")
Back to Journals » International Journal of Nanomedicine » Volume 15
Lipid-Based Gliclazide Nanoparticles for Treatment of Diabetes: Formulation, Pharmacokinetics, Pharmacodynamics and Subacute Toxicity Study
Authors Nazief AM, Hassaan PS , Khalifa HM, Sokar MS, El-Kamel AH
Received 19 October 2019
Accepted for publication 12 January 2020
Published 18 February 2020 Volume 2020:15 Pages 1129—1148
DOI https://doi.org/10.2147/IJN.S235290
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Alaa Mohamed Nazief,1 Passainte Saber Hassaan,2 Hoda Mahmoud Khalifa,3 Magda Samir Sokar,1 Amal Hassan El-Kamel1
1Department of Pharmaceutics, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt; 2Department of Medical Physiology, Faculty of Medicine, Alexandria University, Alexandria, Egypt; 3Department of Histology and Cell Biology, Faculty of Medicine, Alexandria University, Alexandria, Egypt
Correspondence: Amal Hassan El-Kamel
Department of Pharmaceutics, Faculty of Pharmacy, Alexandria University, Alexandria 21521, Egypt
Tel +201005080510
Email [email protected]
Introduction: Solid lipid nanoparticles (SLNs) are considered a promising system in enhancing the oral bioavailability of poorly water-soluble drugs; owing to their intrinsic ability to increase the solubility together with protecting the incorporated drugs from extensive metabolism.
Objective: Exploiting such properties, SLNs loaded with gliclazide (GLZ) were developed in an attempt to improve the oral bioavailability and the anti-diabetic action of GLZ, together with prolonging its duration of action for better glycemic control.
Methods: SLNs were prepared by ultra-sonication technique using glyceryl behenate (Compritol® 888 ATO) as a lipid matrix and poloxamer 188 (PLX) as a stabilizer. A 2*3 asymmetrical factorial design was adopted to study the effect of different stabilizer concentrations at different sonication times on the shape, and size of the particles, PDI and drug loading. The selected optimum formulation was then freeze dried using trehalose di-hydrate as a cryo-protectant in different ratios with respect to glyceryl behenate concentration. After freeze drying, the formulation was tested for in-vitro drug release, pharmacokinetics, and pharmacodynamics. Safety of the selected formula was established after carrying out a subacute toxicity study.
Results: The factorial design experiment resulted in an optimum formulation coded 10F2 (150 mg PLX/10 min sonication). Scanning electron micrographs showed spherical particles with smooth surface, whereas a ratio of 2:1 cryo-protectant:lipid was found to be optimum with particle size of 245.9 ± 26.2 nm, polydispersity index of 0.482 ± 0.026, and biphasic in-vitro release with an initial burst effect, followed by a prolonged release phase. On the other hand, the selected SLNs exhibited prolonged drug release when compared with the GLZ commercial immediate release (IR) tablets (Diamicron®). Pharmacokinetics study showed about 5-fold increase in GLZ oral bioavailability loaded in SLNs when compared with raw GLZ powder. Pharmacodynamics study on a diabetic rat model confirmed the better anti-diabetic action of GLZ loaded SLNs when compared to raw GLZ powder. Subacute toxicity study indicated the safety of SLNs upon repetitive oral administration.
Keywords: gliclazide, lipid nanoparticles, bioavailability, safety
Introduction
Solid lipid nanoparticles (SLNs) are colloidal particles of a lipid matrix that is solid at the body temperature. Lipid nanoparticles have many advantages such as stability, feasibility of carrying both lipophilic and hydrophilic drugs, biodegradability, biocompatibility. Moreover, small particles ranging between 120 and 200 nm rarely undergo blood clearance by the reticuloendothelial system (liver and spleen filtrations), thus skipping first pass metabolism.1 In addition, the scaling up of the formulation technique to industrial production level is feasible at low cost and in a relatively simple way.2
Gliclazide is an oral hypoglycemic useful for the treatment of non-insulin-dependent diabetes mellitus. Gliclazide exhibits slow GI absorption rate and wide inter individual variations in absorption and bioavailability. A single oral dose of gliclazide resulted in a Tmax ranged from 2 to 8 h due to its pH-dependent and low solubility.3,4 The variability in absorption of gliclazide could be related to its early dissolution in the stomach leading to more variability in the intestinal absorption.5 The absorption half-life in man is 1.3 h. Gliclazide has a low volume of distribution, with strong plasma protein binding property, with about 94.2% of the drug being protein bound. Moreover, gliclazide undergoes extensive hepatic metabolic biotransformation to give several inactive metabolites with no circulating active metabolite.6
Different nano-carrier models for gliclazide were addressed and evaluated in-vitro with complete disregard for in-vivo behavior.7–11 Other studies were satisfied with either the pharmacokinetics evaluation,12 or both pharmacokinetics and pharmacodynamics appraisal.13 However, they did not tackle the safety of the prepared systems. The commonly reported lipid-based carrier systems for gliclazide are self-emulsifying delivery systems.
To the best of our knowledge, no SLNs for gliclazide have been developed except for that reported by Dhome et al14 who characterized the prepared nanoparticles only by in-vitro techniques. They also developed the nanoparticles using micro-emulsion technique and very high concentrations of surfactants and co-surfactants raising serious safety issues.
On the other hand, the current work focused not only on in-vitro characterization but also on the in-vivo behavior and safety of the prepared particles. Additionally, FDA approved lipid carrier (glyceryl behenate) and ultra-sonication, as a sole method of preparation, were utilized. Furthermore, low surfactant concentration was used to stabilize the prepared nanoparticles.
The aim of this work was to develop safe and effective gliclazide lipid nanosystem to prolong the release and improve the oral bioavailability of gliclazide thus sustaining its anti-diabetic action.
Materials and Methods
Materials
Gliclazide and phenytoin were provided as a gift from El Amryia Pharmaceutical Co. (Egypt). Poloxamer 188, trehalose di-hydrate and Streptozotocin were purchased from Sigma-Aldrich Chemie GmbH (Germany). Glyceryl behenate (Compritol®888 ATO) was purchased from Gattefossé (France). Commercial immediate release tablets (Diamicron® 80 mg), Servier laboratories (Canada), batch number 22182, and expiry date 6/2019. All other reagents and chemicals were of analytical grade.
Methods
HPLC Assay of Gliclazide
The drug was assayed using HPLC method reported by Rouini et al15 with slight modifications (Agilent technologies, 1260 infinity series, California, USA). Mobile phase composed of acetonitrile: 0.4 µm Millipore filtered distilled water (45%:55%) of pH 3 adjusted by ortho-phosphoric acid. An aliquot of 20 μL sample was injected into HPLC column (kinetex® core-shell C18 column, 5μm, 100°A, 15cm, Phenomenex, California, USA) at a flow rate of 0.9 mL/min. The drug peak was UV detected at λ max = 230 nm at room temperature.
Preparation of GLZ Loaded Solid Lipid Nanoparticles
Ultra-sonication technique was used to prepare SLNs. GLZ was added to molten lipid at 80°C. Millipore filtered distilled water and surfactant (Poloxamer 188) were heated to the same temperature. Then, the aqueous contents were added to the lipid phase and ultra-sonicated (ultrasonic homogenizer, Bandelin Sonopuls 3100, Germany) for 5 min at a 60% amplitude energy output. The formed primary emulsion was then rapidly added to 10 mL of cold Millipore filtered distilled water at 2–3°C (in an ice bath), and ultra-sonicated for an additional 10 or 20 min. The final volume of the prepared emulsion was adjusted to 25 mL using Millipore filtered distilled water.
To study the effect of different surfactant concentrations together with sonication time, a 2*3 factorial design experiment was employed. Three different surfactant concentrations (100, 150 and 200 mg) and 2 different sonication times (10, 20 min) were studied.
Each of the prepared formulations was evaluated for the particle size (PS), polydispersity index (PDI), zeta potential (ZP), and entrapment efficiency (%EE). The results of which were statistically analyzed using factorial ANOVA, with two independent variables (surfactant concentration and sonication time) and four dependent variables (particle size, PDI, zeta potential and entrapment efficiency) in order to choose the optimum formulation.
Particle Size (PS), Polydispersity Index (PDI) and Zeta Potential (ZP) Measurement
PS and PDI analyses were conducted by photon correlation spectroscopy technique using Malvern Zetasizer (Malvern instruments, Malvern, UK). SLNs dispersions were appropriately diluted using Millipore filtered water (0.4µm) and measurements were conducted at a scattering angle of 173º.
Determination of GLZ Entrapment Efficiency (%EE)
In a vivaspin® tube (Sartorius AG, Goettingen, Germany), 1 mL of the SLNs dispersion was mixed with 1 mL of methanol. Centrifugation (Cooling high-speed centrifuge, Sigma laborzentrifugen GmbH, Osterode, Germany) was then done at 20°C, for 15 mins at speed of 2500 rpm. The solution that passed from the ultrafilter after centrifugation was taken and appropriately diluted by the mobile phase and analyzed using the HPLC method.
The percentage entrapment efficiency was calculated by the following equation:
(1)
Scanning Electron Microscopy (SEM)
Scanning electron microscope (JOEL Ltd, Tokyo, Japan) was used to study the morphology of the prepared SLNs. One drop of the SLNs dispersion was poured onto a cover glass to evaporate the aqueous medium. After evaporation, standard coating by gold-palladium sputtering was done and scans were performed at an acceleration voltage of 20 kV.
Freeze Drying of SLNs Formulation
Freeze drying of the chosen optimum formula was carried out after addition of a cryo-protectant; trehalose di-hydrate, which was reported as the cryo-protectant of choice for glyceryl behenate nanoparticles.16
Trehalose was added to the SLNs dispersion, sonicated and the formulas were stored in a freezer at −20°C till the next day. They were then introduced into the freeze dryer (LyoQuest −55 plus, Telstar Life Sciences, Terrassa, Spain) at −50°C and vacuum was operated till the samples were completely dry.
Different amounts of cryo-protectant were added in different ratios (1:1, 2:1, 3:1 and 4:1) with respect to the amount of glyceryl behenate in the particles. The optimum concentration of the cryo-protectant was chosen depending on the particle size and PDI of the reconstituted powder in 0.4µm filtered distilled water.
In-vitro Release Study of Gliclazide
The in-vitro release behavior was studied using USP dissolution apparatus II. The dissolution medium was phosphate buffer pH 7.4 as reported in BP 2013. Gliclazide doses equivalent to 30 mg were added to 500 mL dissolution medium and stirred at 50 rpm. Temperature was kept throughout the experiment at 37°C ±1°C.
The chosen formula was compared to the immediate release GLZ commercial product (whole and ground tablets), and the raw powder drug.
Samples were withdrawn at different time intervals, appropriately diluted by mobile phase, and the amount of gliclazide was analyzed by HPLC. The withdrawn samples were compensated with fresh medium.
To study the release kinetics of gliclazide (GLZ), DD Solver (2010), an add-in program for Microsoft Excel, for modeling and comparison of drug release profile was used.17
Differential Scanning Calorimetry (DSC)
Thermal behavior was studied using DSC (Perkin Elmer Instruments, USA). The samples studied were: lyophilized SLNs, raw gliclazide, glyceryl behenate, poloxamer 188, trehalose di-hydrate, and physical mixture of all the ingredients were of the same ratio as in the prepared SLNs.
