")
Back to Journals » Journal of Inflammation Research » Volume 13
“High Treg” Inflammations Promote (Most) Non-Hematologic Cancers While “Low Treg” Inflammations Promote Lymphoid Cancers
Authors Elkoshi Z
Received 11 February 2020
Accepted for publication 29 April 2020
Published 21 May 2020 Volume 2020:13 Pages 209—221
DOI https://doi.org/10.2147/JIR.S249384
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Zeev Elkoshi
Research and Development Department, Taro Pharmaceutical Industries Ltd, Haifa, Israel
Correspondence: Zeev Elkoshi Email [email protected]
Abstract: In an earlier publication, a binary classification of chronic diseases has been proposed. Chronic diseases were classified as “high Treg” or “low Treg” diseases depending on whether the pro-inflammatory or the anti-inflammatory arms of the immune response are deficient. The present work uses this model to analyze the interplay between cancer and the immune system, based on published literature. The work leans upon the etiology of alcohol and tobacco-related malignancies. The main conclusions are: triggers of specific “high Treg” immune reaction promote most non-hematologic cancers, whereas triggers of “low Treg” immune reaction promote lymphomas. The opposite is also true: triggers of specific “high Treg” immune reaction suppress lymphoma, whereas triggers of “low Treg” immune reaction suppress non-hematologic cancers. Both lymphoma and autoimmune diseases are “low Treg” conditions. For this reason, both are promoted by the same panel of “low Treg” bacteria and parasites and are inhibited by “high Treg” triggers. For example, alcohol consumption, a “high Treg” trigger, protects against lymphoma and autoimmune hypothyroidism. In addition, the same immune-modulatory drugs are effective in the treatment of both lymphoma and autoimmune diseases. Like other cancers, lymphoma transforms from a “low Treg” type at early stage of the disease into a “high Treg” type at advanced stages. However, lymphoma is distinguished from most other cancers by the length of time it dwells at an indolent “low Treg” state (many years) before lymphoma cells sensitivity to transforming growth factor-beta is impaired. This impairment stimulates the switch from “low Treg” into “high Treg” response and results in immune escape. The application of this analysis to the pharmacological activity of checkpoint inhibitors forecasts that checkpoint inhibitors would not be effective in low-grade, indolent lymphomas. As of now, checkpoint inhibitors are approved for the treatment of advanced lymphoma only.
Keywords: regulatory T cells, lymphoma, cancer, alcohol consumption, cigarette smoking, checkpoint inhibitors, high Treg, low Treg, inflammation, autoimmune diseases, immune escape
Introduction
In an earlier publication, a binary classification of chronic diseases was proposed.1 Chronic diseases were classified as “high Treg” or “low Treg” diseases depending on whether the pro-inflammatory or the anti-inflammatory arms of the immune response are deficient. Regulatory T cells (Treg) are considered the main anti-inflammatory cellular agents.
Shortly, the immune system elicits two types of reactions: a pro-inflammatory reaction and an anti-inflammatory reaction. Following an acute insult, these two types of response operate in a timely-coordinated manner where the pro-inflammatory arm attacks the invader (in the case of infection) and eliminates damaged cells and debris (in the case of infection and physical insults) while the anti-inflammatory arm drives the immune response back to its steady-state. Following an acute insult, these two arms act simultaneously and in parallel till resolution is achieved.2 In contrast to acute disease, chronic diseases are characterized by an impaired immune response. Generally, chronic diseases may be divided into two classes: (a) “high Treg” diseases where the inflammatory response is deficient and unable to eliminate the insult, with the result of insult persistence, and, (b) “low Treg” diseases where the anti-inflammatory arm is impaired and inflammatory response lasts after the insult has been eliminated, with the result of a collateral damage. Alternatively, “high Treg” diseases can be defined as conditions which can be alleviated by drugs that suppress Treg activity, while “low Treg” diseases can be defined as conditions which can be alleviated by drugs that promote Treg activity.
This binary division explains the association of certain pathogens with cancer and of other pathogens with autoimmune diseases. In addition, the efficacy or inefficacy of immunotherapy by drugs and bacteria, in cancer infections and autoimmunity may be predicted by the binary model.1
The present work utilizes this model to explain how “high Treg” inflammations induce non-hematologic cancers while “low Treg” inflammations induce lymphomas. Published literature data are used for this purpose.
It is widely and almost equivocally asserted that inflammation drives most cancers (see Refs. [3 or 4] for example). It must be realized however that inflammation is not a pre-requisite for the development of cancer, as much as cancer is not a pre-requisite for the development of inflammation. For example, spontaneous tumors may develop in rodents.5
The present article argues that following a prolonged inflammation, most non-hematologic cancers are promoted by a dysfunction of some components of the immune system, while other components elicit a strong pro-inflammatory effect. In contrast, low-grade lymphomas (and possibly few other cancers) are driven by a pro-inflammatory response (i.e., by “inflammation”), with no dysfunction of the pro-inflammatory arm. To be more specific, following prolonged inflammation, most non-hematologic cancers are promoted (mainly) by repressed antigen-specific response (specific “high Treg” response). Lymphoid cancers, on the other hand, are promoted by fully activated adaptive and innate responses (“low Treg” response) during the long period of indolent disease (before immune resistance develops). The implications of this observation are discussed below.
At the outset of this work, Treg cells modes of action under “high Treg” and “low Treg” conditions will be described. Then, literature data concerning malignancies related to alcohol consumption and tobacco smoking will be used to present the immunosuppressive effect of these agents as a cause for the development of non-hematologic malignancies. The next 3 sections will present the immunological similarities between lymphoma and autoimmune diseases. Then, an immune escape mechanism unique to lymphoma will be displayed. This unique escape mechanism will be proposed as an explanation for the uncommon response of lymphoma to inflammation. Lastly, the implication of the binary model to the pharmacological activity of checkpoint inhibitors will be discussed. A discussion section will summarize the findings.
“High Treg” and “Low Treg” Cells Modus Operandi
Regulatory T cells may develop in the thymus following the stimulation of naïve T cells by self-antigens. These regulatory T cells are termed natural regulatory T cells (nTregs). Alternatively, regulatory T cells may be differentiated upon stimulation of naïve T cells in the periphery, in the presence of certain cytokines. These regulatory T cells are termed inducible regulatory T cells (iTreg). nTreg and iTreg cells are phenotypically similar, and both have comparable function in suppressing immune responses.6 Due to this phenotypic similarity, this work refers to regulatory T cells as “Treg cells”, irrespective of their site of origin.
Autoimmune diseases are typical “low Treg” diseases.1 It may be stated that most (if not all) autoimmune diseases are characterized by an impaired Treg activity.7
It is of note that several autoimmune diseases with reduced Tregs activity present normal Treg cells frequency. In the broad sense, an impaired Treg function may be due to either: (a) inadequate number of Treg cells, (b) an impaired suppression mechanism, or (c) a development of T cells resistance to Treg suppressive effect.7 One such impaired mechanism is an insufficient secretion of TGFβ by Treg cells. An animal model involving TGFβ knockout mice demonstrated multifocal inflammation lesions that reminded systemic lupus erythematosus (SLE).8 Moreover, SLE-like autoantibodies were observed in TGFβ deficient mice.9
Solid cancers, on the other hand, are typical “high Treg” diseases.1 Treg cells infiltrate into tumor microenvironment (TME), with the result of Tregs frequency which is ten times higher in TME relative to their frequency in peripheral blood.10 The accumulation of Tregs in TME generates an immune suppressive environment, rich in TGFβ, with the result of tumor proliferation and spread. At an early stage of tumor development, however, TGFβ directly represses tumor growth.11 At this stage, Treg promoters (i.e., drugs that promote Treg cells frequency or function) suppress tumor growth by promoting TGFβ excretion in TME. The effect is similar to the ameliorating effect of Treg promoters (like corticosteroids) on “low Treg” diseases such as autoimmune disorders. Hence, at early stage of development, tumor may be regarded as a “low Treg” condition. This point will be elaborated later in this work.
