")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 7
Linking obesity with type 2 diabetes: the role of T-bet
Authors Wali J, Thomas H, Sutherland APR
Received 3 March 2014
Accepted for publication 29 April 2014
Published 22 July 2014 Volume 2014:7 Pages 331—340
DOI https://doi.org/10.2147/DMSO.S51432
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jibran A Wali,1,2 Helen E Thomas,1,2 Andrew PR Sutherland1
1Immunology and Diabetes Unit, St Vincent's Institute, 2Department of Medicine, St Vincent's Hospital, University of Melbourne, Fitzroy, Victoria, Australia
Abstract: Obesity is a major predisposing factor for the development of type 2 diabetes (T2D) and is an escalating public health issue around the world. The transition from obesity to T2D is preceded by the induction of a state of insulin resistance, which occurs in response to genetic factors and environmental influences, such as diet. Recent advances have implicated inflammatory immune cells and cytokines as critical pathogenic mediators of insulin resistance and T2D. In particular proinflammatory T helper (Th)1 cells and M1 macrophages are recruited to adipose tissue in response to high fat diet and directly promote the development of insulin resistance. The function of these two cell types is linked by the actions of the cytokine interferon (IFN)γ and one of its major transcriptional regulators T-bet. Recent studies in animal models of T2D demonstrate that T-bet is critical for the development of insulin resistance in response to high fat diet as T-bet-deficient animals are protected from the development of insulin resistance. These data indicate that T-bet and type 1 immunity may constitute novel sites of therapeutic intervention for the treatment of insulin resistance and T2D, in obese human patients.
Keywords: type 2 diabetes, obesity, insulin resistance, immune system, T-bet, Th1, IFNγ
Introduction
The prevalence of obesity has steadily increased in recent decades and currently, more than 10% of the world’s adult population is obese, as determined by a body mass index (BMI) equal to or greater than 30 kg/m2.1 In the USA, the prevalence of obesity is 34%, and this is forecast to increase to 42% by 2030.2 Obesity is caused by an imbalance between intake and expenditure of energy and is particularly associated with increased adiposity. It is an important risk factor for the development of type 2 diabetes (T2D), cardiovascular disorders, musculoskeletal problems, such as osteoarthritis, and increased risk of certain cancers, including colon and breast cancer.1 According to US statistics, more than 80% of people with T2D are obese or overweight.3 Thus, obesity predisposes to a range of debilitating health problems and will represent a significant burden of the public health system for the foreseeable future.
Study of the pathogenesis of obesity and T2D has identified important roles for the immune system in the induction of insulin resistance and development of T2D. As we more fully elucidate the underlying immunological mechanism, it is possible that we will identify new therapeutic strategies for the treatment of T2D based on modulation of immune responses. Our aim is to review the current literature elucidating mechanisms of immune regulation of insulin resistance and T2D, with particular focus on the last decade of work defining the role for the transcription factor T-bet for control of type 1 immunity, and its relevance in T2D.
Progression from obesity to T2D
Obesity can lead to the impairment of insulin sensitivity in sites of glucose disposal, such as skeletal muscle, liver, and adipose tissue – a condition known as “insulin resistance”. The normal actions of insulin include stimulation of glucose uptake and glycogen synthesis in the skeletal muscle and liver, and inhibition of gluconeogenesis in the liver. Insulin also causes inhibition of lipolysis and increases lipogenesis in adipose tissue. Insulin resistance is characterized by reduced glucose uptake by skeletal muscle, increased release of glucose from the liver, and increased release of lipids from adipose tissue into the blood.4 Overall, this results in glucose intolerance and relative hyperglycemia in obese insulin-resistant individuals.
