative. C3, C4, antineutrophil cytoplasmic antibody panel, glomerular basement antibody, hepatitis B and C serology, human immunodeficiency virus testing, and cryoglobulin were all negative. Hemoglobin A1C was 5.6%. Serum protein electrophoresis showed no monoclonal protein. Fasting lipid profile showed total cholesterol (TC) 8.82 mmol/L, low-density lipoprotein (LDL) 6.16 mmol/L, high-density lipoprotein (HDL) 1.12 mmol/L, and triglycerides (TG) 3.38 mmol/L. ApoE level was ordered but never processed by the laboratory. The patient’s perindopril was increased to 16 mg PO daily, but ACR remained elevated (173.8 mmol/L).
Renal biopsy contained up to 20 glomeruli by light microscopy. The biopsy showed that most glomeruli were enlarged and had markedly dilated capillary lumina filled with amorphous to mesh-like material that stained pale with hematoxylin and eosin, Periodic Acid Schiff, Jones’, and Masson trichrome (Figure 1A), but positive with Oil-Red-O (Figure 1B). No foam cells were seen. There was mild-to-moderate mesangial expansion with hypercellularity (segmentally up to 8–9 nuclei). Segmental sclerosis was noted within up to 2 glomeruli in a perihilar distribution. Silver stained sections showed variable thickening and duplication of the glomerular basement membrane in some areas with no epimembranous spike formation. Chronic tubulointerstitial injury (fibrosis and atrophy) was seen in 5% of sampled cortex. There were no red blood cell casts. Interlobular-sized arteries and arterioles were unremarkable, and peritubular capillaries were unaffected. Direct immunofluorescence showed no significant staining for IgG, IgM (trace), IgA, C1q, C3, kappa, lambda, fibrin/fibrinogen, or albumin. Immunofluorescence on paraffin-embedded tissue after pronase digestion showed segmental mesangial and capillary wall staining for IgM (1+), kappa (1+), and lambda (1+) in segmentally sclerosed glomeruli. The intracapillary material did not stain for any of the immunoreactants tested. On electron microscopy, there were large extracellular vacuolated particles that focally showed a laminated structure (Figure 1C). Glomerular basement membranes surrounding this material were thickened, with flocculent material expanding the subendothelial space and focally containing granules and vacuoles. There was mild-to-moderate podocyte foot process effacement. A segment without intracapillary material was unremarkable with normal basement membrane thickness, preserved podocyte foot processes, and no immune deposits.
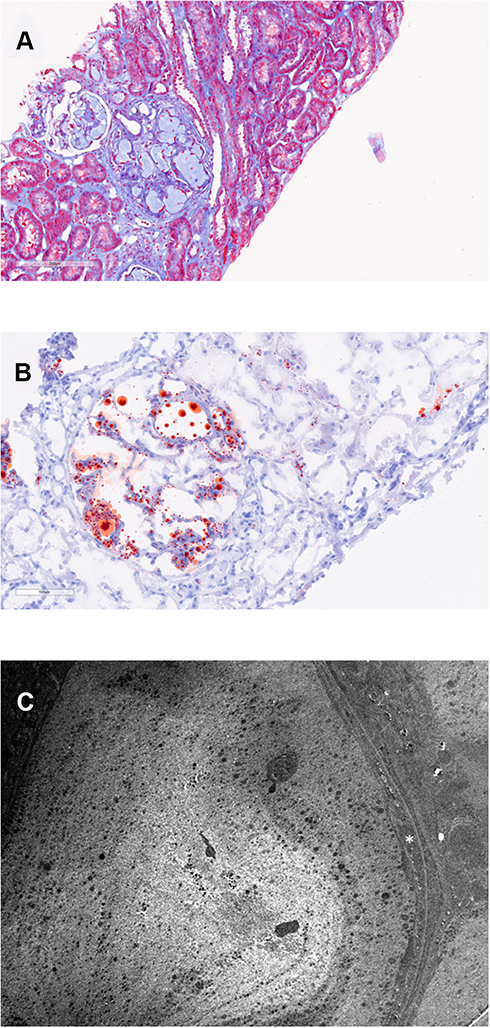 |
Figure 1 Original biopsy. The length of each bar represents the distance in microns. (A) shows dilated glomerular capillaries filled with mesh-like acellular material (Masson trichrome). (B) shows capillary lumina that are dilated with lipid droplets (Oil-Red-O stain). (C) shows a fingerprint-like structure composed of granules and vacuoles within a capillary lumen. There is expansion (asterisk) of the subendothelial space (electron microscopy, 5000x). |
Genotyping with polymerase chain reaction showed apo E3/E3 genotype. Genetic sequencing was done which revealed p.Leu162_Lys164del, known in the literature as the Tokyo/Maebashi mutation of the apolipoprotein E gene (APOE) (Blueprint Genetics, Finland).