It must be added that Tregs are not the sole pro-tumor agents in advanced cancer. In addition to Tregs, TME contains abundance of myeloid-derived suppressor cells (MDSCs), tumor‐associated macrophages (TAMs), and immune checkpoint molecules, all contributing to the suppression of the immune response.12 The cross-talk between Tregs, MDSCs, and TAMs is described by several works.13–15 The function of dendritic cells (DC) within TME is complex. In general, DCs maintain the balance between pro- and anti-inflammatory responses of the immune system. A cross-talk between DCs and Treg cells governs this balance whereby DCs induce Tregs differentiation, while Tregs control DCs phenotype and function.16 In the setting of cancer, different DC subsets play different roles. Within TME, plasmacytoid DCs evoke pro-tumor effect by the induction of T cell tolerance whereas conventional DCs (especially cDC1) promote anti-tumor effect by priming cytotoxic T cells immunity.17 Even though all aforementioned immune cells affect cancer, this work focuses on the role of Treg cells only.
Specific “High Treg” Inflammation Promotes Non-Hematologic Tumors
Non-microbial external triggers of the immune response, and in particular continuous stimuli like alcohol abuse or frequent cigarette smoking, differ from triggers elicited by live forms such as microbes or cancer cells, in at least two respects: (a) when the source is external, the immune system is unable to eliminate it, and (b) an external lifeless insult does not transform into a new phenotype in order to manipulate (or “highjack”) the immune response. As a result, the immunological response to lasting environmental stimuli is expected to be a pro-inflammatory “low Treg” response with the result of collateral tissue damage, much like an autoimmune reaction. However, a lasting external trigger may be directly harmful to live tissues (an effect not mediated by the immune system), while an auto-antigen is detrimental only indirectly, by the autoimmune response it elicits. Sometimes when an external insult is continuous or repetitive for a long period of time, the prolonged intensive inflammation may harm the immune system components themselves. When this happens the immunological response is dual: an over-activated response by part of the immune system and an impaired response by other parts. This is the state of a specific “high Treg” response, when only part of the immune system is suppressed, while other parts are fully-activated or even over-activated. The term “high Treg” is preserved here even though the impaired inflammatory response is not merely due to regulatory T cells activity. As will be discussed below, both, heavy alcohol drinking and cigarette smoking induce such a scenario.
Alcohol Abuse
Alcohol drinking directly affects four organs: the oral cavity, the pharynx, the digestive system, and the liver. Other organs are involved as well. Mutational signatures in alcohol-driven carcinomas of the esophagus and liver have been reported.18,19 Alcohol is metabolized into acetaldehyde in hepatocytes mainly by alcohol dehydrogenase and by cytochrome P450 2E1 (CYP2E1). The extensive metabolism of alcohol generates excessive oxidative stress.20 In an increasing order of severity, steatosis, steatohepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma are all alcohol-related diseases that may develop one following the other. The switch from steatosis to hepatitis occurs as Kupffer cells, the resident liver macrophages, differentiate into the pro-inflammatory M1 phenotype. Endotoxins (lipopolysaccharides) translocated from the guts into the liver through the portal vein are the main stimulus for the generation of the pro-inflammatory phenotype of Kupffer cells. Excess of alcohol induces endotoxemia by promoting bacterial growth and by increasing intestinal permeability.20 In their M1 type, Kupffer cells secrete TNFα, IL-1β, IL-6, all pro-inflammatory cytokines. During fibrosis, a process by which activated hepatic stellate cells (HSC) induce collagen deposition in hepatic extracellular matrix, the pro-inflammatory signature prevails.21 The cytokine TGFβ participates in the process of fibrosis, including the trans-differentiation of HSC into myofibroblasts and the maintenance of the myofibroblastic phenotype.22 The pro-inflammatory reaction of the immune system continues during the stage of cirrhosis.23 Even at the initial state of tumor development, a low TGFβ level drives-on the inflammatory process22 by inducing Th17 differentiation.24 In addition, at low TGFβ levels, tumor-associated neutrophils attain their anti-tumor N1 phenotype.25 It is only later, as the tumor progresses and excretes large amounts of TGFβ, that the reaction turns into immunosuppressive and tumorigenic one, since at high TGFβ levels naïve T cells differentiate into immunosuppressive regulatory T cells (Treg).22 High TGFβ serum levels correlate with a poor prognosis of hepatocellular carcinoma.26 It should be realized that alcohol-related liver disease (as well as liver disease which is unrelated to alcohol) progresses from a basically pro-inflammatory state, characterized by high levels of cytokines such as IL-1, IL-6, TNFα, increased phagocytosis by mononuclear cells, and upregulation of HLA-DR and co-stimulatory molecules expression on monocytes and macrophages, to a predominately immunosuppressed state expressed by high levels of cytokines such as IL-10 and TGFβ, impaired phagocytosis by mononuclear cells and down-regulation of HLA-DR on monocytes and macrophages. In this immune-impaired state, neutrophils, B-lymphocytes, and T-lymphocytes effector activities are hampered. However, even at this immune-impaired state, a pro-inflammatory reaction is still highly activated.27 This coexistence between immune exhaustion (mainly toward endotoxins) and an increased level of pro-inflammatory cytokines is also found in hepatocellular carcinoma (HCC).28
A transition from a predominately pro-inflammatory state in steatosis to immune-suppressed state in HCC is also reflected by the increase in Treg cells frequency. Mou et al. reported that Treg percentage evaluated in blood continuously increased from healthy controls to hepatitis B patients to cirrhosis patients, a change which was correlated with a decrease in liver function.29 A meta-analysis of 23 studies demonstrated that Treg cells blood frequency in HCC patients is significantly higher compared to their frequency in blood of healthy subjects.30 At the initial stage of a liver disease, i.e., at steatosis, circulating Tregs frequency is lower than the values observed in healthy subjects.31 Therefore, Tregs systemic frequency increases from steatosis to HCC. In light of the fact that liver disease develops from an inflammatory (“low Treg”) phenotype to an immunosuppressed (“high Treg”) phenotype, and the fact that the risk of HCC increases with the severity of liver disease,32 it can be concluded that “high Treg” conditions (i.e., an immunosuppressive milieu with high TGFβ levels as a hallmark) drive HCC development. In line with this, hepatocellular carcinoma presents an impaired IFNγ production by tumor-associated antigen-specific CD8+T cells.33 This indicates that HCC is a specific “high Treg” disease.
Alcohol consumption induces an increase in the systemic level of TGFβ. Mean TGFβ plasma level in patients with alcohol dependence was more than double the value observed in healthy subjects.34 Patients with alcohol dependence and liver pathology demonstrated TGFβ levels similar to those observed in drinkers without a liver pathology.34 Hence, alcohol is probably the cause for the increasing levels of blood TGFβ and not liver disease.
An association between alcohol abuse and malignancies other than HCC is reported in the literature. The relative risk of 23 different types of cancers as a function of daily alcohol consumption is presented by Bagnardi et al.35 The relative risk is positively-, zero-, or negatively-related to alcohol daily consumption, depending on the specific site of cancer. Out of 21 non-hematologic cancers, the relative risks of 16 were positively dependent on the daily dose of alcohol consumption, while the relative risks of 5 were not affected by alcohol consumption. However, the relative risk of Hodgkin’s lymphoma and Non-Hodgkin’s lymphoma decreased with the daily dose of alcohol, in a statistically significant manner.