β-cells in the pancreatic islets adapt to hyperglycemia with compensatory responses, such as expansion of the total β-cell mass and increased insulin secretion.5 The resultant hyperinsulinemia is able to maintain normoglycemia to some extent; however, chronic progressive insulin resistance and compensatory hypersecretion of insulin can induce β-cell stress and eventually, β-cell failure, resulting in progression to frank hyperglycemia and T2D.6 Poor glycemic control in individuals with T2D can lead to various downstream complications that include renal failure, blindness, neuropathy, and cardiovascular disorders, such as myocardial infarction and stroke.7
Animal models of obesity
Understanding the mechanisms of obesity-induced insulin resistance has been an area of intensive research in recent years. These mechanisms have been elucidated primarily using animal models, including the high fat diet (HFD) model, ob/ob mice, and Zucker fatty rats.8 In the HFD model, male mice or rats are made obese, insulin resistant, and glucose intolerant by feeding them a lipid- and calorie-rich diet for several weeks. Most studies use either a 45% or 60% HFD (denoting the percentage of total digestible calories obtained from lipid sources).8 The ob/ob mouse is homozygous for a recessive inactivating mutation in leptin, a satiety hormone, and this mutation causes hyperphagia, obesity, and hyperglycemia.8 Zucker fatty rats are homozygous for a recessive mutation in the leptin receptor, which causes leptin resistance secondary to a hypofunctional leptin receptor. Similar to ob/ob mice, these rats are hyperphagic and obese and become insulin resistant and glucose intolerant.8 The intake of an HFD or hyperphagia in these animal models causes increased accumulation of white adipose tissue, which is classified either subcutaneous or visceral. Increased deposition of visceral adipose tissue is generally more deleterious for glucose homeostasis and insulin sensitivity in these animal models.9 Ectopic lipid accumulation is also observed in the liver (steatosis) and muscle of these rodents, and this is a strong predictor of hepatic and skeletal muscle insulin resistance.10
Insulin signaling and insulin resistance
Insulin signaling is a complex process involving several key factors (these have been reviewed more extensively elsewhere).11 Important steps in the insulin signaling cascade include the binding of insulin to its receptor, followed by its autophosphorylation and tyrosine phosphorylation of insulin receptor substrate (IRS)-1 and IRS-2. IRS-1 and IRS-2 then bind to phosphatidylinositol 3-kinase (PI3K), and this induces a series of downstream events resulting in activation of Akt. Activation of Akt in insulin-sensitive cells has various effects, including the translocation of the insulin-sensitive glucose transporter (GLUT-4) to the cell membrane, activation of glycogen synthesis, and inhibition of gluconeogenic genes in the liver.4,6 Impairments of this signaling network can result in insulin resistance, including modulation of the activity of the key signaling molecules, such as IRS-1, IRS-2, and Akt. The kinases IKK (IkB kinase) and JNK-1 (c-Jun N-terminal kinase 1) negatively regulate IRS-1 and IRS-2 by serine phosphorylation, inducing degradation of IRS proteins and inhibition of insulin receptor signaling.11,12 Increased IRS-1 serine phosphorylation is observed in insulin-resistant states.13,14 SOCS (suppressor of cytokine signaling) proteins compete with IRS proteins for binding sites on the insulin receptor, inhibit IRS tyrosine phosphorylation, and induce IRS-1 and IRS-2 degradation.15,16 The protein phosphatases PP2A (protein phosphatase 2A) and PHLPP (PH domain and leucine rich repeat protein phosphatase) inhibit Akt activity via dephosphorylation of the activating sites.17,18 Further, the factor TRB3 (tribbles homolog 3) inhibits Akt activity by sequestering the unphosphorylated form, thereby blocking its phosphorylation and activation.19 These mechanisms are regulated by metabolic and inflammatory factors that induce insulin resistance and predispose to the development of T2D.
Lipid-derived molecules are mediators of insulin resistance
Increased plasma concentration of saturated fatty acids such as palmitate is an important cause of insulin resistance in obesity.20 Palmitate stimulation of cultured skeletal muscle myotubes has been shown to inhibit insulin signaling and glucose uptake in rat soleus muscle preparations.21,22 These inhibitory effects are mediated, in part, by the sphingolipid ceramide, which is synthesized as a consequence of palmitate-induced impairments of mitochondrial lipid oxidation20 and inhibits insulin signaling by blocking Akt activation.21,23 Palmitate also induces insulin resistance via engagement of toll-like receptor 2 (TLR2) and subsequent activation of JNK and IKKβ kinases.24 Myriocin, a ceramide synthesis inhibitor, prevents palmitate-induced impairment of insulin signaling in skeletal muscle cells and diabetes, in rodent models.23,25 Conversely, inhibition of acid ceramidase, an enzyme that reduces ceramide levels by converting to sphingosine, restores the palmitate-induced suppression of insulin signaling in skeletal muscle.26
Unsaturated fatty acids also induce insulin resistance in key metabolic tissues. Linoleate induces increased cellular accumulation of diacylglycerols, and linoleate-rich infusions and diacylglycerols induce insulin resistance in rodent and human skeletal muscles, independently of ceramides.25,27,28 Diacylglycerols induce the activation of PKC (protein kinase C) isoforms, and these can inhibit the activity of IRS-1, leading to insulin resistance.20,29 Thus, lipid-derived molecules have direct effects on insulin-sensitive tissues and can induce insulin resistance.
Mechanisms of inflammation-induced insulin resistance
Appreciation of the role of inflammatory processes in the development of insulin resistance has increased over the last 15 years. It is now recognized that obesity induces an inflammatory state that promotes the development of insulin resistance.9,30 Adipose tissue has high levels of resident macrophages, and these cells are critical players that mediate the development of obesity-associated insulin resistance.31 Adipose tissue macrophages (ATMs) are classified into classically or alternatively activated macrophages, based on their pro- or anti-inflammatory functions respectively and can be identified by their differential expression of cell surface markers and cytokines.
