")
Back to Journals » OncoTargets and Therapy » Volume 13
LHX6 Affects Erlotinib Resistance and Migration of EGFR-Mutant Non-Small-Cell Lung Cancer HCC827 Cells Through Suppressing Wnt/β-Catenin Signaling
Authors Wang Q, Liao J, He Z , Su Y , Lin D, Xu L, Xu H , Lin J
Received 19 April 2020
Accepted for publication 8 October 2020
Published 28 October 2020 Volume 2020:13 Pages 10983—10994
DOI https://doi.org/10.2147/OTT.S258896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Qiang Wang, 1 Jinrong Liao, 2 Zhiyong He, 1, 3 Ying Su, 2 Dong Lin, 1 Ling Xu, 1 Haipeng Xu, 1 Jinghui Lin 1
1 Department of Thoracic Medical Oncology, Fujian Cancer Hospital, Fujian Medical University Cancer Hospital, Fuzhou 350014, People’s Republic of China; 2Department of Radiobiology, Fujian Cancer Hospital, Fujian Medical University Cancer Hospital, Fuzhou 350014, People’s Republic of China; 3Fujian Provincial Key Laboratory of Translation Cancer Medicine, Fuzhou 350014, People’s Republic of China
Correspondence: Zhiyong He
Department of Thoracic Medical Oncology, Fujian Cancer Hospital, Fujian Medical University Cancer Hospital, No. 420 Fuma Road, Fuzhou City, Fujian Province 350014, People’s Republic of China
Email [email protected]
Background: miR-214 has been reported to contribute to erlotinib resistance in non-small-cell lung cancer (NSCLC) through targeting LHX6; however, the molecular mechanisms underlying the involvement of LHX6 in mediating the resistance to EGFR-TKIs in erlotinib-resistant NSCLC HCC827 (HCC827/ER) cells remain unknown. This study aimed to investigate the mechanisms responsible for the contribution of LHX6 to EGFR-TKIs resistance in HCC827/ER cells.
Materials and Methods: HCC827/ER cells were generated by erlotinib treatment at a dose-escalation scheme. LHX6 knockout or overexpression was modeled in HCC827 and HCC827/ER cells, and then erlotinib IC 50 values were measured. The cell migration ability was evaluated using a transwell migration assay, and the TCF/LEF luciferase activity was assessed with a TCF/LEF reporter luciferase assay. LHX6, β-catenin and Cyclin D1 expression was quantified using qPCR and Western blotting assays. In addition, the LHX6 expression was detected in lung cancer and peri-cancer specimens using immunohistochemical staining, and the associations of LHX expression with the clinicopathological characteristics of lung cancer were evaluated.
Results: Lower LHX6 expression was detected in HCC827/ER cells than in HCC827 cells (P < 0.0001), while higher β-catenin expression was seen in HCC827/ER cells than in HCC827 cells (P < 0.001). LHX6 knockout increased erlotinib resistance and cell migration ability in HCC827 cells, and LHX6 overexpression inhibited erlotinib resistance and cell migration ability in HCC827/ER cells. In addition, LHX6 mediated erlotinib resistance and cell migration ability in HCC827/ER cells via the Wnt/β-catenin pathway. Immunohistochemical staining showed lower LHX6 expression in lung cancer specimens relative to peri-cancer specimens, and there were no associations of LHX6 expression with pathologic stage, gender, age or tumor size in lung cancer patients (P > 0.05).
Conclusion: LHX6 down-regulation may induce EGFR-TKIs resistance and increase the migration ability of HCC827/ER cells via activation of the Wnt/β-catenin pathway.
Keywords: non-small-cell lung cancer, epidermal growth factor receptor tyrosine kinase inhibitor, EGFR-TKI, LIM homeobox domain 6, LHX6, erlotinib, chemotherapy resistance, Wnt/β-catenin signaling
Corrigendum for this paper has been published
Further corrigendum has been published
Introduction
Lung cancer leads global cancer morbidity and mortality.1 In 2018, there were 2.09 million new cases suffering from this malignancy (11.6% of total new cancer cases) and 1.76 million deaths attributed to lung cancer (18.4% of total cancer deaths).2 There are two main types of lung cancer, small-cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC),3,4 and NSCLC, which accounts for approximately 85% of all lung cancers, is characterized by relative insensitivity to chemotherapy, high recurrence and poor prognosis.5
The discovery of epidermal growth factor receptor (EGFR) gene mutation has changed the treatment paradigm for advanced NSCLC,5 and EGFR tyrosine kinase inhibitors (EGFR-TKIs) have become the standard care for advanced NSCLC harboring EGFR mutations.6–8 Despite an initial satisfactory response to TKIs, however, development of acquired resistance is inevitable in most NSCLC patients with EGFR mutations after 10–14 months of treatment.9–11 Multiple mechanisms underlying the acquired resistance to EGFR-TKIs have been hypothesized, including secondary EGFR mutation in threonine 790 (T790M), MET amplification, human EGFR 2 (HER2) amplification, transformation from NSCLC to SCLC or conferred epithelial to mesenchymal transition, and loss of phosphatase and tensin homolog (PTEN);12 however, the exact mechanisms have not been fully understood. Recently, there is increasing evidence proving that non-coding RNA contributes to the mechanisms of acquired resistance.12 Our previous study showed the involvement of miR-214 in the acquired resistance to erlotinib in NSCLC through targeting LIM homeobox domain 6 (LHX6).13
LHX6, a member of the LIM-homeobox transcription factor family that plays a critical role in tumor development and progression,14 has shown potential as a novel cancer biomarker as well as a promising therapeutic target for several types of cancers.15 Previous studies have identified LHX6 as a sensitive methylation marker for head and neck squamous cell carcinoma,16 and a putative tumor suppressor gene with epigenetic silencing in lung cancer.17 Further studies showed that LHX6 may serve as a favorable prognostic biomarker for lung adenocarcinoma.18 In addition, LHX6 was found to suppress breast cancer cell growth and invasion through inhibiting Wnt/β‑catenin signaling.19 Our previous study showed that CRISPR-Cas9 system-induced miR-214 knockdown inhibited the resistance to EGFR-TKIs in erlotinib-resistant NSCLC HCC827 (HCC827/ER) cells via its direct target gene LHX6.13 However, the molecular mechanisms underlying the involvement of LHX6 in mediating the resistance to EGFR-TKIs remain unknown in HCC827/ER cells. The present study was therefore designed to investigate the mechanisms responsible for the contribution of LHX6 to the EGFR-TKIs resistance in HCC827/ER cells.
Materials and Methods
Cell Line and Culture
NSCLC HCC827 cell line harboring a deletion mutation at exon 19 of the EGFR gene was purchased from American Type Culture Collection (Manassas, VA, USA) and incubated in RPMI 1640 medium (Gibco; Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA), 100 U/mL penicillin and 100 µg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Generation of HCC827/ER Cells
Erlotinib-resistant HCC827/ER cells were generated as described previously.13 Briefly, HCC827 cells were exposed to erlotinib (Selleckchem; Houston, TX, USA) at dose escalation from 10 to 1600 nM in RPMI 1640 medium containing 10% FBS for 6 months, and then were transferred to erlotinib-free RPMI 1640 medium for a further incubation for 2 months (HCC827/ER cells). Subsequently, both HCC827/ER and HCC827 cells were exposed to erlotinib at concentrations of 100, 200, 400, 800 and 1600 nM in RPMI 1640 medium supplemented with 10% FBS, while erlotinib-free RPMI 1640 medium supplemented with 10% FBS served as controls. The viability of HCC827 and HCC827/ER cells was measured with the MTS assay (Promega; Madison, WI, USA) 72 h post-treatment, and the half maximal inhibitory concentration (IC50) of erlotinib was estimated for HCC827 and HCC827/ER cells using the software SPSS version 17.0 (SPSS, Inc.; Chicago, IL, USA).20 All measurements were repeated in triplicate. Finally, the experimentally induced HCC827/ER cells were identified erlotinib resistant based on the drug IC50 values.