Based on the renal biopsy results, the patient was treated with fenofibrate 160 mg PO daily in addition to rosuvastatin 5 mg PO daily. The patient also remained on perindopril 16 mg PO daily. Just two weeks after starting fenofibrate, the patient’s fasting lipid profile normalized (TC 4.39 mmol/L, LDL 2.43 mmol/L, HDL 1.30 mmol/L, and TG 1.46 mmol/L) and ACR had dropped to 75.0 mg/mmol. ACR continued to decrease and was down to 2.7 mg/mmol two years after fibrate initiation. However, creatinine had increased to 123 µmol/L. This was thought to be most likely from the effect of fibrates on creatinine secretion,4 but a second renal biopsy was ordered to rule out any disease progression. Repeat biopsy done 27 months after the initial biopsy contained up to 17 glomeruli. Capillary lumina were no longer dilated or filled with acellular material (Figure 2A). Occasional glomeruli still had mild-to-moderate mesangial expansion with hypercellularity. Two glomeruli showed global glomerulosclerosis (11%) and three showed focal segmental glomerulosclerosis: two perihilar and one within an indeterminate location. There was no intracapillary material seen and Oil-Red-O stain was negative (Figure 2B), consistent with the resolution of LPG. On electron microscopy, no intracapillary material was seen (Figure 2C). The subendothelial expansion seen in the previous biopsy was notably decreased, with rare persistent flocculent material with a vacuolized appearance, consistent with markedly resolved LPG (Figure 2C).
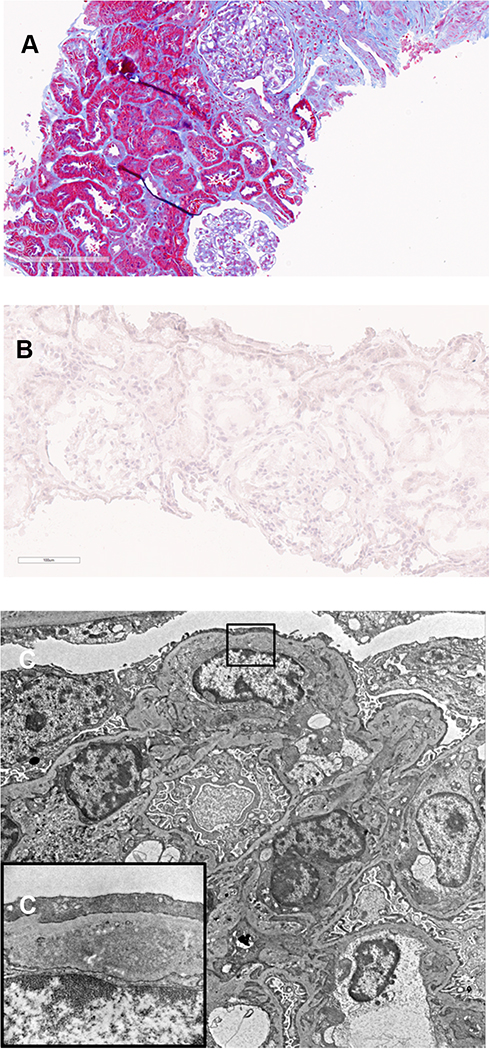 |
Figure 2 Follow up biopsy. The length of each bar represents the distance in microns. (A) shows glomerular capillaries are no longer dilated or filled with acellular material (Masson trichrome). (B) confirms the absence of lipid droplets (Oil-Red-O stain). (C) shows electron micrograph of capillary lumina that no longer contain acellular material (5000x). However, rare subendothelial spaces are expanded with lipoprotein material [osmiophilic granules (protein) and vacuoles (lipid)] as depicted in the inset (C) (15000x, corresponding to the rectangle on (C)). |
The patient continues to do well 3 years after initial diagnosis of LPG and continues to take fenofibrate, rosuvastatin, and perindopril. Creatinine remains stable (106 µmol/L) and ACR is 3.4 mg/mmol.
