")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
LINC00612/miR-31-5p/Notch1 Axis Regulates Apoptosis, Inflammation, and Oxidative Stress in Human Pulmonary Microvascular Endothelial Cells Induced by Cigarette Smoke Extract
Authors Luo J, Li L, Hu D, Zhang X
Received 2 April 2020
Accepted for publication 21 July 2020
Published 26 August 2020 Volume 2020:15 Pages 2049—2060
DOI https://doi.org/10.2147/COPD.S255696
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Jun Luo,1,* Li Li,2,* Die Hu,2 Xian Zhang1
1Department of Laboratory Medicine, Chengdu Second People’s Hospital, Chengdu 610000, Sichuan, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, Dujiangyan People’s Hospital, Dujiangyan 611830, Sichuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Li Li
Department of Respiratory and Critical Care Medicine, Dujiangyan People’s Hospital, No. 622 Baolian Road, Dujiangyan 611830, Sichuan, People’s Republic of China
Tel +86-28-87121111
Email [email protected]
Background: Long non-coding RNAs (lncRNAs) have been reported as key regulators in chronic obstructive pulmonary disease (COPD). However, the precise role of LINC00612 remains unclear.
Methods: The real-time quantitative polymerase chain reaction (RT-qPCR) was used to quantify the expression levels of LINC00612, miR-31-5p, and Notch homolog 1 (Notch1) in lung tissues and cells. Under a cigarette smoke extract (CSE) stimulation condition, the apoptosis was analyzed by flow cytometry assay. Caspase-3 activity was examined with a caspase-3 activity assay kit; besides, inflammation and oxidative stress were assessed by measuring interleukin-6, tumor necrosis factor-α, glutathione/oxidized glutathione, reactive oxygen species, malondialdehyde, and superoxide dismutase activity. The interaction relationship between miR-31-5p and LINC00612 or Notch1 was predicted by bioinformatics databases, while dual-luciferase reporter, RNA immunoprecipitation, and RNA pull-down assays were performed to confirm prediction. Eventually, the related protein expression was estimated with western blot assay.
Results: LINC00612 was downregulated in COPD tissues when compared with controls. Consistently, CSE inhibited LINC00612 expression in HPMECs with a dose/time-dependent method. Gain-of-function experiments indicated that the upregulation of LINC00612 could repress cell apoptosis, inflammation, and oxidative stress in HPMECs induced by CSE. In addition, miR-31-5p was negatively regulated by LINC00612 in HPMECs treated with CSE. The overexpression of miR-31-5p could abolish LINC00612-induced effects on HPMECs exposed to CSE. Importantly, LINC00612 could weaken CSE-induced cell apoptosis, inflammation, and oxidative stress in HPMECs by regulating the miR-31-5p/Notch1 signaling pathway.
Conclusion: Current findings suggest that CSE-mediated cell apoptosis, inflammation, and oxidative stress in HPMECs were abolished by upregulation of LINC00612. Furthermore, the LINC00612/miR-31-5p/Notch1 axis may represent a novel regulator of apoptosis, inflammation, and oxidative stress of HPMECs, which may be a potential therapeutic target for COPD in the future.
Keywords: LINC00612, miR-31-5p, Notch1, COPD, CSE
Introduction
Chronic obstructive pulmonary disease (COPD), a type of pulmonary disease involving the respiratory system, is a common reason for death all over the world.1 According to the prediction by the World Health Organization, COPD will remain a challenge for clinicians in the future.2 COPD is characterized by airflow limitation and chronic inflammation, leading to persistent airflow restriction and destruction of lung parenchyma.3 In addition, the pathogenesis of COPD has not been investigated thoroughly. Epithelial to mesenchyme transition,4 autophagy,5 inflammation,6 cell apoptosis, and oxidative stress7 were reported as significant factors contributing to the destruction of pulmonary tissue in COPD. Therefore, it is meaningful to identify the molecular mechanism of COPD.
Long non-coding RNAs (lncRNAs) are non-coding RNAs ~200 nucleotides in the length.8 In previous studies, lncRNAs were widely concerned on account of their important function in human diseases.9 LINC00612, a non-coding RNA, was decreased in acute myocardial infarction.10 In addition, Miao et al revealed that LINC00612 was significantly increased in bladder cancer samples when compared with controls, revealing that LINC00612 might be carcinostatic in bladder cancer.11 Interestingly, Qian et al analyzed and identified dysregulated genes in smoking patients with COPD disease, including LINC00612, and results indicated the diagnostic value of LINC00612 in COPD.12
Furthermore, microRNAs (miRNAs) are a class of small non-coding RNAs regulating gene expression by binding to target mRNAs.13 As post-transcriptional regulators of gene expression, miRNAs play a critical role in numerous biological behaviors.14 Abnormal expression of miR-31 was found in a variety of cancers, and its function has been described as carcinostatic factors in oral squamous cell carcinoma,15 colorectal cancer,16 and lung cancer.17 Interestingly, a systematic analysis showed that miR-31 could act as a reliable biomarker for lung cancer diagnostics.18
In addition, Notch homolog 1 (Notch1) was a member of receptor proteins (Notch1–Notch 4) in the Notch signaling pathway involved in cell growth, differentiation, survival, and apoptosis of diverse cell types.19 Importantly, downregulation of the Notch signaling was reported to be associated with smoking and COPD.20 Therefore, the function of Notch1 was investigated in this study.
Based on the above findings, lncRNAs have emerged as crucial biomarkers and regulators in many cancers. Therefore, lncRNA biomarkers in COPD urgently need to be investigated in regard to function and regulatory mechanisms. Moreover, we hypothesized that the association relationship among LINC00612, miR-31-5p, and Notch1 existed in COPD.
Materials and Methods
Patient Specimens
The clinical samples of lung tissue were collected from Chengdu Second People’s Hospital from 2015 to 2019, including NS: non-smokers (n=10), SM: smokers (n=10), and smokers with COPD (n=22). Collection and use of samples were authorized by the Ethics Committee of Chengdu Second People’s Hospital. The removed tissue samples were stored at −80 °C for subsequent study. The written informed consent was offered from each participant, and it was conducted in accordance with the Declaration of Helsinki.
Cell Lines
Human pulmonary microvascular endothelial cells (HPMECs) were acquired from the Chinese Academy of Sciences (Beijing, China). HPMECs were cultured in a humidified atmosphere containing 5% CO2 at 37 °C in Dulbecco’s modified Eagle medium (DMEM; GIBCO BRL, Grand Island, NY, USA) medium. In addition, 10% (v/v) fetal bovine serum (FBS; GIBCO BRL) and 1% penicillin/streptomycin (GIBCO BRL) were added to DMEM. To establish an in vitro model for COPD, HPMECs were incubated with 2.5% cigarette smoke extract (CSE; Murty Pharmaceuticals, Lexington, KY, USA) for different times or treated with increasing doses of CSE for 24 h.
RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated from samples or cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Concentration and integrity of total RNA were tested under Nanodrop 2000c (Thermo Fisher Scientific, Carlsbad, CA, USA). Complementary DNA (cDNA) was synthesized using a reverse transcription kit (Takara, Dalian, China) or All-in-One miRNA cDNA Synthesis Kit (Invitrogen), with isolated RNA as the template. Afterward, SYBR Premix Ex Taq II (Takara) was used to quantify the expression level of RNA under the CFX96™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) with the 2−ΔΔCt method. The expression levels of LINC00612 and Notch1 were standardized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), while small nuclear RNA U6 was regarded as the internal control for miR-31-5p.
The primers were presented:
LINC00612 (sense, 5ʹ-GGCAGAGCCATGTGTTGGATA-3ʹ; antisense, 5ʹ-GTGCTCCCTAATGGCTCACA-3ʹ);
miR-31-5p (sense, 5ʹ-GCCGAGAGGCAAGATGCTG-3ʹ; antisense, 5ʹ-CTCAACTGGTGTCGTGGA-3ʹ);
Notch1 (sense, 5ʹ-GAGGCGTGGCAGACTATGC-3ʹ; antisense, 5ʹ-CTTGTACTCCGTCAGCGTGA-3ʹ);
GAPDH (sense, 5ʹ-TCCCATCACCATCTTCCAGG-3ʹ; antisense, 5ʹ-GATGACCCTTTTGGCTCCC-3ʹ);
U6 (sense, 5ʹ-AACGCTTCACGAATTTGCGT-3ʹ; antisense, 5ʹ-CTCGCTTCGGCAGCACA-3ʹ).
Transfection Assay
miR-31-5p mimic (miR-31-5p), miR-NC, miR-31-5p inhibitor (anti-miR-31-5p), and anti-NC were acquired from Biossci Company (Wuhan, China). Small interfering RNA (siRNA) objecting LINC00612 (si-LINC00612: sense, 5ʹ-AUCUUUCAAGCUAUUUCACAA3ʹ; antisense, 5ʹ-GUGAAAUAGCUUGAAAGAUCU-3ʹ), siRNA scrambled control (si-NC: sense, 5ʹ-UUCUAAGAAGCGGUCACGUTT-3ʹ; antisense, 5ʹ-ACGACACAUAGUCGGUUAATT-3ʹ), and LINC00612-overexpression vector (LINC00612) were synthesized by RiboBio Corporation (Guangzhou, China). For transfection assay, HPMECs were seeded in 24-well plates at the density of approximately 1×105 cells/well. After 48 h of incubation, HPMECs were transfected with the 40 nM of the above-mentioned oligonucleotides or 1 μg of plasmids with Lipofectamine 2000 reagent (Invitrogen) referring to the supplier’s recommendations.
Flow Cytometer Assay
The Annexin V–fluorescein isothiocyanate (FITC) Apoptosis Analysis Kit I (KeyGen, Nanjing, China) was used for apoptosis assay. HPMECs were collected by trypsin and then washed three times with ice-cold phosphate saline buffer (PBS). Then, 100 μL of cell suspension (1×106/mL) was added into a tube and incubated with 5 μL of propidium iodide (PI) and 5 μL of FITC Annexin V in dark conditions. After incubation for 30 min, stained cells were monitored and analyzed by flow cytometry (Applied Biosystems, Foster City, CA, USA) and Flowjo V10 software, respectively.
Caspase-3 Activity Assay
The caspase-3 activity assay was assessed by the enzyme-linked immunosorbent assay (ELISA) kit (Sigma, St. Louis, MO, USA) as described by Wu et al.21 After incubation for 48 h, HPMECs were treated with 2 mM MPP+ for 48 h. After that, cell lysates were collected by centrifuging at 10,000×g for 1 min. The caspase-specific peptide conjugated with the color molecule ρNA was added to lysates from each sample and then incubated for 1 h at 37 °C to measure protease activity. The wavelength of samples was read at 405 nm under a microplate reader (Thermo Fisher Scientific). The bicinchoninic acid (BCA) protein assay (Bio-Rad) was used to quantify the protein level.
ELISA Assay for Interleukin-6 (IL-6), Tumor Necrosis Factor-α (TNF-α), Glutathione (GSH), and Oxidized Glutathione (GSSG)
Cell media were collected from wells and then centrifuged at low speed to remove cell debris. The levels of IL-6 and TNF-α were analyzed with the IL-6 kit and TNF-α kit (Beyotime, Shanghai, China), respectively. In addition, cells were collected and then lysed in lysis buffer. The GSH and GSSG Assay kit (Beyotime) was used to quantify GSH content and GSSG content according to the manufacturer’s protocol.
Measurement of Reactive Oxygen Species (ROS), Malondialdehyde (MDA) Level, and Superoxide Dismutase (SOD) Activity
The ROS Fluorescent Probe kit (Solarbio, Beijing, China) was used to measure intracellular ROS generation. Briefly, cells were lysed and subjected to BCA protein assay. Then, 20 μM of 2ʹ,7ʹ-dichlorofluorescein diacetate (DCFDA) was added into cell lysates at 37 °C for 1 h. After washing by PBS and then high-centrifuging, the fluorescence intensity was detected by microplate reader (Applied Biosystems; excitation: 488 nm wavelength and emission: 525 nm wavelength). Furthermore, the activities of SOD and the level of MDA intracellularly were assessed using SOD and MDA assay kits (Solarbio) referring to the manufacturer’s instruction.
Dual-Luciferase Reporter Assay
Targeting sites between LINC00612 and miR-31-5p were presented by bioinformatics database miRcode (http://www.mircode.org/). Starbase (http://starbase.sysu.edu.cn/) was used to predict binding sites between miR-31-5p and Notch1. Additionally, the fragment sequence of LINC00612 and Notch1 3ʹUTR were cloned into the Dual-Luciferase miRNA Target Expression Vector (Promega, Madison, WI, USA), named as LINC00612-wt and Notch1 3ʹUTR-wt, respectively. Mutations of miR-31-5p-binding sites in LINC00612 and Notch1 3ʹUTR were produced by the Mut Express II Fast Mutagenesis Kit (Vazyme, Nanjing, China) and then cloned into vector, named as LINC00612-mut and Notch1 3ʹUTR-mut, respectively. HPMECs were seeded in 24-well plates (5×104 cells/well) and incubated overnight, then co-transfected with indicated vector and miR-31-5p mimic or control. After 48 h, luciferase activity was measured and normalized to Renilla luciferase activity with the Dual-Luciferase Reporter Assay System (Promega).
RNA Immunoprecipitation (RIP) and RNA Pull-Down Assays
RIP assay was conducted as described by Chen et al.22 The Imprint® RNA immunoprecipitation kit (Sigma) was used for RIP assay in compliance with recommendations. Antibodies used in the RIP assay were bought from Millipore (Billerica, MA, USA), including Argonaute-2 (Ago2; Millipore) or IgG (Millipore) antibodies. Furthermore, HPMECs were transfected with bio-miR-31-5p-wt, bio-miR-31-5p-mut, or bio-miR-NC (RiboBio) at a final concentration of 50 nM for 48 h before harvest. Cell lysates were treated with streptavidin-coated magnetic beads overnight at 4 °C. After centrifugation at 10,000×g for 10 min, RNA complexes bound to the beads were extracted for RT-qPCR assay.
