")
Back to Journals » Journal of Inflammation Research » Volume 14
LCC-09, a Novel Salicylanilide Derivative, Exerts Anti-Inflammatory Effect in Vascular Endothelial Cells
Authors Angom RS, Zhu J, Wu ATH , Sumitra MR, Pham V, Dutta S, Wang E, Madamsetty VS , Perez-Cordero GD , Huang HS , Mukhopadhyay D, Wang Y
Received 3 February 2021
Accepted for publication 7 August 2021
Published 8 September 2021 Volume 2021:14 Pages 4551—4565
DOI https://doi.org/10.2147/JIR.S305168
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Ramcharan Singh Angom,1,* Jian Zhu,1,2,* Alexander TH Wu,3 Maryam Rachmawati Sumitra,4 Victoria Pham,1 Shamit Dutta,1 Enfeng Wang,1 Vijay Sagar Madamsetty,1 Gabriel D Perez-Cordero,1 Hsu-Shan Huang,4 Debabrata Mukhopadhyay,1 Ying Wang5,6
1Department of Biochemistry and Molecular Biology, College of Medicine and Science, Mayo Clinic, Jacksonville, FL, 32224, USA; 2Department of Cardiology, The First Affiliated Hospital of Bengbu Medical College, Bengbu, Anhui, 233004, People’s Republic of China; 3The Ph.D. Program for Translational Medicine, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan; 4Graduate Institute for Cancer Biology & Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei, 110, Taiwan; 5Department of Cardiovascular Medicine, College of Medicine and Science, Mayo Clinic, Rochester, MN, USA; 6Department of Biochemistry and Molecular Biology, College of Medicine and Science, Mayo Clinic, Rochester, MN, 55905, USA
*These authors contributed equally to this work
Correspondence: Ying Wang
Department of Cardiovascular Medicine, Department of Biochemistry and Molecular Biology, Mayo Clinic College of Medicine and Science, 200 1st ST SW, Rochester, MN, 55905, USA
Email [email protected]
Objective: Endothelial cell (EC) activation facilitates leukocyte adhesion to vascular walls, which is implicated in a variety of cardiovascular diseases and is a target for prevention and treatment. Despite the development of anti-inflammatory medications, cost-effective therapies with significant anti-inflammatory effects and lower organ toxicity remain elusive. The goal of this study is to identify novel synthetic compounds that inhibit EC inflammatory response with minimal organ toxicity.
Methods and Results: In this study, we discovered LCC-09, a salicylanilide derivative consisting of the functional fragment of magnolol, 2,4-difluorophenyl, and paeonol moiety of salicylate, as a novel anti-inflammatory compound in cultured ECs and zebrafish model. LCC-09 was shown to inhibit pro-inflammatory cytokine tumor necrosis factor-α (TNFα)-induced expression of adhesion molecules and inflammatory cytokines, leading to reduced leukocyte adhesion to ECs. Mechanistically, LCC-09 inhibits the phosphorylation of signal transducer and activator of transcription 1 (STAT1), TNFα-induced degradation of NF-κ-B Inhibitor-α (IκBα) and phosphorylation of NFκB p65, resulting in reduced NFκB transactivation activity and binding to E-selectin promoter. Additionally, LCC-09 attenuated TNFα-induced generation of reactive oxygen species in ECs. Molecular docking models suggest the binding of LCC-09 to NFκB essential modulator (NEMO) and Janus tyrosine kinase (JAK) may lead to dual inhibition of NFκB and STAT1. Furthermore, the anti-inflammatory effect of LCC-09 was validated in the lipopolysaccharides (LPS)-induced inflammation model in zebrafish. Our results demonstrated that LCC-09 significantly reduced the LPS-induced leukocyte recruitment and mortality of zebrafish embryos. Finally, LCC-09 was administered to cultured ECs and zebrafish embryos and showed minimal toxicities.
Conclusion: Our results support that LCC-09 inhibits EC inflammatory response but does not elicit significant toxicity.
Keywords: endothelial cells, inflammation, salicylanilide derivative, tumor necrosis factor-α, lipopolysaccharides, toxicity
Introduction
Vascular endothelial cells (ECs) form a non-adhesive, highly selective physical barrier between blood flow and vessel wall, and regulate vascular tone and remodeling through secretion of multiple vasoactive substances.1 Upon stimulation with diverse numbers of pro-inflammatory factors, EC activation is induced to mediate leukocyte adhesion, increase vascular permeability and thrombosis, all of which have been recognized as key early events of a variety of cardiovascular diseases.2,3 Although current anti-inflammatory therapies including statins and human anti-interleukin-1β monoclonal antibody have been convincingly shown to improve disease outcomes, they are costly and are associated with significant side-effects.4–8 Thus, the development of a more cost-effective anti-inflammatory therapeutic strategy with minimal organ toxicity is still necessary for the prevention and treatment of cardiovascular diseases.