M1 or classically activated macrophages express CD11c, secrete proinflammatory cytokines, such as tumor necrosis factor (TNF)α, interleukin (IL)-6, and IL-1β, and their differentiation is promoted by interferon (IFN)γ and lipopolysaccharide.31,32 M2 or alternatively activated macrophages express MGL-1 (macrophage galactose lectin 1), secrete anti-inflammatory molecules, such as IL-10, transforming growth factor (TGF)β, and IL-1 receptor antagonist, and differentiate in response to IL-4 and IL-13.31,32 Under normal homeostatic conditions, ATMs have a predominantly M2 phenotype; however, obesity induces a shift in the ATM population, from the M2 to M1 phenotype. This occurs via two main mechanisms: phenotypic conversion of M2 to M1 and recruitment of M1 macrophages into adipose tissue.6,31–34 This phenotypic conversion can be mediated by multiple stimuli, such as lipotoxicity and adipocyte dysfunction, including hypertrophy, hypoxia, and necrosis. Necrotic adipocytes also stimulate recruitment and the accumulation of M1 macrophages and secretion of proinflammatory cytokines and chemokines, including monocyte chemoattractant protein (MCP)-1.6,33,35,36
M1 ATMs produce proinflammatory cytokines, such as TNFα, IL-6, and IL-1β, in response to obesity, and circulating levels of these cytokines are increased in human patients and mouse models of T2D.30 These cytokines have several effects that could link obesity to insulin resistance, most notably, the induction of local inflammatory pathways. Engagement of TNFα receptors activates the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) and MAP (mitogen-activated protein) signaling pathways via IKK and JNK1 kinases.10 Liver-specific deletion of IKK protects from hepatic insulin resistance in both the HFD and ob/ob mouse models.37 Similarly, JNK1-deficient mice are protected from obesity-induced insulin resistance. Compared with wild type controls, these mice gain less weight and have a relatively higher body temperature, indicating increased energy expenditure.14 IL-6 suppresses insulin receptor signal transduction by inhibiting IRS-1, IRS-2, and Akt in a SOCS3-dependent manner.15,38 These cytokines also cause increased lipolysis in adipose tissue, by reducing the expression of stabilizing lipid droplet proteins, including perilipin and FSP27 (fat specific protein 27).39,40 This could promote ectopic lipid deposition in other insulin-sensitive tissues.
NLRP (Nucleotide-binding oligomerization domain Leucine-rich Repeat and Pyrin domain-containing family) inflammasome-dependent IL-1β, secretion from macrophages and adipocytes plays an important role in the pathogenesis of insulin resistance.41 The NLRP3 inflammasome mediates the activation of caspase 1, which is required for the processing of pro-IL-1β to the active IL-1β form.42 IL-1β is directly toxic to pancreatic β-cells and induces insulin resistance via inflammatory pathways.41 The saturated fatty acid palmitate and ceramide activate the inflammasome complex in macrophages, while the unsaturated fatty acid oleate does not.43,44 Increased inflammasome activity has been observed in monocyte-derived macrophages from T2D patients.45 Inflammasome activation promotes insulin resistance via inhibitory serine phosphorylation of IRS-1 in liver and adipocytes, and inhibition of Akt activity.44 Mice deficient in NLRP3 or caspase 1 are protected from HFD-induced insulin resistance and glucose intolerance.44,46 Thus, inflammasome activation and the production of inflammatory cytokines by adipose tissue macrophages can inhibit insulin signaling and cause insulin resistance.
The adaptive immune system regulates insulin sensitivity in obesity
The adaptive immune system is composed of a vast array of differing lymphocyte subsets. The predominant lymphocytes include CD4+ T cells, regulatory T cells (Tregs), CD8+ T cells, and B cells, and these cells perform numerous critical functions, such as secretion of cytokines, the limiting of self-reactivity, killing of infected cells, and production of antibodies. CD4+ T cells are central regulators of the adaptive immune system, and these can diverge along multiple lineages, such as T helper (Th)1, Th2, Th17, and Tregs, in response to environmental cues.47 These lineages exhibit unique cytokine signatures and functional properties.47
Lymphocytes play a critical role in the control of adiposity and glucose homeostasis in response to a HFD, in mouse models.31,48 When fed a normal diet, adipose tissue resident Tregs and Th2 T cells maintain an anti-inflammatory environment via the production of anti-inflammatory cytokines and induction of alternative macrophage activation.31,49 In response to a HFD, Th1 type T cells are recruited to subcutaneous and visceral adipose tissue, in animal models. This recruitment increases the ratio of Th1 cells to Tregs and leads to a proinflammatory state in visceral adipose tissue. This is temporally correlated with an increased number of M1-type macrophages in adipose tissue, induction of insulin resistance, and impaired glucose homeostasis, in mice.34,50,51 There is some evidence that this mechanism may be relevant to humans as analysis of a small cohort of obese patients revealed that the Th1/Treg ratio in visceral adipose tissue was positively correlated with BMI.52
Lymphocyte-deficient RAG2−/− mice have exacerbated weight gain and insulin resistance in response to a HFD, which can be partially corrected by the transfer of CD4+ T cells.52 These results highlight the function of anti-inflammatory CD4+ T cells, and loss of these cells in HFD mice contributes to insulin resistance.50,53 Treatment of obese mice with immune therapies, such as immunosuppressive anti-CD3 monoclonal antibodies, was shown to result in the induction of anti-inflammatory responses in adipose tissue, including increased FoxP3+ Tregs and alternatively activated macrophages, increased IL-10, and reduced TNFα. This was associated with reduced insulin resistance and improved glucose tolerance in the anti-CD3 treated animals.52 Thus, distinct CD4+ T cell subsets have opposing proinflammatory and anti-inflammatory functions in obesity. Th1 responses are central to the recruitment of M1 macrophages and the induction of insulin resistance in models of obesity-induced diabetes. These effects are counterbalanced by the function of Tregs and Th2 cells, which maintain an anti-inflammatory state and enhance insulin sensitivity.