Western Blot Assay
The radio-immunoprecipitation assay (RIPA) buffer (Beyotime) was used to lyse cells or tissues. Samples of extracted protein were separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electroblotted to nitrocellulose filter membranes (Millipore). Membranes were immersed in 5% non-fat milk for 2 h prior to incubation with the primary antibodies, including Notch1 (ab8925; 1:1000 dilution; Abcam, Cambridge, MA, USA), Hes1 (ab108937; 1:1000 dilution; Abcam), Hey2 (ab86010; 1:1000 dilution; Abcam), B-cell lymphoma-2 (Bcl-2; #4223S; 1:1000 dilution; Cell Signaling Technology, Danvers, MA, USA), or Bcl-2-Associated X (Bax; #5023S; 1:1000 dilution; Cell Signaling Technology), with GAPDH (ab181602; 1:2500 dilution; Abcam) as control. Horseradish peroxidase-conjugated secondary antibodies (ab150077; 1:1000 dilution; Abcam) were added to membranes at a dilution ratio of 1:4000, and then incubated for 1 h. The antibody binding was visualized using the Clarity™ Western ECL Substrate Kit (Bio-Rad) under the Bio-Rad ChemiDoc MP system (Bio-Rad).
Statistical Analysis
The data were exhibited as mean±standard deviation, and all statistical analyses were conducted with GraphPad Prism 7 (GraphPad, La Jolla, CA, USA). Student’s t-test or one-way analysis of variance was used to assess the statistical analysis in two treatment groups or multiple groups, respectively. P value less than 0.05 meant a significant difference. Associations between miR-31-5p and LINC00612 or Notch1 expression were analyzed using Pearson’s correlation analysis in COPD tissues.
Results
LINC00612 Was Downregulated in COPD Tissues and HPMECs Exposed to CSE
Initially, the results of the RT-qPCR data suggested that LINC00612 was lower in lung tissues from smokers when compared with non-smokers, and smokers with COPD showed the lowest expression level of LINC00612 in all groups (Figure 1A). Furthermore, when HPMECs were treated with 2.5% CSE for indicated times, LINC00612 was reduced in HPMECs with a time-dependent method (Figure 1B). Surely, treatment with CSE induced the downregulation of LINC00612 in HPMECs with a dose-dependent method (Figure 1C). Therefore, LINC00612 played a critical role in the development of COPD.
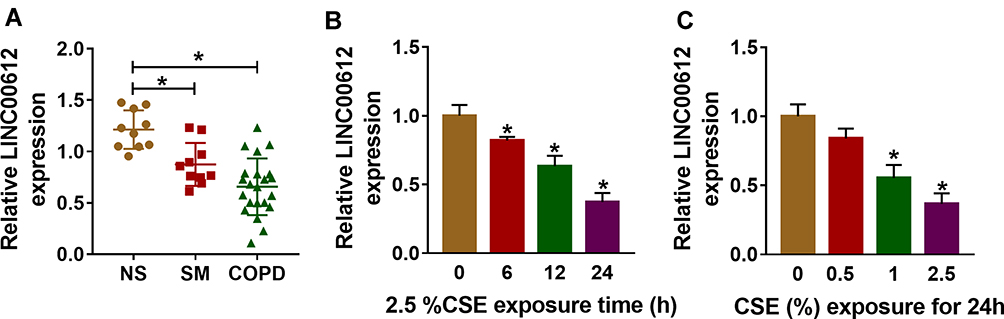 |
Figure 1 The expression level of LINC00612 in lung tissues and HPMECs exposed to CSE. (A) RT-qPCR assay was applied to measure the expression level of LINC00612 in lung tissues (NS: non-smokers, n=10, SM: smokers, n=10, and smokers with COPD, n=22). (B, C) The expression level of LINC00612 was assessed by RT-qPCR assay in HPMECs exposed to 2.5% CSE for indicated times or treated with CSE at different concentrations for 24 h. Data shown are mean±SD and from three independent experiments. *P<0.05. |
CSE-Induced Effects on Apoptosis, Inflammation, and Oxidative Stress in HPMECs Were Weakened by Overexpression of LINC00612
Based on the above results, HPMECs treated with 2.5% CSE for 24 h were used for an in vitro model for COPD. To figure out the potential role of LINC00612 in COPD, overexpression of LINC00612 in HPMECs was conducted by transfection with LINC00612 into HPMECs. As shown in Figure 2A, LINC00612 was upregulated in HPMECs transfected with LINC00612 when compared with the control group. In addition, treatment with 2.5% CSE induced cell apoptosis, which was mitigated by overexpression of LINC00612 in HPMECs (Figure 2B). Consistently, CSE-induced enhancement of caspase-3 activity was abolished in HPMECs by the upregulation of LINC00612 (Figure 2C). Moreover, the increased levels of IL-6 and TNF-α in HPMECs induced by CSE were abolished by treatment with LINC00612 (Figure 2D and E). ROS production and MDA level were increased in HPMECs after treatment with 2.5% CSE, which was overturned by overexpression of LINC00612 (Figure 2F and G). We also noticed that CSE induced the downregulation of SOD activity and the GSH/GSSG ratio was reversed by overexpression of LINC00612 in HPMECs (Figure 2H and I). All results suggested that the upregulation of LINC00612 inhibited CSE-induced apoptosis, inflammation, and oxidative stress in HPMECs, indicating the critical role of LINC00612 in the development of COPD.