In this study, we discovered LCC-09 as a novel synthetic compound with anti-inflammatory effects. LCC-09 is a 5-(2ʹ,4ʹ-difluorophenyl)-salicylanilide derivative consisting of the functional fragment of magnolol, 2,4-difluorophenyl, and paeonol moiety of salicylate, with the International Union of Pure and Applied Chemistry (IUPAC) name N-(3-cyanophenyl)-2ʹ,4ʹ-difluoro-4-hydroxy[1,1ʹ-biphenyl]-3-carboxamide.9,10 LCC-09 was initially designed and synthesized to block multiple signaling pathways to inhibit osteoclastogenesis and growth of glioma stem cells.9,10 It was recently observed to relieve disease severity and chronic pain in the mouse rheumatoid arthritis model, indicating it has a potential anti-inflammatory effect.11
To study the role of LCC-09, we utilized a tumor necrosis factor-α (TNFα)-induced EC inflammation model in vitro. TNFα is widely accepted as a major inducer of EC inflammatory response during the pathogenesis of multiple cardiovascular diseases;12,13 it binds to membrane receptors to elicit multiple downstream signaling pathways including NFκB and Janus tyrosine kinase (JAK)/signal transducer and activator of transcription (STAT).14–16 Additionally, we have examined the effect of LCC-09 on leukocyte recruitment and extravasation in the zebrafish lipopolysaccharide (LPS)-induced inflammation model. To further evaluate the cytotoxic effect of LCC-09, we studied how LCC-09 affected cell viability of cultured ECs and development of zebrafish embryos.
Zebrafish have been widely used for high-throughput drug screening studies for human inflammatory diseases because of the similarity of immune cell lineages to mammals, high fecundity, rapid ex utero development, and optical transparency of embryos and larvae.17,18 Our results collectively support that LCC-09 is a novel anti-inflammatory compound with minimal cardiac toxicity which suppresses EC inflammatory response in vitro and in vivo through dual inhibition of NFκB and STAT1 activation.
Materials and Methods
Cells, Chemicals and Reagents
HUVECs (Lonza) were passaged in endothelial cell basal medium supplemented with EGM-MV SingleQuots (Lonza). In all experiments, HUVECs within five passages were used and randomized for groups in the experiments. Group sizes are equal by design. THP-1 (ATCC) were grown in RPMI 1640 containing 10% FBS. LCC-09 (Figure 1A) was synthesized as previously described.10 Stock solution of LCC-09 were dissolved in dimethyl sulfoxide (20 mM) (DMSO; Sigma Aldrich) and stored at −20°C away from light, then diluted in sterile culture medium just before use. Recombinant human TNFα protein was purchased from R&D. Adenoviruses bearing an NF-κB luciferase reporter were used as previously described19 and luciferase activity was examined with a luciferase assay kit according to the manufacturer’s instructions (Promega). Cellrox Green and Hoechst 33342 were obtained from Thermo Fisher Scientific. In-situ staining of FTIC Annexin V and PI (BioLegend) was used to analyze cell apoptosis. Cell viability was analyzed with a CellTiter Aqueous One Solution Cell Proliferation Assay (MTS) from Promega.
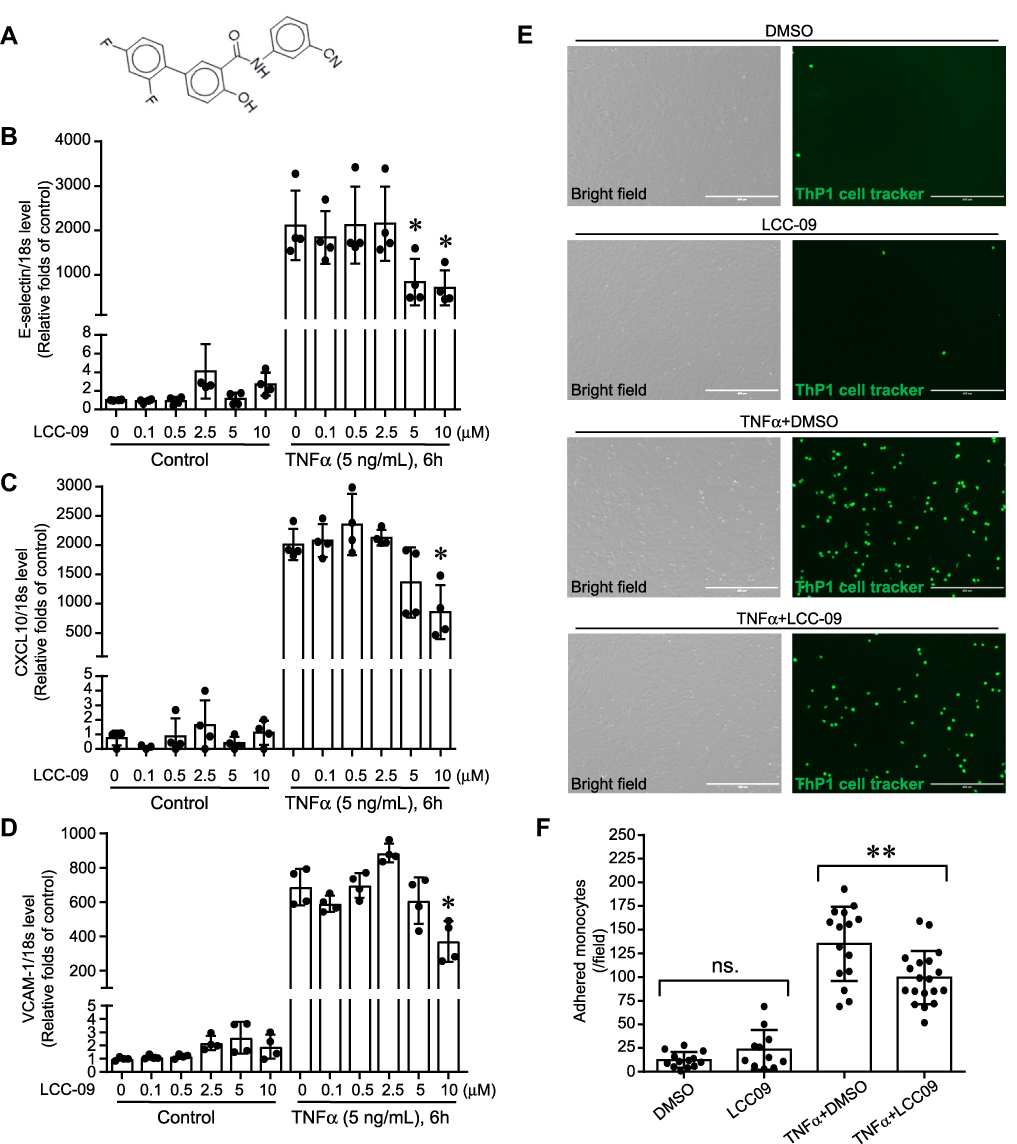 |
Figure 1 LCC-09 reduces TNFα-induced leukocyte adhesion on endothelial cells. (A) Chemical structure of LCC-09. (B–D) HUVECs were pre-treated with LCC-09 or control DMSO vehicle control for 30 mins at the indicated concentrations and then stimulated with TNFα (5 ng/mL) for 6 h. qPCR was performed (N=4 per group) and expressed as relative folds of control group, which was normalized to 1. (E and F) HUVECs were co-cultured with LCC-09 (5 μM) or DMSO vehicle control for 30 mins and then stimulated with TNFα (5 ng/mL) for 20 h. Then HUVECs were co-cultured with CMFDA-labeled monocytic THP1 cells for 30 mins and adhered monocytes were imaged (D) and counted (E). *p<0.05; **p<0.01. |
Leukocyte Adhesion Assay