Vector Construction and Viral Infection
The first exon of LHX6 was selected as the target of sgRNA, and a pair of oligonucleotides was designed for sgRNA synthesis using the online CRISPR software (http://crispr.mit.edu/), while AAV-sg served as a control. The synthesized oligonucleotides were annealed and ligated to BsmBI (Thermo Fisher Scientific; Waltham, MA, USA) digested CRISPR/Cas9 vector lentiCRISPRv2. The CDS sequences of LHX6 and β-actin were retrieved in The University of California Santa Cruz (UCSC) Genome Browser Database (http://genome.ucsc.edu/),21 and the sequences of clones containing EcoRI/BamHI and XbaI/BamHI restriction digestion sites were designed, respectively (Table 1). The LHX6 fragment was cloned, ligated to the pLVX-IRES-ZsGreen1 vector, transformed in the Stlb3, and plated into the agar plate containing ampicillin. Positive clones were selected and subjected to Sanger sequencing. Then, the clones with correct sequencing were amplified, and plasmid extraction was done with the Plasmid Mini Preparation Kit (Fermentas, Inc.; Burlington, Canada). The plasmid concentration was quantified using a Nanodrop® ND-1000 spectrophotometer (NanoDrop Technologies, Inc.; Wilmington, DE, USA).
 |
Table 1 Oligo Nucleotide and PCR Primers |
The expression vectors lentiCRISPRv2-LHX6-sg1, lentiCRISPRv2-LHX6-sg2, lentiCRISPRv2-AAV-sg, pLV, pLV-LHX6 and pLV-CNTTB1 were co-transfected with the packaging plus envelope plasmid PsPAX2 and the packaging plasmid pCMV-VSVG into HEK293T cells. At 24 and 48 h post-transfection, the cell culture supernatant was collected and centrifuged at 21,600 r/min at 4°C for 90 min. The sediment was re-suspended in serum-free RPMI 1640 medium, and the viral titer was measured at 107 TU/mL. HCC827 and HCC827/ER cells were seeded onto 6-well plates at a density of 5 × 105 cells per well for 24 h, and the multiplicity of infection (MOI) for each cell line and lentiviral vector combination was all determined at 10. Cells were infected with 6 µg/mL polybrene for 48 h, and screened with 2 µg/mL puromycin for 2 weeks to generate stably transfected HCC827/AAV-sg, HCC827/LHX6-sg1, HCC827/LHX6-sg2, HCC827/ER/pLV, HCC827/ER/pLV-LHX6 and HCC827/ER/pLV-LHX6/CNTTB1 cell lines.
MTS Assay
HCC827, HCC827/AAV-sg, HCC827/LHX6-sg1, HCC827/LHX6-sg2, HCC827/ER, HCC827/ER/pLV and HCC827/ER/pLV-LHX6 cells were seeded onto 96-well plates (Corning, Inc.; Corning, NY, USA) at a density of 5 × 103 cells per well for 24 h. Then, log-phase cells were treated with erlotinib at concentrations of 100, 200, 400, 800 and 1600 nM in RPMI 1640 medium supplemented with 10% FBS, while erlotinib-free RPMI 1640 medium supplemented with 10% FBS served as controls. Following 72 h treatment, the supernatant was removed, and 100 μL of RPMI 1640 medium and 20 μL of MTS solution (Promega; Madison, WI, USA) were added to each well for 2 h further incubation at 37°C containing 5% CO2. The cell viability was measured 72 h post-treatment, and the erlotinib IC50 values were calculated. All measurements were repeated in triplicate.
Transwell Migration Assay
HCC827, HCC827/AAV-sg, HCC827/LHX6-sg1, HCC827/LHX6-sg2, HCC827/ER, HCC827/ER/pLV and HCC827/ER/pLV-LHX6 cells digested in 0.25% trypsin-EDTA (Gibco; Rockville, MD, USA), and re-suspended in serum-free RPMI 1640 medium at a density of 5 × 105 cells/mL. The lower transwell chamber was coated with 1 mg/mL fibronectin (Millipore; Bedford, CA, USA), and added with 200 µL of cells, while the lower chamber was immersed in the RPMI 1640 medium supplemented with 20% FBS. The transwell chamber was inserted in a 24-well plate, which was incubated at 37°C in a humidified atmosphere containing 5% CO2 for 24 h. Then, the chamber was collected, and cells were gently removed from the upper chamber using cotton swabs, washed twice in PBS, fixed in methanol for 15 min and stained with 0.1% crystal violet. Five fields of vision were randomly selected, and the number of cells in each field was counted under an inverted microscope at a magnification of × 200. All assays were repeated in triplicate.
Dual-Luciferase Reporter Assay
HCC827/ER, HCC827/ER/pLV and HCC827/ER/pLV-LHX6 cells were seeded onto 96-well plates at a density of 5 × 103 cells per well for 24 h. Then, the supernatant was discarded, and cells were transfected with 0.1 μg TCF/LEF1 luciferase reporter plasmid (Qiagen GmbH; Hilden, Germany) using the X-treme GENE HP DNA Transfection Reagent (Roche NimbleGen; Madison, WI, USA) following the manufacturer’s instructions. The Fly and Renilla luciferase activities were measured with the Dual-Glo Luciferase Assay System (Promega; Madison, WI, USA) on a Synergy H4 Hybrid Multi-Mode Microplate Reader (BioTeK; Winooski, VT, USA) 24 h post-transfection. All measurements were repeated in triplicate.
qPCR Assay
HCC827 and HCC827/ER cells were seeded onto 6-well plates (Corning, Inc.; Corning, NY, USA) at a density of 4 ×105 cells per well for 24 h. Then, log-phase cells were digested in 0.25% trypsin-EDTA (Gibco; Rockville, MD, USA), and washed twice in PBS. Total RNA was extracted with an RNeasyPlus Mini Kit (Qiagen GmbH; Hilden, Germany), and the RNA concentration was determined using a Nanodrop® ND-1000 spectrophotometer. Subsequently, approximately 1 µg of total RNA was reversely transcribed into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific; Waltham, MA, USA). LHX6 expression was quantified with the Lightcycler 480 SYBR Green I Master (Roche Applied Science; Indianapolis, IN, USA) under the following conditions: at 95°C for 15 min, followed by 40 cycles of at 95°C for 15 s, at 55°C for 30 s and at 72°C for 1 s, while β-actin was used as an internal control. Relative quantity of LHX6 expression was estimated using the 2−ΔΔCT method. All experiments were repeated in triplicate.