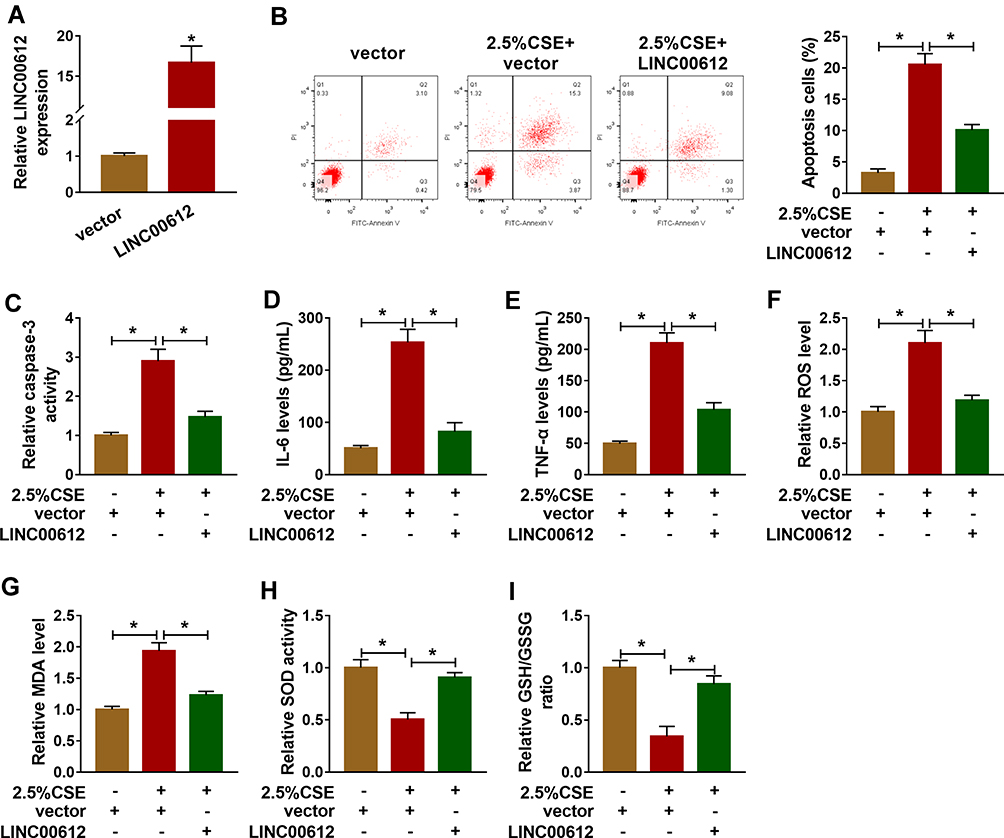 |
Figure 2 LINC00612 regulated apoptosis, inflammation, and oxidative stress in HPMECs treated with CSE. (A) The expression level of LINC00612 was quantified by RT-qPCR in HPMECs transfected with LINC00612 or vector. (B–I) HPMECs were divided into three groups: vector, 2.5% CSE+vector, and 2.5% CSE+LINC00612. (B) The percentage of apoptotic cells was shown by flow cytometry assay in HPMECs. (C) The caspase-3 assay kit was used to examine the caspase-3 activity in HPMECs. (D–E) The expression levels of IL-6 and TNF-α were assessed by ELISA kits in supernatant. (F–I) The oxidative stress was analyzed in HPMECs by measuring ROS production, MDA level, SOD activity, and GSH/GSSG ratio with colorimetric assay kits. Data shown are mean±SD and from three independent experiments. *P<0.05. |
miR-31-5p Was a Target of LINC00612 in HPMECs
As it had been confirmed that LINC00612 played a critical role in the development of COPD, the possible target gene of LINC00612 was searched by bioinformatics tools. As shown in Figure 3A, we identified LINC00612 within the complementary sequence in miR-31-5p. We also found that miR-31-5p was overexpressed in HPMECs after transfection with miR-31-5p mimic (Figure 3B). Afterward, dual-luciferase reporter assay suggested that miR-31-5p mimic could inhibit the luciferase signal of LINC00612-wt when compared with the control group, while the luciferase signal of LINC00612-mut was not affected by miR-31-5p mimic in HPMECs (Figure 3C). By performing the RIP assay, we noticed that LINC00612 and miR-31-5p were enriched in the Ago2 group compared with the IgG group, indicating that LINC00612 and miR-31-5p could bind to Ago2 protein (Figure 3D). In addition, LINC00612 was increased in the bio-miR-31-5p-wt group when compared with the bio-miR-31-5p-mut group and control group, which indicated that miR-31-5p could interact with LINC00612 in HPMECs (Figure 3E). As displayed in Figure 3F, LINC00612 was decreased in HPMECs after silencing of LINC00612. Furthermore, miR-31-5p was overexpressed in HPMECs after knockdown of LINC00612, while upregulation of LINC00612 repressed miR-31-5p expression, suggesting miR-31-5p was negatively regulated by LINC00612 in HPMECs (Figure 3G). Importantly, miR-31-5p was upregulated in COPD tissues and HPMECs exposed to 2.5% CSE for 24 h when compared with matched control groups (Figure 3H–I). Eventually, Pearson’s correlation analysis revealed a negative correlation relationship between LINC00612 and miR-31-5p in COPD tissues (Figure 3J). Interestingly, silencing of miR-31-5p abolished 2.5% CSE-induced effects on HPMECs (Supplementary Figure 1). Conclusively, miR-31-5p might play a key role in apoptosis, inflammation, and oxidative stress in HPMECs treated with CSE, and miR-31-5p was a direct target gene of LINC00612 in HPMECs treated with CSE.
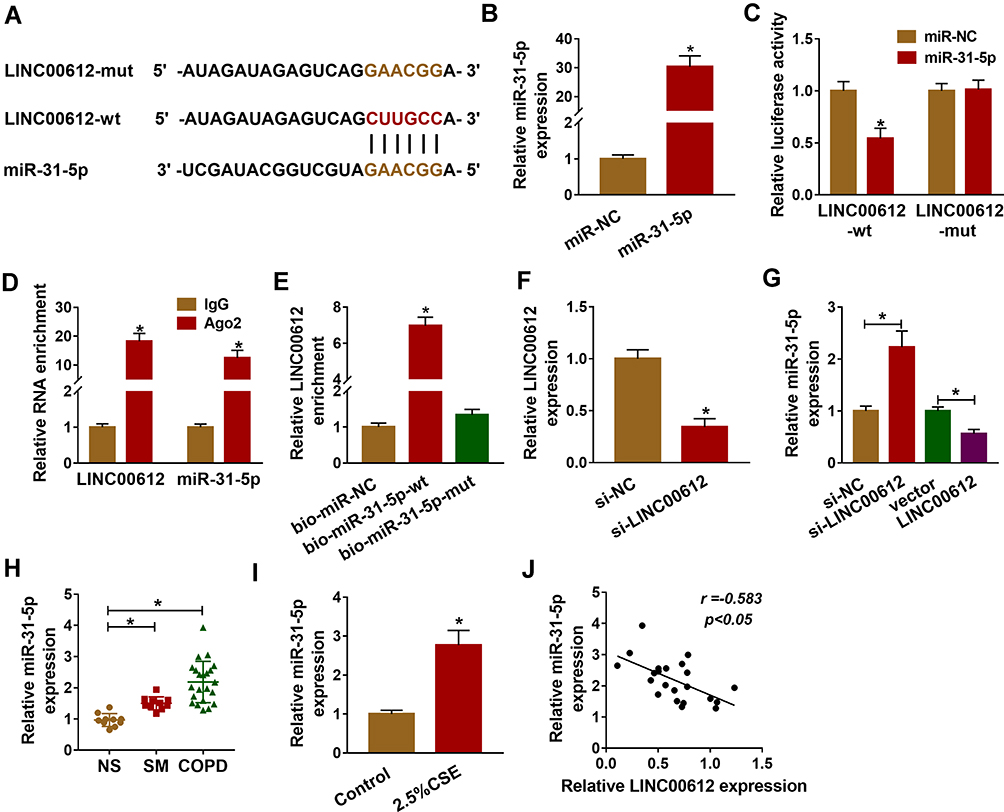 |
Figure 3 miR-31-5p was upregulated in COPD tissues and was a target of LINC00612. (A) LINC00612 had the complementary sequence in miR-31-5p by bioinformatics analysis. (B) Transfection efficiency of miR-31-5p was assessed by RT-qPCR assay in HPMECs, with miR-NC as control. (C) The relative luciferase activity was analyzed by dual-luciferase reporter assay in HPMECs co-transfected with miR-31-5p mimic or miR-NC and indicated luciferase reporter vectors. (D) The enrichments of LINC00612 and miR-31-5p were detected by RT-qPCR assay after RIP assay. (E) The biotinylated bio-miR-31-5p-wt or bio-miR-31-5p-mut was transfected into HPMECs for RNA pull-down assay, with bio-miR-NC as control. (F, G) RT-qPCR assay was used to quantify the expression levels of LINC00612 and miR-31-5p in HPMECs transfected with si-NC, si-LINC00612, vector, or LINC00612. (H, I) The expression level of miR-31-5p was estimated by RT-qPCR assay in lung tissues (NS: non-smokers, n=10, SM: smokers, n=10, and smokers with COPD, n=22) and HPMECs exposed to 2.5% CSE for 24 h. (J) The relationship between LINC00612 and miR-31-5p was analyzed by Pearson’s correlation analysis in COPD tissues. Data shown are mean±SD and from three independent experiments. *P<0.05. |