Other lymphocyte subsets, including CD8+ T cells and B cells, also regulate glucose homeostasis in models of obesity. CD8+ T cells are recruited to adipose tissue after an HFD, and activation of CD8+ T cells was shown to be enhanced in the epididymal adipose tissue from animals on an HFD compared with a normal diet.48 These activated CD8+ T cells support the recruitment and expansion of macrophages expressing high levels of TNFα. Similarly, B cells, particularly class-switched immunoglobulin (Ig)G+ B cells, are recruited to adipose tissue after an HFD, accompanied by a specific enhancement in the production of IgG2c isotype antibodies.54 These antibodies were shown to be pathogenic as transfer from obese mice into B cell-deficient recipients caused increased numbers of M1 macrophages in visceral adipose tissue, increased TNFα production, increased fasting insulin levels, and exacerbated glucose intolerance.52 The pathogenic roles of CD8+ T cells and B cells were further illustrated by improved glucose tolerance in CD8+ T cell- and B cell-deficient mice on an HFD.48,52 Thus several lymphocyte lineages contribute to the development of insulin resistance in response to obesity.
IFNγ promotes insulin resistance
IFNγ is the signature Th1 cytokine, and its expression in adipose tissue increases in obesity.55 IFNγ receptor engagement and activation of the downstream JAK (Janus kinase) and STAT1 (signal transducer and activator of transcription 1) pathway causes decreased insulin-stimulated glucose uptake, inhibited differentiation, and reduced triglyceride content in adipocytes.56 IFNγ stimulates the expression of macrophage and T cell chemoattractants and secretion of TNFα from cultured adipocytes and visceral adipose tissue preparations.55 Moreover, IFNγ acts on adipose tissue macrophages to facilitate M2 to M1 phenotypic conversion in obesity.57 In addition, IFNγ directly impairs insulin signaling by reducing insulin receptor IRS-1 and GLUT-4 expression and Akt-1 phosphorylation.56 IFNγ-deficient mice had improved insulin sensitivity and glucose tolerance after an HFD, consistent with the role of IFNγ as a proinflammatory factor in adipose tissue.55,58 This favorable metabolic phenotype was accompanied by increased physical activity and lower food intake58 and was likely due, in part, to the reduced recruitment of M1 macrophages and other inflammatory cells to adipose tissue, in IFNγ−/− mice.55,57
T-bet is a central regulator of IFNγ production and immune function
T-bet is a Tbox family transcription factor that was first identified as a transcriptional inducer of IFNγ in CD4+ T cells.59 Its expression is induced in naïve CD4+ T cells by T cell receptor (TCR) and cytokine stimulation, particularly STAT1-inducing cytokines, such as IFNγ. Expression of T-bet leads to the expression of a transcriptional program that stabilizes Th1 cell differentiation, including IFNγ, IL-12Rβ2 and Hlx-1,60 which promote further T-bet expression in a positive feedback loop, and other molecules that are required for Th1 function, such as CXCR3 (C-X-C motif chemokine receptor type 3).61 In addition T-bet directly antagonizes the development of the Th2 and Th17 lineages, by suppressing the function of the lineage defining transcription factors GATA3 (GATA binding protein 3) and RORγt (retinoid acid receptor-related orphan receptor gamma t) respectively.62,63 Overexpression of T-bet is sufficient to reprogram Th2 cells to express IFNγ, and T-bet-deficient CD4+ T cells fail to differentiate into Th1 cells under polarizing conditions.59 Thus T-bet regulates the expression of a Th1 transcriptional program in CD4+ T cells and is necessary and sufficient to promote Th1 polarization.