HUVECs were pretreated with LCC-09 and then exposed to TNFα for 20h, after which the medium was removed and replaced with fresh medium. Then, HUVECs were co-cultured with THP-1 cells (ATCC) labeled with CellTracker™ Green CMFDA Dye (0.6 μM, Thermo Fisher Scientific) for 30 min. The adhered cells were fixed with 4% paraformaldehyde for 10 minutes and unbound THP-1 cells were removed by washing. The adhered cells were imaged with EVOS Imaging System (Thermo Fisher Scientific) and blindly counted.
Zebrafish Model
Zebrafish were maintained and raised according to the protocol described by Dr. Westerfield.20 Embryos were collected after natural spawning and the unfertilized eggs were removed. Fertilized embryos were cultured at 28.5 °C in clean Petri dishes in egg water containing 60 mg/L “Instant Ocean” salt and 2 mg/L methylene blue (Sigma) and randomized to groups. To inhibit skin pigment formation, 0.003% 1-phenyl-2-thiourea (PTU) (Sigma) was added to the egg water after 12 h. All fish experiments have been approved by IACUC at Mayo Clinic. The fish water qualities including pH, conductivity, hardness, and ammonium level were monitored daily. The fish are housed in 2.5L or 6L transparent polycarbonate tanks according to their population densities. The zebrafish housing units are fully integrated with filter systems, germicidal irradiation (UVC) and light, temperature control units, and a recirculating water system which pumps feed water into the tanks. The wastewater is partially purified before being recirculated. Humane endpoints include morphological abnormalities such as swollen abdomen, skin discoloration, frayed fins, etc., and physical abnormalities, such as difficult swimming and reaching food, lack of balance, etc. At the experimental endpoint, fish were euthanized using a chemical overdose of 0.04% tricaine as approved by the Mayo Clinic IACUC.
To examine the role of LCC-09 in vivo, we used the transgenic zebrafish myeloperoxidase promoter (MPO):GFP line,21 a generous gift from Dr. Stephen C Ekker’s laboratory at Mayo Clinic, Rochester, which expresses green fluorescent protein in neutrophils under the control of the myeloperoxidase promoter. At 48 hours post fertilization (hpf), the MPO:GFP zebrafish larvae were anesthetized using 0.015% tricaine and LPS (0.4 mg/mL, 2 nL) was injected either into the yolk or the blood circulation via the caudal vein. The control group was injected with phosphate-buffered saline (PBS). Microinjection (PLI-100, Harvard Apparatus, Inc., Massachusetts, MA, USA) was performed at a volume of 2 nL per larva. The injected larvae were then cultured at 28.5 °C and observed for signs of disease and mortality. Prior to the LPS injection, larvae were treated with 5 μM LCC-09 by static immersion in the egg water for 3 h in a total volume of 1 mL in a 12 well plate. Control larvae were exposed to DMSO in regular egg water. The larvae were observed under the fluorescence microscope to visualize the GFP-labelled neutrophil accumulation at the specified time points. To quantify neutrophil accumulation after LPS injection in yolk sac, the fluorescent intensity was measured. Group sizes are equal by design in all the zebrafish experiments. However, more embryos were included in the LPS injection groups as we expected mortality induced by the LPS, which resulted in unequal group sizes. Additionally, zebrafish embryos which were not properly imaged due to improper orientation were excluded from the data analysis, and we have taken into consideration every fish in each group which was in similar orientation at the time of data acquisition. Residual dark region (Figure 2B, bright field image) and fluorescent signal foci (Figure 2B, fluorescent image) in the zebrafish yolk region were observed after LPS injection, which is likely caused by necrosis of neutrophil and yolk tissues as previously reported,22 but was not quantified in the current study.