Upregulation of miR-31-5p Abolished LINC00612-Induced Effects in HPMECs Exposed to CSE
To further confirm the association between miR-31-5p and LINC00612 in HPMECs exposed to CSE, the overexpression vector of LINC00612 and miR-31-5p mimic were used. As shown in Figure 4A, overexpression of LINC00612 induced a reduction in apoptosis in HPMECs exposed to CSE, which was abolished by miR-31-5p overexpression. Overexpression of LINC00612 overturned the CSE-induced enhancement effect on caspase-3 activity, which was reversed by the upregulation of miR-31-5p in HPMECs (Figure 4B). Moreover, the upregulation of miR-31-5p overturned the decrease of IL-6 and TNF-α in HPMECs treated with CSE induced by overexpression of LINC00612 (Figure 4C and D). ELISA indicated that overexpression of miR-31-5p could rescue the ROS and MDA decrease induced by LINC00612 overexpression in CSE-induced HPMECs (Figure 4E and F). Transfection with LINC00612 into HPMECs weakened the inhibitory effects on SOD activity and the GSH/GSSG ratio in HPMECs caused by CSE, which was overturned by co-transfection with miR-31-5p mimic (Figure 4G and H). In summary, the LINC00612/miR-31-5p axis regulated apoptosis, inflammation, and oxidative stress in CSE-induced HPMECs, suggesting the application value of LINC00612miR-31-5p in COPD.
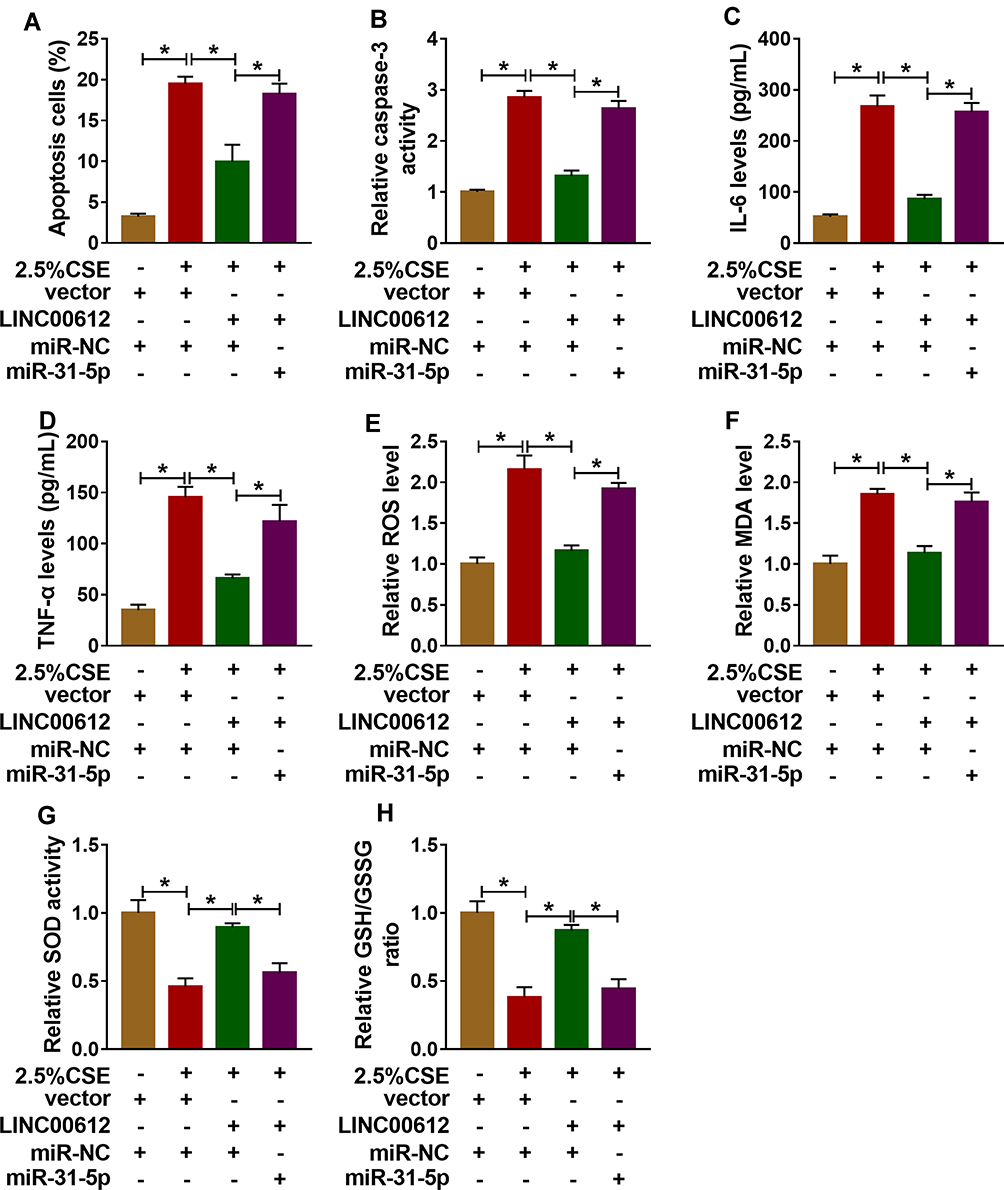 |
Figure 4 Overexpression of LINC00612 abolished CSE-induced effects on apoptosis, inflammation, and oxidative stress in HPMECs by regulation miR-31-5p. (A–H) HPMECs were treated with vector+miR-NC, 2.5% CSE+vector+miR-NC, 2.5% CSE+LINC00612+miR-NC, or 2.5% CSE+LINC00612+miR-31-5p. (A) The flow cytometry assay was performed to monitor cell apoptosis in HPMECs. (B) The caspase-3 activity in HPMECs was assessed by colorimetric assay kit. (C, D) ELISA kits were applied to assess the expression levels of IL-6 and TNF-α in supernatant. (E–H) ROS production, MDA level, SOD activity, and GSH/GSSG ratio were evaluated with assay kits. Data shown are mean±SD and from three independent experiments. *P<0.05. |
miR-31-5p Regulated Notch1 Expression in HPMECs Exposed to CSE
By performing bioinformatics analysis, binding regions between miR-31-5p and 3ʹUTR of Notch1 are displayed in Figure 5A. Using dual-luciferase reporter assay, we found that miR-31-5p mimic inhibited luciferase activity of Notch1 3ʹUTR-wt instead of Notch1 3ʹUTR-mut, implying that miR-31-5p could directly interact with the predicted binding sites of 3ʹUTR of Notch1 (Figure 5B). Additionally, miR-31-5p was decreased in HPMECs treated with anti-miR-31-5p compared with the anti-NC group (Figure 5C). We also noticed that overexpression of miR-31-5p repressed Notch1 expression in HPMECs, and Notch1 was increased in HPMECs after silencing of miR-31-5p (Figure 5D). Importantly, Notch1 was decreased in COPD tissues compared with other groups (Figure 5E). Consistently, CSE induced the downregulation of Notch1 in HPMECs by western blot assay (Figure 5F). Moreover, Notch1 was negatively related to miR-31-5p expression in COPD tissues (Figure 5G). Synthetically, Notch1 was a target of miR-31-5p in HPMECs, and it was downregulated in COPD tissues and HPMECs exposed to CSE.