Taken together, alcohol consumption induces both, a pro-inflammatory effect and a specific immunodeficiency. Non-hematologic malignancies develop under the immunosuppressive specific “high Treg” response and as a result of high TGFβ levels, even though inflammatory response by unimpaired components of the immune system is highly activated concurrently. The relative risks of most non-hematologic cancers are positively related to the daily dose of alcohol, while few are unaffected (or only slightly affected) by alcohol. Outstandingly, the relative risk of developing lymphoid cancer (Hodgkin’s or non-Hodgkin’s lymphomas) has a statistically significant inverse association with the daily dose of alcohol.
Cigarette Smoking
Cigarette smoke contains about 60 carcinogens, and smoking increases cancer risk by increasing the somatic mutation load.36 As asserted above, a long-lasting external insult is expected to induce a pro-inflammatory reaction. This is indeed the case with cigarette smoking. Cigarette smoke activates aryl hydrocarbon nuclear receptor in airway epithelial cells to overexpress IL-1β, IL-6, and TNFα, all pro-inflammatory cytokines.37 However, similar to the effect of alcohol on the liver tissue, the pro-inflammatory reaction to cigarette smoking not only inflicts local damage to the lungs but also impairs the function of certain members of the immune system. Lugade et al. have demonstrated this dual effect in a mice model, as part of an effort to investigate the relation between cigarette smoke and bacterial infection.38 In this study, mice were first exposed to cigarette smoke and then infected with non-typeable Haemophilus influenzae (NTHI) virus. As a result, macrophages and neutrophil numbers increased in mice airways while IL-1b, IL-6, TNF-a, and IL-17 levels increased in their bronchoalveolar lavage (BAL) fluid. At the same time, an impaired adaptive specific immunity against NTHI was observed. Lower secretion of IL-4 and INF-γ by lung and splenic NTHI-specific T lymphocytes has been reported. The decreased secretion was accompanied by a decrease in the number and frequency of these NTHI-specific T lymphocytes, compared to air-exposed controls. In addition, NTHI-specific B cell responses were impaired in the smoke-exposed mice.38 The impaired T and B lymphocytes specific function was observed in the lungs, BAL, spleen, serum, and bone marrow of the mice.38 Indeed, prolonged smoking history is often associated with an increased prevalence of respiratory infections. Ostadkarampour et al. have reported an increased frequency of a potent suppressor Treg subset and a decreased frequency of Th17 cells in the BAL of young healthy moderate smokers with normal lung function, relative to healthy never-smokers.39
Chronic obstructive pulmonary disease (COPD) is a lung disorder highly associated with cigarette smoking. About 90% of COPD patients are current smokers or ex-smokers, and about 50% of lifelong smokers develop COPD.40 Kalathil et al. reported increased frequencies of Treg cells, myeloid-derived suppressor cells (MDSC), and PD-1+ exhausted effector T cells (PD-1 is an immune checkpoint that promotes self-tolerance to Tregs) in the blood of COPD patients, compared to healthy controls.41 These findings imply a high suppressive activity by the adaptive immune system. The last topic is presented in an excellent review by Bhat et al.42
These observations imply an induction of effector T cell control by tobacco smoking (a “high Treg” effect). The observations are supported by an analysis that compares the somatic mutational load in lungs and head and neck squamous cell carcinomas.43 In this work, Wang et al. analyzed RNA and DNA sequencing data from cases studied as part of The Cancer Genome Atlas (TCGA), as well as two independent gene expression datasets of lung squamous cell carcinoma (LUSC) and head and neck squamous cell (HNSC) tumors.43 The authors pointed to a strong immunosuppressive effect of smoking on local tumor environment (assessed by evaluating the degree of immune cell infiltration in the tumor microenvironment), an effect that was observed also in non-cancerous airway epithelial tissue of smokers and less in ex-smokers.
Similar to HCC, tumor-specific CD8+T cells cytotoxic activity is hampered in lung cancer.44 In addition, dendritic cell function in lung cancer is impaired.45
Collectively, tobacco smoking is a trigger of a “high Treg” effect (and, at the same time, a trigger of a strong pro-inflammatory effect). This immunosuppressive effect hampers the adaptive immune system function.
A meta-analysis of site-specific cancer risks in smokers, carried out on 216 study reports by Gandini et al., reveals 13 cancer sites which are involved with smoking. The relative risk (RR) values for solid tumors in current smokers span from RR=8.96 for lung cancer, to RR=1.51 for liver cancer.46 Lymphomas do not appear in this meta-analysis. However, in another meta-analysis evaluating RR for Hodgkin’s lymphoma (HL) and non-Hodgkin’s lymphoma (NHL), the pulled RR values in ever smokers were 1.05 for NHL and 1.15 for HL.47
These values for HL and NHL are smaller than any pulled RR value observed for non-hematologic tumors in Gandini et al.’s meta-analysis.46
Collectively, it may be stated that (a) cigarette smoking elicits a specific “high Treg” response which impairs the specific adaptive immune system function, (b) in parallel, a pro-inflammatory response is elicited as well, and (c) within smoking population, the risk of any non-hematologic cancer is higher than the risk of a lymphoid cancer.
“Low Treg” Inflammation Promotes Both Lymphoid Cancers and Autoimmune Diseases
The number of tumor-infiltrating foxp3+Treg cells was found to correlate with an improved prognosis of several lymphomas, including follicular lymphoma, germinal center-like diffuse large B-cell lymphoma (DLBCL), and classical Hodgkin’s lymphoma (CHL).48 This is in contrast with the negative effect on the disease outcomes of Tregs infiltration into most solid tumors (Ref. [78], see below). The beneficial effect of Treg tumor infiltration on classical Hodgkin’s lymphoma (CHL) was supported by another study demonstrated that low infiltration of foxp3(+) cells in conjunction with high infiltration of cytotoxic T cells (TIA-1(+) cells) was a predictor of poor CHL prognosis.49 It should be added that in the first aforementioned study,48 the beneficial effect of tumor-infiltrating Tregs was highly statistically significant in CHL, but the statistical significance was borderline in germinal center-like DLBCL and follicular lymphoma. In addition, a negative effect (also a statistical borderline) was observed for non-germinal center–like DLBCL.
There are three groups of infectious agents that are etiologically related to non-Hodgkin lymphoma (NHL): (a) viruses (such as Epstein-Barr virus) that transform lymphatic cells into lymphoma cells (b) human immunodeficiency virus (HIV) that induces a major depletion in CD4+T lymphocytes, impairing this way the control on lymphomas development, and (c) agents that increase NHL risk by an immune-stimulating effect.50 The last group includes Helicobacter pylori, Campylobacter jejuni, Chlamydia psittaci, Borrelia afzelii, Hepatitis C virus, and Plasmodium falciparum.50
The first four members of this group (or bacteria of the same genus) are “low Treg” agents also involved in autoimmune diseases.1 As stated above, autoimmune diseases can be regarded as “low Treg” diseases since most autoimmune diseases are characterized by an impaired Tregs function.7 In addition, Campylobacter jejuni induces a strong pro-inflammatory (“low Treg”) effect in human host with an overexpression of IFNγ, IL-22, IL-17A.51 Moreover, another “low Treg” bacterial genus, streptococcus, which is involved in rheumatic fever,1 is also associated with NHL.52 Cellulitis, a skin infection elicited by staphylococcus or streptococcus bacteria was found to be associated with all NHL subtypes, following an analysis of a large US elderly population database.52 It is of note that rheumatic heart disease, a complication which affects 50% of rheumatic fever patients, has a possible autoimmune etiology.53
Hepatitis C virus (HCV), however, is not known to elicit a “low Treg” immune response. Instead, it may induce NHL by a direct effect on B cells. Three theories supporting this possibility have been proposed.54 Hence, HCV probably belongs to group (a) aforementioned.