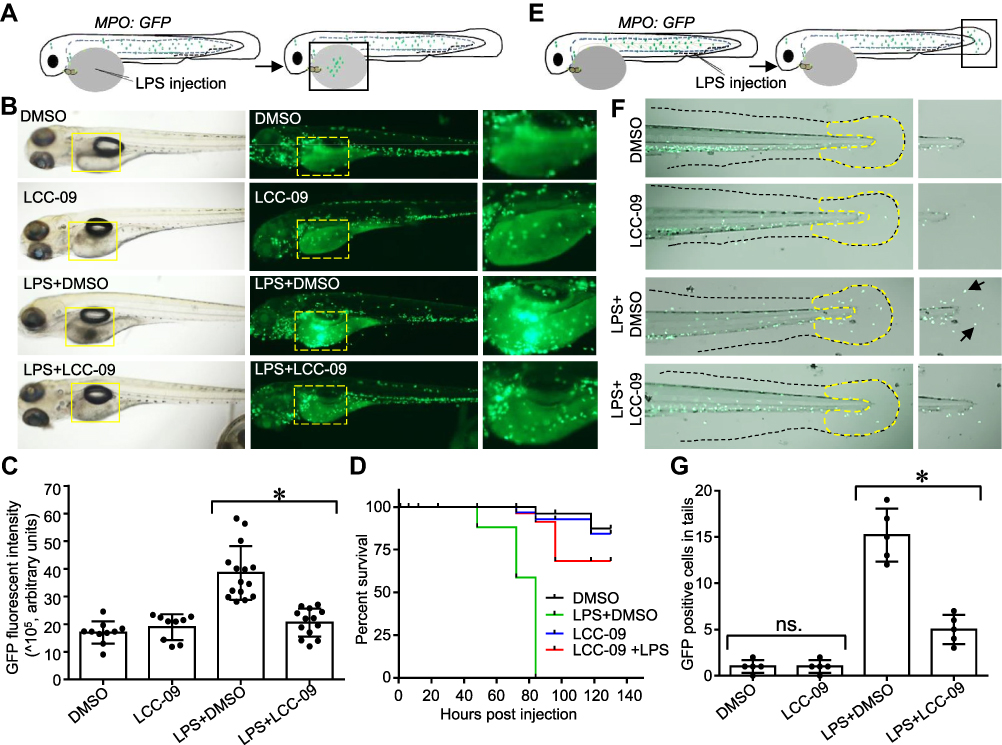 |
Figure 2 LCC-09 inhibits LPS-induced inflammatory response in zebrafish. (A–D) Zebrafish (MPO;GFP) embryos at 48 hpf were treated with LCC-09 (5 μM) and DMSO as control for 3 h and then injected with LPS (0.4 mg/mL, 2 nL) through yolk sac. Images were collected 12 h after injection (B). Local accumulation of neutrophils was quantified using GFP fluorescent intensity and compared (C). Zebrafish survival was monitored (N=8 per group) (D). (E–G) LPS (0.4 mg/mL, 2 nL) was intravenously injected to the caudal vein of zebrafish (MPO;GFP) embryos at 48 hpf. Images were collected 6 h post-injection. Neutrophil accumulation in tail fins (outlined by yellow dotted dash lines) was counted and compared. Arrow denotes infiltration of GFP positive neutrophils induced by LPS injection. *p<0.05. |
In the cardiotoxicity analysis, WT AB zebrafish embryos received from Zebrafish International Resource Center at 6 hpf were incubated with 10 μM LCC-09 in the water for 48 h and 72 h, respectively. Five zebrafish embryos from each group were randomly selected for visual observation and imaging acquisition of specific phenotype under the microscope (Zeiss Observer Z.1). The occurrence of pericardial edema and abnormal circulation were thereby evaluated by blinded observation. The heart rate was counted for 60 seconds, and heart rhythm was also monitored for any potential arrhythmia, as described previously.23
Western Blotting
HUVECs were lysed with RIPA lysis buffer supplemented with proteinase inhibitor and phosphatase inhibitor cocktails (Thermo Fisher Scientific). Protein extracts were resolved in 10% SDS-PAGE gels and electroblotted to PVDF membranes (BioRad), which was performed by an investigator who is blinded to the group information. Membranes were blocked for 1 hour at room temperature in TBST containing 5% (weight/vol) non-fat dry milk and incubated overnight with primary antibodies. After TBST washing, membranes were incubated with a Horseradish Peroxidase-conjugated secondary antibody (Santa Cruz) diluted at 1:10000 for 1 hour and scanned using ChemiDoc Image System. To control unwanted source of variation, the intensity of the protein bands was quantified using Image Lab Software (BioRad) and expressed as relative folds of control group which was normalized to 1 for comparison (Figure 3B–D). Antibodies against NFκB p65 (#4764, 1:1000), phosphorylated p65 S536 (#3031, 1:1000), STAT1 (#9172, 1:1000), phosphorylated STAT1 Y701 (#9167, 1:1000), STAT3 (#4904, 1:1000), phosphorylated STAT3 Y705 (#9131, 1:1000), JNK (#9258, 1:1000), phosphorylated JNK T183/Y185 (#4668, 1:1000), ERK (#9102, 1:1000), phosphorylated ERK T202/Y204 (#9101, 1:1000), p38 (#8690, 1:1000) and phosphorylated p38 T180/Y182 (#4511, 1:1000), and IκBα (#2682, 1:1000), were purchased from Cell Signaling Technology. The antibody against β-Actin (A2228; 1:5000) was purchased from Sigma-Aldrich.