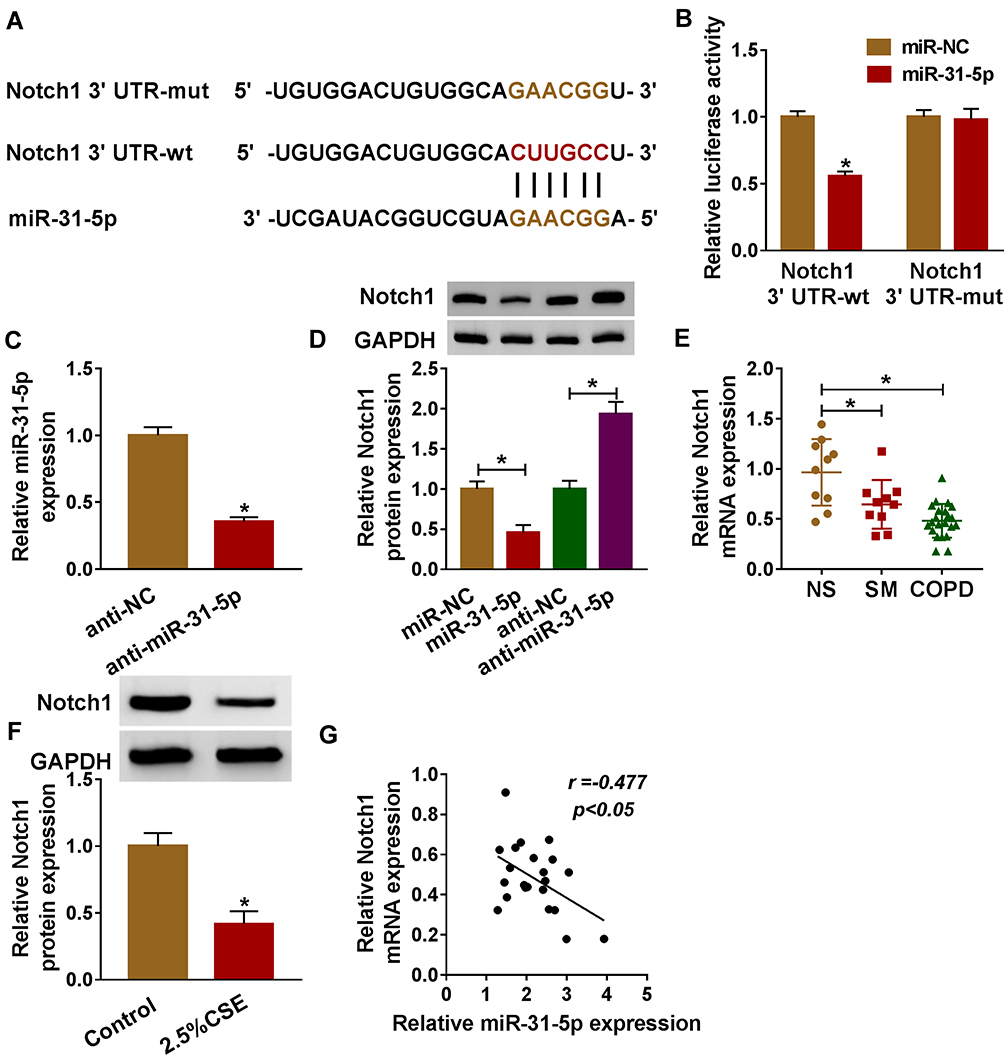 |
Figure 5 Notch1 was a target of miR-31-5p in HPMECs. (A) Binding region between miR-31-5p and 3ʹUTR of Notch1, as well as mutated nucleotides of Notch1 3ʹUTR are shown. (B) Dual-luciferase reporter assay was carried out to examine the luciferase activity in HPMECs. (C) RT-qPCR assay was used to show the expression level of miR-31-5p in HPMECs transfected with anti-miR-31-5p or anti-NC. (D) The protein expression level of Notch1 was measured by western blot assay in HPMECs transfected with anti-miR-31-5p, anti-NC, miR-NC, or miR-31-5p. (E, F) RT-qPCR and western blot assays were conducted to assess Notch1 levels in tissues (NS: non-smokers, n=10, SM: smokers, n=10, and smokers with COPD, n=22) and HPMECs exposed to 2.5% CSE. (G) The relationship between miR-31-5p and Notch1 was analyzed by Pearson’s correlation analysis in COPD tissues. Data shown are mean±SD and from three independent experiments. *P<0.05. |
Notch Signaling Pathway Was Regulated by the LINC00612/miR-31-5p Axis
Firstly, overexpression of Notch1 abolished 2.5% CSE-induced effects on HPMECs (Supplementary Figure 2), which implied that Notch1 regulated apoptosis, inflammation, and oxidative stress in HPMECs exposed to CSE. In addition, western blot assay suggested that suppression effects on the Notch signaling pathway by decreasing Notch1, Hes1, and Hey2 expression induced by CSE were weakened by overexpression of LINC00612, while co-transfection with miR-31-5p counteracted LINC00612-induced effects (Figure 6A–D). We also assessed apoptosis-related protein expression in CSE-induced HPMECs. Bcl-2 was decreased while Bax was increased in CSE-induced HPMECs, which was abrogated by transfection with LINC00612; besides, LINC00612-induced effects on Bcl-2 and Bax expression were overturned by upregulation of miR-31-5p (Figure 6E and F). Therefore, the LINC00612/miR-31-5p/Notch1 axis might regulate COPD development by regulation of the Notch signaling pathway.