Samples were hermetically sealed in an aluminum pan. DSC traces were recorded between 30 and 250ºC, at a heating rate of 10°C/minute. Inert atmosphere was maintained by purging nitrogen at a flow rate of 10 mL/min. An empty pan was subjected to the same conditions and used as control.
Fourier Transform Infrared Spectroscopy (FT-IR)
FT-IR spectroscopy (Perkin Elmer Instruments, USA) was performed for lyophilized SLNs, gliclazide, glyceryl behenate, poloxamer 188, trehalose di-hydrate, and physical mixture of all the ingredients were of the same ratio as in the prepared SLNs. Samples were finely ground in a mortar and mixed with potassium bromide then pressed for 5 min at high pressure to form a disc. IR spectrum were then scanned over the range of 400 to 4000 cm−1 using a resolution of 1 cm−1.
In-vivo Testing for the Optimum SLN Formulation
To further evaluate the optimum SLN formulation, several in-vivo tests were adopted, including measuring the pharmacokinetic parameters after single oral dose, and pharmacodynamic evaluation of SLNs on high-fat diet – streptozotocin-induced diabetic rats. Moreover, a subacute toxicity study was done to test for the safety of the particles upon repetitive oral administration.
Animals
Adult male Wistar rats were provided by the animal facility of the Faculty of Medicine, Alexandria University, and kept in the animal house of Faculty of Medicine, Al-Mowasah, Alexandria University. Rats were housed at ambient temperature, fed standard chow diet, and given tap water ad libitum. Experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (Faculty of Pharmacy, Alexandria University, Project no. ACCUC17/15). Suffering of animals was minimized in accordance with the internationally accepted principles for laboratory use and care of the European Community.
Pharmacokinetics Study Protocol
GLZ concentration in rat plasma was determined using the same HPLC method used in the in-vitro quantification, with the use of phenytoin (PHY) as an internal standard.
Healthy male Wistar rats weighing 160–190 g were fasted for 18 hrs but allowed water ad libitum. Ten rats were divided into 2 groups, comprising 5 rats each, in a parallel design. Group 1 administered lyophilized Formula (10F2), and group 2 administered raw gliclazide powder, all in a dose corresponding to 10 mg/kg gliclazide; such GLZ dose was previously reported in literature.18
Doses were dispersed in 2 mL distilled water, and vortexed for 10 seconds not more than 1 min before oral administration directly followed by 1 mL of glucose solution in a dose of 1 g/kg to prevent severe hypoglycemia that might occur as a result of gliclazide administration.19
Doses were orally administered using an oral gavage. During the experiment, animals were allowed to drink water ad libitum. Food was withdrawn for the first 6 h of the experiment (till the 6 h sample was withdrawn) then animals were allowed to eat.
A sample of 0.5 mL blood, from every rat, was collected in EDTA tube by retro-orbital vein puncture at the following intervals: 0 (pre-dose), 0.5, 1, 2, 3, 4, 6 and 24 h.
Blood samples were centrifuged at 8000 rpm for 20 mins and the separated plasma was collected and stored at −20°C for further analysis. Phenytoin working standard (100 µL) of concentration 12 µg/mL was added to 100 µL of plasma, vortexed for 30 seconds, and centrifuged at 10,000 rpm for 15 mins at 20°C to ensure plasma protein precipitation. The supernatant was filtered by 0.2 µm PTFE syringe filter, and then injected into HPLC system. GLZ/PHY peak area ratio of each sample was recorded, and GLZ concentration was determined using the constructed calibration curve of GLZ in plasma.
Pharmacokinetic analysis of the data was performed using PKSolver (2010), an add-in program for Microsoft Excel for pharmacokinetic and pharmacodynamic data analysis.20 The area under the plasma concentration-time profile (AUC 0–24 and AUC 0-inf) was calculated using the linear trapezoidal method. The maximum plasma concentration (Cmax) and the time to reach Cmax (Tmax) were subsequently determined.
Pharmacodynamics Study Protocol
Adult male Wistar rats weighing 160–190 g were used for the study. Rats were housed in polypropylene cages at ambient temperature, fed standard chow diet and given tap water ad libitum prior to dietary manipulation. They were given 2 weeks to adapt to their environment before the experiment.
Animals were assigned into 2 groups; Group I (control) which received commercially available normal pellet diet (NPD), and group II which received in house prepared high-fat diet (HFD),21 for three weeks, at the end of which, changes in body weight, plasma cholesterol, and plasma triglycerides were recorded and compared. Each of the 2 groups followed the assigned diet till the end of the experiment.
After three weeks of the dietary manipulation, the rats of the HFD group were injected intraperitoneally (IP) with a low dose of STZ equivalent to 35 mg/kg dissolved just prior to the injection in 0.1 M citrate buffer adjusted to pH 4.5 by 1N NaOH.22 The doses of STZ were individualized for each rat depending on its body weight.
One week after STZ injection, fasting and non-fasting blood glucose levels (BSL) were measured using the commercially available Accu-Chek® glucometer (Roche Diagnostics GmbH, Germany). Rats with fasting BSL ≥ 200 mg/dL or non-fasting BSL ≥ 250 mg/dL were considered diabetic and were grouped for the pharmacodynamics study.
Diabetic male Wistar rats were fasted for 18 h but allowed water ad libitum. Ten rats were divided into 2 groups, comprising 5 rats each, in a parallel design. Group I administered lyophilized GLZ loaded SLNs (10F2), and group II administered raw gliclazide powder all of which in an oral dose corresponding to 10 mg/kg gliclazide.
During the experiment, animals were allowed to drink water ad libitum. Food was withdrawn for the first 6 h of the experiment (till the 6 h sample was withdrawn) then animals were allowed to eat.
A drop of blood from the tail vein was taken at the following intervals: 0 (pre-dose), 1, 2, 3, 4, 6 and 24 h and BSL was measured using Accu-Chek® glucometer.
In-vivo 14-Day Subacute Toxicity Study
Male Wistar rats weighing 160–220 g were randomly selected and assigned to three test groups (five animals per each group). Group I “the negative control group” received 3 mL of distilled water, Group II “the formula group” received SLNs loaded with the drug (10F2) in a dose equivalent to 20 mg/kg/d (20 times the human daily dose) suspended in 3 mL distilled water, and Group III “the placebo group” received approximately 50 mg of blank SLNs (equivalent to 20 mg/kg from GLZ loaded SLNs) suspended in 3 mL distilled water. Animals were dosed daily by gavage for 14 consecutive days. Individual animal dosages were based on individual body weights recorded on the first day of treatment and were adjusted according to each intermediate body weight determination.
Sub-acute toxicity was measured by monitoring mortality, survival time, clinical picture of intoxication and behavioral reactions. Animals on the study were observed for any adverse reactions during the first 24 h of treatment and then daily till the end of the study. Body weight of each rat was determined at the initiation of the experiment and then once weekly. The amounts of daily food (g/d) and water (mL/d) consumption were also recorded every other day.
Animals were sacrificed on day 15 of the experiment, 24 h after the last dose was administered. Animals were fasted for 18 h before sacrifice. Animals were anesthetized using di-ethyl ether, then they were sacrificed by making a puncture in the descending aorta and collecting the blood of the animal from it. After animals were sacrificed, they were necropsied, and the organs (liver, kidneys, stomach and small intestine) were collected, washed with 0.9% saline, weighed to monitor any enlargement or atrophy that might happen, and were subjected to gross examination to monitor any significant change in texture and shape.
Blood samples collected from the sacrificed animals were divided into 2 groups; one was allowed to clot, while the other was collected in EDTA tubes. Next, blood samples were centrifuged at 8000 rpm for 20 mins. The recovered serum and plasma were stored at −80°C for determination of liver functions (SGPT and SGOT kinetic UV kits, Vitro Scient, MDSS GmbH, Germany) and kidney functions (Urea, Berthelot enzymatic colorimetric kit, and Creatinine, Jaffé colorimetric kinetic kit, Diamond Diagnostics, USA) using serum samples, and determination of cholesterol (cholesterol oxidase/peroxidase kit, Biosystems S.A, Spain) and triglycerides (triglycerides, glycerol phosphate oxidase/peroxidase kits, Biosystems S.A, Spain) level using plasma samples.
After animals’ sacrifice, the stomach, small intestine, liver and kidney were collected and fixed in 10% formalin phosphate buffer pH 6.8 then subjected to histopathological examination. The tissues of organ samples were embedded in paraffin blocks, then 5–6 μm sections were cut and stained with hematoxylin and eosin (H & E) for microscopic evaluation, the slides were observed and photos were taken using light microscope and histopathological examination was performed.23
Statistical Analysis
Data were analyzed using SPSS Statistical Package (Version 22, SPSS Inc, 2013, USA). All data were statistically compared for any significant difference using analysis of variance, ANOVA test. P values ≤ 0.05 were considered statistically significant while p values ≤0.001 were considered highly statistically significant. Post Hoc Duncan test was done to compare the results when required.
Results and Discussion
Preparation of GLZ-Loaded Solid Lipid Nanoparticles
One of the fundamental principles for the design of SLNs is the decrease of the size of the particles, which allows for an efficient uptake in the intestine particularly in the lymphoid tissue thus bypassing the first pass metabolism and resulting in an improvement of drug oral bioavailability.24 Ultra-sonication technique was selected for the preparation of SLNs, as it includes no addition of organic solvents. The high-energy output decreases the size of the lipid to a uniform range with narrow polydispersity, while the presence of the surfactants prevents the re-aggregation of the formed particles.25
Preliminary trials were done and showed that poloxamer alone was sufficient to stabilize the formed SLNs. Factorial design was constructed to study the effect of both surfactant concentration; 100 (F1), 150 (F2), and 200 (F3) mg PLX-188, together with sonication time during the cooling phase (10 min vs 20 min) on the particle size (PS), polydispersity index (PDI), zeta potential (ZP) and percentage entrapment efficiency (%EE) of the prepared GLZ loaded SLNs. Data for PS, PDI, ZP and %EE are recorded in Table 1.