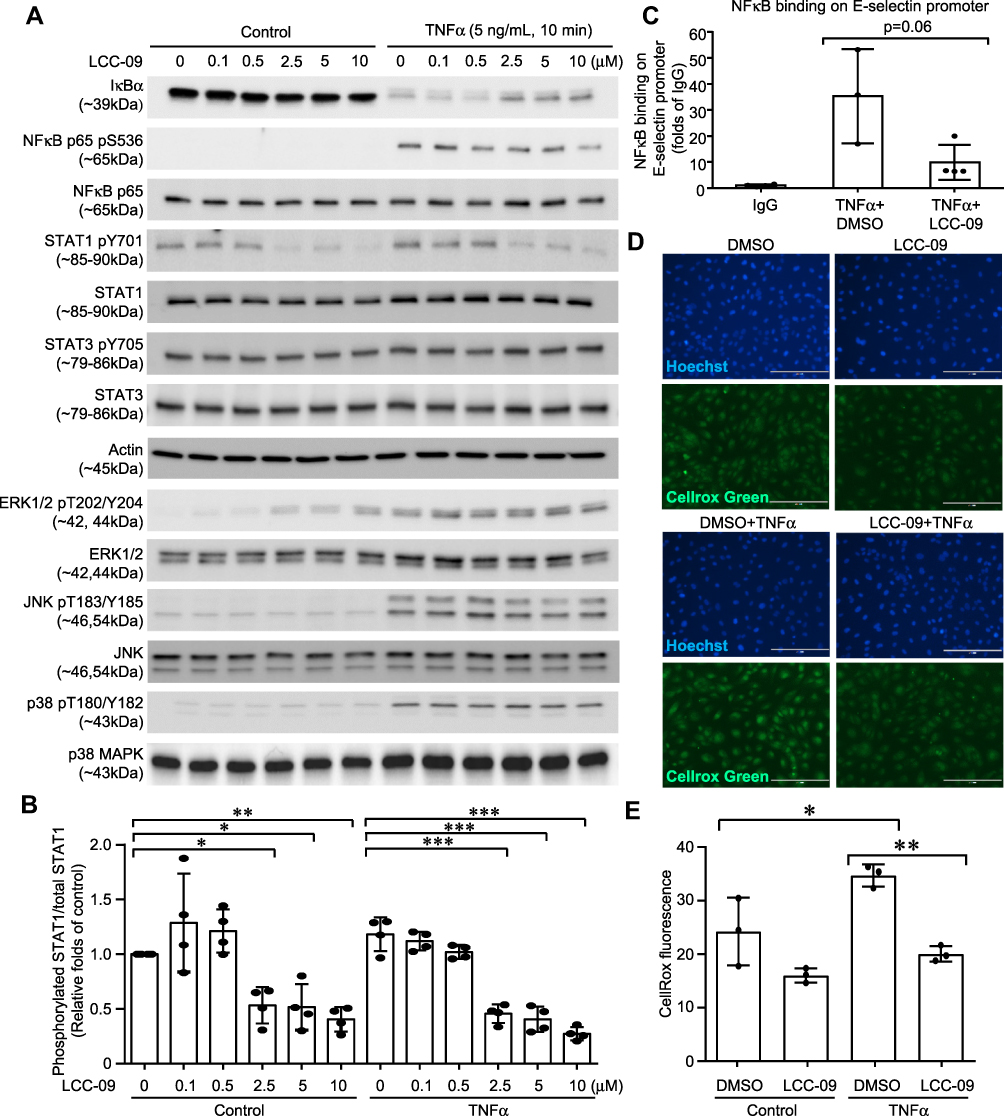 |
Figure 3 LCC-09 inhibits activation of NFκB and STAT1, and ROS generation in endothelial cells. HUVECs were pre-treated with LCC-09 and control DMSO for 30 mins at the indicated concentrations and then stimulated with TNFα (5 ng/mL). (A and B) Cell lysates were subjected to Western blotting. (C) ChIP was performed to examine the binding of NFκB to E-selectin promoter 2 h after TNFα stimulation. (D and E) Cells were stained with Cellrox Green and imaged (D). Fluorescent intensity was measured with a SpectraMax plate reader and compared (E) 30 mins after TNFα stimulation. *p<0.05; **p<0.01; ***p<0.001. |
qPCR
Total RNA was isolated from HUVECs 6 h post TNFα stimulation with the RNeasy Mini Kit (Qiagen) by an investigator who is blinded to the group information. Previous studies suggest that TNFα-induced E-selectin peaked at 2–4 hours, while VCAM-1 peaked at later times (12–24h).24 Thus, 6 h time point has been used in many previous studies to evaluate the expression of adhesion molecules and inflammation cytokines after TNFα stimulation.25–27 RNA was reverse-transcribed by using the iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed with a TaqMan SYBR Green Master Mix (Applied Biosystems). The comparative cycle threshold method was used to calculate the relative abundance of E-selectin mRNA to 18s ribosome RNA expression. E-selectin forward primer: 5ʹ- CTGTGCACTGGAAAGCTTCA-3ʹ, reverse primer: 5ʹ- AGCCCAGGTTGAATGCAC-3ʹ; CXCL10 forward primer: 5ʹ-AACCAGAGGGGAGCAAAATC-3ʹ, reverse primer: 5ʹ-CTGTGTGGTCCATCCTTGG-3ʹ; VCAM-1 forward primer: 5ʹ-ACT CCG CGG TAT CTG CAT-3ʹ, reverse primer: 5ʹ-TTT GTG TCC CAC CTG TGT GT-3ʹ; 18s forward primer: 5ʹ-GTAACCCGTTGAACCCCATT-3ʹ, 18s reverse primer: 5ʹ-CCATCCAATCGGTAGTAGCG-3ʹ. Technical replicates were used in qPCR experiments to ensure reliability of single values, but single values from each experimental sample were presented and analyzed. To control unwanted source of variation, the qPCR data was expressed as relative folds of control group which was normalized to 1 (Figures 1B–D and 3C; Supplemental Figure 1).