The data regarding the immune response to Plasmodium falciparum are conflicting with some studies indicating a “low Treg” response, and other indicating a “high Treg” response. However, malaria, caused by infection with Plasmodium parasites, has been associated with the development of autoimmunity in patients and mouse models.55 It has been shown that Plasmodium DNA induces autoimmunity against erythrocytes by activating a certain population of B cells, which become major producers of autoantibodies that promote malarial anemia.56 This association of Plasmodium falciparum with autoimmunity indicates a “low Treg” signature.
It should be added that a large-scale Scandinavian study demonstrated a highly statistically significant increased-risk of Hodgkin lymphoma in people with personal or family history of certain autoimmune conditions.57
Taken together, all the microbial agents known to induce lymphoma by modulating the immune response are “low Treg” agents which are also associated with autoimmune diseases, “low Treg” conditions by themselves.
The Same Immune-Modulating Drugs are Effective in the Treatment of Both Lymphoma and Autoimmune Diseases
Since both, indolent lymphoma and autoimmune diseases are “low Treg” diseases, Treg promoters are expected to be effective in treating both conditions. Here are few examples:
- Corticosteroids (CS): CS were used for decades as the mainstay for the treatment of autoimmune diseases.58 As demonstrated by Bereshchenko et al,59 glucocorticoid-induced leucine zipper drives Treg cell proliferation and enhances Treg signaling. Hence, CS are Treg promoters. Indeed, CS are very effective in the treatment of lymphoid cancers.60
- Vorinostat: Vorinostat is a histone deacetylase inhibitor approved in the US for the treatment of cutaneous manifestations in patients with cutaneous Tcell lymphoma (CTCL). Histone deacetylase inhibitors promote Treg generation and function.61 It has been shown that vorinostat ameliorates autoimmune encephalomyelitis.62 In addition, vorinostat revert diabetes in the nonobese diabetic (NOD) mouse model of type 1 diabetes.63 Hence, vorinostat is effective in the treatment of both, lymphoma and autoimmunity.
- Sirolimus: Sirolimus inhibits T-cell activation, by blocking calcium-dependent and calcium-independent intracellular signal transduction via the inhibition of mammalian target of rapamycin (mTOR), a critical kinase for cell cycle progression. Even though sirolimus selectively depletes effector T cells, it promotes Treg cells expansion and activity.64 The drug was found effective in patients with systemic lupus erythematosus,65 autoimmune lymphoproliferative syndrome,66 and immune thrombocytopenia.67 At the same time, improved survival was observed in lymphoma patient receiving sirolimus as a prophylaxis for the prevention of graft versus host disease after allogeneic hematopoietic stem-cell transplantation.68
Alcohol Consumption, a “High Treg” Trigger, Confers Protection Against Both Lymphoma and Autoimmune Hypothyroidism
As mentioned above, the risk of lymphoma was found to be lower in people who regularly drink alcohol, relative to alcohol abstainers. The protecting effect observed was positively related to the daily dose of alcohol consumption.35 Similarly, Carle´ et al. have found that moderate alcohol consumption confers considerable protection against development of overt autoimmune hypothyroidism irrespective of sex and type of alcohol consumed.69 This finding is not surprising since alcohol consumption afflicts an increase in TGFβ plasma levels34 (a “high Treg” effect), which is expected to protect against “low Treg” conditions such as indolent lymphoma and autoimmune hypothyroidism.
The Very Late Transformation of Lymphomas from an Indolent Disease to an Aggressive Disease Is the Clue for Their Unique Behavior
As delineated above, at early stages of tumor development, TGFβ controls tumor growth, whereas at advanced stages, TGFβ acquires a pro-oncogenic and pro-metastatic role.11,70 A direct interaction between TGFβ and tumor cells is responsible for this functional switch.
The addition of exogenous TGFβ to cultures of indolent Ki-1 lymphoma cells suppressed IL-2-dependent cells mitosis. However, no suppression was observed in a cell line derived from a case of advanced nodular sclerosis CHL. This last cell line lacked expression of TGFβ and IL-2 receptors. It was suggested that in this way the tumor evades suppression by TGFβ.71 Due to the lack of direct tumor suppression at advanced stage, lymphoma transforms from a disease, where TGFβ induces an anti-tumor “low Treg” effect to a disease where TGFβ exerts a pro-tumor “high Treg” effect. Kadin et al. demonstrated that TGFβ (directly) inhibits tumor propagation of indolent lymphoma but not of advanced lymphoma. Cells of indolent type in cutaneous T cell lymphoma (CTCL) were found to express TGFβRI and TGFβRII, which rendered indolent CTLC cell line responsive to TGFβ inhibitory effect. In contrast, receptors I and II were not detected in advanced CTCL. In accordance with this observation, cell line of advanced CTLC demonstrated resistance to TGFβ inhibitory effect.72 It is important to note that indolent lymphomas, a disease of a “low Treg” type, may last for many years.73
On the other hand, some cancers transform very early to their more resistant type. In lung cancer, for example, this transformation occurs even at the premalignant stage.74 Similarly, SCC transforms very early. An aggressive SCC may develop from verrucous carcinoma, a relatively unaggressive cancer. Verrucous carcinoma expresses TGFβRII (TGFβ receptor II) on the membrane of the neoplastic keratinocytes, which make it vulnerable to the inhibiting effect of TGFβ. Upon the transformation to the more aggressive SCC, TGFβRII receptors are relocated into the cytoplasm where they are protected from TGFβ effect.75 In addition, canonical TGFβ signaling is mediated by a number of downstream proteins including Smad family proteins. Smad4 loss or reduction is a common event even in the early stage of SCC and Smad4 mainly plays a suppressive role in SCC progression.11 These events result in a pro-tumor effect of TGFβ and a “high Treg” character as soon as aggressive SCC forms and even before. Breast cancer is another example where the loss of TGFβRII expression appears at a pre-malignant stage, i.e., at epithelial hyperplastic lesions lacking atypia (EHLA). Moreover, the percentage loss of TGFβRII expression in epithelial cells of EHLA positively correlates with the risk of invasive breast cancer.76
It may be concluded that lymphoma is a type of cancer where the switch from a “low Treg” into a “high Treg” state occurs late. In contrast to other cancers, lymphoma may dwell in a “low Treg” state for many years.
Checkpoint Inhibitors are Not Expected to Be Effective in the Treatment of Indolent Lymphomas
The aforementioned switch in TGFβ direct effect may explain the efficacy and inefficacy of checkpoint inhibitors in different types of cancer.
Nivolumab, a PD-1 immune checkpoint inhibitor is indicated for the treatment of several non-hematologic cancers as well as for the treatment of relapsed or refractory classical Hodgkin lymphoma. PD-1 receptor is a negative regulator of T-cell activity that has been shown to be involved in the control of T-cell immune responses. Inhibiting the PD-1 receptor up-regulates T effector cell responses. Nivolumab (Opdivo®) is therefore a pro-inflammatory, anti-cancer, Treg suppressor. In fact, nivolumab drug label restricts its use to relapsed or refractory CHL (Opdivo®, summary of product characteristics, UK), which is a “high Treg” state of the disease. Drug labels of other PD-1 inhibitors include the same use restriction. In line with that, immune checkpoint inhibitors are not indicated in any type of non-Hodgkin lymphoma, which are all considered “low Treg” cancers, for a long period of time after they emerge.