Western Blotting Assay
HCC827, HCC827/ER, HCC827/AAV-sg, HCC827/AAV-sg1, HCC827/AAV-sg2, HCC827/ER/pLV, HCC827/ER/pLV-LHX6 and HCC827/ER/pLV-LHX6/pLV-CNTTB1 cells onto 6-well plates at a density of 5 ×105 cells per well for 24 h, and then log-phase cells were digested in 0.25% Trypsin-EDTA, washed twice in PBS, and lysed in cell lysis buffer (1 ×; Cell Signaling Technology; Beverly, MA, USA) containing 1 mM phenylmethylsulfonyl fluoride (PMSF) on ice for 20 min. The solution was centrifuged at 12,000 r/min at 4°C for 15 min and the supernatant was collected. The concentration of total protein was determined with the BCA Protein Assay Kit (Thermo Fisher Scientific; Waltham, MA, USA) and total protein was separated with SDS-PAGE. Subsequently, the blots were transferred to nitrocellulose (NC) membranes and blocked in 3% bovine serum albumin (BSA) at 25°C for 1 h. Subsequently, the blots were incubated in mouse monoclonal anti-LHX6 antibody (1:500 dilution; Santa Cruz Biotechnology; Santa Cruz, CA, USA) at 4°C overnight, while β-actin served as a loading control. The blots were then washed three times in TBST (20 mM Tris-HCl, 150 mM NaCl and 0.05% Tween-20; pH 7.4), of 10 min each time, incubated in anti-mouse/rabbit HRP-conjugated IgG antibody (1:4000 dilution; Cell Signaling Technology; Beverly, MA, USA) at 25°C for 3 h, and washed three times in TBST, of 10 min each time. The protein bands were visualized using an ECL Kit (Thermo Fisher Scientific; Waltham, MA, USA), and the expression level of LHX6 was normalized to that of β-action. All determinations were repeated in triplicate.
Immunohistochemistry (IHC)
The tissue microarray that is comprised of 15 pairs of pathologically confirmed lung cancer and peri-cancer specimens was purchased from Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). Immunohistochemical staining of LHX6 was performed using the ElivisionTM plus Polymer HRP IHC kit (Fuzhou MXB Biotechnologies; Fuzhou, China) according to the manufacturer’s protocol with the primary mouse monoclonal anti-LHX6 antibody (1:200 dilution; Santa Cruz Biotechnology; Santa Cruz, CA, USA). The cytoplasm was stained, and nuclear staining was defined positive. The intensity of staining was scored as following: 0, no obvious staining; 1, weak staining; 2, moderate staining; and 3, strong staining. In addition, the positively stained specimens were scored based on the proportion of the stained cells: 0, < 5% of positively stained cells; 1, 5% to 25% of positively stained cells; 2, 26% to 50% of positively stained cells; 3, 51% to 75% of positively stained cells; 4, 76% to 100% of positively stained cells. The final score was calculated by multiplying the staining intensity score by the percentage score. All scores were independently read by two well-experienced pathologists. A score of 0 to 7 was defined as negative LHX6 expression, and a score of 8 to 12 was defined as positive expression.
Ethical Consideration
This study was approved by the Ethical Review Committee of Fujian Provincial Cancer Hospital (approval no. FJZLYY2015-00179). All experimental procedures were performed in accordance with the Declaration of Helsinki.
Statistical Analysis
All measured data were described as mean ± standard deviation (SD), and all statistical analyses were performed using the software GraphPad Prism version 6.0 (GraphPad Software, Inc.; La Jolla, CA, USA). Comparisons of means between groups were done with Student t-test, with a P value of < 0.05 indicative of statistical significance.
Results
LHX6 Expression Negatively Correlates with β-Catenin Expression in HCC827/ER Cells
Our previous study identified LHX6 as a downstream target gene of miR-214.13 Hereby, the LHX6 expression was found to be significantly lower in HCC827/ER cells than in HCC827 cells at both translational and transcriptional levels (P < 0.0001). Western blotting assay determined significantly higher β-catenin expression in HCC827/ER cells than in HCC827 cells (P < 0.001) (Figure 1). These data indicated a correlation between LHX6 and β-catenin expression in HCC827/ER cells. In addition, the activity of the Wnt/β-catenin signaling pathway was greater in HCC827/ER cells than in HCC827 cells.
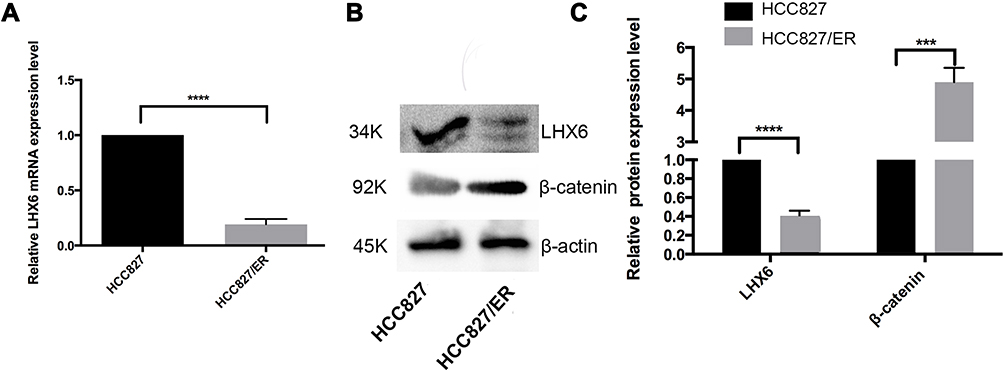 |
Figure 1 LHX6 expression negatively correlates with β-catenin expression in HCC827/ER cells. (A) qPCR assay determines LHX6 gene expression in HCC827 and HCC827/ER cells; (B and C) Western blotting quantifies the LHX6 and β-catenin protein expression in HCC827 and HCC827/ER cells, while β-actin serves as a loading control. ***P < 0.001; ****P < 0.0001. |
LHX6 Knockout Increases the Resistance to Erlotinib and Cell Migration Ability in HCC827 Cells
We used the CRISPR/Cas9 system to knockout LHX6 expression, and HCC827/LHX6-sg1 and HCC827/LHX6-sg2 cells were generated following puromycin screening, while HCC827/AAV-sg cells served as controls. Western blotting detected significantly lower LHX6 expression in HCC827/LHX6-sg1 and HCC827/LHX6-sg2 cells than in HCC827/AAV-sg cells (P < 0.0001), with a more remarkable decline seen in HCC827/LHX6-sg2 cells (Figure 2A and B). MTS assay measured 0.24 μM erlotinib IC50 for HCC827 cells, 0.19 μM for HCC827/AAV-sg cells and 1.34 μM for HCC827/LHX6-sg1 cells, respectively. In addition, the viability of HCC827/LHX6-sg2 cells was significantly greater than that of HCC827/AAV-sg cells post-treatment with erlotinib (P < 0.05) (Figure 2C). Transwell migration assay showed much more HCC827/LHX6-sg2 cells penetrating through the polyester membrane relative to HCC827/AAV-sg cells (P < 0.0001) (Figure 2D).