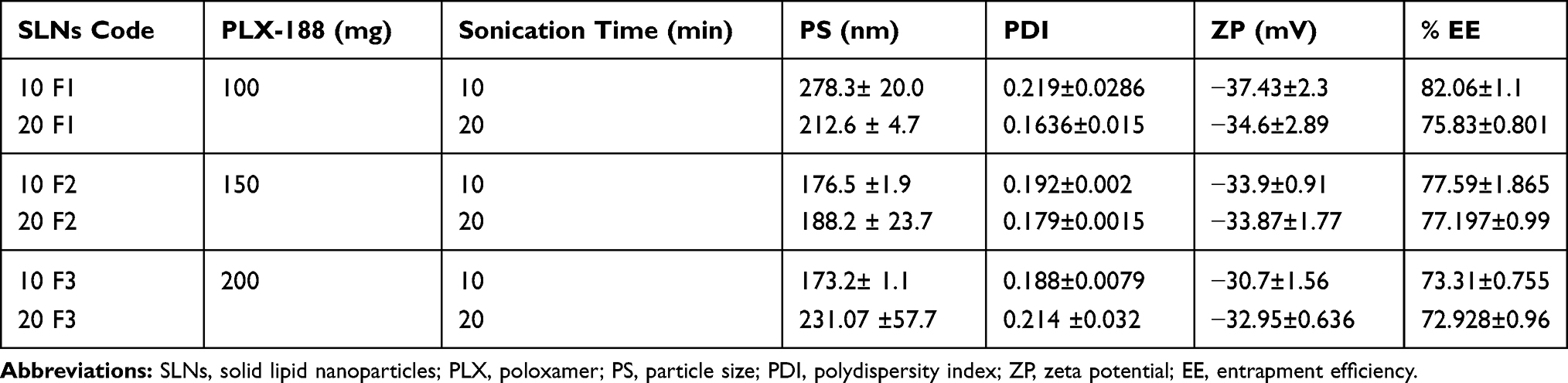 |
Table 1 Particle Size (PS), Polydispersity Index (PDI), Zeta Potential (ZP), and Percentage Entrapment Efficiency (%EE) of the Prepared Gliclazide (GLZ)-Loaded SLNs |
The factorial ANOVA can be used to estimate the main effect of each factor and the interactions between the factors. The main effect is the difference between the mean response at one level of a particular factor and the mean response at the other level of the same factor, collapsing over the levels of all remaining factors.26 An interaction between 2 factors means that the effect that one factor has on the dependent variable depends on which level of the other factor is being administered, and if the interaction is significant, main effects in this situation are generally not meaningful, and the interpretation of the main effects is often incomplete or misleading.27
Measurement of Particle Size (PS), Polydispersity Index (PDI) and Zeta Potential (ZP)
The particle size of GLZ loaded SLNs ranged from 173 nm to 278 nm, showing homogenous dispersions of PDI 0.219 or less. Factorial ANOVA followed by post hoc test indicated that PS was mainly affected by the stabilizer concentration. It also showed a significant dis-ordinal interaction (p ≤ 0.05) between PLX-188 concentration and sonication time affecting the PS of the prepared SLNs. This indicated that the effect of each factor depends on the level of the other.
However, at both 10- and 20 min sonication, PS decreased by increasing the concentration of PLX-188 (from 100 to 150 mg). This may be due to insufficient coverage of nanoparticles by the surfactant at lower concentration which resulted in less stabilization of the particle dispersion. This suggested that higher concentrations of poloxamer were needed to prevent particle aggregation.28 Also, Poloxamer 188 was known to decrease the surface tension and stabilize newly developed surfaces during sonication and thus producing smaller particles.29 PS was then observed to stay unaffected by further increase in surfactant concentration to 200 mg. This indicated that optimal surface coverage of the particles by the surfactant was achieved at 150 mg PLX-188, where the particles were fully stabilized and a further increase in the surfactant concentration was not needed to further reduce the particle size.
Also, it was noticed that at 150 mg PLX-188, changes in the sonication time had insignificant effect on the particle size of the prepared SLNs (176.5 ±1.9 nm for 10F2, and 188.2 ± 23.7 nm for 20F2). This indicated that 150 mg PLX-188 provided sufficient stabilization for the nanoparticles.
As far as the polydispersity index (PDI) was concerned, all the formulations showed PDI less than 0.2 except for the short sonication time (10 min) at low surfactant level (100 mg) (0.219±0.0286) and long sonication time (20 min) at high surfactant level (200 mg) (0.214 ±0.032). Statistical analysis showed that the main effects of both sonication time and surfactant concentration were insignificant (p > 0.05); however, there was a significant dis-ordinal interaction between both of them where the response due to each factor depended on the level of the other factor.
Also, it was observed that at 150 mg PLX-188, changing the sonication time did not affect the PDI of the produced SLNs dispersion significantly (0.192 for 10 F2, and 0.179 for 20 F2).
From particle size and polydispersity index analysis, it could be concluded that 150 mg PLX-188 were enough to produce a homogenous SLNs dispersion with low PDI value (0.19 for 10 F2, and 0.18 for 20 F2) and small PS (188 nm for 10 F2, and 176 nm for 20 F2).
As far as zeta potential (ZP) was concerned, all the prepared gliclazide loaded SLNs showed absolute zeta potential of ≥30 mV, indicating a very good dispersion stability.
Measurement of Percentage Entrapment Efficiency (%EE)
All the prepared formulations showed high entrapment efficiency ranged from 72% to 82%. This was in agreement with previously published literature, where glyceryl behenate as the lipid matrix, had good drug entrapment ability, due to its complex nature and less-perfect orientation thus leaving more space for the drug to be loaded.30 Also, the long chain length of behenic acid in glyceryl behenate enhanced the intermolecular entrapment of the drug by inter-chain intercalation.31
Both surfactant concentration and the sonication time affected the amount of the drug entrapped inside the particles significantly (p ≤ 0.05). Also, a significant (p ≤ 0.05) interaction between the two factors was observed.
At 10 min sonication time, the increase in the PLX-188 concentration significantly (p ≤ 0.05) decreased %EE, while at 20 min the increase in the PLX-188 concentration led to insignificant (p > 0.05) initial increase in %EE (from ≈ 76% for 20 F1 to ≈ 77 for 20 F2), followed by a significant decrease in %EE (down to ≈ 73% for 20 F3). The decrease in %EE as the concentration of PLX-188 increased could be explained by partition phenomenon. High surfactant level in the external phase might increase the partition of drug. This increased partition was due to the increased solubilization of the drug in the external aqueous phase.30
Increasing the sonication time at 100 mg PLX-188 led to a decrease in the percentage of drug entrapped inside the particles. The decrease in %EE might be due to the effect of higher sheer forces that forced the drug out of the lipid particles and hindered its incorporation into the lipid matrix, or the formation of small particles which had larger surface area to volume ratio causing more drug loss into the aqueous phase.28 However, at higher PLX concentrations (150 and 200 mg) the decrease in %EE with increasing the sonication time was insignificant (p > 0.05).
Similar to the particle size and polydispersity index data, %EE at 150 mg PLX-188 was insignificantly (p > 0.05) affected by the changes in the sonication time.
From the previous results, and as a final conclusion from the factorial design experiment, the best formulations regarding PS, PDI and %EE would be 10 F2 (10 min sonication, 150 mg PLX-188), and 20 F2 (20 min sonication, 150 mg PLX-188), with average PS ≈ 182 nm, average PDI ≈ 0.185, and average %EE ≈ 77%. GLZ loaded 10 F2 was chosen as the optimum formulation since less time for sonication was supplied and thus less output energy was required to prepare SLNs dispersion.
Scanning Electron Microscopy (SEM)
SEM micrograph of 10 F2, Figure 1, demonstrated the formation of homogeneous nano-sized spherical particles with almost smooth surface characteristic for glyceryl behenate SLNs, as reported by Bhalekar et al.32 The spherical and smooth particles are often favored for the uptake through the cells and to the lymphatic tissue.
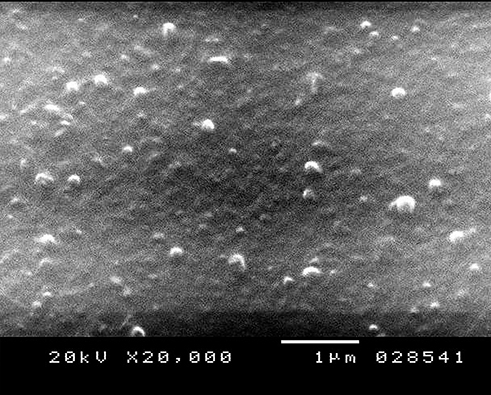 |
Figure 1 Scanning electron microscopy (SEM) micrograph of GLZ-loaded SLNs formulation 10 F2. Abbreviations: SEM, scanning electron microscope; GLZ, gliclazide; SLNs, solid lipid nanoparticles. |
Freeze Drying of the Chosen Optimum SLNs Formulation
The effect of freeze drying of the selected GLZ loaded SLNs (10F2) with different concentrations of cryo-protectant trehalose di-hydrate was studied with respect to PS and PDI after redispersion of the dried samples in 0.45µm filtered distilled water. Results are recorded in Figure 2.
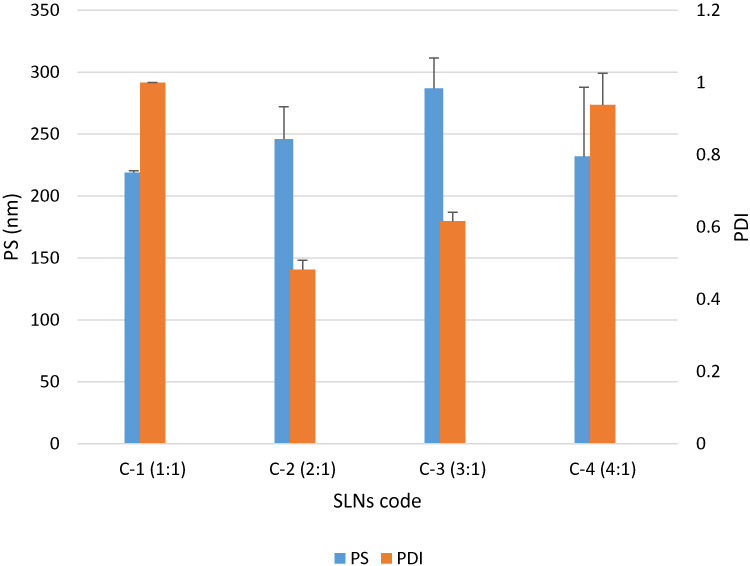 |
Figure 2 Particle size (PS) and polydispersity index (PDI) for the lyophilized GLZ-loaded SLNs (10 F2) after reconstitution. Abbreviations: PS, particle size; PDI, polydispersity index; GLZ, gliclazide; SLNs, solid lipid nanoparticles. |
At all evaluated ratios of glyceryl behenate: trehalose, lyophilized SLNs showed an increase in the particle size and a decrease in the homogeneity of the dispersion, where before lyophilization 10 F2 showed PS of 176.5 ±1.9 nm and PDI of 0.192 ±0.00265. These values increased massively after lyophilization where the lowest PDI (≈0.4) was obtained when using cryo-protectant in a concentration double that of the lipid, while the size increased to 245.9 ± 26.2 nm.
However, such results have been previously reported, where freeze-dried SLNs usually resulted in larger particles with wider size distribution even in the presence of cryo-protectants, probably due to the presence of aggregates between the nanoparticles, as a result of the conditions of freeze-drying process and the removal of water which probably promoted the aggregation among SLNs.33
It was found that reconstituted particles containing a 1:2 (lipid: cryoprotectant) had the best PDI; 0.482, and particle size (246 nm). Consequently, GLZ loaded SLNs 10 F2 lyophilized with cryo-protectant ratio of 1:2 (lipid: cryoprotectant) was chosen for further studies.