Discussion
LPG is a renal lipidosis characterized by abnormal lipoprotein metabolism causing lipoprotein thrombi, proteinuria, and renal insufficiency.5 It is usually caused by a mutation in apolipoprotein E (apoE) and is inherited in an autosomal dominant fashion with incomplete penetrance.3 It was first reported in 1989.1 Since then, there have been approximately 200 cases reported worldwide, 145 of which include confirmatory gene sequencing.6–20 Most of these reports come from China and Japan. Only 10 cases of LPG (including this one) have been reported from North America,2,18,21–26 and previously only 5 of these included gene sequencing: 3 APOE Kyoto,18,23 1 APOE Chicago,22 1 APOE Las Vegas.25 The case we present here is the first in North America to be identified with APOE Tokyo/Maebashi. It is also the first case report of LPG from Canada.
LPG can affect any age group.3 It is usually found in patients of East Asian descent, with only 18 cases identified in patients of non-Asian descent,2,10,12,20,22,23,25–33 of which only 9 include confirmatory gene sequencing: 1 APOE Chicago,22 3 APOE Kyoto,12,23 1 APOE Las Vegas,25 2 APOE Modena,27,29 and 2 APOE Osaka/Kurashiki.10 It has a male predilection and usually presents with proteinuria found incidentally on routine laboratory testing.3 Creatinine clearance is usually normal at the time of diagnosis.5 It is associated with elevated apoE serum levels and hypertriglyceridemia, althoug this is not always the case.3 The majority of patients have hypertension.34 Stigmata of lipidosis such as xanthomas are usually absent.3
To our knowledge, there have been only 13 LPG patients in the world (including this patient) that have been identified with the APOE Tokyo/Maebashi mutation. 5 are from Japan,13,35–37 6 are from China,38,39 and 1 is from Switzerland in a man of Indonesian descent.11 APOE Tokyo/Maebashi appears to be fairly rare in frequency in the general population. In one study, 200 Chinese individuals with normal lipid profiles were screened for this mutation and all tested negative.38 This same study also demonstrated that some family members carrying the APOE Tokyo/Maebashi mutation did not have LPG, suggesting incomplete penetrance. This finding is consistent with other mutations linked to LPG. The APOE Tokyo/Maebashi mutation is found at the LDL receptor binding site of apoE,13 which is where most mutations associated with LPG occur.35 A change in binding affinity of the apoE to the LDL receptor impairs catabolism of triglyceride-rich lipoproteins.3 These lipoproteins then accumulate in glomeruli and form lipoprotein thrombi. Studies have shown that proteins with mutations causing LPG such as APOE Sendai, APOE Chicago, APOE Osaka/Kurashiki,40 APOE Kyoto, APOE Tsukuba, and APOE Las Vegas have a tendency to aggregate.41 It is unclear why these lipoprotein thrombi are not found in any structure other than the glomeruli. However, there is speculation that mutations that increase the positive charge of apoE would increase its affinity for the negatively charged glomerular basement membrane, causing them to localize and aggregate in that area.3 It is also postulated that precursors of lipoprotein thrombi assemble in the mesangium and/or subendothelium, and then build up in the glomerular capillaries as a downstream effect.42 Our post-treatment biopsy findings provide evidence to support this hypothesis.