ChIP Assay
HUVECs were pretreated with LCC-09 or DMSO as control for 30 mins and then stimulated with TNFα (5 ng/mL) for 2 h. Cells were then cross-linked with 1% formaldehyde, lysed in buffer (Tris-HCl 50 mM, SDS 1%, EDTA 5 mM, pH 7.0), and then sonicated. The sheared chromatin was immunoprecipitated with Dynabeads (R&D) conjugated with control IgG or NFκB p65 primary antibodies (#sc-109, Santa Cruz). The eluted immunoprecipitates were incubated at 65 °C for 6 h for reverse formaldehyde cross-linking. DNA was extracted with a Qiagen PCR purification kit and subjected to qPCR with specific primers which amplify a region in human E-selectin promoter containing a known NFκB binding site required for TNFα-induced maximum expression of E-selectin.28 Forward primer: 5ʹ-GGCATGGACAAAGGTGAAGT-3ʹ, reverse primer: 5ʹ- GGAGGATATTGTCCACATCCAG-3ʹ.
Protein Docking Model
The LCC-09 ligand was prepared by drawing on Avogadro software. The ligand was converted into pdb format using Pymol and converted into pdbqt format using AutoDock Tools 1.5.6. All the protein targets; JAK1 (PDB ID: 6W8I), JAK2 (PDB ID: 6AAJ), JAK3 (PDB ID: 1YVJ), STAT1 (PDB ID: 1BF5) and NEMO (PDB ID: 3BRT) were retrieved from Protein Data Bank as pdb format file, and converted into pdbqt format. The molecular docking experiments were performed by using AutoDock Vina, which is more accurate than Autodock 4.2, has a shorter running time and the ability to process many rotatable bonds. The grid box of 40Å × 40Å × 40Å was generated on defined binding site residues of the protein targets. The protein targets were prepared through deleting water, adjusting hydrogen (polar only) and charges (Kollman charges). All parameters of software were set as default, and all bonds in the ligand are rotated freely, presuming the receptor as rigid. The final visualization of the molecular docking was performed using Pymol.
Statistics
All analyses were performed using GraphPad Prism 5 (GraphPad Software). All the values are expressed as means ± SD. In the figures, the value of each individual sample was represented as a single dot in the scatter graph. Statistical significance was determined using 2-sided Student’s t-test (2 groups) and one-way ANOVA with Tukey’s multiple comparisons test (more than 2 groups). A value of p < 0.05 was considered significant.
Results
LCC-09 Reduced TNFα-Induced Leukocyte Adhesion to Endothelial Cells
We first examined the effect of LCC-09 (Figure 1A) on TNFα-induced adhesion molecules and inflammatory cytokines of cultured HUVECs. HUVECs were pretreated with LCC-09 at 0, 0.1, 0.5, 2.5, 5, 10 μM for 30 mins and then stimulated with TNFα (5 ng/mL) for 6 h. Our results showed that LCC-09 reduced TNFα-induced expression of E-selectin, C-X-C motif chemokine ligand 10 (CXCL10) and vascular cell adhesion molecule 1 (VCAM-1) at 10 μM (Figure 1B–D). Adhesion molecules and inflammatory cytokines of endothelial cells mediate the recruitment and accumulation of leukocyte to vascular wall,29–31 thus we evaluated the effect of LCC-09 on TNFα-induced leukocyte adhesion. Since 5 μM is the lowest dose of LCC-09 which decreased TNFα-induced adhesion molecules and chemokines, we validated the effect of 5 μM LCC-09 on leukocyte adhesion. As shown in Figure 1E and F, LCC-09 (5 μM) did not alter the leukocyte adhesion to control HUVECs, which barely bound any monocytes. However, it significantly reduced the numbers of attached monocytes to HUVECs that were primed with TNFα (135.1±39.3 vs 99.5±28.2. p<0.05). Additionally, LCC-09 (5 μM) was administered 30 minutes after TNFα stimulation to examine its therapeutic effect. As shown in the Supplemental Figure 1, post-treatment LCC-09 also effectively reduced TNFα-induced expression of CXCL-10 and E-selectin. These results demonstrate LCC-09 attenuates both TNFα-induced upregulation of adhesion molecules and leukocyte adhesion.