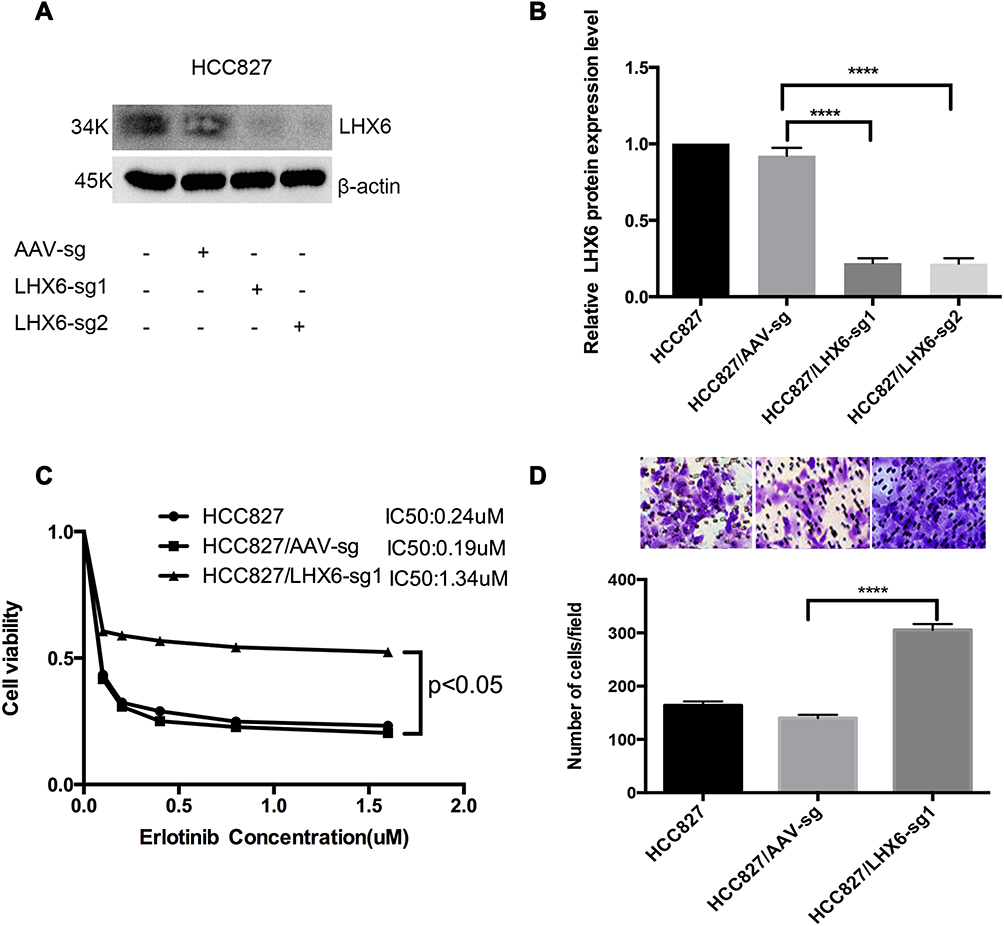 |
Figure 2 LHX6 knockout increases the resistance to erlotinib and cell migration ability in HCC827 cells. (A and B) Western blotting quantifies LHX6 expression in HCC827, HCC827/AAV-sg, HCC827/LHX6-sg1 and HCC827/LHX6-sg2 cells; (C) HCC827, HCC827/AAV-sg1 and HCC827/LHX6-sg2 cells are seeded onto 96-well plates at a density of 5000 cells per well for 24 h, and then cells are exposed to erlotinib at concentrations of 0, 0.1, 0.2, 0.4, 0.8 and 1.6 μM for 72 h. The cell viability and erlotinib IC50 values are measured using an MTS assay; (D) HCC827, HCC827/AAV-sg and HCC827/LHX6-sg2 cells are re-suspended in serum-free RPMI 1640 medium and transferred to the upper chamber of the transwell insert at 1 × 105 cells, and the lower chamber is inserted into the serum-free RPMI 1640 medium supplemented with 20% fetal bovine serum for 24 h. Then, cells in the lower chamber of the Tranwell insert are observed under a microscope. ****P < 0.0001. |
LHX6 Overexpression Inhibits the Resistance to Erlotinib and Cell Migration Ability in HCC827/ER Cells
HCC827/pLV-LHX6 cells were generated by overexpressing LXH6 and puromycin screening in HCC827/ER cells, while HCC827/ER/pLV cells served as a negative control. Western blotting determined significantly higher LHX6 expression in HCC827/ER/pLV-LHX6 cells than in HCC827/ER/pLV cells (P < 0.001) (Figure 3A and B), and MTS assay measured 1.94 μM erlotinib IC50 for HCC827/ER cells, 1.6 μM for HCC827/ER/pLV cells and 0.56 μM for HCC827/ER/pLV-LHX6 cells, respectively. In addition, the viability of HCC827/ER/pLV-LHX6 cells was significantly lower in relative to HCC827/ER/pLV cells post-treatment with erlotinib (P < 0.05) (Figure 3C). Transwell migration assay showed significantly less HCC827/ER/pLV-LHX6 cells penetrating through the polyester membrane than HCC827/ER/pLV cells (P < 0.001) (Figure 3D).
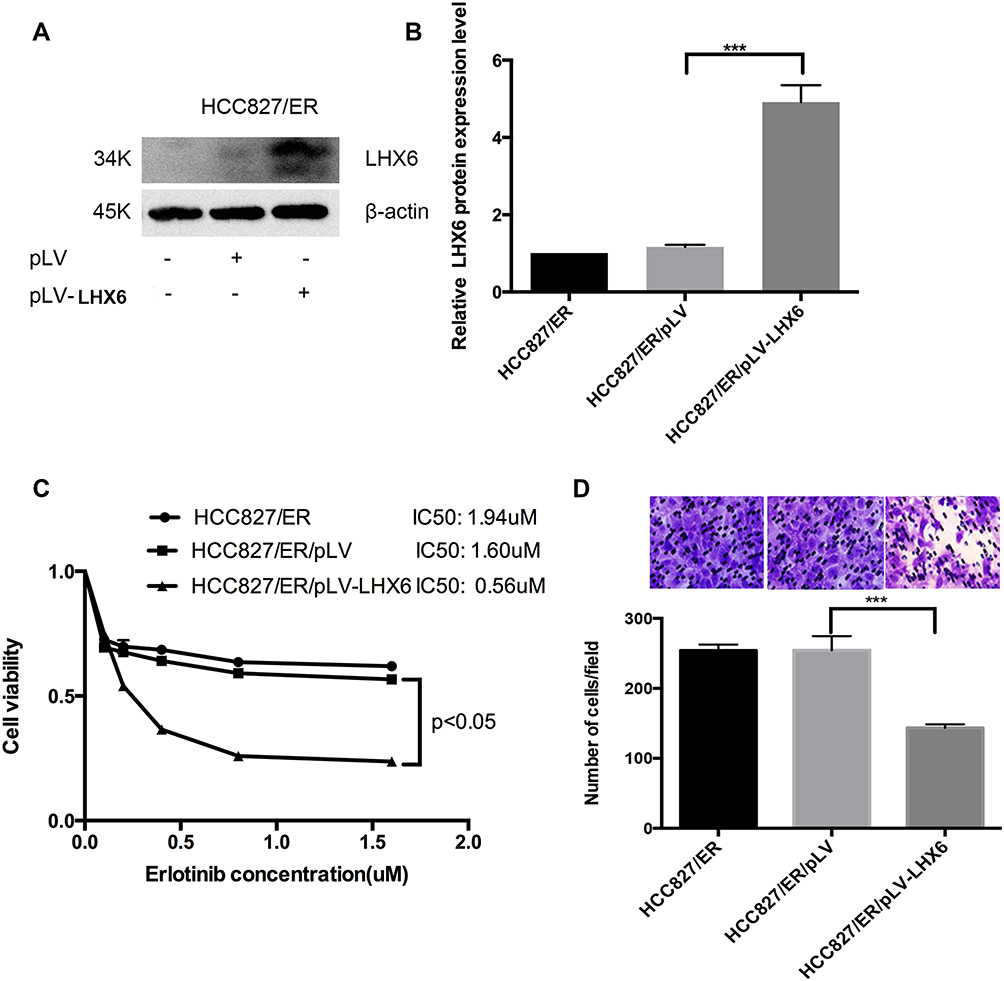 |
Figure 3 LHX6 overexpression inhibits the resistance to erlotinib and cell migration ability in HCC827/ER cells. (A and B) Western blotting determines LHX expression in HCC827/ER, HCC827/ER/pLV and HCC827/ER/pLV-LHX6 cells; (C) HCC827, HCC827/AAV-sg1 and HCC827/LHX6-sg2 cells are seeded onto 96-well plates at a density of 5000 cells per well for 24 h, and then cells are exposed to erlotinib at concentrations of 0, 0.1, 0.2, 0.4, 0.8 and 1.6 μM for 72 h. The cell viability and erlotinib IC50 values are measured using an MTS assay; (D) HCC827, HCC827/AAV-sg and HCC827/LHX6-sg2 cells are re-suspended in serum-free RPMI 1640 medium and transferred to the upper chamber of the transwell insert at 1 × 105 cells, and the lower chamber is inserted into the serum-free RPMI 1640 medium supplemented with 20% fetal bovine serum for 24 h. Then, cells in the lower chamber of the Tranwell insert are observed under a microscope. ***P < 0.001. |
LHX6 Mediates the Resistance to Erlotinib and Cell Migration Ability in HCC827/ER Eells via the Wnt/β-Catenin Signaling Pathway
As shown above, LHX6 expression was significantly lower in HCC827/ER cells than in HCC827 cells, and β-catenin expression was higher in HCC827/ER cells than in HCC827 cells, suggesting a negative correlation between LHX6 expression and the activation of the Wnt/β-catenin signaling pathway. Western blotting determined lower nuclear β-catenin and Cyclin D1 expression in HCC827/ER/pLV-LHX6 cells than in HCC827/ER/pLV cells (P < 0.001) (Figure 4A and B), and the TCF/LEF reporter luciferase assay revealed a lower relative TCF/LEF luciferase activity in HCC827/ER/pLV-LHX6 cells than in HCC827/ER/pLV cells (P < 0.01) (Figure 