On kidney biopsy, the most striking feature of LPG is enlarged glomerular capillaries filled with lipoprotein thrombi. These thrombi stain pale with H&E, PAS, and Jones’, but positive for Oil-Red-O.42 There is often associated mesangial proliferation, but no foam cells. Sometimes there is membrane reduplication in capillary walls. Segmental sclerosis is also a common finding. There is no significant staining on immunofluorescence. On electron microscopy, the lipoprotein thrombi are shown to consist of lipid granules and vacuoles of various sizes that form a lamellated “fingerprint” appearance. Several biopsies also demonstrate osmiophilic substances in the subendothelium and/or mesangium, even in capillary lumina that do not contain lipoprotein thrombi.42 Thus, it is hypothesized that these osmiophilic substances could represent an early step in the formation of lipoprotein thrombi, although it was unclear whether these densities could just be a non-specific finding.3 The glomerular subendothelium is an ideal nesting spot for LPG-mutated lipoprotein to accumulate since mutated apoE has a higher affinity to both endothelial cells and the glomerular basement membrane. One study showed that triglyceride-rich lipoproteins containing apoE3 affected by the APOE Kyoto mutation had greater binding affinity to endothelial cells than triglyceride-rich lipoproteins bound to wild-type apoE3.43 It is also thought that changes in the electrical charge of mutated apoE increase its affinity for the negatively charged glomerular basement membrane.3 Our discoveries corroborate previous pathological findings of LPG, and add to the existing knowledge of the disease. We are the first to report a repeat biopsy showing that after resolution of lipoprotein thrombi, there is a marked decrease in these subendothelial granular densities, suggesting that the presence of this subendothelial material is a feature specific to LPG that correlates with disease severity. Additionally, this flocculent subendothelial material did not disappear completely despite the complete resolution of lipoprotein thrombi, which supports the idea that this substance is a milder morphologic manifestation of this condition and possibly a precursor in the formation of lipoprotein thrombi.
Worldwide, only 22 cases of LPG including this case have reported the results of more than one native kidney biopsy.14,15,29,32,44–49 Of these repeat biopsies, 20 (including this one) showed improvement or resolution of lipoprotein thrombi, and 2 cases showed no improvement. Our repeat biopsy findings were consistent with previous cases which all reported persistent mesangial changes, foot process effacement, segmental sclerosis, interstitial fibrosis, and tubular atrophy even after lipoprotein thrombi have disappeared. This case is the first to report a repeat biopsy with improvement in, but not complete disappearance of, osmiophilic granules in the subendothelium, even when lipoprotein thrombi have resolved, supporting the theory that these substances represent an early step in the formation of lipoprotein thrombi.
Of the 19 cases with repeat biopsy that improved, 1 was treated with probucol and 6 were treated with a fibrate with or without other lipid-lowering agents. One was treated with LDL apheresis, and 12 were treated with protein A immunoadsorption. Both cases that showed no improvement were treated with fibrates but did not respond. Various treatments have been used for LPG, with variable success. Fibrates are currently the mainstay of treatment for LPG, even for LPG patients who do not have dyslipidemia. One study of 35 LPG patients showed that patients who received fibrate therapy for at least 12 months had 93.7% renal survival versus only 30% renal survival after 3 years in those that did not receive a fibrate.34 Response to fibrate therapy is variable and does not seem to be predicted by apoE mutation type. Less consistent results have been seen with other-lipid lowering therapies. There is one case report that suggests niceritol could be effective.37 Probucol, icosapent ethyl,37 and statins have all had mixed success.17 Most reports suggest that even though statins normalize patient lipid profiles, they usually do not lead to improvement in proteinuria.5 This is consistent with our findings. Our patient was on rosuvastatin 10 mg PO daily for over 6 months prior to starting fibrate therapy, and even though his lipid profile improved, his proteinuria did not. Similarly, reports suggest that angiotensin converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) alone do not improve LPG. Our patient received four months of perindopril prior to starting fibrate therapy and proteinuria remained unchanged until fibrates were started. When patients are refractory to fibrate therapy, alternative treatments include LDL apheresis29 and staphylococcal protein A immunoadsorption,48 although a need for repeat treatments might be required.