T-bet is expressed in a variety of other immune cell types and regulates type 1 immunity more generally. This is achieved by promoting the differentiation of multiple immune cell lineages that contribute to cell-mediated immunity and by suppressing the function of counterregulatory lineages.64,65 T-bet is required for the development of optimal CD8+ T cell responses, where it transactivates the expression of IFNγ and cytotoxic molecules, such as perforin and granyzme B.66 T-bet-deficient CD8+ T cells fail to upregulate activation markers, express lower levels of IFNγ and increased levels of Tc2 cytokines, including IL-4 and IL-10, and fail to develop into functional cytotoxic T lymphocytes (CTLs) in vitro and in vivo.67
B cell antibody responses are regulated by cytokines, particularly differentiation and isotype class switching. The production of the IgG2a/c isotype is associated with activation of cell-mediated immunity and is induced in response to IFNγ stimulation of B cells. T-bet-deficient B cells have impaired production of class-switched IgG2b and IgG3, and almost complete absence of IgG2a downstream of IFNγ signaling.68,69
T-bet also controls adaptive immune responses by modulating the function of innate immune cells, including dendritic cells. T-bet is induced, in response to IFNγ, in dendritic cells via a STAT1-dependent mechanism70 and is required for IFNγ production in response to IL-12 and IL-18 stimulation. Immunization of mice with T-bet-deficient dendritic cells resulted in reduced IFNγ in responding CD4+ T cells, most likely the result of reduced IFNγ production from the dendritic cells. Furthermore the adjuvant properties of CpG (unmethylated DNA of microbial origin) in the clearance of Listeria monocytogenes are dependent on the IFNγ-induced expression of T-bet in dendritic cells.71 T-bet-deficient mice also show profound defects in the frequencies of natural killer (NK) and natural killer T (NKT) cell lineages72 and functional impairments in NK cells, in the context of metastatic cancer.73
The importance of T-bet for the development of optimal immune responses has been demonstrated in multiple in vivo models of infection and autoimmunity. T-bet-deficient mice are more susceptible to a range of intracellular pathogens, including Mycobacterium tuberculosis, Leishmania major, Staphylococcus aureus, and Salmonella typhimurium. In keeping with generalized deficiencies in type 1 immunity, T-bet-deficient mice are less susceptible to autoimmune disorders, including inflammatory bowel disease, experimental autoimmune encephalomyelitis, collagen-induced arthritis, systemic lupus erythematosus, and type 1 diabetes.64 Strikingly, T-bet-deficient mice also develop spontaneous allergic airway inflammation that is reminiscent of human asthma,74 in keeping with their inability to suppress the expression of Th2 cytokines during Th1 polarization.75 Thus T-bet is a central transcriptional regulator for type 1 immunity and is required for optimal function of multiple innate and adaptive immune cell lineages.
Role of T-bet in obesity and T2D
A recent study has demonstrated that T-bet plays a critical role in the development of insulin resistance in animal models of obesity (Figure 1). T-bet knockout mice fed an HFD showed increased weight gain and adiposity; however, they were refractory to the induction of insulin resistance.76 The authors argue that the uncoupling of weight gain and insulin resistance in the absence of T-bet was mediated by the adaptive immune system and impaired IFNγ production.
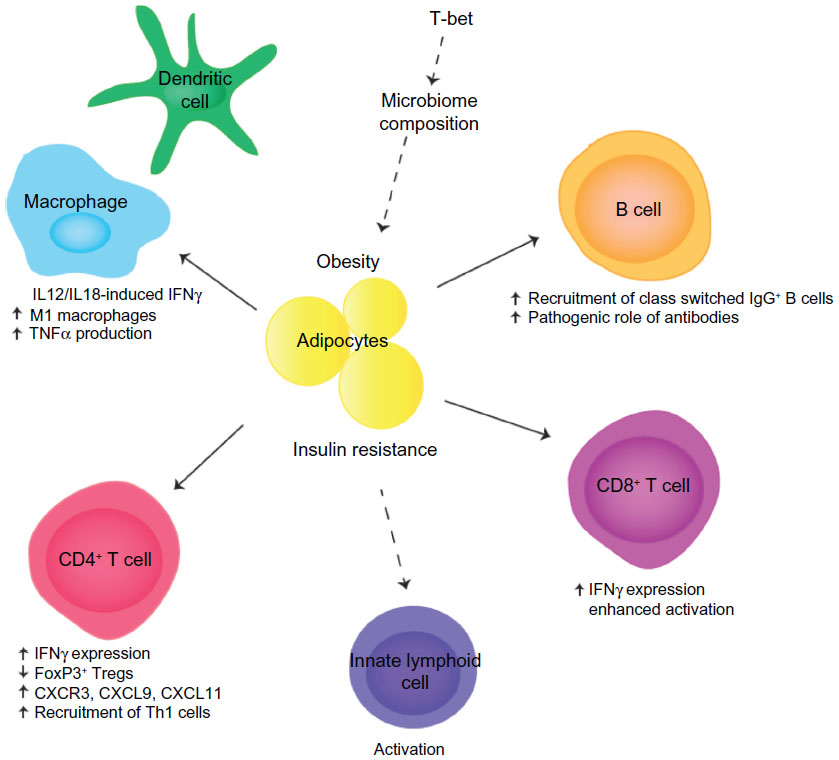 | Figure 1 T-bet/IFNγ effects in obesity and insulin resistance. |
A detailed analysis revealed that T-bet-deficient mice had increased body weight and perigonadal and mesenteric fat deposits on both normal diet and HFD.76 This was correlated with improved insulin sensitivity and glucose tolerance on both diets, suggesting that T-bet regulates insulin sensitivity in the basal state as well as in response to an HFD. Quantification of lymphocytes from adipose tissue demonstrated reduced numbers of CD4+ T cells, CD8+ T cells, and NK cells, and a reduced production of inflammatory cytokines, including IFNγ, TNFα, IL-1β, and IL-6. Generation of doubly deficient T-bet−/− × RAG2−/− mice implicated the adaptive immune system as these lymphocyte-deficient mice were no longer protected from the development of insulin resistance.76 These data indicate that the T-bet-dependent effects on insulin sensitivity reside within the adaptive immune system.