4C), indicating that overexpressing LHX6 suppresses the Wnt/β-catenin signaling pathway in HCC827/ER cells. In addition, overexpression of β-catenin led to activation of the Wnt/β-catenin signaling pathway in HCC827/ER/pLV-LHX6 cells (P < 0.05), and higher β-catenin expression was determined in HCC827/ER/pLV-LHX6/pLV-CNTTB1 cells than in HCC827/ER/pLV-LHX6 cells (Figure 4D and E). MTS assay measured 0.56 μM erlotinib IC50 for HCC827/ER/pLV-LHX6 cells and 1.6 μM for HCC827/ER/pLV-LHX6/pLV-CNTTB1 cells, respectively, and the viability of HCC827/ER/pLV-LHX6/pLV-CNTTB1 cells was significantly higher than that of HCC827/ER/pLV-LHX6 cells (P < 0.05) (Figure 4F). Transwell migration assay showed more HCC827/ER/pLV-LHX6/pLV-CNTTB1 cells penetrating through the polyester membrane than HCC827/ER/pLV-LHX6 cells (P < 0.001) (Figure 4G). These data demonstrate that down-regulation of LHX6 expression may induce the resistance to erlotinib in HCC827/ER cells through activating the Wnt/β-catenin signaling pathway.
 |
Figure 4 LHX6 mediates the resistance to erlotinib and cell migration ability in HCC827/ER cells via the Wnt/β-catenin signaling pathway. (A and B) Western blotting detects nuclear β-catenin expression in HCC827/ER, HCC827/ER/pLV and HCC827/ER/pLV-LHX6 cells, with PCNA serving as a loading control, and quantifies Cyclin D1 expression in HCC827/ER, HCC827/ER/pLV and HCC827/ER/pLV-LHX6 cells, with β-actin as a loading control; (C) the TCF/LEF reporter luciferase assay measures the relative TCF/LEF luciferase activity in HCC827/ER, HCC827/ER/pLV and HCC827/ER/pLV-LHX6 cells; (D and E) Western blotting quantifies β-catenin expression in HCC827/pLV, HCC827/pLV-LHX6 and HCC827/pLV-LHX6/pLV-CNTTB1 cells; (F) HCC827/ER/pLV, HCC827/ER/pLV-LHX6 and HCC827/ER/pLV-LHX6/pLV-CNTTB1 cells are exposed to erlotinib at concentrations of 0, 0.1, 0.2, 0.4, 0.8 and 1.6 μM for 72 h, and then the cell viability and erlotinib IC50 values are measured using an MTS assay; (G) HCC827/ER/pLV, HCC827/ER/pLV-LHX6 and HCC827/ER/pLV-LHX6/pLV-CNTTB1 cells are re-suspended in serum-free RPMI 1640 medium and transferred to the upper chamber of the transwell insert at 1 × 105 cells, and the lower chamber is inserted into the serum-free RPMI 1640 medium supplemented with 20% fetal bovine serum for 24 h. Then, cells in the lower chamber of the Tranwell insert are observed under a microscope. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
Associations of LHX6 Expression with Clinicopathological Characteristics of Lung Cancer
IHC was performed to detect LHX6 expression in lung cancer and peri-cancer specimens, and positive staining showed apparently brown. LHX6 was found to be predominantly expressed in cytoplasm in both lung cancer specimens and peri-cancer specimens, with less than 5% expression in the nucleus (Figure 5). We detected lower LHX6 expression in lung cancer specimens relative to peri-cancer specimens (Table 2). In addition, a significantly lower staining intensity of LHX6 was measured in cancer specimens than in peri-cancer specimens (P < 0.05), and a low staining intensity was seen in interstitial cells and lymphocytes of peri-cancer specimens. If the mean score of 8 was defined as the cut-off value for the intensity of staining in each field of vision, the percentage of negative LHX6 expression was 86.7% in lung cancer specimens than in peri-cancer specimens (P < 0.05). If an IHC score of 8 was defined as the cut-off value for negative and positive LHX6 expression, and there were no associations of LHX6 expression with pathologic stage (P = 0.299) or age (P = 0.231) in lung cancer patients (Table 3).