There have been no cases reported of LPG that resolve spontaneously without treatment. Almost all patients with LPG who receive a kidney transplant have a recurrence of LPG on kidney biopsy within several months.3
LPG is a rare kidney disease mostly seen in China and Japan. To our knowledge, we are the first to report a case of LPG in Canada. Considering the diverse ethnic population found in Canada, and in particular the large Asian communities in Canada, it is likely that the absence of case reports of LPG represents under-diagnosis or under-reporting of this entity. By reporting our findings, we hope to raise awareness of this disease in non-Asian countries. Indeed, the clinical presentation can be very subtle and patients with this disease are likely under-biopsied due to the presumption that their underlying kidney disease must be from more common conditions, such as hypertension, diabetes, or IgA nephropathy. However, as in this case, patients with proteinuria who are not responding to usual treatments such as ACEI or ARB should receive a kidney biopsy to assess for LPG, especially if they are of East Asian descent and have hypertriglyceridemia. This diagnosis is a particularly critical one to make since the treatment used for this disease is very different from other glomerular diseases, and the prognosis without treatment is quite poor. In addition, we offer insight into the pathological evolution of LPG, with novel biopsy findings to support the hypothesis that precursors to lipoprotein thrombi are found in the subendothelium of glomeruli. Meta-analysis of existing reports and studies is required to target therapies more selectively.
Conclusion
LPG is a disease that is difficult to clinically differentiate from other proteinuric kidney diseases associated with dyslipidemias. While appropriate treatment results in significant recovery, lack of treatment can often lead to significant loss of kidney function, further highlighting the importance of accurate diagnosis and appropriate treatment. Considering the diversity of population in many countries across the world, it is likely that this disease is under-recognised outside of China and Japan. By reporting the first case of LPG in Canada, we hope to raise awareness of this rare disease. We also offer insight into the pathogenesis of LPG through repeat biopsy that showed improvement in, but not complete disappearance of, subendothelial lipoprotein material even after resolution of lipoprotein thrombi and complete clinical remission of disease. This supports growing evidence that precursors to lipoprotein thrombi are found in the subendothelial space of glomeruli in LPG.
Abbreviations
ACEI, angiotensin-converting enzyme inhibitor; ACR, urine albumin-to-creatinine ratio; APOE, apolipoprotein E gene; apoE, apolipoprotein E; ARB, angiotensin receptor blocker; HDL, high-density lipoprotein; LDL, low-density lipoprotein; LPG, lipoprotein glomerulopathy; TC, total cholesterol; TG, triglycerides.
Data Sharing Statement
The data referred to in this case report are available from the corresponding author on reasonable request.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. Institutional approval was not required to publish the case details.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval for the version to be published; and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Saito T, Sato H, Kudo K, et al. Lipoprotein glomerulopathy: glomerular lipoprotein thrombi in a patient with hyperlipoproteinemia. Am J Kidney Dis. 1989;13(2):148–153. doi:10.1016/s0272-6386(89)80134-9