HPLC Assay of Gliclazide
The retention time of gliclazide was 7.6 mins. The peak appeared well separated, symmetrical, with no fronting or tailing. Linearity was established (r2 = 0.9998) over concentration range of 25–400 µg/mL. Accuracy was depicted by % GLZ recovery and ranged from 97.50 to 106.80. Inter- and intra-day precision were presented by CV %, which ranged from 0.350 to 0.809. Reproducibility of the calibration curve was performed in 3 different days. CV % values ranged from 0.15 to 0.92%.
In-vitro Release Study of Gliclazide
It is worth mentioning that gliclazide degraded after 10 hrs in the gastric pH (1.2) and showed to be highly stable at the alkaline pH values as reported by Bansal et al.34 The drug, however, stays in the stomach for only 2 h; a duration of time during which the drug proved to be stable.35–38 Consequently, phosphate buffer pH 7.4 was chosen as dissolution medium for gliclazide as documented in BP.39
The optimum lyophilized GLZ loaded formulation (10 F2) was compared to the raw GLZ powder, and GLZ commercial tablets (immediate release (IR)). Tablet disintegration test was done and revealed that GLZ commercial tablets did not disintegrate over 2 h in phosphate buffer pH 7.4 under the applied experimental conditions, so in-vitro dissolution test was done using both the intact and ground tablets. In-vitro release profiles are shown in Figure 3.
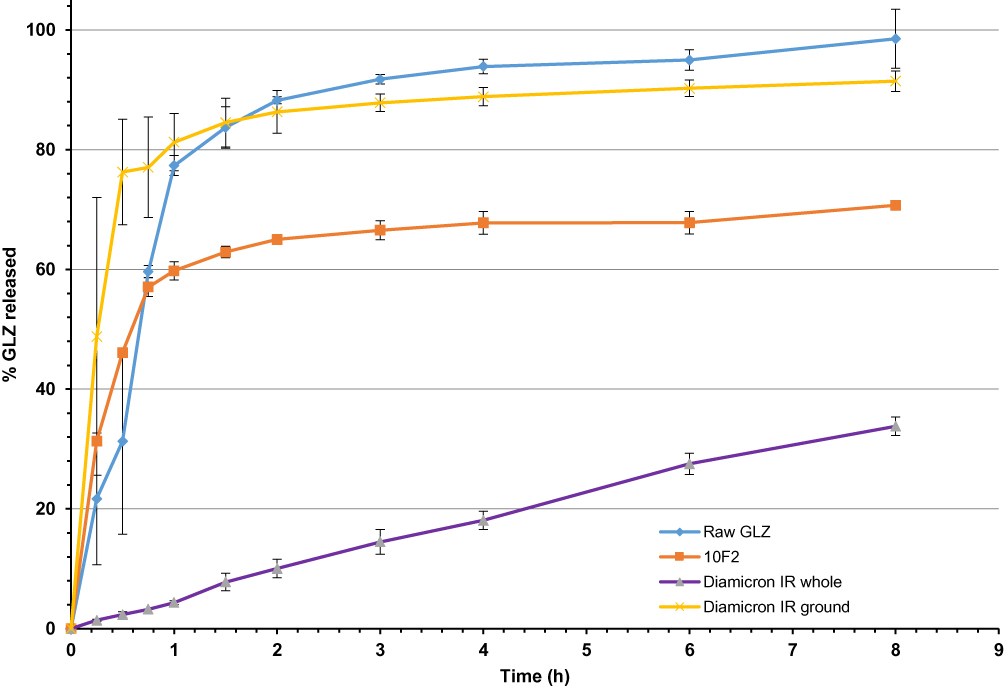 |
Figure 3 In-vitro release profile of GLZ from GLZ raw powder, 10 F2 formulation and the GLZ commercial IR whole and ground tablets, in phosphate buffer pH=7.4 for 8 hrs (n=3). Abbreviations: GLZ, gliclazide; SLN, solid lipid nanoparticles; IR, immediate release. |
For raw GLZ powder, the dissolution of GLZ was almost immediate with about 90% drug released within the first 2 h. This can be attributed to the high solubility of GLZ in phosphate buffer pH=7.4, which was supported by previously published data by Grbic et al,4 where the dissolution process at pH 7.4 was expected to be fast and complete giving a very rapid dissolution profile.
GLZ commercial whole IR tablets, however, showed very slow dissolution rate, where after 8 h, only about 33% of the drug was released. GLZ commercial IR tablets are composed of GLZ and excipients: lactose (diluent), magnesium stearate (lubricant), talc (glidant), corn starch, and pre-gelatinized corn starch (filler and binder). The slow dissolution rate might be due to the high binding strength of the used starch which prevented the disintegration of the tablets in the medium, and thus the dissolution medium should first diffuse into the tablet matrix, dissolve the drug and then the solubilized drug would diffuse out of the tablets.
On the other hand, once the tablets were ground, the drug was free to be in direct contact with the dissolution medium, and it acted similarly to raw GLZ powder, where about 86% of the drug was released during the first 2 h of the test.
GLZ loaded formulation 10 F2 showed an initial burst release of the drug of around 55% released in the first hour, followed by a prolonged release phase, where after 8 h, only 70% of GLZ was released. The initial burst release of the drug could be due to drug-enriched shell of SLNs together with the solubilizing effect of the surfactant surrounding the nanoparticles. On the other hand, prolonged release of drug could be due to increased diffusional distance and hindering effects by the surrounding solid lipid shell, as reported by Kushwaha et al.28 The prolonged release effect could be also attributed to the hydrophobic long chain fatty acids of glyceryl behenate that retain the lipophilic drug resulting in more prolonged release effect.40 The drug enriched shell SLNs model could be explained by the effect of temperature and surfactant on the drug partitioning between the lipid core and the aqueous phase. The aqueous solubility of the drug – and, consequently, its amount in the aqueous phase – increased with increasing temperature of the aqueous phase during the preparation of SLNs and the presence of surfactant. On cooling, the solubility of the drug in the aqueous phase decreased forcing the drug to partition to the lipid phase and, consequently, to be entrapped in the core of the SLNs. However, when the formed solid lipid core was no more reachable by the drug, the later concentrated in the outer shells of the produced SLNs and/or on their surfaces leading to a burst effect.41 GLZ loaded SLNs managed to enhance the dissolution rate when compared to GLZ commercial IR whole tablets, while it prolonged the drug release when compared to raw GLZ powder.
Different models have been applied for elucidation of drug release mechanism and characterization of drug release profile of different formulations under test. Drug release data of GLZ loaded SLNs were fitted into Weibull’s model with high R2 value of 0.9803 and Korsmeyer-Peppas model with R2 = 0.9804. The release was governed by Fickian diffusion mechanism, where the drug diffused through the lipid matrix to the solution due to the concentration gradient.
Differential Scanning Calorimetry (DSC)
Raw GLZ powder thermogram showed a sharp endothermic peak corresponding to its melting point at 172.3°C. However, this peak disappeared in both the physical mixture and SLNs as shown in Figure 4. The disappearance of the peak in SLNs could be attributed to the homogenous dispersion of GLZ in an amorphous state incorporated inside SLNs.41,42 For the physical mixture, the reason for drug endotherm disappearance could be that the crystalline state drug became amorphous due to the heat generated by the mixing force during preparation of the physical mixture.
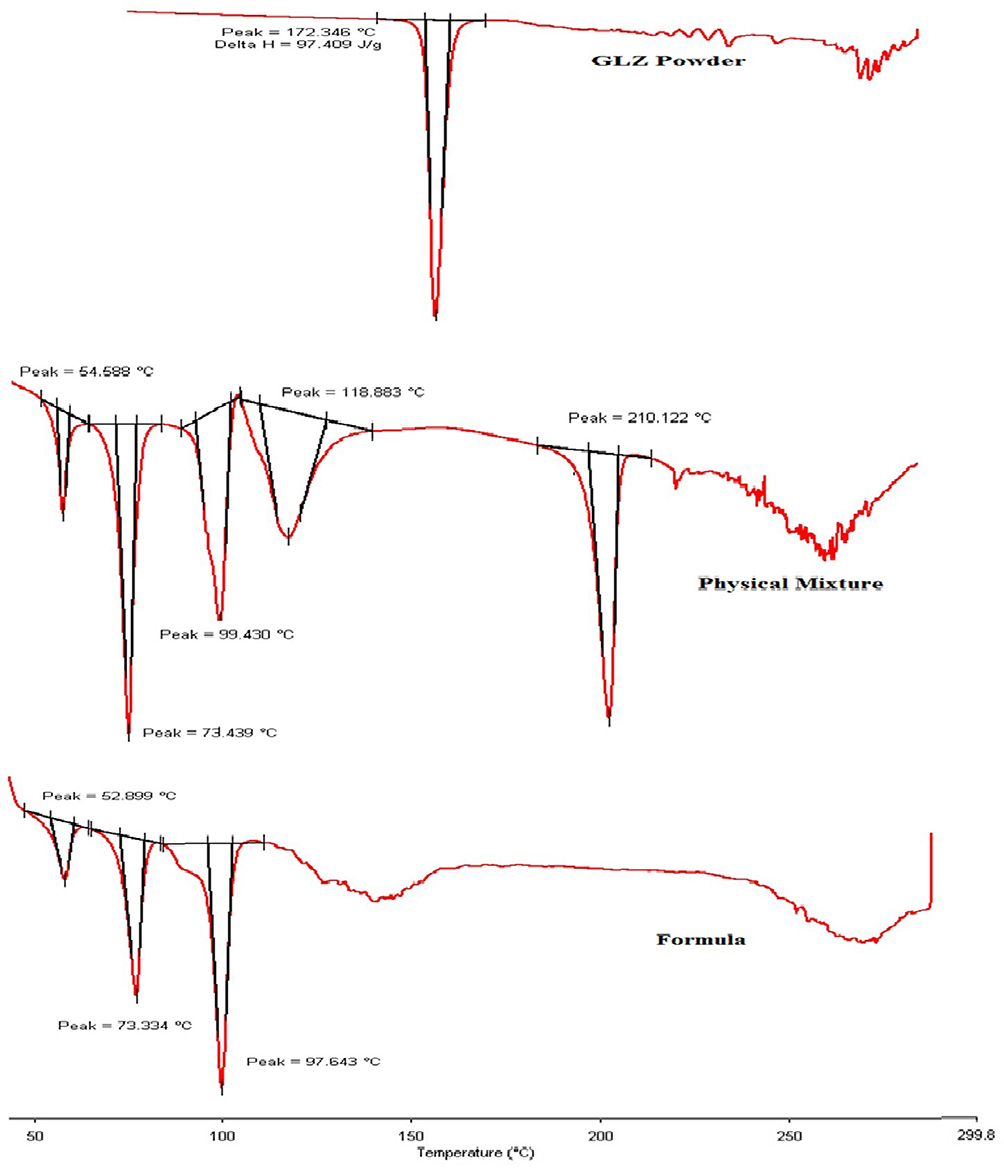 |
Figure 4 DSC thermograms of GLZ powder, physical mixture of glyceryl behenate, GLZ powder, poloxamer 188 and trehalose di-hydrate, and GLZ-loaded SLNs (10 F2). Abbreviation: GLZ, gliclazide. |
Peaks for other ingredients appeared in the physical mixture and GLZ loaded SLNs; peaks around 100, 120 and 210°C are characteristic peaks of trehalose di hydrate. While peaks at 73°C and 53°C are characteristic peaks of glyceryl behenate and poloxamer 188, respectively.