The T-bet-deficient mice displayed increased percentages of FoxP3+ Tregs in perigonadal fat and reduced expression of CXCR3 and its ligands CXCL9 and CXCL11, which are required for the recruitment of Th1 T cells into inflammatory sites. It is likely that reduced levels of inflammation and insulin resistance are due to reduced recruitment of Th1 T cells into visceral fat, although the numbers of Th1 or Th2 cells were not directly quantified. The predicted downstream consequences of decreased Th1 recruitment are reduced activation of M1 inflammatory macrophages. This is consistent with the observed reduction in inflammatory cytokine production, although the authors did not quantify the frequencies of M1 and M2 macrophages.76
The generation of doubly deficient T-bet−/− × IFNγ−/− mice indicates that IFNγ is also a key mediator of these processes. The phenotypes of IFNγ−/− mice and T-bet−/− × IFNγ−/− mice were found to be almost identical,76 indicating that IFNγ deficiency is likely to be a primary cause of the observed protection from insulin resistance in T-bet-deficient mice. Additionally T-bet directly antagonizes the function of the Th2 lineage defining transcription factor GATA3.62 Thus, increases in Th2 cells, in combination with reduced Th1 cells, may also contribute to the observed protection. T-bet also suppresses the production of IL-2 in activated T cells,77 and thus, T-bet-deficient T cells may produce elevated levels of IL-2 in adipose tissue. Since IL-2 supports the survival of Tregs in vivo, this may sustain increased levels of Tregs in adipose tissue, maintain an anti-inflammatory environment, and prevent induction of insulin resistance.
While the transfer of wild type CD4+ T cells into T-bet−/− × RAG2−/− doubly deficient mice was found to partially restore insulin resistance, indicating an important role for CD4+ T cells, complete restoration was not observed.76 These data indicate that other T-bet-expressing lymphocyte lineages may also contribute to the development of insulin resistance. Other potential candidates include CD8+ T cells and B cells, which are involved in the pathogenic processes leading to insulin resistance and express T-bet. T-bet is required for optimal activation, cytokine production, and CTL effector function in CD8+ T cells66,67 and thus may make an important contribution to the development of insulin resistance in obesity. Similarly, T-bet is required for the optimal function of B cells, particularly class switching to IgG2a/c. As there is a specific enhancement of pathogenic IgG2a/c antibodies in response to an HFD and IgG2a/c production is controlled by T-bet,68,69 the T-bet deficiency in B cells may also contribute to the protection from insulin resistance in obese T-bet-deficient mice. Thus, it is likely that T-bet regulates insulin sensitivity by controlling the function of multiple lymphocyte lineages, including CD4+ and CD8+ T cells and B cells.
Recently, the role of the microbiome in the pathogenesis of obesity and T2D has been further elucidated. Obese and T2D patients have been found to have alterations in the composition of their microbiome,78–80 and gut microflora regulates the development of obesity in animal models.81,82 The microbiome composition changes in mouse models during the development of obesity, and fecal transplantation transfers an obese phenotype to wild type recipients.83,84 These data indicate that microflora play a causative role in the development of obesity. T-bet regulates mucosal T cell activation, and T-bet deficiency alters the composition of microflora, in mice, via T-bet actions in dendritic cells and innate lymphoid cells.85–87 Thus, it is tempting to speculate that T-bet deficiency may also alter the microbiome in the context of an HFD and obesity and that this may also contribute to the inflammatory and metabolic processes that regulate the T2D.
Whilst the data from mouse models demonstrating a role for the immune system and T-bet in the regulation of insulin sensitivity during obesity is compelling, much less convincing data exists from human patients. Small clinical studies in T2D patients have identified increased ratios of inflammatory Th1 to Th2 or Th1 to Tregs in adipose tissue and increased expression of major histocompatibility complex (MHC) molecules in adipocytes.50,52,88 These data are suggestive of ongoing CD4+ T cell-mediated inflammation within adipose tissue in T2D patients.
Data from genome-wide association studies indicate that the majority of genetic linkages to T2D are associated with genes that regulate β-cell function.89 Strikingly, there is little evidence of linkage to immune or inflammatory pathways, such as MHC or cytokine signaling, as is observed for classical autoimmune diseases, such as type 1 diabetes.90 Peroxisome proliferator-activated receptor (PPAR)γ polymorphisms are associated with T2D,89,91 and PPARγ agonists improve insulin sensitivity in T2D patients.92 Whilst the therapeutic effects of these agonists were assumed to be due to a direct action on adipocytes, the recent discovery that PPARγ is preferentially expressed by adipose tissue resident Tregs indicates that the primary mechanism of PPARγ agonists may instead be immune regulation.53,93 Similarly, other genes identified by genome-wide association studies in T2D may have hitherto unappreciated roles in the immune system.
There have been promising studies demonstrating that blockade of inflammatory cytokines and pathways can be used to reduce insulin resistance in T2D.9,30 The discovery that T-bet is required for the development of insulin resistance in preclinical models indicates that immune therapies based on antagonism of Th1 immunity or induction of regulatory T cells may also be of therapeutic utility in T2D. Suppression of type 1 immunity could potentially be achieved by direct pharmacological antagonism of T-bet function or alternatively, reduction of T-bet expression using gene silencing technologies. Alternatively, blockade of cytokines that induce T-bet expression, such as IFNγ and/or IL-12, may also provide a means to suppress T-bet and type 1 immunity in the context of T2D. Such therapeutic strategies must be weighed against the potential side effects of T-bet inhibition, such as reduced immunity to intracellular pathogens and tumors and/or increased susceptibility to airway inflammation, as suggested by the phenotype of T-bet-deficient mice.74 Finally, expansion of Tregs via low-dose IL-2 treatment may also serve to dampen inflammation generally, which may prove an effective means of increasing insulin sensitivity in T2D patients.