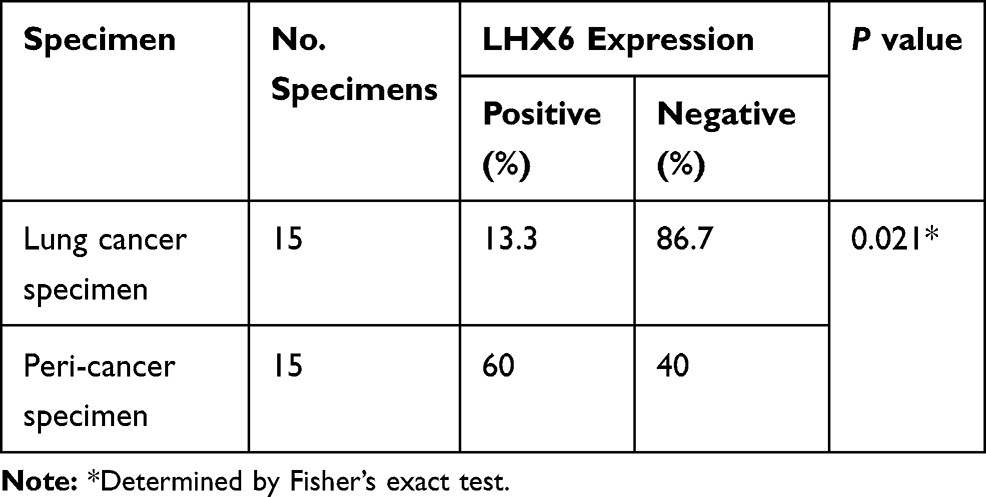 |
Table 2 LHX6 Expression in Lung Cancer and Paired Peri-Cancer Specimens |
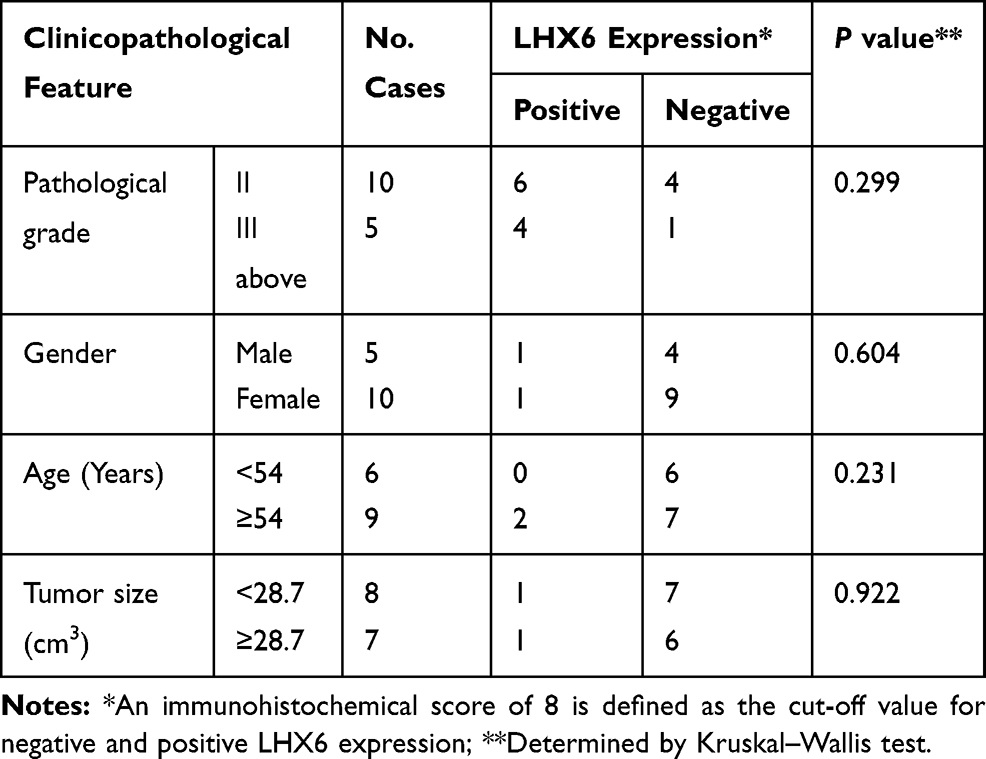 |
Table 3 Associations of LHX6 Expression with the Clinicopathological Characteristics in Patients with Lung Cancer |
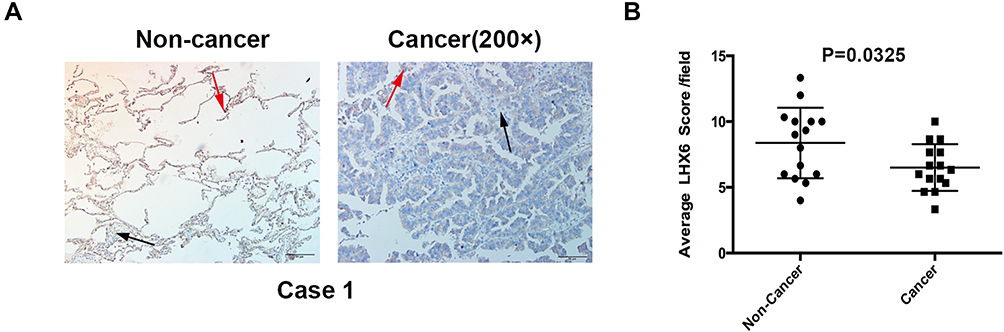 |
Figure 5 Immunohistochemical staining of LHX6 in lung cancer and peri-cancer specimens. (A) immunohistochemical detection of LHX6 expression in lung cancer and peri-cancer specimens (200 ×). The red arrows indicate high LHX6 expression, and the black arrows indicate low LHX6 expression; (B) comparison of LHX6 expression between lung cancer and peri-cancer specimens (P = 0.0325). |
Discussion
Currently, acquired resistance to EGFR-TKIs is the biggest challenge affecting the therapeutic efficacy in patients with EGFR-mutant NSCLC.12,22,23 To date, the known underlying mechanisms mainly include EGFR T790M secondary mutation, MET amplification, HER2 amplification, and loss of PTEN;12 however, the exact mechanisms underlying acquired resistance to EGFR-TKIs have not been fully illustrated. Our previous study demonstrated that overexpressing miR-214 induced the resistance to erlotinib in HCC827/ER cells through targeting LHX6.13 In this study, we detected lower LHX6 expression in HCC827/ER cells than in HCC827 cells at both translational and transcriptional levels, and higher β-catenin expression was seen in HCC827/ER cells than in HCC827 cells. We also found that LHX6 affected the resistance to erlotinib and the cell migration ability in HCC827/ER cells via the Wnt/β-catenin signaling pathway. In addition, IHC detected negative nuclear LHX6 expression in lung cancer specimens and lower LHX6 expression in lung cancer specimens than in peri-cancer specimens.