Fourier Transform Infrared Spectroscopy (FT-IR)
Fourier-transform infrared (FT-IR) spectroscopy was used to characterize possible interactions between the drug and other components of SLNs in solid state, as shown in Figures 5 and 6.
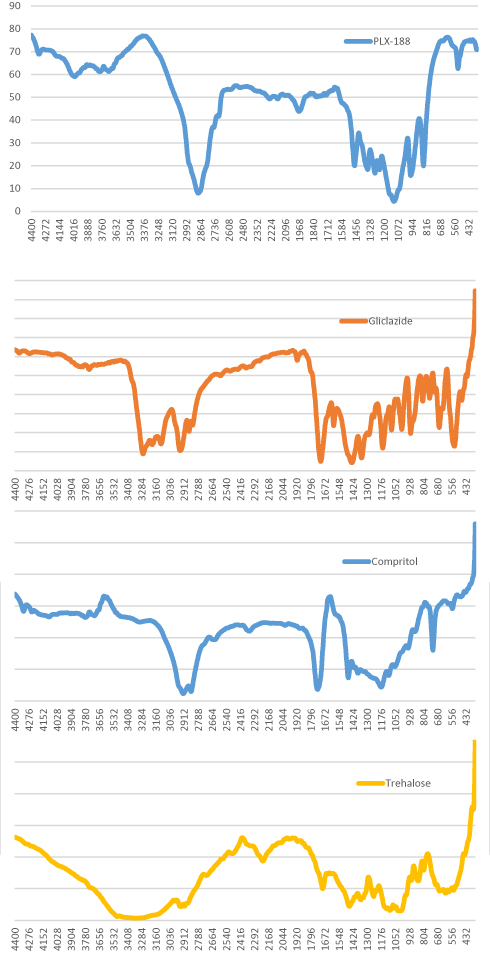 |
Figure 5 FT-IR spectra of Compritol®888 ATO, gliclazide (GLZ), poloxamer 188 (PLX-188) and trehalose. |
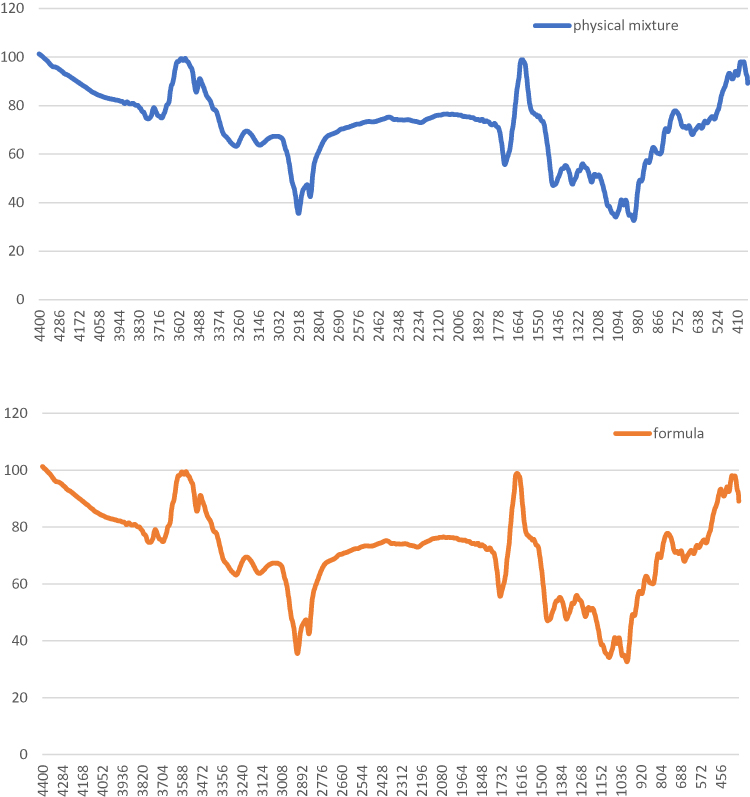 |
Figure 6 FT-IR spectra of the physical mixture of the components, and the optimum formula (10 F2). |
Glyceryl behenate showed IR absorption frequency at 1737 cm−1, characteristic of C=O stretching, similar to the findings reported by Dandagi et al.43 Also, C-H stretching due to alkanes appeared as previously reported by Jose et al44 at 2918 cm−1.
The IR spectrum of gliclazide was characterized by the absorption of carbonyl (C=O) and NH group at 1708 cm−1 and 3271cm−1, respectively. The sulfonyl group (S=O) bands were located at 1349 cm−1 and 1163 cm−1, for the asymmetrical and symmetrical vibration, respectively. Similar spectra for gliclazide were reported by Febriyenti et al.45
The physical mixture showed all the characteristic peaks of the components. Thus, it is concluded that there was no significant physicochemical interaction between the different components in the physical mixture.
The FT-IR spectrum of SLNs (10 F2) showed the absence of the characteristic O-H stretching at 3499 cm−1 and 3305 cm−1 of trehalose, together with the decrease in C=O peak of both glyceryl behenate and gliclazide, and decrease in S=O vibrational bands, and N-H band of gliclazide. This could be attributed to strong hydrogen bonding interactions between the functional groups of different components in the prepared SLNs. An intermolecular hydrogen bond was expected to occur between the hydrogen atom of the NH group of gliclazide and one of the lone pair electrons of C=O group of glyceryl behenate and between C=O group, and/or S=O group of gliclazide and one of the hydrogen atoms of poloxamer 188, and trehalose di-hydrate. Similar interactions were detected by Biswal et al46 when formulating solid dispersions of gliclazide with components of multiple OH groups. However, absence of any additional peaks in SLNs indicated that no further interaction between gliclazide and other ingredients in SLNs was detectable.47
In-vivo Testing of the Optimum SLN Formulation
Pharmacokinetics Study
The drug concentration in the collected plasma samples at different time intervals was determined by HPLC assay. The calibration curve of GLZ was linear over the range of 0.5–40 µg/mL with correlation coefficient (R2) = 0.9999. Accuracy was depicted by % GLZ recovery and ranged from 92.8–116.5%. Intra-day and inter-day precision were presented by CV % which ranged from 2.02–9.91%. PHY and GLZ were well separated with retention times of around 3.2 and 7.6 min, respectively.
The mean plasma drug concentration–time profile was plotted in Figure 7. The essential pharmacokinetic parameters (Cmax, Tmax, AUC 0-t, AUC 0-∞) of GLZ resulted after the oral administration of lyophilized 10F2 and raw GLZ were calculated and summarized in Table 2. Maximum concentration (Cmax) achieved after administration of GLZ powder was 2.30 ± 1.27 μg/mL compared to 14.50 ± 3.51 μg/mL for lyophilized GLZ loaded SLNs (10 F2). This was about 6.5-fold increase in maximum plasma concentration, which indicated a marked increase in the amount of drug absorption after oral administration of GLZ loaded SLNs. Statistical analysis showed a highly significant (p ≤ 0.001) increase in Cmax after the administration of GLZ loaded SLNs when compared to raw GLZ powder.
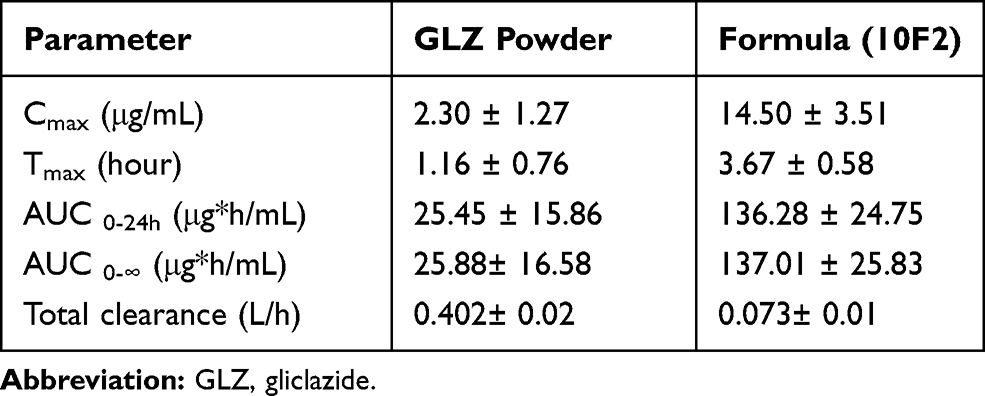 |
Table 2 Pharmacokinetic Parameters of the Lyophilized Formula (10F2) and Raw GLZ Powder After Oral Administration to Rats in a Dose Corresponding to 10 mg/kg GLZ. Data are Expressed as Mean ±SD |
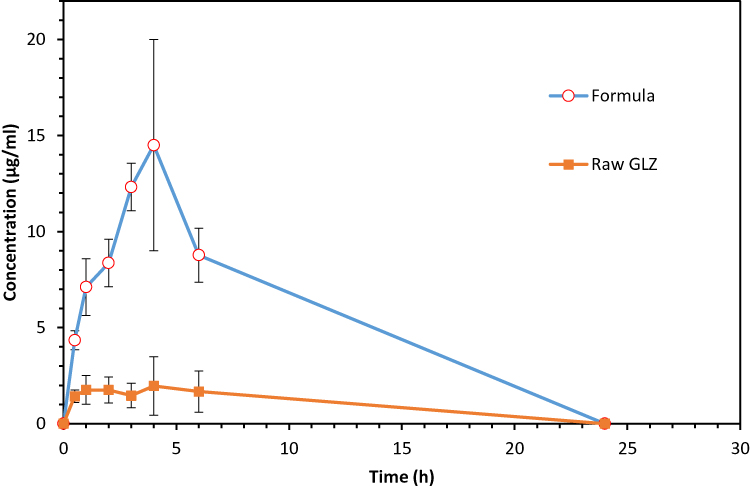 |
Figure 7 Mean plasma concentration-time profile of GLZ after oral administration of lyophilized 10 F2 and raw GLZ to 5 rats in a dose corresponding to 10 mg/kg. Abbreviation: GLZ, gliclazide. |
AUC0-t and AUC0-∞ are a measure for the total drug exposure and are considered as a measurement for bioavailability.48 GLZ loaded SLNs showed an AUC0-∞ ≈ 137 μg h/mL, while raw GLZ had an AUC0-∞ ≈ 26 μg h/mL, giving a 5.4-fold increase in the drug bioavailability, which is a highly significant increase.