2. Majeed NK, McLaughlin J, Gonzalez M. Lipoprotein glomerulopathy in a Hispanic female: a case report and literature review. Can J Kidney Heal Dis. 2019;6:2054358119859576. doi:10.1177/2054358119859576
3. Tsimihodimos V, Elisaf M. Lipoprotein glomerulopathy. Curr Opin Lipidol. 2011;22(4):262–269. doi:10.1097/MOL.0b013e328345ebb0
4. Sica DA. Fibrate therapy and renal function. Curr Atheroscler Rep. 2009;11(5):338–342. doi:10.1007/s11883-009-0051-5
5. Saito T, Matsunaga A, Oikawa S. Impact of lipoprotein glomerulopathy on the relationship between lipids and renal diseases. Am J Kidney Dis. 2006;47(2):199–211. doi:10.1053/j.ajkd.2005.10.017
6. Saito T, Matsunaga A. Lipoprotein glomerulopathy may provide a key to unlock the puzzles of renal lipidosis. Kidney Int. 2014;85(2):243–245. doi:10.1038/ki.2013.404
7. Li W, Wang Y, Han Z, Luo C, Zhang C, Xiong J. Apolipoprotein e mutation and double filtration plasmapheresis therapy on a new Chinese patient with lipoprotein glomerulopathy. Kidney Blood Press Res. 2014;39(4):330–339. doi:10.1159/000355810
8. Yang M, Weng Q, Pan X, et al. Clinical and genetic analysis of lipoprotein glomerulopathy patients caused by APOE mutations. Mol Genet Genomic Med. 2020;8(8). doi:10.1002/mgg3.1281
9. Yang Z, Wu H, Hu Z. [Discovery of a Chinese Tibetan patient with lipoprotein glomerulopathy due to APOE Osaka/Kurashiki variant]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2020;37(2):166–169. Chinese. doi:10.3760/cma.j.issn.1003-9406.2020.02.017
10. da Silveira-Neto JN, de Oliveira Ahn GJ, de Menezes Neves PDM, et al. Lipoprotein glomerulopathy associated with the Osaka/Kurashiki APOE variant: two cases identified in Latin America. Diagn Pathol. 2021;16(1):65. doi:10.1186/s13000-021-01119-x
11. Kollbrunner L, Hirt-Minkowski P, Sanz J, et al. Case report: lipoprotein glomerulopathy complicated by atypical hemolytic uremic syndrome. Front Med. 2021;8. doi:10.3389/fmed.2021.679048
12. Marinaki S, Kalaitzakis E, Kolovou K, et al. A case of lipoprotein glomerulopathy in a Greek Caucasian male. Int Urol Nephrol. 2021;54(4):969–970. doi:10.1007/s11255-021-02930-7
13. Takasaki S, Maeda K, Joh K, et al. Macrophage infiltration into the glomeruli in lipoprotein glomerulopathy. Case Rep Nephrol Dial. 2015;5(3):204–212. doi:10.1159/000441715
14. Usui R, Takahashi M, Nitta K, Koike M. Five-year follow-up of a case of lipoprotein glomerulopathy with APOE Kyoto mutation. CEN Case Rep. 2016;5(2):148–153. doi:10.1007/s13730-016-0214-5
15. Kodera H, Mizutani Y, Sugiyama S, et al. A case of lipoprotein glomerulopathy with apoE Chicago and apoE (Glu3Lys) treated with fenofibrate. Case Rep Nephrol Dial. 2017;7(2):112–120. doi:10.1159/000478902
16. Wu H, Yang Y, Hu Z. The novel apolipoprotein E mutation ApoE Chengdu (c.518T>C, p.L173P) in a Chinese patient with lipoprotein glomerulopathy. J Atheroscler Thromb. 2018;25(8):733–740. doi:10.5551/jat.41996
17. Lui DTW, Lee ACH, Yap DYH, Chan GSW, Tan KCB. A young Chinese man with nephrotic syndrome due to lipoprotein glomerulopathy. J Clin Lipidol. 2018;13(2):251–253. doi:10.1016/j.jacl.2018.12.004