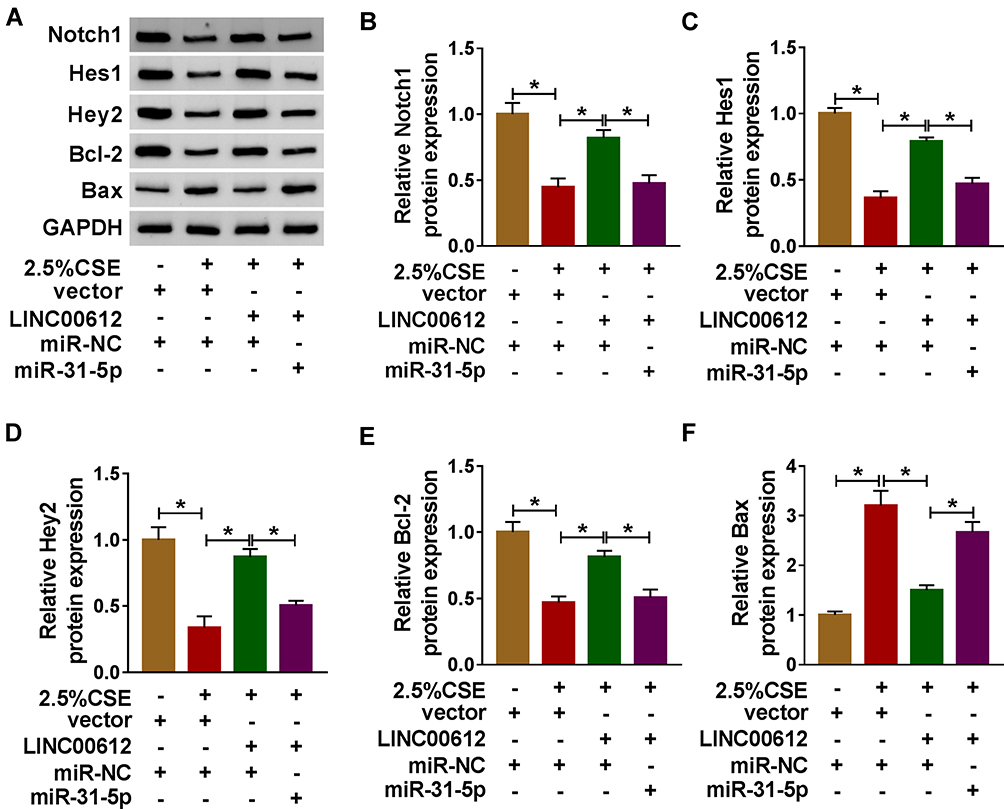 |
Figure 6 The expression levels of Notch signaling pathway-related proteins in CSE-induced HPMECs. (A–F) HPMECs were treated with vector+miR-NC, 2.5% CSE+vector+miR-NC, 2.5% CSE+LINC00612+miR-NC, or 2.5% CSE+LINC00612+miR-31-5p. (A) The representative pictures of western blot assay are presented. (B–F) The protein expression levels of Notch1, Hes1, Hey2, Bcl-2, and Bax were quantified by western blot assay in CSE-induced HPMECs. Data shown are mean±SD and from three independent experiments. *P<0.05. |
Discussion
A large number of research studies had shown the close relationship between cigarette smoke and the development of COPD.23,24 As a common inducement for COPD, cigarette smoke could induce apoptosis, inflammatory response, and oxidative stress of endothelial cells, which aggravates lung injury.25,26 Consistent with previous reports, our research indicated that CSE- induced apoptosis of HPMECs with a dose/time-dependent method. Analogously, Chen et al also revealed that CSE induced apoptosis of vascular endothelial cells dose-dependently and time-dependently, which could lead to lung injury.27
Recently, research studies displayed the several functions of lncRNAs in human diseases, and interactions of miRNA–mRNA–lncRNA might play a key role in treating COPD.12 Currently, LINC00612 was significantly decreased in COPD tissues and CSE-treated HPMECs when compared with matched control groups. The following experiments exactly elucidated the upregulation of LINC00612-inhibited cell apoptosis, inflammation, and oxidative stress in HPMECs induced by CSE, indicating the potential therapeutic value of LINC00612 in COPD. Mechanistically, LINC00612 abolished CSE-induced endothelial cell apoptosis, inflammation, and oxidative stress by mediating the miR-31-5p/Notch1 axis in HPMECs. Bioinformatics analysis and dual-luciferase report assays manifested that miR-31-5p was a direct target of LINC00612. The rescue experiments suggested that LINC00612 was involved in CSE-induced effects of HPMECs, which contributed to the regulation of miR-31-5p. We also found miR-31-5p was upregulated in HPMECs after treatment with CSE. Consistently, Xi et al revealed that CSE notably reinforced the miR-31 level in normal respiratory epithelia and lung cancer cells when compared with controls.28 Furthermore, the knockdown of miR-31-5p abolished CSE-induced effects on HPMECs.
Importantly, Notch signaling modulated the inflammatory response through multiple mechanisms. Inflammatory response and immune dysfunction were associated with the development of COPD.29 Coincidentally, Notch signaling regulated and participated in the differentiation of T lymphocytes.30 Not surprisingly, Notch signaling played a significant role in asthma, allergic pulmonary inflammation, and other lung diseases.31,32 Paul et al confirmed a novel regulatory signal path by which nuclear transcription factor 2 depended on the regulation of Notch1 signaling to control the intracellular oxidation level.33 Additionally, Notch1 has been shown to protect multiple cells against apoptosis, including epithelial cells.34 According to the analysis results of western blot assay, the expression levels of Notch1, Hes1, and Hey2 in CSE-induced HPMECs were obviously lower than those in the control group, and then greatly enhanced after transfection with LINC00612, while miR-31-5p mimic weakened the effects of LINC00612 in CSE-induced HPMECs. All data demonstrated that the expression of Notch1 might be correlated with LINC00612 and miR-31-5p in CSE-induced HPMECs.
Currently, overexpression of LINC00612 markedly inhibits apoptosis, inflammation, and oxidative stress in HPMECs caused by CSE. The present study revealed that the LINC00612/miR-31-5p/Notch1 axis could be a key regulator of apoptosis, inflammation, and oxidative stress in HPMECs induced by CSE, which provided a new perspective to the function of LINC00612 in COPD.
Conclusion
In summary, we revealed the function of LINC00612 in COPD, which could regulate cell apoptosis, inflammation, and oxidative stress in HPMECs exposed to CSE by targeting the miR-31-5p/Notch1 axis, indicating that LINC00612 could function as a potential biomarker and treatment target for COPD in the future.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Siganaki M, Koutsopoulos AV, Neofytou E, et al. Deregulation of apoptosis mediators’ p53 and bcl2 in lung tissue of COPD patients. Respir Res. 2010;11(1):46. doi:10.1186/1465-9921-11-46
2. López‐Campos JL, Tan W, Soriano JB. Global burden of COPD. Respirology. 2016;21(1):14–23. doi:10.1111/resp.12660
3. Barbu C, Iordache M, Man M. Inflammation in COPD: pathogenesis, local and systemic effects. Rom J Morphol Embryol. 2011;52(1):21–27.