Depending on the previous results, it could be noticed that GLZ loaded SLNs successfully enhanced the oral bioavailability of GLZ by approximately 5 folds when compared to raw GLZ powder. Glyceryl-behenate-based SLNs could noticeably improve the oral bioavailability of different drugs as reported by Bhalekar et al32 and Negi et al.49 This increase in bioavailability indicated higher GI uptake of GLZ loaded SLNs in comparison to raw GLZ powder. This can be explained by the ability of SLNs to reach the oral lymphatic region and undergo lymphatic uptake bypassing the hepatic first pass metabolism and reach the blood circulation at high amounts, thus increasing Cmax and AUC.32,49
Another interesting point of view was the ability of glyceryl behenate SLNs coated with PLX-188 to protect SLNs from the action of lipases in GIT, where the use of the sterically stabilizing PLX-188 could prevent the degradation of SLNs.50 This was attributed to the lack of anchoring of the lipase to the particle surface prerequisite for enzymatic degradation. Protection of the particles against enzymatic degradation is important as it allows the lipid particles to be taken up by the lymphatic uptake together with protecting the incorporated drug from GIT conditions.50
Increasing the bioavailability could be also attributed to several other reasons. Mainly, the nanoscale dimensions of the particles combined with the almost molecular dispersion of the drug molecules into the lipid carrier and the presence of surfactants, which strongly increased the drug gastrointestinal solubilization, and the in vivo absorption.51 Another interpretation was that due to their small particle size, SLNs might exhibit bio-adhesion to the gastrointestinal tract wall or enter the inter-villar spaces thus increasing their residence time in the gastrointestinal tract. This increased adhesion would result in enhanced bioavailability.51,52
Time to reach maximum plasma concentration (Tmax) is a measurement for the rate of drug absorption. Raw GLZ powder showed erratic absorption, with high inter-subject variation regarding Tmax ranging from as low as half an hour, up to 4 h. Such behavior was already reported, and could be related to its early dissolution in the stomach leading to more variability in the absorption in the intestine.53
Moreover, raw GLZ showed bimodal release, with 2 peaks one at 1 h and the other at 4 hrs, which was also reported by Davis et al53 who attributed it to that GLZ underwent enterohepatic circulation, together with its absorption from a second more distal site in the GIT.
Statistical analysis was used to compare Tmax of GLZ loaded SLNs to raw GLZ powder. GLZ loaded SLNs showed a prolonged release with Tmax ≈ 3.6 h, which was a significant difference (P ≤ 0.05) when compared with raw GLZ powder (Tmax = 1.1 h).
Increasing Tmax in SLNs formula indicated prolonging the release of the drug. The apparent total clearance of the drug from plasma after oral administration of the nanoparticles was 0.073 L/h, while that of raw gliclazide powder was 0.402 L/h. Therefore, the total clearance of gliclazide from the body decreased by 5.5-fold after administration of nanoparticles. These results confirmed the slow release pattern of drug from the prepared nanoparticles that resulting in prolonged circulation time and decreased drug clearance.
Prolonged drug release in SLNs had several explanations. First, the mobility of incorporated drugs was reduced due to the solid state of the particles at room temperature. Second, slow release of drug could be achieved when the drug was homogenously dispersed in the lipid matrix. Moreover, there was an inverse relationship between drug release and the partition coefficient of the drug, if the partition coefficient of a drug was not high (having high water solubility), partitioning of drug to the aqueous phase became rapid and a rapid release could be observed (gliclazide has a partition coefficient of 2.1).54 Such results were in agreement with the in-vitro release data that showed a prolonged release behavior for drug release from SLNs.
Pharmacodynamics Study
Combining high-fat diet with a small dose of streptozotocin (STZ) was reported in literature as a way to induce type II diabetes mellitus.55
After 3 weeks of dietary manipulation of the 2 groups; control which received normal chow diet, and the high-fat fed group, both cholesterol and triglycerides increased significantly in the high-fat fed group which indicated the successful induction of the metabolic syndrome, predisposing insulin resistance and type II diabetes, as reported by Veerapur et al.56 STZ was then injected to the high-fat fed rats. One week after the injection, rats with blood sugar level ≥250 mg/dL were assigned for the pharmacodynamics study.
Results obtained from measuring blood sugar level (BSL) of the tested groups (raw GLZ powder and GLZ loaded SLNs) were recorded as percentage decrease in BSL, shown in Figure 8, and the areas under the curve (AUC) of the percentage decrease in BSL were calculated and statistically compared.
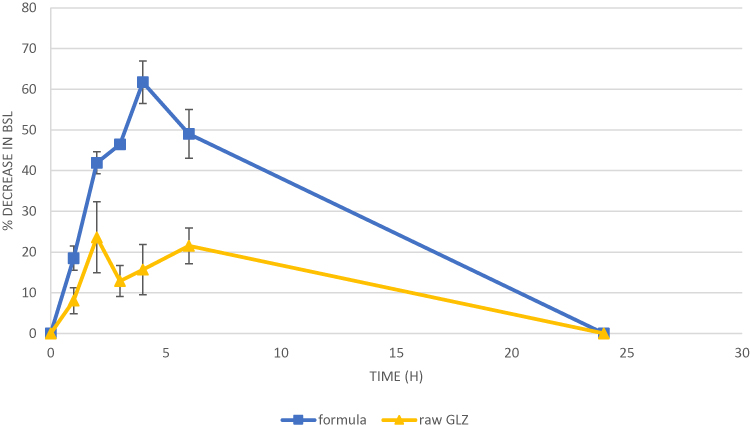 |
Figure 8 Mean percentage decrease in (BSL) after oral administration of lyophilized 10F2 (formula), and raw GLZ to 5 rats in a dose corresponding to 10 mg/kg. Abbreviations: BSL, blood sugar level; GLZ, gliclazide. |
The pharmacodynamics results confirmed the enhancement of the oral bioavailability of GLZ when administered as SLNs and complemented the pharmacokinetics study results. Also, raw GLZ showed a bi-modal action corresponding to its blood concentration peaks at 2 and 6 h. GLZ loaded SLNs showed the highest percentage decrease at 4 h. AUC of percentage decrease in BSL represents the body response after the dose administration. AUC in case of GLZ loaded SLNs was increased by about 2.5 times when compared to raw GLZ powder.
Glyceryl behenate-based SLNs with poloxamer coating seemed to be effectively absorbed through the lymphatic system, protected the incorporated drug against metabolism and prolonged the circulation of drug in the blood by acting as a reservoir.57
In-vivo Subacute Toxicity Study
Nanoparticle toxicity refers to the ability of the particles to interfere with the physiology of the body and damage the normal structure of organs and tissues of humans and animals.58
It is generally believed that lipids are biocompatible. However, it is known that materials structured in nanoscale might have their intrinsic physicochemical properties modified. Glyceryl behenate is a physiological lipid that has a GRAS (generally regarded as safe) status; however, it was reported by Weyhers et al59 that high doses of glyceryl behenate can lead to accumulation in the liver, and subsequent pathological alterations, despite being slight and reversible. Subacute toxicity tests evaluate the probability of the drug together with the lipid in the form of SLNs to generate collective hematological, biochemical, and physiological changes following multiple administrations.60
During the sub-acute toxicity study, no mortality or adverse reactions in the condition of the eye, nose and motor activity were observed till the end of the study. Animals’ nutritional state was assessed. Body weights showed an increase over the period of 2 weeks in the control, blank SLNs and GLZ loaded SLNs-treated groups. The amounts of daily food (g/day) and water (mL/day) consumption were also determined. No significant variations (p > 0.05) between the three groups were found concerning food intake, water intake and the percentage change in body weight. The food consumption of the different groups followed a similar pattern indicating normal metabolism of the animals. These data indicated that food intake and utilization of proteins and other nutrients were not affected by nanoparticles intake precluding the potential toxicity of the GLZ loaded SLNs on the GIT.61
After animals were sacrificed and necropsied, no significant change in texture and shape of the animals’ organs (stomach, intestine, liver, and kidneys) was noticed. Those organs were chosen since the stomach and intestine are the main absorption sites of the drug, the liver is the main organ of metabolism for the drug and the site where accumulation of glyceryl behenate may occur,59 and the kidney is the excretory organ for the drug and its metabolites. Different organs were weighed and percentage organ weight relative to body weight was calculated. Statistical analysis showed no significant difference between the three groups (p >0.05) concerning the organ weights of the liver, kidneys and stomach.
To further confirm or exclude the SLNs potential systemic toxicity, biochemical parameters of liver and kidney functions were assessed in the control and treated groups, as shown in Table 3. Liver functions (ALT and AST) and kidney functions (urea and creatinine) presented no significant difference (p>0.05) between the three groups; hence, no in vivo toxicity to the liver and kidneys was detected. Also, plasma cholesterol and triglycerides showed no significant difference (p>0.05) between the three groups, thus showing that administration of glyceryl behenate SLNs for 14 consecutive days did not affect the blood lipid profile.
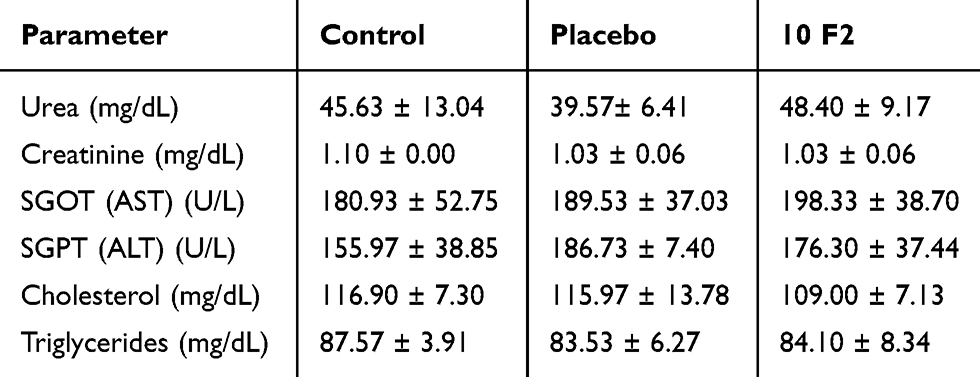 |
Table 3 Liver Function and Kidney Function Parameters, Plasma Cholesterol and Triglycerides of the Three Experimental Groups of Rats (n=5) |
Repetitive administration of blank and GLZ loaded SLNs for 14 consecutive days did not alter the biochemical parameters of the rats, indicating normal metabolic process of the animals.62 These results showed that GLZ loaded SLNs did not cause cumulative and latent biochemical changes following multiple administrations, even at a high dose ≈ 15-20-fold human daily exposure. The results indicated the oral safety of the developed formulation for the study period at least.
Histological examination was performed on tissues that have been fixed after sacrifice of the exposed animal, and changes in the tissue and cell morphology were assessed using light microscopy. Histopathological study was of great importance because Weyhers et al59 reported the accumulation of glyceryl behenate nanoparticles in the liver causing cell toxicity signs. Figures 9–12 show the photomicrographs of sections of rats’ stomach, small intestine, kidney and liver, respectively.