Epigenetic modification of tumor suppressor genes, such as promoter methylation, histone modification and microRNA regulation, may lead to drug resistance in multiple cancers, including NSCLC.24,25 Previous studies have demonstrated the involvement of LHX gene in tumor development and progression.14 LHX2 was reported to promote breast cancer growth and metastasis,26 and promote pancreatic ductal adenocarcinoma cell proliferation in vitro and in vivo.27 LHX3 was identified as an early-stage and radiosensitivity prognostic biomarker, and a novel potential oncogene in lung adenocarcinoma,28 was required to maintain cancer cell development of high-grade oligodendroglioma.29 LHX4 may be involved in the tumorigenesis of colorectal cancer and leukemia.30,31 LHX9 was detected to be frequently silenced in pediatric malignant astrocytomas by hypermethylation and this epigenetic alteration was found to contribute to glioma cell migration and invasiveness.32 LHX6 expression was found to be downregulated or silenced with hypermethylation status in both lung cancer cell lines and tissues, and LHX6 overexpression caused suppression of cell viability, colony formation and migration and induction of apoptosis and G1/S arrest in lung cancer 95D and H358 cell lines, as well as inhibition of lung cancer cell tumorigenicity in nude mice through upregulating p21 and p53 expression, and downregulating Bcl-2, cyclinD1, c-myc, CD44, and MMP7 expression.17 Another study showed that overexpression of LHX6.1 (an isoform of hLHX6) suppressed the proliferation and tumorigenic phenotype of cervical cancer cells.33 There is also evidence showing LHX6 as a tumor suppressor gene.15,17 Our previous study demonstrated that miR-214 up-regulation increased the resistance to erlotinib in HCC827/ER cells through down-regulating its target gene LHX6 expression.13 In this study, IHC showed that LHX6 was predominantly expressed in the cytoplasm with extremely low expression in the nucleus, and lower LHX6 expression was detected in lung cancer specimens than in peri-cancer specimens; qPCR and Western blotting assays determined lower LHX6 expression in HCC827/ER cells than in HCC827 cells at both transcriptional and translational levels. In addition, the CRISPR-Cas9 system-mediated LHX6 knockdown increased the resistance to erlotinib and cell migration ability in HCC827 cells, and overexpressing LHX6 resulted in a reduction in the resistance to erlotinib and cell migration ability in HCC827/ER cells. We also detected a negative correlation between LHX6 expression and the activation of the Wnt/β-catenin signaling pathway. Our data demonstrate that LHX6 may mediate the resistance to erlotinib via the Wnt/β-catenin signaling pathway.
Wnt/β-catenin signaling is critical for mammalian development and maintenance of cell homeostasis.34 Aberrant expression of Wnt/β-catenin signaling has been detected and has been reported to play an important role in therapeutic resistance in multiple cancers.35–39 β-catenin siRNA-induced inhibition of the Wnt/β-catenin signaling resulted in reversal of multi-drug resistance (MDR) to chemotherapeutics in MDR cholangiocarcinoma QBC939/5-FU cells through down-regulating P-glycoprotein (P-gp) expression.40 In chronic myeloid leukemia stem cells, TKIs induced CD70 expression, which mediated the resistance to TKIs through activating the Wnt/β-catenin pathway.41 In chronic myeloid leukemia, activating of the Wnt/β-catenin signaling is required for intrinsic BCR-ABL1 kinase-independent resistance to TKIs.42 In addition, pharmacologic inhibition of β-catenin was found to suppress EGFR-L858R-T790M mutated lung tumor growth, and genetic deletion of the β-catenin gene dramatically reduced lung tumor formation in EGFR-L858R-T790M transgenic mice.43 In gefitinib-resistant PC9M2 cells generated from gefitinib-sensitive lung cancer PC9 cells, either siRNA-induced knockdown of β-catenin or treatment with a β-catenin inhibitor at least partially restored the sensitivity to gefitinib.44 These data link the Wnt/β-catenin signaling pathway to drug resistance in human cancers.
Zhang and colleagues found that LHX6 directly interacted with paired-like homeodomain transcription factor 2 (PITX2) to inhibit PITX2 transcriptional activities.45 In addition, PITX2 was reported to directly interact with β-catenin to synergistically regulate lymphoid enhancer factor (LEF-1) expression.46 In this study, overexpressing LHX6 resulted in nuclearβ-catenin accumulation, down-regulation of CyclinD1 expression and a remarkable reduction in LEF/TCF luciferase activity in HCC827/ER cells than in HCC827/ER/pLV cells, suggesting that up-regulation of LHX6 may inhibit the Wnt/β-catenin pathway. This may be attributed to LHX6-mediated down-regulation of PITX2 transcriptional activity.45 Then, overexpressing β-catenin was found to reactivate the Wnt/β-catenin pathway, which led to a marked increase in the resistance to erlotinib and cell migration ability in HCC827/ER/pLV-LHX6 cells. Our findings show that down-regulation of LHX6 may induce the resistance to TKIs and increase the cell migration ability in HCC827/ER cells through activating the Wnt/β-catenin pathway.
Conclusions
In summary, we detect lower LHX6 expression in lung cancer specimens than in peri-cancer specimens, and our data demonstrate that LHX6 down-regulation may induce EGFR-TKIs resistance and increase the migration ability of HCC827/ER cells via activation of the Wnt/β-catenin signaling pathway. Our findings suggest that LHX6 may be a promising potential target to overcome erlotinib resistance in HCC827/ER cells, and our data provide new insights to overcome the resistance to EGFR-TKIs in NSCLC.
Funding
This study was supported by grants from the Natural Science Foundation of Fujian Province (grant nos. 2017J01261 and 2016JD1488).
Disclosure
The authors declare no conflicts of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet. 2011;378(9804):1741–1755. doi:10.1016/S0140-6736(11)60165-7
4. Goldstraw P, Ball D, Jett JR, et al. Non-small-cell lung cancer. Lancet. 2011;378(9804):1727–1740. doi:10.1016/S0140-6736(10)62101-0
5. da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. 2011;6(1):49–69. doi:10.1146/annurev-pathol-011110-130206
6. Russo A, Franchina T, Ricciardi GRR, et al. Third generation EGFR TKIs in EGFR-mutated NSCLC: where are we now and where are we going. Crit Rev Oncol Hematol. 2017;117:38–47. doi:10.1016/j.critrevonc.2017.07.003
7. Tan CS, Cho BC, Soo RA. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor -mutant non-small cell lung cancer. Lung Cancer. 2016;93:59–68. doi:10.1016/j.lungcan.2016.01.003
8. Liao BC, Lin CC, Lee JH, Yang JC. Optimal management of EGFR-mutant non-small cell lung cancer with disease progression on first-line tyrosine kinase inhibitor therapy. Lung Cancer. 2017;110:7–13. doi:10.1016/j.lungcan.2017.05.009
9. Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. doi:10.1158/1078-0432.CCR-12-2246
10. Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–5769. doi:10.1158/1078-0432.CCR-06-0714
11. Shih JY, Gow CH, Yang PC. EGFR mutation conferring primary resistance to gefitinib in non–small-cell lung cancer. N Engl J Med. 2005;353(2):207–208. doi:10.1056/NEJM200507143530217
12. Zhang K, Yuan Q. Current mechanism of acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and updated therapy strategies in human nonsmall cell lung cancer. J Cancer Res Ther. 2016;12(7):C131–C137. doi:10.4103/0973-1482.200613
13. Liao J, Lin J, Lin D, et al. Down-regulation of miR-214 reverses erlotinib resistance in non-small-cell lung cancer through up-regulating LHX6 expression. Sci Rep. 2017;7(1):781. doi:10.1038/s41598-017-00901-6
14. Wang X, He HX, Hu X. LIM homeobox transcription factors, a novel subfamily which plays an important role in cancer. Oncol Rep. 2014;31(5):1975–1985. doi:10.3892/or.2014.3112
15. Nathalia E, Theardy MS, Elvira S, et al. Downregulation of tumor-suppressor gene LHX6 in cancer: a systematic review. Rom J Intern Med. 2018;56:135–142.