It is now appreciated that inflammation and the immune system play a critical role in the development of T2D, in the context of obesity. Immune cells, including M1 macrophages and Th1 T cells, induce insulin resistance in response to environmental factors, such as dietary lipids, doing so via the action of immune mediators, including cytokines, such as IFNγ. With the demonstration that T-bet-deficient mice are protected from the development of insulin resistance in the context of obesity, it is now clear that T-bet is a central regulator of insulin resistance via its ability to promote IFNγ production, Th1 T cell function, and type 1 immunity. These effects are likely to be involved in the development of insulin resistance and T2D in human patients, and thus, a new generation of therapeutics that antagonize the function of T-bet, its upstream regulators, or downstream mediators may prove efficacious for the alleviation of insulin resistance and T2D, in obese patients.
Review criteria
The articles in this Review were identified by a literature search in PubMed for articles published from 2000 to 2013. The keywords used were “type 2 diabetes”, “obesity”, “insulin resistance”, “immune system”, “T-bet”, “Th1”, “IFNγ”. Additional relevant articles selected from the authors’ database were also included.
Disclosure
The authors report no conflicts of interest in this work.
References
who.int [homepage on the Internet]. Obesity and overweight. World Health Organization; 2013 [updated May 2014; cited January 20, 2014]. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/. Accessed May 21, 2014. | |
Finkelstein EA, Khavjou OA, Thompson H, et al. Obesity and severe obesity forecasts through 2030. Am J Prev Med. 2012;42(6):563–570. | |
diabetes.niddk.nih.gov [homepage on the Internet]. Diabetes overview. National Diabetes Information Clearinghouse; 2008 [updated April 2, 2014; cited January 20, 2014]. Available from: http://diabetes.niddk.nih.gov/dm/pubs/overview/. Accessed May 21, 2014. | |
Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118(9):2992–3002. | |
Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004;53 Suppl 3:S16–S21. | |
Kalupahana NS, Moustaid-Moussa N, Claycombe KJ. Immunity as a link between obesity and insulin resistance. Mol Aspects Med. 2012; 33(1):26–34. | |
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. | |
Lutz TA, Woods SC. Overview of animal models of obesity. Curr Protoc Pharmacol. 2012;Chapter 5:Unit5.61. | |
Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. | |
Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. | |
Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006; 7(2):85–96. | |
Paz K, Hemi R, LeRoith D, et al. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem. 1997;272(47):29911–29918. | |
Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11(2):183–190. | |
Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. | |
Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277(44):42394–42398. | |
Hirashima Y, Tsuruzoe K, Kodama S, et al. Insulin down-regulates insulin receptor substrate-2 expression through the phosphatidylinositol 3-kinase/Akt pathway. J Endocrinol. 2003;179(2):253–266. | |
Siddle K. Signalling by insulin and IGF receptors: supporting acts and new players. J Mol Endocrinol. 2011;47(1):R1–R10. | |
Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18(1):13–24. | |
Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003; 300(5625):1574–1577. | |
Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15(5):585–594. | |
Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem. 1999;274(34):24202–24210. | |
Thrush AB, Harasim E, Chabowski A, Gulli R, Stefanyk L, Dyck DJ. A single prior bout of exercise protects against palmitate-induced insulin resistance despite an increase in total ceramide content. Am J Physiol Regul Integr Comp Physiol. 2011;300(5):R1200–R1208. | |
Chavez JA, Knotts TA, Wang LP, et al. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem. 2003;278(12):10297–10303. | |
Senn JJ. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem. 2006;281(37):26865–26875. | |
Holland WL, Brozinick JT, Wang LP, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5(3):167–179. | |
Chavez JA, Holland WL, Bär J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem. 2005;280(20):20148–20153. | |
Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51(7):2005–2011. | |
Qu X, Seale JP, Donnelly R. Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats – effects of feeding. J Endocrinol. 1999;162(2):207–214. | |
Kim JK, Fillmore JJ, Sunshine MJ, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004; 114(6):823–827. | |
Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. | |
Mathis D. Immunological goings-on in visceral adipose tissue. Cell Metab. 2013;17(6):851–859. | |
Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18(3):363–374. | |
Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes. 2008;57(12):3239–3246. | |
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. | |
Kitade H, Sawamoto K, Nagashimada M, et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes. 2012;61(7):1680–1690. | |
Prieur X, Mok CY, Velagapudi VR, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes. 2011; 60(3):797–809. | |
Arkan MC, Hevener AL, Greten FR, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11(2):191–198. | |
Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. 2002;51(12):3391–3399. | |
Bézaire V, Mairal A, Anesia R, Lefort C, Langin D. Chronic TNFalpha and cAMP pre-treatment of human adipocytes alter HSL, ATGL and perilipin to regulate basal and stimulated lipolysis. FEBS Lett. 2009;583(18):3045–3049. | |
Ranjit S, Boutet E, Gandhi P, et al. Regulation of fat specific protein 27 by isoproterenol and TNF-α to control lipolysis in murine adipocytes. J Lipid Res. 2011;52(2):221–236. | |