LCC-09 Attenuated STAT1 Phosphorylation, and TNFα-Induced Activation of NFκB and Generation of ROS in Endothelial Cells
To investigate the molecular mechanism through which LCC-09 reduced TNFα-induced inflammatory response in ECs, we examined the activation of downstream-signaling pathways of TNFα. As shown in Figure 3A and Supplemental Figure 2, pre-treatment of LCC-09 for 30 mins did not significantly affect TNFα-induced activation of JNK, ERK1/2 or p38MAPK, but attenuated TNFα-stimulated IκBα degradation in a dose-dependent manner, which was accompanied with reduced phosphorylation of NFκB p65. Meanwhile, LCC-09 shows inhibitory effect on phosphorylation of STAT1 both at basal level and upon TNFα stimulation but did not affect phosphorylation of STAT3 (Figure 3A and B). Previous studies have identified one NFκB binding site in the human E-selectin promoter which is required for its maximum expression induced by TNFα.28 Thus, ChIP assay was performed to assess how LCC-09 affected NFκB binding on E-selectin gene promoter upon TNFα stimulation. As shown in Figure 3C, LCC-09 significantly reduced the binding of NFκB p65 on E-selectin promoter. Additionally, we assessed the effect of LCC-09 on intracellular ROS levels, signal transducers that are implicated in endothelial dysfunction.32 Our results (Figure 3D and E) show that LCC-09 significantly reduced ROS levels after TNFα stimulation but not under basal conditions.
Furthermore, molecular docking was performed to investigate how LCC-09 regulates TNFα-induced NFκB and STAT1 activation in ECs. In the docking model, LCC-09 was docked as a ligand with several receptors including IKKβ and NEMO, JAK1, JAK2, JAK3 and STAT1. The ligand is shown as green stick model and the interaction bonds are illustrated with