Discussion
In a meta-analysis of 76 articles encompassing 17 types of cancer, and including 15,512 cancer cases, Shang et al. evaluated the prognostic value of infiltrating Treg cells in different cancer types.77 In cervical cancer, renal cancer, melanoma, hepatocellular carcinoma, gastric cancer, breast cancer, and lung cancer, the extent of tumor infiltration by Treg cells was a statistically significant predictor of unfavorable cancer prognosis (P < 0.05). Tumor-infiltrating Tregs were found predictive of poor prognosis in ovarian cancer as well, but the statistical power was low.77 A study not included in this analysis indicates that Tregs infiltration is predictive of poor prognosis also in esophageal cancer.78 Colorectal and endometrial cancers are the only cancers in Shang’s meta-analysis where Tregs tumor infiltration positively correlates with a statistically significant favorable prognosis. It can be stated that in the majority of non-hematologic cancers, Tregs tumor infiltration is a predictor of poor outcomes.
A cancer trigger, whether chemical or pathogen, may elicit its effect by one of the two ways:
- Promoting tumor genesis by inducing DNA damage which, if misreplicated, leads to an increased burden of somatic mutations and hence a higher probability of acquiring “driver” cancer-related mutations.18,19,36
- Promoting tumor growth by a direct effect that does not involve a genetic damage. For example, through an excessive stimulation of cell receptors, leading to cell proliferation.54
Considering the direct effect of TGFβ on tumors (i.e., the effect that is not mediated by immune response), TGFβ may repress tumor growth at early stage or promote it at late stages.11 In contrast, the effect mediated through TGFβ control of the immune response is generally pro-cancer,79 and occurs late, as TGFβ accumulates.25 Other effects on tumor microenvironment are all pro-tumor and include effect on fibroblasts and effect on extracellular matrix (leading to epigenetic changes, hypoperfusion, and hypoxia). In addition, TGFβ affects epithelial–mesenchymal transition (EMT), a process which induces metastasis.70 Therefore, the overall effect of TGFβ on early non-hematologic tumor can be positive, zero, or negative. The effect on advanced non-hematologic tumor is expected to be positive (pro-tumor).
Out of 21 non-hematologic cancers reported in alcohol abusers, the highest relative risk (RR), was observed for pharynx and oral cavity cancers (case control pooled RR>5 in heavy drinkers).35 The high risk of oral cavity cancer may be related to DNA damage following extensive direct exposure of these sites to alcohol. Somatic mutations were reported in oral cavity cancer specimens, with TP53 the most mutated gene in early stages of the disease.80 Mutations in NOTCH1 and PIK3CA were found to be associated with worse overall survival.80
The direct anti-tumor effect of TGFβ becomes impaired at later cancer stages. As explained above, uniquely to lymphoma (and few other cancers) this switch occurs relatively late. TGFβ (directly) inhibits tumor propagation of indolent lymphoma but not of advanced lymphoma.72 Indolent lymphoma, however, may last for many years.73 The present work proposes that in indolent lymphoma, direct inhibition overrides other effects and induces cancer suppression for a long period of time. This assumption is supported by: (1) the lower risk of lymphoma in alcohol consumers relative to alcohol abstainers,35 (2) the negative dependence of this risk on the daily dose of alcohol,35 and (3) the favorable effect of Treg cells intra-tumor infiltration on the prognosis of lymphoma.48,49 It is proposed that in (most) non-hematologic cancers, the opposite effects are observed because the impairment in TGFβ pathway occurs early or even at the pre-malignant stage, as seen in lung cancer, squamous cell carcinoma, and breast cancer.74–76 Therefore, the risk of most solid cancers increases with the daily dose of alcohol consumption35 and Treg intratumor infiltration in most solid cancers correlates with a poor prognosis.77
Urinary bladder cancer is another example where this transformation occurs relatively late. In bladder cancer, TGFβ-RI (TGFβ receptor I) reduced expression has been reported in bladder tumor specimens from patients with bladder transitional cell carcinoma (the most common type of bladder cancer). This reduction in TGFβ-RI expression was statistically significant only for Grade 3 (advanced stage) bladder cancer patients.81 The authors propose that the reduction in TGFβ-RI receptor expression in advanced stage renders bladder carcinoma cells resistant to the direct suppressive effect of TGFβ on their growth (observed in the initial stages of the disease).81 This decreased expression can be regarded as an immune escape mechanism at stage 3. This means that at Grades 1 and 2, the disease is “low Treg” and tumor infiltration by Treg cells should correlate with good prognosis (if estimated before Grade 3). In accordance with this prediction, a clinical study demonstrated a positive correlation between Treg tumor infiltration and a better survival rate in urinary bladder cancer patients.82
In cigarette smoking, lymphoma direct suppression by TGFβ83 is counteracted by tobacco mutagenic effect43 and by TGFβ induced immune suppression. The net effect is a slight increase in the risk of lymphoma in smokers compared to non-smokers.46 However, due to the long-lasting effect of direct tumor suppression,73 the risk of lymphoma, which is estimated over time, is lower than the risk of any non-hematologic malignancy involved with cigarette smoking.46
The “high Treg” response to alcohol or cigarette smoke is dual: a high (specific and systemic) suppressive effect by the adaptive immune system, accompanied by a high pro-inflammatory effect mediated by cytokines such as IL-6, TNFα, IL-1, and IL-17 and by neutrophilic oxidative stress.27,42 On the other hand, “low Treg” microorganisms do not present this dual effect since they do not suppress the adaptive immune system. “Low Treg” bacteria and parasites induce Th17 rather than Treg differentiation. For example, an intranasal infection of mice with Group A Streptococcus induced predominant Th17 differentiation.84 In addition, it was found that memory or effector CD4+ T cells, produced following mice nasal priming with S. pyogenes, are not T-regulatory cells.85
A low Treg activity promotes lymphoma, for the same reason it promotes autoimmune diseases: low TGFβ levels do not suppress the antigen (or auto-antigen)-induced adaptive response.
Adapting the same rationale, “low Treg” microbes are expected to slowdown non-hematologic tumor growth because they promote the pro-inflammatory anti-cancer response. In fact, Streptococcus pyogenes, a pathogen involved with NHL, is among several “low Treg” bacteria that have demonstrated such an anti-tumor activity.1,86
It can be stated that most non-hematologic cancers are promoted by a “high Treg” immunosuppressive reaction of cancer-antigen-specific Tregs.38,44,77 At the same time, the pro-inflammatory arm is activated as well. In this respect, the conventional conception of cancers promoted by inflammation is misleading, since the indispensable role of the immune suppression is ignored.
Of course, inflammation induces cancer. However, without a specific suppression of the immune response, growth and propagation of cancers would not be promoted. The simultaneous activity of both, a pro-inflammatory response and a specific immunosuppressed response is probably the optimal scenario for cancer (and infection) propagation or the worth scenario for host wellbeing.
Summary
The present work proposes an application of the binary classification model of chronic diseases,1 with the aim of analyzing the interplay between cancer triggers, tumor growth, and the immune response.
Key Findings
- The majority of non-hematologic cancers (most cancers) demonstrate a specific “high Treg” phenotype. Lymphoma, on the other hand, starts as “low Treg” disease with an unimpaired inflammatory response and transforms into a “high Treg” phenotype at advanced stages.
- “High Treg” triggers (chemicals or microbes that induce immunodeficiency) promote (most) non-hematologic cancers.
- “High Treg” triggers that promote non-hematologic cancers either repress lymphoma or marginally promote it (less than they promote any other cancer).
- “Low Treg” triggers (bacteria or parasites that induce a pro-inflammatory reaction) promote lymphoma.
- “Low Treg” triggers inhibit non-hematologic cancers.
- Both lymphoma and autoimmune diseases are “low Treg” conditions.
- The same immune-modulating drugs (Treg promoters) are effective in the treatment of both lymphoma and autoimmune diseases.