16. Estécio MR, Youssef EM, Rahal P, et al. LHX6 is a sensitive methylation marker in head and neck carcinomas. Oncogene. 2006;25(36):5018–5026. doi:10.1038/sj.onc.1209509
17. Liu WB, Jiang X, Han F, et al. LHX6 acts as a novel potential tumour suppressor with epigenetic inactivation in lung cancer. Cell Death Dis. 2013;4(10):e882. doi:10.1038/cddis.2013.366
18. Yang J, Han F, Liu W, et al. LHX6, an independent prognostic factor, inhibits lung adenocarcinoma progression through transcriptional silencing of β-catenin. J Cancer. 2017;8(13):2561–2574. doi:10.7150/jca.19972
19. Hu Z, Xie L. LHX6 inhibits breast cancer cell proliferation and invasion via repression of the Wnt/β-catenin signaling pathway. Mol Med Rep. 2015;12(3):4634–4639. doi:10.3892/mmr.2015.3997
20. Guanggang X, Diqiu L, Jianzhong Y, et al. Carbamate insecticide methomyl confers cytotoxicity through DNA damage induction. Food Chem Toxicol. 2013;53:352–358. doi:10.1016/j.fct.2012.12.020
21. Karolchik D, Baertsch R, Diekhans M, et al. The UCSC genome browser database. Nucleic Acids Res. 2003;31(1):51–54. doi:10.1093/nar/gkg129
22. Viloria-Petit AM, Kerbel RS. Acquired resistance to EGFR inhibitors: mechanisms and prevention strategies. Int J Radiat Oncol Biol Phys. 2004;58(3):914–926. doi:10.1016/j.ijrobp.2003.09.091
23. Lim SM, Syn NL, Cho BC, Soo RA. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: mechanisms and therapeutic strategies. Cancer Treat Rev. 2018;65:1–10. doi:10.1016/j.ctrv.2018.02.006
24. Zhang YW, Zheng Y, Wang JZ, et al. Integrated analysis of DNA methylation and mRNA expression profiling reveals candidate genes associated with cisplatin resistance in non-small cell lung cancer. Epigenetics. 2014;9(6):896–909. doi:10.4161/epi.28601
25. Giovannetti E, Erozenci A, Smit J, Danesi R, Peters GJ. Molecular mechanisms underlying the role of microRNAs (miRNAs) in anticancer drug resistance and implications for clinical practice. Crit Rev Oncol Hematol. 2012;81(2):103–122. doi:10.1016/j.critrevonc.2011.03.010
26. Kuzmanov A, Hopfer U, Marti P, Meyer-Schaller N, Yilmaz M, Christofori G. LIM-homeobox gene 2 promotes tumor growth and metastasis by inducing autocrine and paracrine PDGF-B signaling. Mol Oncol. 2014;8(2):401–416. doi:10.1016/j.molonc.2013.12.009
27. Zhou F, Gou S, Xiong J, Wu H, Wang C, Liu T. Oncogenicity of LHX2 in pancreatic ductal adenocarcinoma. Mol Biol Rep. 2014;41(12):8163–8167. doi:10.1007/s11033-014-3716-2
28. Lin X, Li Y, Wang J, et al. LHX3 is an early stage and radiosensitivity prognostic biomarker in lung adenocarcinoma. Oncol Rep. 2017;38(3):1482–1490. doi:10.3892/or.2017.5833
29. Liu H, Liu W, Zhu B, Xu Q, Ni X, Yu J. Lhx3 is required to maintain cancer cell development of high-grade oligodendroglioma. Mol Cell Biochem. 2015;39(1–2):1–5. doi:10.1007/s11010-014-2209-x
30. Cha N, Liu W, Yang N, et al. Oncogenicity of LHX4 in colorectal cancer through Wnt/β-catenin/TCF4 cascade. Tumour Biol. 2014;35(10):10319–10324. doi:10.1007/s13277-014-2210-8
31. Yamaguchi M, Yamamoto K, Miura O. Aberrant expression of the LHX4 LIM-homeobox gene caused by t(1;14)(q25;q32) in chronic myelogenous leukemia in biphenotypic blast crisis. Genes Chromosomes Cancer. 2003;38(3):269–273. doi:10.1002/gcc.10283
32. Vladimirova V, Mikeska T, Waha A, et al. Aberrant methylation and reduced expression of LHX9 in malignant gliomas of childhood. Neoplasia. 2009;11(7):700–711. doi:10.1593/neo.09406
33. Jung S, Jeong D, Kim J, et al. Epigenetic regulation of the potential tumor suppressor gene, hLHX6.1, in human cervical cancer. Int J Oncol. 2011;38:859–869.
34. Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5(9):691–701. doi:10.1038/nrg1427
35. Arqués O, Chicote I, Puig I, et al. Tankyrase inhibition blocks Wnt/β-catenin pathway and reverts resistance to PI3K and AKT inhibitors in the treatment of colorectal cancer. Clin Cancer Res. 2016;22(3):644–656. doi:10.1158/1078-0432.CCR-14-3081
36. Cui J, Jiang W, Wang S, Wang L, Xie K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr Pharm Des. 2012;18(17):2464–2471. doi:10.2174/13816128112092464
37. Nagaraj AB, Joseph P, Kovalenko O, et al. Critical role of Wnt/β-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget. 2015;6(27):23720–23734. doi:10.18632/oncotarget.4690
38. Yao H, Ashihara E, Maekawa T. Targeting the Wnt/β-catenin signaling pathway in human cancers. Expert Opin Ther Targets. 2011;15(7):873–887. doi:10.1517/14728222.2011.577418
39. Pai SG, Carneiro BA, Mota JM, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10(1):101. doi:10.1186/s13045-017-0471-6
40. Shen DY, Zhang W, Zeng X, Liu CQ. Inhibition of Wnt/β-catenin signaling downregulates P-glycoprotein and reverses multi-drug resistance of cholangiocarcinoma. Cancer Sci. 2013;104(10):1303–1308. doi:10.1111/cas.12223
41. Al Baghdadi T, Abonour R, Boswell HS. Novel combination treatments targeting chronic myeloid leukemia stem cells. Clin Lymphoma Myeloma Leuk. 2012;12(2):94–105. doi:10.1016/j.clml.2011.10.003
42. Eiring AM, Khorashad JS, Anderson DJ, et al. β-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia. 2015;29(12):2328–2337. doi:10.1038/leu.2015.196
43. Nakayama S, Sng N, Carretero J, et al. β-catenin contributes to lung tumor development induced by EGFR mutations. Cancer Res. 2014;74(20):5891–5902. doi:10.1158/0008-5472.CAN-14-0184
44. Nakata A, Yoshida R, Yamaguchi R, et al. Elevated β-catenin pathway as a novel target for patients with resistance to EGF receptor targeting drugs. Sci Rep. 2015;5(1):13076. doi:10.1038/srep13076
45. Zhang Z, Gutierrez D, Li X, et al. The LIM homeodomain transcription factor LHX6: a transcriptional repressor that interacts with pituitary homeobox 2 (PITX2) to regulate odontogenesis. J Biol Chem. 2013;288(4):2485–2500. doi:10.1074/jbc.M112.402933
46. Vadlamudi U, Espinoza HM, Ganga M, et al. PITX2, beta-catenin and LEF-1 interact to synergistically regulate the LEF-1 promoter. J Cell Sci. 2005;118(6):1129–1137. doi:10.1242/jcs.01706
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.