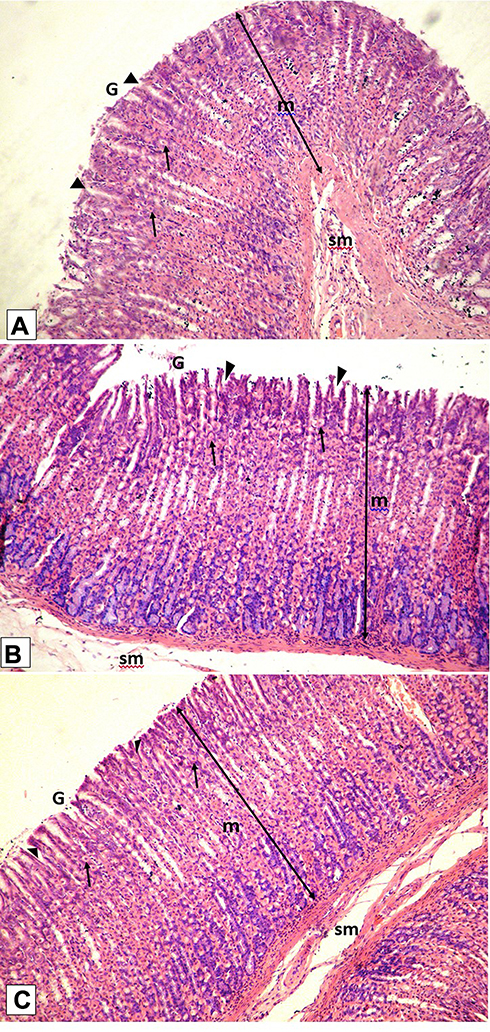





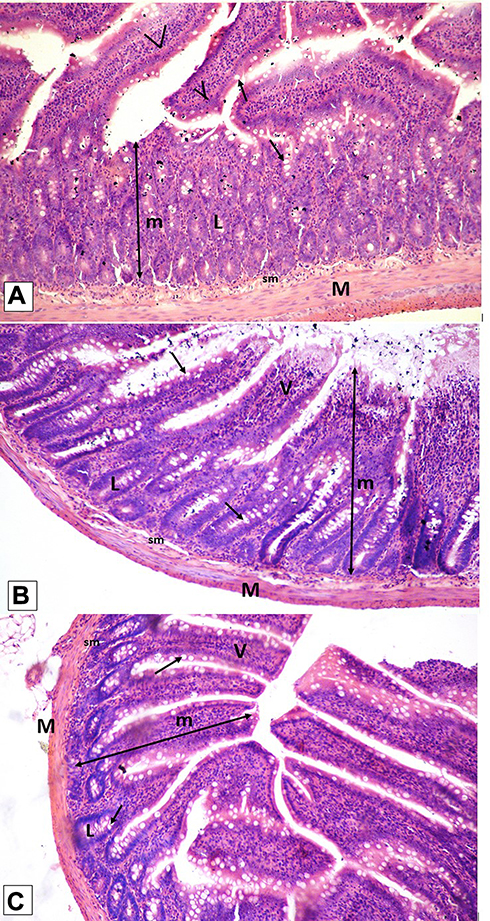

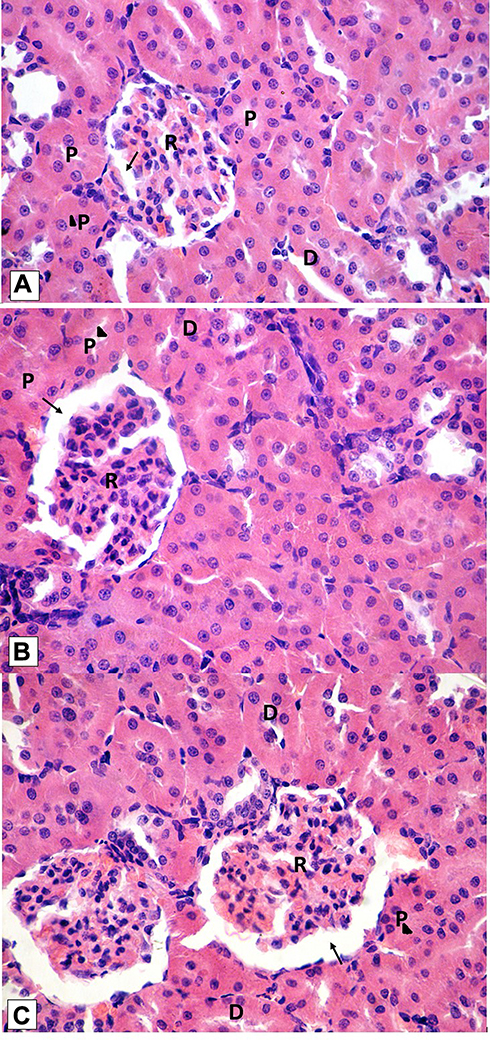

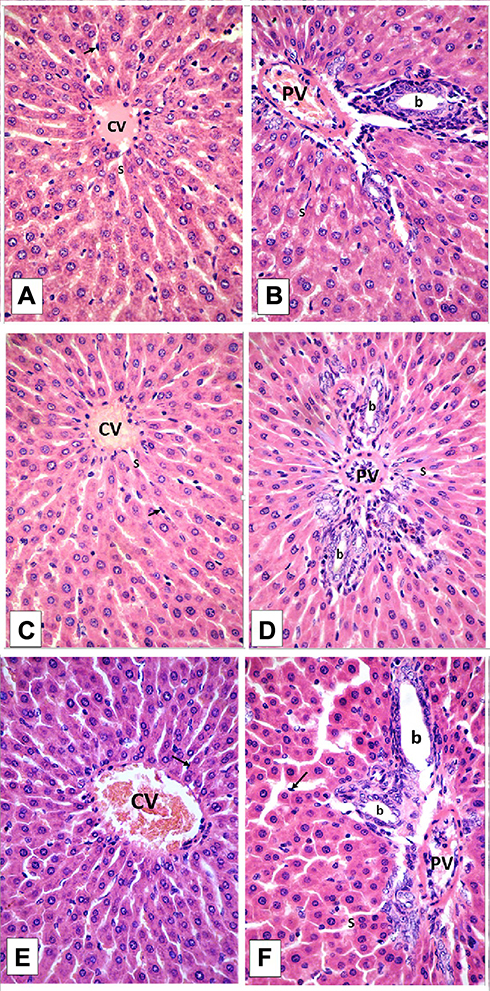

Examination of the control rats revealed the classical histological structures of the examined organs.
Examination of all the treated rats in both placebo and formula treated groups reflected nearly control image. Mucosal ulceration, cytoplasmic vacuolation, nuclear pyknosis, and cellular degeneration were not encountered in any of the tested samples denoting absence of cytotoxic effect.
Such results contradicted those found by Weyhers et al59 where histological analysis of cuts obtained from the liver after repetitive IV injection of glyceryl behenate SLNs showed pathological results. Hepatic tissue had infiltrations of mono-nuclear cells and showed Kupffer cell hyperplasia. In addition, liver cell necrosis had been observed. On the contrary, in this work and after 14-day repetitive oral administration of glyceryl behenate SLNs, no signs of toxicity were observed either on the liver or any other studied organ, indicating the safety of those nanoparticles after repetitive oral administration.
Conclusion
GLZ loaded SLNs were successfully prepared by ultra-sonication technique. These particles enhanced the oral bioavailability of GLZ, together with giving better glycemic control over a longer period of time when tested on high-fat diet streptozotocin-induced diabetic rats. This result was promising in the aspect of better control of diabetes and enhanced patient compliance. Also, upon repetitive oral administration, GLZ loaded SLNs proved to be safe at doses equivalent to 15–20 times the human dose.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Baek J-S, Cho C-W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur J Pharm Biopharm. 2017;117:132–140. doi:10.1016/j.ejpb.2017.04.013
2. Gupta S, Kesarla R, Chotai N, Misra A, Omri A. Systematic approach for the formulation and optimization of solid lipid nanoparticles of efavirenz by high pressure homogenization using design of experiments for brain targeting and enhanced bioavailability. Biomed Res Int. 2017;2017:1–18.
3. Sarkar A, Tiwari A. Pharmacological and pharmaceutical profile of gliclazide: a review. J Appl Pharm Sci. 2011;1(9):11–19.
4. Grbic S, Parojcic J, Ibric S, Djuric Z. In vitro-in vivo correlation for gliclazide immediate-release tablets based on mechanistic absorption simulation. AAPS PharmSciTech. 2011;12(1):165–171. doi:10.1208/s12249-010-9573-y
5. Shaik M, Shaik S, Kilari EK. Population pharmacokinetics of gliclazide in normal and diabetic rabbits. Biopharm Drug Dispos. 2018;39(5):265–274. doi:10.1002/bdd.v39.5
6. Pop DI, Oroian M, Bhardwaj S, et al. Bioequivalence of two formulations of gliclazide in a randomized crossover study in healthy caucasian subjects under fasting conditions. Clin Pharmacol Drug Dev. 2019;8(1):16–21. doi:10.1002/cpdd.445
7. Morrissey H, Ball P, Abdel-Aziz F, Nugroho B. Formulation of gliclazide using self-nano-emulsification technology. Eur J Biomed Pharm Sci. 2018;5(5):29–39.
8. Deshmukh SR, Bakhle SS, Upadhye KP, Dixit GR. Formulation and evaluation of solid self-emulsifying drug delivery system of Gliclazide. Int J Pharm Pharm Sci. 2016;8(11):144–151. doi:10.22159/ijpps.2016v8i11.14104
9. Lo YK, Chen CJ, Tsai TR, Cham TM. Comparison of the solubility and dissolution rate between gliclazide solid complex and its nanospheres. Drug Dev Ind Pharm. 2007;33(3):301–309. doi:10.1080/03639040600920622
10. Wankhade V, Tapar K, Pande S, Bobade N. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for Gliclazide. Der Pharm Lett. 2010;2(4):132–143.
11. Solunke RS, Borge UR, Murthy K, Deshmukh MT, Shete RV. Formulation and evaluation of gliclazide nanosponges. Int J Appl Pharm. 2019;11(6):181–189. doi:10.22159/ijap.2019v11i6.35006
12. Averineni RK, Shavi GV, Ranjan OP, et al. Formulation of gliclazide encapsulated chitosan nanoparticles: in vitro and in vivo evaluation. In: Tiddy GJT, Tan R. Nanoformulation. Cambridge, UK: RCS Publishing; 2012:77–85.
13. Ravouru N, Venna RSA, Penjuri SCB, Damineni S, Kotakadi VS, Poreddy SR. Fabrication and characterization of gliclazide nanocrystals. Adv Pharm Bull. 2018;8(3):419–427. doi:10.15171/apb.2018.049
14. Dhome AG, Deshkar SS, Shirolkar SV. Gliclazide solid lipid nanoparticles: formulation, optimization and in vitro characterization. Pharm Reson. 2018;1(1):8–16.
15. Rouini M-R, Mohajer A, Tahami M-H. A simple and sensitive HPLC method for determination of gliclazide in human serum. J Chromatogr B. 2003;785(2):383–386. doi:10.1016/S1570-0232(02)00951-0
16. Khan A, Mudassir J, Akhtar S, Murugaiyah V, Darwis Y. Freeze-dried lopinavir-loaded nanostructured lipid carriers for enhanced cellular uptake and bioavailability: statistical optimization, in vitro and in vivo evaluations. Pharmaceutics. 2019;11(2):97–115. doi:10.3390/pharmaceutics11020097
17. Zhang Y, Huo M, Zhou J, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010;12(3):263–271. doi:10.1208/s12248-010-9185-1
18. Khalil SS, Ali HA, Al-Saadawy HA, El-Dawy K. Combination of gliclazide drug and lupin seeds powder alleviate hyperglycemia on induced-diabetic rats receiving high-fat high fructose/sucrose diet. Slov Vet Res. 2018;55(20–Suppl):273–281.