- The same panel of immune-modulating bacteria and parasites that induce non-Hodgkin lymphomas are also associated with autoimmune diseases.
The key findings are presented in Figure 1.
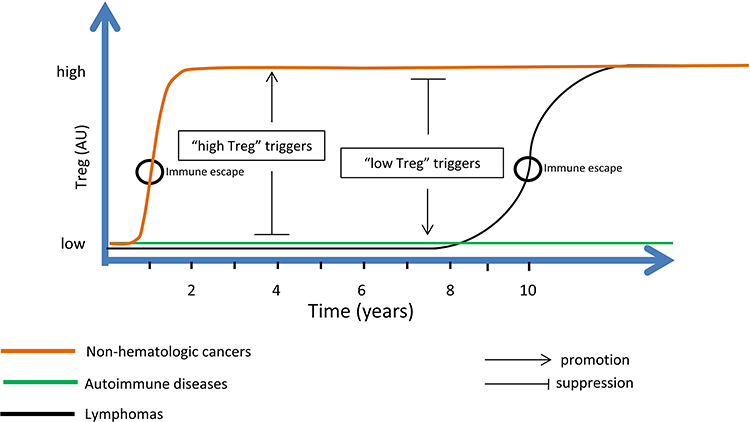 |
Figure 1 TGFβ exerts an anti-tumor effect in early cancer development; however, it evolves into a pro-tumor effect as disease progresses. This functional switch, which may be regarded as an immune escape mechanism, occurs as TGFβ pathways are hampered with the result of a decreased anti-tumor suppressive effect by TGFβ. Other effects of TGFβ, such as immunosuppression, EMT, effect on fibroblasts, and effect on extracellular matrix, are all pro-tumor. Early cancer stages correspond to a “low Treg” disease when immune response is fully active. At advanced cancer stages, specific anti-tumor T cell activity is impaired while other pro-inflammatory components of the immune system are highly activated. This corresponds to a specific “high Treg” state of the disease. Therefore, cancers evolve from a “low Treg” state where tumor proliferation is controlled, into a specific “high Treg” state where this control loosens. Lymphoma is unique among cancers by the length of time it may reside at a “low Treg” state (years). During this indolent low-grade stage, lymphoma treatment is relatively effective. “High Treg” triggers promote “high Treg” states but suppress “low Treg” states. “Low Treg” triggers promote “low Treg” states but suppress “high Treg” states. For example, alcohol abuse, a “high Treg” trigger, increases the risk of most non-hematologic cancers but decreases the risk of lymphomas. Similarly, alcohol consumption confers a protection against autoimmune hypothyroidism. On the other hand, the same set of “low Treg” bacteria and parasites that promotes (indolent) lymphoma also promotes autoimmune diseases, all “low Treg” conditions. Abbreviation: EMT, epithelial–mesenchymal transition; TGFβ, transforming growth factor-beta; Treg, regulatory T cells. |
A Proposed Explanation of the Key Findings
- All cancers start as “low Treg” diseases and transform later into “high Treg” more aggressive diseases.
- At its initial “low Treg” state, the tumor is sensitive to the direct suppressive effect of TGFβ. In addition, the anti-cancer immune response is not impaired at this early stage. Therefore, in its initial “low Treg” state, cancer tumor is more responsive to the treatment.
- Later on, as tumor expands and TGFβ accumulates, some TGFβ pathways are blocked and the tumor becomes insensitive to the suppressive effect of TGFβ. Besides, under these high TGFβ levels, the differentiation of naïve T cells to Treg cells prevails (over the alternative Th17 differentiation route). At this “high Treg” state, the anti-cancer immune response is hampered by Treg’s effect.
- Both properties that characterize the “high Treg” state: the decreased sensitivity of tumor to TGFβ suppressive effect and the impaired immune response, promote cancer propagation and spread.
- Lymphoma is unique among cancers by the long period of time (years) it resides in the “low Treg” indolent state. During this “low Treg” state, lymphoma is more responsive to treatment. In addition, it may be repressed by “high Treg” inflammation due to the high levels of TGFβ involved with this inflammation.
Model Predictions
- Checkpoint inhibitors are expected to be effective at all stages of non-hematologic cancers.
- Checkpoint inhibitors are expected to be effective in lymphoma suppression only at advanced stage, following the development of TGFβ resistance.
Insights Derived from the Etiology of Alcohol and Tobacco-Related Cancers
- Cancer is promoted by a simultaneous engagement of both a high pro-inflammatory response and a hampered specific adaptive response.
- The immunodeficiency elicited by an antigen is antigen-specific (i.e., only the specific immune reaction against a specific antigen is suppressed).
- The conventional conception that cancers are promoted by inflammation is misleading since it ignores the indispensable role of adaptive immunity dysfunction in cancer development.
Disclaimer
The views and opinions expressed, and/or conclusions drawn, in this article are those of the author and do not necessarily reflect those of Taro Pharmaceutical Industries Ltd., its affiliates, directors or employees.
Disclosure
Zeev Elkoshi is employed by Taro Pharmaceutical Industries Ltd. The author reports no other conflicts of interest in this work.
References
1. Elkoshi Z. The binary classification of chronic diseases. J Inflamm Res. 2019;12:319–333. doi:10.2147/JIR.S227279
2. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. 2016;7:160. doi:10.3389/fimmu.2016.00160
3. Todoric J, Antonucci L, Karin M. Targeting inflammation in cancer prevention and therapy. Cancer Prev Res (Phila). 2016;9(12):895–905. doi:10.1158/1940-6207.CAPR-16-0209
4. Ritter B, Greten FR. Modulating inflammation for cancer therapy. J Exp Med. 2019;216(6):1234–1243. doi:10.1084/jem.20181739
5. Son WC, Gopinath C. Early occurrence of spontaneous tumors in CD-1 mice and sprague—dawley rats. Toxicol Pathol. 2004;32(4):371–374. doi:10.1080/01926230490440871
6. Deng G. Tumor-infiltrating regulatory T cells: origins and features. Am J Clin Exp Immunol. 2018;7(5):81–87.
7. Buckner JH. Mechanisms of impaired regulation by CD4+CD25+FOXP3+regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10(12):849–859. doi:10.1038/nri2889
8. Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-β 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi:10.1038/359693a0
9. Dang H, Geiser AG, Letterio JJ, et al. SLE-like autoantibodies and Sjogren’s syndrome-like lymphoproliferation in TGF-β knockout mice. J Immunol. 1995;155:3205–3212.