4. Milara J, Peiró T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68(5):410–420. doi:10.1136/thoraxjnl-2012-201761
5. Hwang J-W, Chung S, Sundar IK, et al. Cigarette smoke-induced autophagy is regulated by SIRT1–PARP-1-dependent mechanism: implication in pathogenesis of COPD. Arch Biochem Biophys. 2010;500(2):203–209. doi:10.1016/j.abb.2010.05.013
6. Rajendrasozhan S, Yang S-R, Edirisinghe I, Yao H, Adenuga D, Rahman I. Deacetylases and NF-κ B in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid Redox Signal. 2008;10(4):799–812. doi:10.1089/ars.2007.1938
7. Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87(3):1047–1082. doi:10.1152/physrev.00048.2006
8. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47. doi:10.1038/nrg.2015.10
9. Liao Q, Bu D, Sun L, Luo H, Zhao Y. Identification and functional annotation of LncRNAs in human disease. Health Inf Data Anal. 2017;51–60.
10. Li L, Cong Y, Gao X, Wang Y, Lin P. Differential expression profiles of long non-coding RNAs as potential biomarkers for the early diagnosis of acute myocardial infarction. Oncotarget. 2017;8(51):88613. doi:10.18632/oncotarget.20101
11. Miao L, Liu HY, Zhou C, He X. LINC00612 enhances the proliferation and invasion ability of bladder cancer cells as ceRNA by sponging miR-590 to elevate expression of PHF14. J Exp Clin Cancer Res. 2019;38(1):143. doi:10.1186/s13046-019-1149-4
12. Qian Y, Mao ZD, Shi YJ, Liu ZG, Cao Q, Zhang Q. Comprehensive analysis of miRNA-mRNA-lncRNA networks in non-smoking and smoking patients with chronic obstructive pulmonary disease. Cell Physiol Biochem. 2018;50(3):1140–1153. doi:10.1159/000494541
13. Lai EC. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30(4):363–364. doi:10.1038/ng865
14. Gebert LF, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20(1):21–37. doi:10.1038/s41580-018-0045-7
15. Lai YH, Liu H, Chiang WF, et al. MiR-31-5p-ACOX1 axis enhances tumorigenic fitness in oral squamous cell carcinoma via the promigratory prostaglandin E2. Theranostics. 2018;8(2):486. doi:10.7150/thno.22059
16. Lei SL, Zhao H, Yao HL, et al. Regulatory roles of microRNA-708 and microRNA-31 in proliferation, apoptosis and invasion of colorectal cancer cells. Oncol Lett. 2014;8(4):1768–1774. doi:10.3892/ol.2014.2328
17. Zhu B, Cao X, Zhang W, et al. MicroRNA-31-5p enhances the warburg effect via targeting FIH. FASEB J. 2019;33(1):545–556. doi:10.1096/fj.201800803R
18. Gao W, Liu L, Xu J, et al. A systematic analysis of predicted MiR-31-targets identifies a diagnostic and prognostic signature for lung cancer. Biomed Pharmacother. 2014;68(4):419–427. doi:10.1016/j.biopha.2014.03.009
19. Borggrefe T, Oswald F. The notch signaling pathway: transcriptional regulation at notch target genes. Cell Mol Life Sci. 2009;66(10):1631–1646. doi:10.1007/s00018-009-8668-7
20. Tilley AE, Harvey BG, Heguy A, et al. Down-regulation of the notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179(6):457–466. doi:10.1164/rccm.200705-795OC
21. Wu N, Wu G-C, Hu R, Li M, Feng H. Ginsenoside Rh2 inhibits glioma cell proliferation by targeting microRNA-128. Acta Pharmacol Sin. 2011;32(3):345–353. doi:10.1038/aps.2010.220
22. Chen S, Wu -D-D, Sang X-B, et al. The lncRNA HULC functions as an oncogene by targeting ATG7 and ITGB1 in epithelial ovarian carcinoma. Cell Death Dis. 2017;8(10):e3118–e3118. doi:10.1038/cddis.2017.486
23. Eisner MD, Anthonisen N, Coultas D, et al. An official American Thoracic Society public policy statement: novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(5):693–718. doi:10.1164/rccm.200811-1757ST
24. Pauwels NS, Bracke KR, Dupont LL, et al. Role of IL-1α and the Nlrp3/caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. 2011;38(5):1019–1028. doi:10.1183/09031936.00158110
25. Comer DM, Kidney JC, Ennis M, Elborn JS. Airway epithelial cell apoptosis and inflammation in COPD, smokers and nonsmokers. Eur Respir J. 2013;41(5):1058–1067. doi:10.1183/09031936.00063112
26. Chen L, Ge Q, Tjin G, et al. Effects of cigarette smoke extract on human airway smooth muscle cells in COPD. Eur Respir J. 2014;44(3):634–646. doi:10.1183/09031936.00171313
27. Chen Y, Luo H, Kang N, et al. Beraprost sodium attenuates cigarette smoke extract-induced apoptosis in vascular endothelial cells. Mol Biol Rep. 2012;39(12):10447–10457. doi:10.1007/s11033-012-1924-1
28. Xi S, Yang M, Tao Y, et al. Cigarette smoke induces C/EBP-β-mediated activation of miR-31 in normal human respiratory epithelia and lung cancer cells. PLoS One. 2010;5(10):e13764. doi:10.1371/journal.pone.0013764
29. Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol. 2016;38(4):497–515. doi:10.1007/s00281-016-0561-5
30. Gu W, Xu W, Ding T, Guo X, Xu J. Fringe controls naïve CD4+T cells differentiation through modulating notch signaling in asthmatic rat models. PLoS One. 2012;7(10):e47288. doi:10.1371/journal.pone.0047288
31. Kang JH, Kim BS, Uhm TG, et al. γ-secretase inhibitor reduces allergic pulmonary inflammation by modulating Th1 and Th2 responses. Am J Respir Crit Care Med. 2009;179(10):875–882. doi:10.1164/rccm.200806-893OC
32. Zong D, Ouyang R, Li J, Chen Y, Chen P. Notch signaling in lung diseases: focus on notch1 and notch3. Ther Adv Respir Dis. 2016;10(5):468–484. doi:10.1177/1753465816654873
33. Paul MK, Bisht B, Darmawan DO, et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent notch signaling. Cell Stem CellCell Stem Cell. 2014;15(2):199–214. doi:10.1016/j.stem.2014.05.009
34. Rangarajan A, Syal R, Selvarajah S, Chakrabarti O, Sarin A, Krishna S. Activated notch1 signaling cooperates with papillomavirus oncogenes in transformation and generates resistance to apoptosis on matrix withdrawal through PKB/Akt. Virology. 2001;286(1):23–30. doi:10.1006/viro.2001.0867
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.