Wen H, Ting JP, O’Neill LA. A role for the NLRP3 inflammasome in metabolic diseases – did Warburg miss inflammation? Nat Immunol. 2012;13(4):352–357. | |
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. | |
Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–415. | |
Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188. | |
Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194–204. | |
Stienstra R, van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108(37):15324–15329. | |
Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–489. | |
Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15(8):914–920. | |
Shu CJ, Benoist C, Mathis D. The immune system’s involvement in obesity-driven type 2 diabetes. Semin Immunol. 2012;24(6):436–442. | |
Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. | |
Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56(1):16–23. | |
Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15(8):921–929. | |
Cipolletta D, Feuerer M, Li A, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486(7404):549–553. | |
Winer DA, Winer S, Shen L, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17(5):610–617. | |
Rocha VZ, Folco EJ, Sukhova G, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103(5):467–476. | |
McGillicuddy FC, Chiquoine EH, Hinkle CC, et al. Interferon gamma attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the JAK/STAT pathway. J Biol Chem. 2009;284(46):31936–31944. | |
O’Rourke RW, White AE, Metcalf MD, et al. Systemic inflammation and insulin sensitivity in obese IFN-γ knockout mice. Metabolism. 2012;61(8):1152–1161. | |
Wong N, Fam BC, Cempako GR, et al. Deficiency in interferon-gamma results in reduced body weight and better glucose tolerance in mice. Endocrinology. 2011;152(10):3690–3699. | |
Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100(6):655–669. | |
Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. | |
Lord GM, Rao RM, Choe H, et al. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106(10):3432–3439. | |
Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science. 2005;307(5708):430–433. | |
Lazarevic V, Chen X, Shim JH, et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nat Immunol. 2011;12(1):96–104. | |
Lazarevic V, Glimcher LH. T-bet in disease. Nat Immunol. 2011;12(7):597–606. | |
Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 2013;13(11):777–789. | |
Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nat Rev Immunol. 2004;4(11):900–911. | |
Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci U S A. 2003;100(26):15818–15823. | |
Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A. 2002;99(8):5545–5550. | |
Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc Natl Acad Sci U S A. 2013;110(34):E3216–E3224. | |
Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, Glimcher LH. T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci U S A. 2003;100(13):7749–7754. | |
Lugo-Villarino G, Ito S, Klinman DM, Glimcher LH. The adjuvant activity of CpG DNA requires T-bet expression in dendritic cells. Proc Natl Acad Sci U S A. 2005;102(37):13248–13253. | |
Townsend MJ, Weinmann AS, Matsuda JL, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20(4):477–494. | |
Werneck MB, Lugo-Villarino G, Hwang ES, Cantor H, Glimcher LH. T-bet plays a key role in NK-mediated control of melanoma metastatic disease. J Immunol. 2008;180(12):8004–8010. | |
Finotto S, Neurath MF, Glickman JN, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 2002;295(5553):336–338. | |
Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002; 295(5553):338–342. | |
Stolarczyk E, Vong CT, Perucha E, et al. Improved insulin sensitivity despite increased visceral adiposity in mice deficient for the immune cell transcription factor T-bet. Cell Metab. 2013;17(4):520–533. | |
Hwang ES, Hong JH, Glimcher LH. IL-2 production in developing Th1 cells is regulated by heterodimerization of RelA and T-bet and requires T-bet serine residue 508. J Exp Med. 2005;202(9):1289–1300. | |
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. | |
Karlsson FH, Tremaroli V, Nookaew I, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498(7452):99–103. | |
Zhang X, Shen D, Fang Z, et al. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS One. 2013; 8(8):e71108. | |
Bäckhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–15723. | |
Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104(3):979–984. | |
Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–11075. | |
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. | |
Neurath MF, Weigmann B, Finotto S, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med. 2002;195(9):1129–1143. | |
Garrett WS, Lord GM, Punit S, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131(1):33–45. | |
Powell N, Walker AW, Stolarczyk E, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity. 2012;37(4):674–684. | |
Deng T, Lyon CJ, Minze LJ, et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab. 2013;17(3):411–422. | |
Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS? Ann N Y Acad Sci. 2010;1212:59–77. | |
Barrett JC, Clayton DG, Concannon P, et al; Type 1 Diabetes Genetics Consortium. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–707. | |
Sanghera DK, Blackett PR. Type 2 diabetes genetics: beyond GWAS. J Diabetes Metab. 2012;3(198):6948. | |
Olefsky JM. Treatment of insulin resistance with peroxisome proliferator–activated receptor γ agonists. J Clin Invest. 2000;106(4):467–472. | |
Cipolletta D, Kolodin D, Benoist C, Mathis D. Tissular T(regs): a unique population of adipose-tissue-resident Foxp3+CD4+ T cells that impacts organismal metabolism. Semin Immunol. 2011;23(6):431–437. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.