10. Tada Y, Togashi Y, Kotani D, et al. Targeting VEGFR2 with Ramucirumab strongly impacts effector/activated regulatory T cells and CD8+ T cells in the tumor microenvironment. J Immunother Cancer. 2018;6(1):106. doi:10.1186/s40425-018-0403-1
11. Wu F, Weigel KJ, Zhou H, Wang XJ. Paradoxical roles of TGF-β signaling in suppressing and promoting squamous cell carcinoma. Acta Biochim Biophys Sin (Shanghai). 2018;50(1):98–105. doi:10.1093/abbs/gmx127
12. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 2019;110(7):2080–2089. doi:10.1111/cas.14069
13. Fujimura T, Kambayashi Y, Aiba S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology. 2012;1(8):1433–1434. doi:10.4161/onci.21176
14. Zhang C, Wang S, Yang C, Rong R. The crosstalk between myeloid derived suppressor cells and immune cells: to establish immune tolerance in transplantation. J Immunol Res. 2016;2016:4986797. doi:10.1155/2016/4986797
15. Singh S, Mehta N, Lilan J, Budhthoki MB, Chao F, Yong L. Initiative action of tumor-associated macrophage during tumor metastasis. Biochim Open. 2017;4:8–18. doi:10.1016/j.biopen.2016.11.002
16. Kornete M, Piccirillo CA. Functional crosstalk between dendritic cells and Foxp3(+) regulatory T cells in the maintenance of immune tolerance. Front Immunol. 2012;3:165. doi:10.3389/fimmu.2012.00165
17. Wylie W, Macri M, Mintern M, Waithman W. Dendritic cells and cancer: from biology to therapeutic intervention. Cancers (Basel). 2019;11(4):521. doi:10.3390/cancers11040521
18. Li XC, Wang MY, Yang M, et al. A mutational signature associated with alcohol consumption and prognostically significantly mutated driver genes in esophageal squamous cell carcinoma. Ann Oncol. 2018;29(4):938–944. doi:10.1093/annonc/mdy011
19. Li X, Xu W, Kang W, et al. Genomic analysis of liver cancer unveils novel driver genes and distinct prognostic features. Theranostics. 2018;8(6):1740–1751. doi:10.7150/thno.22010
20. Osna NA, Donohue TM
21. Holt AP, Salmon M, Buckley CD, Adams DH. Immune interactions in hepatic fibrosis. Clin Liver Dis. 2008;12(4):861–82, x. doi:10.1016/j.cld.2008.07.002
22. Fabregat I, Moreno-Càceres J, Sánchez A, et al. TGF-β signalling and liver disease. FEBS J. 2016;283(12):2219–2232. doi:10.1111/febs.13665
23. Prystupa A, Kiciński P, Sak J, et al. Proinflammatory cytokines (IL-1α, IL-6) and hepatocyte growth factor in patients with alcoholic liver cirrhosis. Gastroenterol Res Pract. 2015;2015:532615. doi:10.1155/2015/532615
24. Zhang G. The role of transforming growth factor β in T helper 17 differentiation. Immunology. 2018;155(1):24–35. doi:10.1111/imm.12938
25. Sagiv JY, Michaeli J, Assi S, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10(4):562–573. doi:10.1016/j.celrep.2014.12.039
26. Lin TH, Shao YY, Chan SY, Huang CY, Hsu CH, Cheng AL. High serum transforming growth factor-β1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib. Clin Cancer Res. 2015;21(16):3678–3684. doi:10.1158/1078-0432
27. Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol. 2014;61(6):1385–1396. doi:10.1016/j.jhep.2014.08.010
28. Yu LX, Ling Y, Wang HY. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis Oncol. 2018;2(1):6. doi:10.1038/s41698-018-0048-z
29. Mou H, Wu S, Zhao G, Wang J. Changes of Th17/Treg ratio in the transition of chronic hepatitis B to liver cirrhosis and correlations with liver function and inflammation. Exp Ther Med. 2019;17(4):2963–2968. doi:10.3892/etm.2019.7299
30. Zhao HQ, Li WM, Lu ZQ, Yao YM. Roles of tregs in development of hepatocellular carcinoma: a meta-analysis. World J Gastroenterol. 2014;20(24):7971–7978. doi:10.3748/wjg.v20.i24.7971
31. Van Herck MA, Weyler J, Kwanten WJ, et al. The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Front Immunol. 2019;10:82. doi:10.3389/fimmu.2019.00082
32. Sakurai T, Kudo M. Molecular link between liver fibrosis and hepatocellular carcinoma. Liver Cancer. 2013;2(3–4):365–366. doi:10.1159/000343851
33. Flecken T, Schmidt N, Hild S, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59(4):1415–1426. doi:10.1002/hep.26731
34. Kim YK, Lee BC, Ham BJ, et al. Increased transforming growth factor-beta1 in alcohol dependence. J Korean Med Sci. 2009;24(5):941–944. doi:10.3346/jkms.2009.24.5.941
35. Bagnardi V, Rota M, Botteri E, et al. Alcohol consumption and site-specific cancer risk: a comprehensive dose-response meta-analysis. Br J Cancer. 2015;112(3):580–593. doi:10.1038/bjc.2014.579
36. Alexandrov LB, Ju YS, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354(6312):618–622. doi:10.1126/science.aag029
37. Chiba T, Chihara J, Furue M. Role of the Arylhydrocarbon Receptor (AhR) in the pathology of asthma and COPD. J Allergy (Cairo). 2012;2012:372384. doi:10.1155/2012/372384
38. Lugade AA, Bogner PN, Thatcher TH, Sime PJ, Phipps RP, Thanavala Y. Cigarette smoke exposure exacerbates lung inflammation and compromises immunity to bacterial infection. J Immunol. 2014;192(11):5226–5235. doi:10.4049/jimmunol.1302584
39. Ostadkarampour M, Müller M, Öckinger J, et al. Distinctive regulatory T cells and altered cytokine profile locally in the airways of young smokers with normal lung function. PLoS One. 2016;11(10):e0164751. doi:10.1371/journal.pone.0164751
40. Ward H. Oxford Handbook of Epidemiology for Clinicians. Oxford University Press;2012:289–290. ISBN 978-0-19-165478-7
41. Kalathil SG, Lugade AA, Pradhan V, et al. T-Regulatory cells and programmed death 11 T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(1):40–50. doi:10.1164/rccm.201312-2293OC
42. Bhat TA, Panzica L, Kalathil SG, Thanavala Y. Immune dysfunction in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2015;12(Suppl 2):S169–S175. doi:10.1513/AnnalsATS.201503-126AW
43. Wang J, Linxweiler M, Yang W, Chan TA, Morris LGT. Immunomodulatory and immunotherapeutic implications of tobacco smoking in squamous cell carcinomas and normal airway epithelium. Oncotarget. 2019;10(39):3835–3839. doi:10.18632/oncotarget.26982
44. Prado-Garcia H, Romero-Garcia S, Aguilar-Cazares D, Meneses-Flores M, Lopez-Gonzalez JS. Tumor-induced CD8+ T-cell dysfunction in lung cancer patients. Clin Dev Immunol. 2012;2012:741741. doi:10.1155/2012/741741
45. Wang JB, Huang X, Li FR. Impaired dendritic cell functions in lung cancer: a review of recent advances and future perspectives. Cancer Commun. 2019;39(1):43. doi:10.1186/s40880-019-0387-3
46. Gandini S, Botteri E, Iodice S, et al. Tobacco smoking and cancer: a meta‐analysis. Int J Cancer. 2008;122(1):155–164. doi:10.1002/ijc.23033
47. Sergentanis TN, Kanavidis P, Michelakos T, Petridou ET. Cigarette smoking and risk of lymphoma in adults: a comprehensive meta-analysis on hodgkin and non-hodgkin disease. Eur J Cancer Prev. 2013;22(2):131–150. doi:10.1097/CEJ.0b013e328355ed08
48. Tzankov A, Meier C, Hirschmann P, Went P, Pileri SA, Dirnhofer S. Correlation of high numbers of intratumoral FOXP3+ regulatory T cells with improved survival in germinal center-like diffuse large B-cell lymphoma, follicular lymphoma and classical hodgkin’s lymphoma. Haematologica. 2008;93(2):193–200. doi:10.3324/haematol.11702
49. Alvaro T, Lejeune M, Salvadó MT, et al. Outcome in hodgkin’s lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res. 2005;11(4):1467–1473. doi:10.1158/1078-0432.CCR-04-1869
50. Engels EA. Infectious agents as causes of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2007;16(3):401–404. doi:10.1158/1055-9965.EPI-06-1056
51. Edwards LA, Nistala K, Mills DC, et al. Delineation of the innate and adaptive T-cell immune outcome in the human host in response to Campylobacter jejuni infection. PLoS One. 2010;5(11):e15398. doi:10.1371/journal.pone.0015398
52. Anderson LA, Atman AA, McShane CM, et al. Common infection-related conditions and risk of lymphoid malignancies in older individuals. Br J Cancer. 2014;110(11):2796–2803. doi:10.1038/bjc.2014.173
53. Cunningham MW. Rheumatic fever, autoimmunity, and molecular mimicry: the streptococcal connection. Int Rev Immunol. 2014;33(4):314–329. doi:10.3109/08830185.2014.917411
54. Peveling-Oberhag J, Arcaini L, Hansmann ML, Zeuzem S. Hepatitis C-associated B-cell non-hodgkin lymphomas. Epidemiology, molecular signature and clinical management. J Hepatol. 2013;59(1):169–177. doi:10.1016/j.jhep.2013.03.018
55. Rivera-Correa JRA. In Malaria: Immune Response to Infection and Vaccination (Eds Rodriguez, A. & Mota, M.). Switzerland: Springer; 2016.