18. Batal I, Fakhoury G, Groopman E, D’Agati VD, Morris H. Unusual case of lipoprotein glomerulopathy first diagnosed in a protocol kidney allograft biopsy. Kidney Int Rep. 2019;4(2):350–354. doi:10.1016/j.ekir.2018.09.020
19. Xie W, Xie Y, Lin Z, Xu X, Zhang Y. A novel apolipoprotein E mutation caused by a five amino acid deletion in a Chinese family with lipoprotein glomerulopathy: a case report. Diagn Pathol. 2019;14(1):41. doi:10.1186/s13000-019-0820-6
20. Morris CS, Bois MC, Aust CH, Thomas R, Sethi S, Maleszewski JJ. Intravascular cardiac lipoproteinosis: extrarenal manifestation of lipoprotein glomerulopathy. Cardiovasc Pathol. 2019;42:6–9. doi:10.1016/j.carpath.2019.04.006
21. Zhang P, Matalon R, Kaplan L, Kumar A, Gallo G. Lipoprotein glomerulopathy: first report in a Chinese male. Am J Kidney Dis. 1994;24(6):942–950. doi:10.1016/s0272-6386(12)81066-3
22. Sam R, Wu H, Yue L, et al. Lipoprotein glomerulopathy: a new apolipoprotein E mutation with enhanced glomerular binding. Am J Kidney Dis. 2006;47(3):539–548. doi:10.1053/j.ajkd.2005.12.031
23. Rovin BH, Roncone D, McKinley A, Nadasdy T, Korbet SM, Schwartz MM. APOE Kyoto mutation in European Americans with lipoprotein glomerulopathy. N Engl J Med. 2007;357(24):2522–2524. doi:10.1056/NEJMc072088
24. Sethi S. Renal failure with intracapillary thrombi. Lipoprotein glomerulopathy. Kidney Int. 2008;73(9):1097–1098. doi:10.1038/ki.2008.13
25. Bomback AS, Song H, D’Agati VD, et al. A new apolipoprotein E mutation, apoE Las Vegas, in a European-American with lipoprotein glomerulopathy. Nephrol Dial Transplant. 2010;25(10):3442–3446. doi:10.1093/ndt/gfq389
26. Boumendjel R, Papari M, Gonzalez M. A rare case of lipoprotein glomerulopathy in a white man: an emerging entity in Asia, rare in the white population. Arch Pathol Lab Med. 2010;134(2):279–282. doi:10.5858/134.2.279
27. Russi G, Furci L, Leonelli M, et al. Lipoprotein glomerulopathy treated with LDL-apheresis (Heparin-induced extracorporeal lipoprotein precipitation system): a case report. J Med Case Rep. 2009;3(1):9311. doi:10.1186/1752-1947-3-9311
28. Pasquariello A, Pasquariello G, Innocenti M, et al. Lipoprotein glomerulopathy: first report of 2 not consanguineous Italian men from the same town. J Nephrol. 2011;24(3):381–385. doi:10.5301/JN.2011.7772
29. Magistroni R, Bertolotti M, Furci L, et al. Lipoprotein glomerulopathy associated with a mutation in apolipoprotein e. Clin Med Insights Case Rep. 2013;6:189–196. doi:10.4137/CCRep.S12209
30. Pêgas KL, Rohde R, Garcia CD, et al. Lipoprotein glomerulopathy: a case report of a rare disease in a Brazilian child. J Bras Nefrol. 2014;36(1):93–95. doi:10.5935/0101-2800.20140015
31. Sipovskii VG, Klemina IK, Zverkov RV, Dobronravov VA, Smirnov AV. [A case of diagnosing lipoprotein glomerulopathy in Russia]. Arkh Patol. 2016;78(6):52–57. Russian. doi:10.17116/patol201678652-57
32. Meyrier A, Dairou F, Callard P, Mougenot B. Lipoprotein glomerulopathy: first case in a white European. Nephrol Dial Transplant. 1995;10(4):546–549. doi:10.1093/ndt/10.4.546
33. Mourad G, Djamali A, Turc-Baron C, Cristol JP. Lipoprotein glomerulopathy: a new cause of nephrotic syndrome after renal transplantation. Nephrol Dial Transplant. 1998;13(5):1292–1294. doi:10.1093/ndt/13.5.1292
34. Hu Z, Huang S, Wu Y, et al. Hereditary features, treatment, and prognosis of the lipoprotein glomerulopathy in patients with the APOE Kyoto mutation. Kidney Int. 2014;85(2):416–424. doi:10.1038/ki.2013.335