19. Nipun TS, Ashraful Islam SM. SEDDS of gliclazide: preparation and characterization by in-vitro, ex-vivo and in-vivo techniques. Saudi Pharm J. 2014;22(4):343–348. doi:10.1016/j.jsps.2013.06.001
20. Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. doi:10.1016/j.cmpb.2010.01.007
21. Woods SC, Seeley RJ, Rushing PA, D’Alessio D, Tso P. A controlled high-fat diet induces an obese syndrome in rats. J Nutr. 2003;133(4):1081–1087. doi:10.1093/jn/133.4.1081
22. Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005;52(4):313–320. doi:10.1016/j.phrs.2005.05.004
23. Menshawy M, Sharaf W, Esmail RE, Farrag AH. Histopathological and histochemical effects of nicotine on the liver and kidney of adult male rats. J Arab Soc Med Res. 2019;14(1):7–13. doi:10.4103/jasmr.jasmr_39_18
24. Banerjee S, Pillai J. Solid lipid matrix mediated nanoarchitectonics for improved oral bioavailability of drugs. Expert Opin Drug Metab Toxicol. 2019;15(6):499–515. doi:10.1080/17425255.2019.1621289
25. Ekambaram P, Sathali AAH, Priyanka K. Solid lipid nanoparticles: a review. Sci Rev Chem Commun. 2012;2(1):80–102.
26. Collins LM, Dziak JJ, Kugler KC, Trail JB. Factorial experiments: efficient tools for evaluation of intervention components. Am J Prev Med. 2014;47(4):498–504. doi:10.1016/j.amepre.2014.06.021
27. Smith CE, Cribbie R. Factorial ANOVA with unbalanced data: a fresh look at the types of sums of squares. J Data Sci. 2014;12:385–404.
28. Kushwaha AK, Vuddanda PR, Karunanidhi P, Singh SK, Singh S. Development and evaluation of solid lipid nanoparticles of raloxifene hydrochloride for enhanced bioavailability. Biomed Res Int. 2013;2013:1–9. doi:10.1155/2013/584549
29. Gade S, Patel KK, Gupta C, et al. An ex vivo evaluation of moxifloxacin nanostructured lipid carrier enriched in situ gel for transcorneal permeation on goat cornea. J Pharm Sci. 2019;108(9):2905–2916. doi:10.1016/j.xphs.2019.04.005
30. Khalil RM, Abd El-bary A, Kassem MA, Ghorab MM, Ahmed MB Solid lipid nanoparticles for topical delivery of meloxicam: development and in vitro characterization. In:
31. Aburahma MH, Badr-Eldin SM. Compritol 888 ATO: a multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin Drug Deliv. 2014;11(12):1865–1883. doi:10.1517/17425247.2014.935335
32. Bhalekar MR, Upadhaya PG, Madgulkar AR. Fabrication and efficacy evaluation of chloroquine nanoparticles in CFA-induced arthritic rats using TNF-α ELISA. Eur J Pharm Sci. 2016;84:1–8. doi:10.1016/j.ejps.2016.01.009
33. Singh B, Diwan A. Effect of process parameters on formulation of solid lipid nanoparticles of protease inhibitor, Atazanavir. Pharma Res. 2005;7(2):1–15.
34. Bansal G, Singh M, Jindal KC. Forced degradation study on gliclazide and application of validated stability-indicating HPLC-UV method in stability testing of gliclazide tablets. Chromatographia. 2007;66(9–10):751–755. doi:10.1365/s10337-007-0394-4
35. Patil MP, Gaikwad NJ. Characterization of gliclazide-polyethylene glycol solid dispersion and its effect on dissolution. Brazilian J Pharm Sci. 2011;47(1):161–166.
36. El-Sabawi D, Hamdan II. Improvement of dissolution rate of gliclazide through sodium salt formation. Dissolut Technol. 2014;2:49–55. doi:10.14227/DT210414P49
37. Barzegar-Jalali M, Valizadeh H, Shadbad M-RS, et al. Cogrinding as an approach to enhance dissolution rate of a poorly water-soluble drug (gliclazide). Powder Technol. 2010;197(3):150–158. doi:10.1016/j.powtec.2009.09.008
38. Lakka NS, Goswami N. Solubility and dissolution profile studies of gliclazide in pharmaceutical formulations by RP-HPLC. Int Res J Pharm. 2012;3(6):126–129.
39. British Pharmacopoeia Commission. British Pharmacopoeia. London, UK: The Stationery Office; 2013.
40. Sangsen Y, Likhitwitayawuid K, Sritularak B, Wiwattanawongsa K, Wiwattanapatapee R. Novel solid lipid nanoparticles for oral delivery of oxyresveratrol effect of the formulation parameters on the physicochemical properties and in vitro release. Int J Med Sci Eng. 2013;7(12):873–880.
41. Aboul-Einien MH, Mahrouq GM E, Elkasabgy NA. Development and in-vitro evaluation of olanzapine-loaded solid lipid nanoparticles in solid matrix. IJPI’s J Pharm Cosmetol. 2012;2(8):19–36.
42. Tran TH, Choi JY, Ramasamy T, et al. Hyaluronic acid-coated solid lipid nanoparticles for targeted delivery of vorinostat to CD44 overexpressing cancer cells. Carbohydr Polym. 2014;114:407–415. doi:10.1016/j.carbpol.2014.08.026
43. Dandagi P, Dessai G, Gadad A, Desai V. Formulation and evaluation of nanostructured lipid carrier (NLC) of lornoxicam. Int J Pharm Pharm Sci. 2014;6(2):73–77.
44. Jose S, Anju SS, Cinu TA, Aleykutty NA, Thomas S, Souto EB. In vivo pharmacokinetics and biodistribution of resveratrol-loaded solid lipid nanoparticles for brain delivery. Int J Pharm. 2014;474(1):6–13. doi:10.1016/j.ijpharm.2014.08.003
45. Febriyenti F, Rahmi S, Halim A. Study of gliclazide solid dispersion systems using PVP K-30 and PEG 6000 by solvent method. J Pharm Bioallied Sci. 2019;11(3):262–267. doi:10.4103/jpbs.JPBS_87_18
46. Biswal S, Sahoo J, Murthy PN. Physicochemical properties of solid dispersions of gliclazide in polyvinylpyrrolidone K90. AAPS PharmSciTech. 2009;10(2):329–334. doi:10.1208/s12249-009-9212-7
47. Lu Z, Yang Y, Covington RA, Bi Y, Dürig T, Fassihi R. Amorphous-based controlled-release gliclazide matrix system. AAPS PharmSciTech. 2017;18(5):1699–1709. doi:10.1208/s12249-016-0642-8
48. Vishwanathan K, So K, Thomas K, Bramley A, English S, Collier J. Absolute bioavailability of osimertinib in healthy adults. Clin Pharmacol Drug Dev. 2019;8(2):198–207. doi:10.1002/cpdd.v8.2
49. Negi JS, Chattopadhyay P, Sharma AK, Ram V. Development and evaluation of glyceryl behenate based solid lipid nanoparticles (SLNs) using hot self-nanoemulsification (SNE) technique. Arch Pharm Res. 2014;37(3):361–370. doi:10.1007/s12272-013-0154-y
50. Müller RH, Rühl D, Runge SA. Biodegradation of solid lipid nanoparticles as a function of lipase incubation time. Int J Pharm. 1996;144(1):115–121. doi:10.1016/S0378-5173(96)04731-X
51. Gonçalves LMD, Maestrelli F, Di Cesare Mannelli L, Ghelardini C, Almeida AJ, Mura P. Development of solid lipid nanoparticles as carriers for improving oral bioavailability of glibenclamide. Eur J Pharm Biopharm. 2016;102:41–50. doi:10.1016/j.ejpb.2016.02.012
52. Kalepu S, Nekkanti V. Insoluble drug delivery strategies: review of recent advances and business prospects. Acta Pharm Sin B. 2015;5(5):442–453. doi:10.1016/j.apsb.2015.07.003
53. Davis TME, Daly F, Walsh JP, et al. Pharmacokinetics and pharmacodynamics of gliclazide in Caucasians and Australian Aborigines with type 2 diabetes. Br J Clin Pharmacol. 2001;49(3):223–230. doi:10.1046/j.1365-2125.2000.00162.x
54. Üner M. Preparation, characterization and physico-chemical properties of solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC): their benefits as colloidal drug carrier systems. Pharmazie. 2006;61(5):375–386.
55. Al-Trad B, Alkhateeb H, Alsmadi W, Al-Zoubi M. Eugenol ameliorates insulin resistance, oxidative stress and inflammation in high fat-diet/streptozotocin-induced diabetic rat. Life Sci. 2019;216:183–188. doi:10.1016/j.lfs.2018.11.034
56. Veerapur VP, Prabhakar KR, Thippeswamy BS, Bansal P, Srinivasan KK, Unnikrishnan MK. Antidiabetic effect of Ficus racemosa Linn. stem bark in high-fat diet and low-dose streptozotocin-induced type 2 diabetic rats: a mechanistic study. Food Chem. 2012;132(1):186–193. doi:10.1016/j.foodchem.2011.10.052
57. Alex A, Paul W, Chacko AJ, Sharma CP. Enhanced delivery of lopinavir to the CNS using Compritol®-based solid lipid nanoparticles. Ther Deliv. 2011;2(1):25–35. doi:10.4155/tde.10.96
58. Yildirimer L, Thanh NTK, Loizidou M, Seifalian AM. Toxicology and clinical potential of nanoparticles. Nano Today. 2011;6(6):585–607. doi:10.1016/j.nantod.2011.10.001
59. Weyhers H, Ehlers S, Hahn H, Souto EB, Müller RH. Solid lipid nanoparticles (SLN) – effects of lipid composition on in vitro degradation and in vivo toxicity. Pharmazie. 2006;61(6):539–544.
60. Thakkar A, Chenreddy S, Thio A, Khamas W, Wang J, Prabhu S. Preclinical systemic toxicity evaluation of chitosan-solid lipid nanoparticle-encapsulated aspirin and curcumin in combination with free sulforaphane in BALB/c mice. Int J Nanomedicine. 2016;11:3265–3276. doi:10.2147/IJN.S106736
61. van der Zande M, Vandebriel RJ, Groot MJ, et al. Sub-chronic toxicity study in rats orally exposed to nanostructured silica. Part Fibre Toxicol. 2014;11(8):1–19. doi:10.1186/1743-8977-11-8
62. Dhar S, Mali V, Bodhankar S, Shiras A, Prasad BLV, Pokharkar V. Biocompatible gellan gum-reduced gold nanoparticles: cellular uptake and subacute oral toxicity studies. J Appl Toxicol. 2011;31(5):411–420. doi:10.1002/jat.v31.5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.