56. Rivera-Correa J, Guthmiller JJ, Vijay R, et al. Plasmodium DNA-mediated TLR9 activation of T-bet+ B cells contributes to autoimmune anaemia during malaria. Nat Commun. 2017;8(1):1282. doi:10.1038/s41467-017-01476-6
57. Landgren O, Engels EA, Pfeiffer RM, et al. Autoimmunity and susceptibility to hodgkin lymphoma: a population-based case-control study in Scandinavia. J Natl Cancer Inst. 2006;98(18):1321–1330. doi:10.1093/jnci/djj361
58. Li P, Zheng Y, Chen X. Drugs for autoimmune inflammatory diseases: from small molecule compounds to anti-TNF biologics. Front Pharmacol. 2017;8:460. doi:10.3389/fphar.2017.00460
59. Bereshchenko O, Coppo M, Bruscoli S, et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-b signaling. Cell Rep. 2014;7(2):464–475. doi:10.1016/j.celrep.2014.03.004
60. Pufall MA. Glucocorticoids and cancer. Adv Exp Med Biol. 2015;872:315–333. doi:10.1007/978-1-4939-2895-8_14
61. Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–1307. doi:10.1038/nm1652
62. Ge Z, Da Y, Xue Z, et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp Neurol. 2013;241:56–66. doi:10.1016/j.expneurol.2012.12.006
63. Christensen DP, Gysemans C, Lundh M, et al. Lysine deacetylase inhibition prevents diabetes by chromatin-independent immunoregulation and β-cell protection. Proc Natl Acad Sci U S A. 2014;111(3):1055–1059. doi:10.1073/pnas.1320850111
64. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177(12):8338–8347. doi:10.4049/jimmunol.177.12.8338
65. Eriksson P, Wallin P, Sjöwall C. Clinical experience of sirolimus regarding efficacy and safety in systemic lupus erythematosus. Front Pharmacol. 2019;10:82. doi:10.3389/fphar.2019.00082
66. Teachey DT, Greiner R, Seif A, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145(1):101–106. doi:10.1111/j.1365-2141.2009.07595.x
67. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans Syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol. 2017;39(6):420–424. doi:10.1097/MPH.0000000000000818
68. Armand P, Gannamaneni S, Kim HT, et al. Improved survival in lymphoma patients receiving sirolimus for graft-versus-host disease prophylaxis after allogeneic hematopoietic stem-cell transplantation with reduced-intensity conditioning. J Clin Oncol. 2008;26(35):5767–5774. doi:10.1200/JCO.2008.17.7279
69. Carlé A, Pedersen IB, Knudsen N, et al. Moderate alcohol consumption may protect against overt autoimmune hypothyroidism: a population-based case-control study. Eur J Endocrinol. 2012;167(4):483–490. doi:10.1530/EJE-12-0356
70. Papageorgis P, Stylianopoulos T. Role of TGFβ in regulation of the tumor microenvironment and drug delivery (Review). Int J Oncol. 2015;46(3):933–943. doi:10.3892/ijo.2015.2816
71. Newcom SR, Kadin ME, Ansari AA. Production of transforming growth factor-beta activity by Ki-1 positive lymphoma cells and analysis of its role in the regulation of Ki-1 positive lymphoma growth. Am J Pathol. 1988;131(3):569–577.
72. Kadin ME, Cavaille-Coll MW, Gertz R, Massagué J, Cheifetz S, George D. Loss of receptors for transforming growth factor beta in human T-cell malignancies. Proc Natl Acad Sci U S A. 2019;10(39):3835–3839. doi:10.1073/pnas.91.13.6002
73. Lockmer S, Wahlin BE, Smedby KE, Kimby E. Chemotherapy-free initial treatment of advanced indolent lymphoma has durable effect with low toxicity: results from two nordic lymphoma group trials with more than 10 years of follow-up. J Clin Oncol. 2018;36(33):3315–3323. doi:10.1200/JCO.18.00262
74. Muñoz-Antonia T, Muro-Cacho C, Sharma S, Cantor A, Bepler G. Expression of TGFbeta type-II receptor in association with markers of proliferation and apoptosis in premalignant lung lesions. Cancer. 2007;110(7):1527–1531. doi:10.1002/cncr.22937
75. Anderson M, Muro-Cacho C, Cordero J, Livingston S, Muñoz-Antonia T. Transforming growth factor beta receptors in verrucous and squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 1999;125(8):849–854. doi:10.1001/archotol.125.8.849
76. Gobbi H, Dupont WD, Simpson JF, et al. Transforming growth factor-beta and breast cancer risk in women with mammary epithelial hyperplasia. J Natl Cancer Inst. 1999;91(24):2096–2101. doi:10.1093/jnci/91.24.2096
77. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. 2015;5:15179. doi:10.1038/srep15179
78. Vacchelli E, Semeraro M, Enot DP, et al. Negative prognostic impact of regulatory T cell infiltration in surgically resected esophageal cancer post-radiochemotherapy. Oncotarget. 2015;6(25):20840–20850. doi:10.18632/oncotarget.4428
79. Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-beta and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13(18 Pt 1):5262–5270. doi:10.1158/1078-0432.CCR-07-1157
80. Nakagaki T, Tamura M, Kobashi K, et al. Profiling cancer-related gene mutations in oral squamous cell carcinoma from Japanese patients by targeted amplicon sequencing. Oncotarget. 2017;8(35):59113–59122. doi:10.18632/oncotarget.19262
81. Tokunaga H, Lee DH, Kim IY, Wheeler TM, Lerner SP. Decreased expression of transforming growth factor b receptor type I Is associated with poor prognosis in bladder transitional cell carcinoma patients. Clin Cancer Res. 1999;5(9):2520–2525.
82. Winerdal ME, Marits P, Winerdal M, et al. FOXP3 and survival in urinary bladder cancer. BJU Int. 2011;108:1672–1678. doi:10.1111/j.1464-410X.2010.10020.x
83. Bakkebø M, Huse K, Hilden VI, Smeland EB, Oksvold MP. TGF-b-induced growth inhibition in B-cell lymphoma correlates with Smad1/5 signalling and constitutively active p38 MAPK. BMC Immunol. 2010;11:
84. Wang B, Dileepan T, Briscoe S, et al. Induction of TGF-β1 and TGF-β1–dependent predominant Th17 differentiation by group A streptococcal infection. Proc Natl Acad Sci U S A. 2010;107(13):5937–5942. doi:10.1073/pnas.0904831107
85. Costalonga M, Cleary PP, Fischer LA, et al. Intranasal bacteria induce Th1 but not Treg or Th2. Mucosal Immunol. 2009;2(1):85–95. doi:10.1038/mi.2008.67
86. Linnebacher M, Maletzki C, Klier U, Klar E. Bacterial immunotherapy of gastrointestinal tumors. Langenbecks Arch Surg. 2012;397(4):557–568. doi:10.1007/s00423-011-0892-6
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.