35. Konishi K, Saruta T, Kuramochi S, et al. Association of a novel 3-amino acid deletion mutation of apolipoprotein E (apo E Tokyo) with lipoprotein glomerulopathy. Nephron. 1999;83(3):214–218. doi:10.1159/000045513
36. Ogawa T, Maruyama K, Hattori H, et al. A new variant of apolipoprotein E (apo E Maebashi) in lipoprotein glomerulopathy. Pediatr Nephrol. 2000;14(2):149–151. doi:10.1007/s004670050032
37. Hamatani H, Hiromura K, Kobatake K, et al. Successful treatment of lipoprotein glomerulopathy in a daughter and a mother using niceritrol. Clin Exp Nephrol. 2010;14(6):619–624. doi:10.1007/s10157-010-0333-9
38. Han J, Pan Y, Chen Y, et al. Common apolipoprotein E gene mutations contribute to lipoprotein glomerulopathy in China. Nephron Clin Pract. 2010;114(4):c260–c267. doi:10.1159/000276578
39. Cheung CY, Chan AOK, Chan GPT, Iu HYP, Shek CC, Chau KF. Long-term outcome of kidney transplantation in a patient with coexisting lipoprotein glomerulopathy and fibrillary glomerulonephritis. Clin Kidney J. 2014;7(4):396–398. doi:10.1093/ckj/sfu058
40. Georgiadou D, Stamatakis K, Efthimiadou EK, et al. Thermodynamic and structural destabilization of apoE3 by hereditary mutations associated with the development of lipoprotein glomerulopathy. J Lipid Res. 2013;54(1):164–176. doi:10.1194/jlr.M030965
41. Katsarou M, Stratikos E, Chroni A. Thermodynamic destabilization and aggregation propensity as the mechanism behind the association of apoE3 mutants and lipoprotein glomerulopathy. J Lipid Res. 2018;59(12):2339–2348. doi:10.1194/jlr.M088732
42. Saito T, Oikawa S, Sato H, Sato T, Ito S, Sasaki J. Lipoprotein glomerulopathy: significance of lipoprotein and ultrastructural features. Kidney Int. 1999;56:S37–S41. doi:10.1046/j.1523-1755.1999.07110.x
43. Murano T, Matsumura R, Misawa Y, et al. Interaction of endothelial cells and triglyceride-rich lipoproteins with apolipoprotein E (Arg25[rarr]Cys) from a patient with lipoprotein glomerulopathy. Metabolism. 2002;51(2):201–205. doi:10.1053/meta.2002.29990
44. Amenomori M, Haneda M, Morikawa J, et al. A case of lipoprotein glomerulopathy successfully treated with probucol. Nephron. 1994;67(1):109–113. doi:10.1159/000187897
45. Arai T, Yamashita S, Yamane M, et al. Disappearance of intraglomerular lipoprotein thrombi and marked improvement of nephrotic syndrome by bezafibrate treatment in a patient with lipoprotein glomerulopathy. Atherosclerosis. 2003;169(2):293–299. doi:10.1016/s0021-9150(03)00194-1
46. Ieiri N, Hotta O, Taguma Y. Resolution of typical lipoprotein glomerulopathy by intensive lipid-lowering therapy. Am J Kidney Dis. 2003;41(1):244–249. doi:10.1053/ajkd.2003.50016
47. Kinomura M, Sugiyama H, Saito T, et al. A novel variant apolipoprotein E Okayama in a patient with lipoprotein glomerulopathy. Nephrol Dial Transplant. 2008;23(2):751–756. doi:10.1093/ndt/gfm675
48. Xin Z, Zhihong L, Shijun L, et al. Successful treatment of patients with lipoprotein glomerulopathy by protein A immunoadsorption: a pilot study. Nephrol Dial Transplant. 2008;24(3):864–869. doi:10.1093/ndt/gfn555
49. Matsunaga A, Furuyama M, Hashimoto T, Toyoda K, Ogino D, Hayasaka K. Improvement of nephrotic syndrome by intensive lipid-lowering therapy in a patient with lipoprotein glomerulopathy. Clin Exp Nephrol. 2009;13(6):659–662. doi:10.1007/s10157-009-0207-1
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.