yellow dash lines in the ligand-receptor interaction model (Figure 4). After visualizing in Pymol, it was found that NEMO is the most promising protein target, which is explained by lowest binding energy of −10.6 kcal/mol with LCC-09 as a ligand, following with JAK1 and JAK3 with binding energy of −10.1 and −10.0 kcal/mol respectively, while IKKβ revealed the lowest binding energy of −7.3 kcal/mol (Table 1). Specifically, in the docking model with NEMO, LCC-09 acquired the central pocket of NEMO which leads to several hydrogen bond-mediated interactions between LCC-09 and the amino acid residues of the protein target. The hydrogen in the hydroxyl group and hydrogen in the amine group (in ketone group) shows significant hydrogen bonding of LCC-09 with GLU-89 of NEMO.
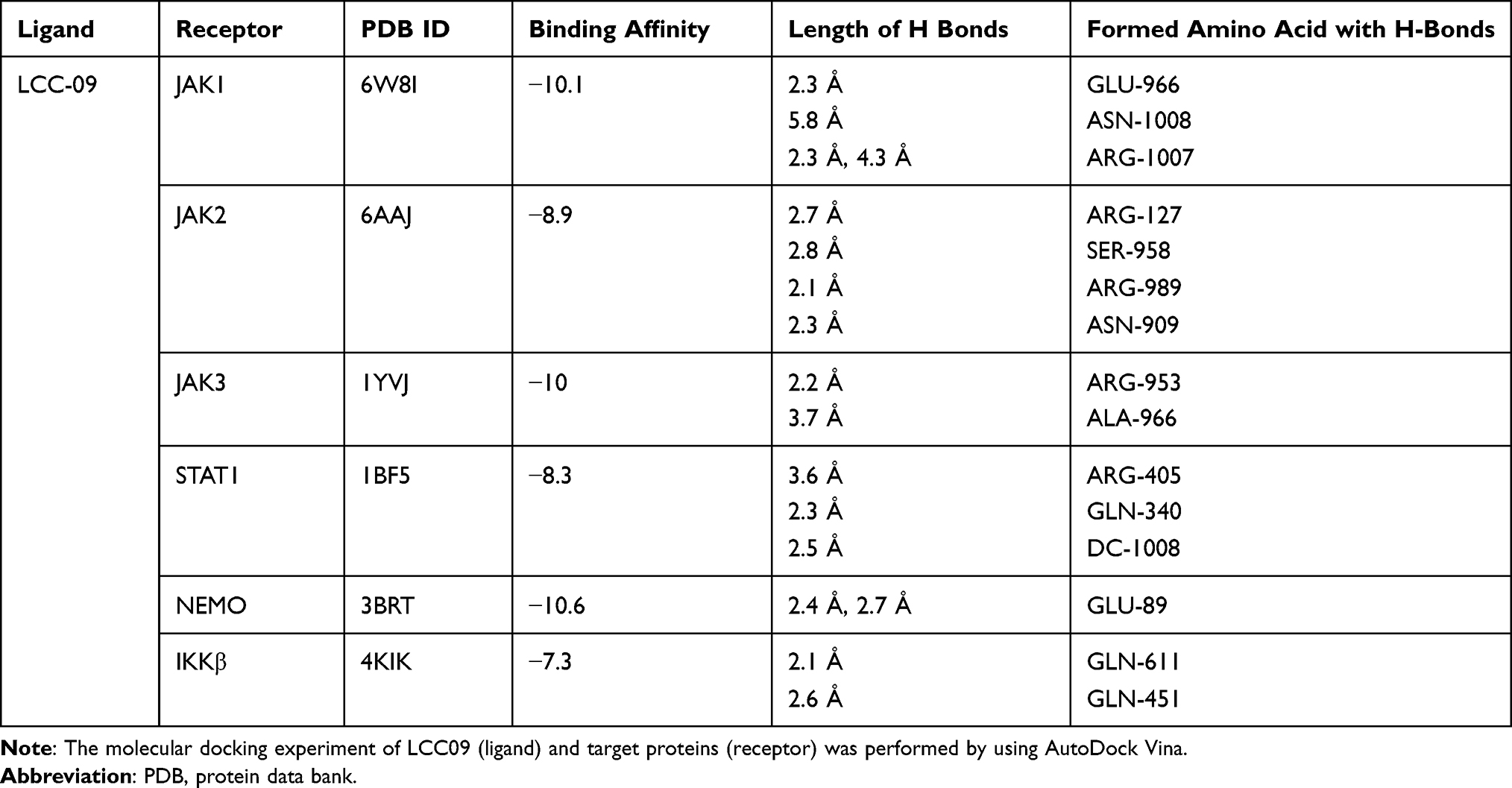 |
Table 1 Molecular Docking of LCC-09 (Ligand) and Candidate Proteins (Receptor) |
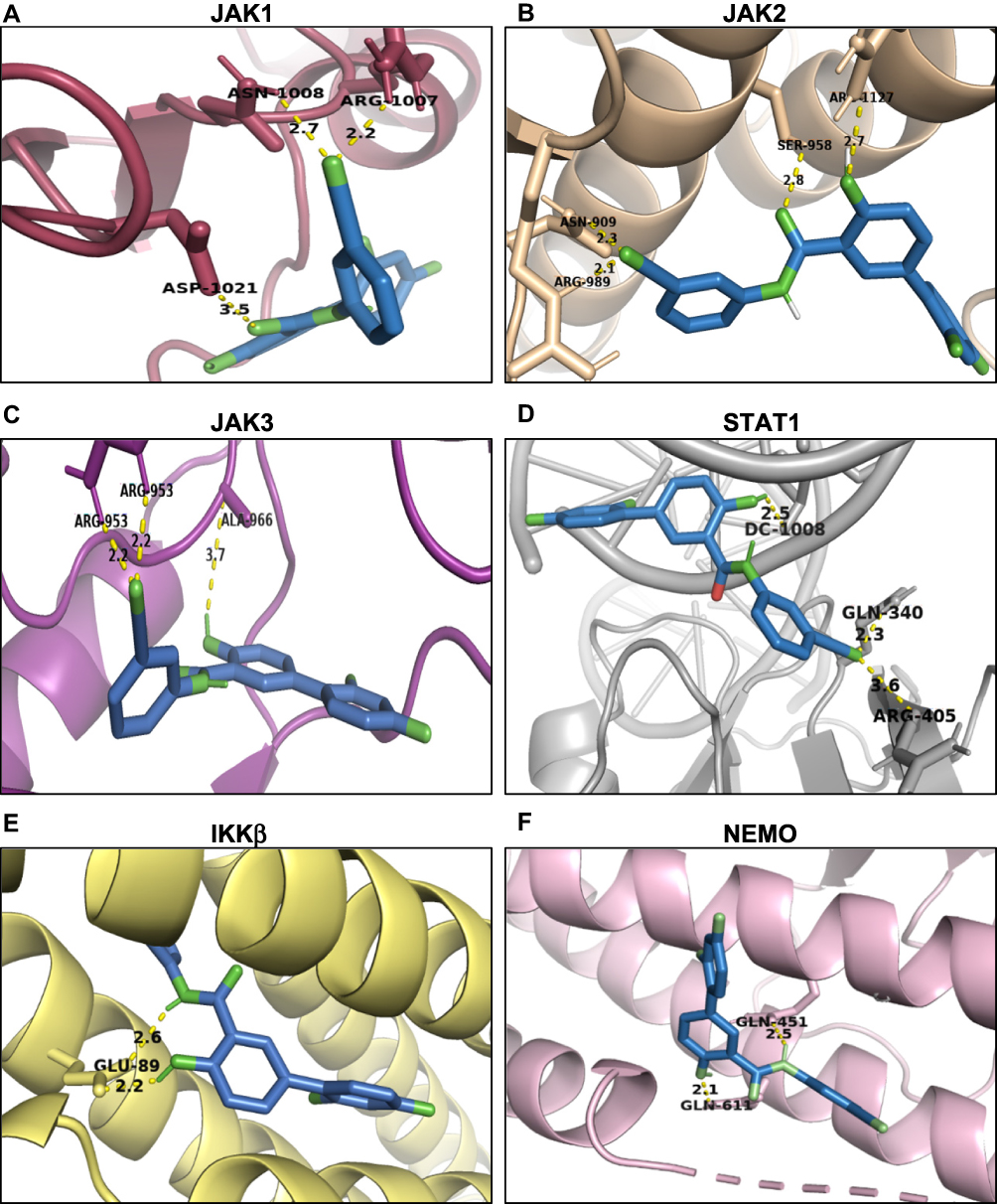 |
Figure 4 Molecule docking model of LCC-09. LCC-09 (green color sticks) was docked in JAK1 (PDB ID 6W8I) (A), JAK2 (PDB ID 6AAJ) (B), JAK3 (PDB ID 1YVJ) (C), STAT1 (PDB ID 1BF5) (D), IKKβ (PDB ID 4KIK) (E) and NEMO (PDB ID 3BRT) (F). |
LCC-09 Inhibited LPS-Induced Inflammatory Response in vivo
To further define the role of LCC-09 in vivo, we utilized a previously established LPS-induced zebrafish inflammation model, where LPS induces systemic inflammatory response with the release of several inflammatory cytokines, including TNFα, and results in organ dysfunction, multiple organ failure, and death.22,33,34 At 48 hpf, transgenic zebrafish Tg (MPO:EGFP), expressing GFP under the neutrophil-specific myeloperoxidase promoter,21 were pre-treated with LCC-09 (5 μM) for 3 h before receiving a LPS injection (0.4 mg/mL, 2 nL) (Figure 2A). Consistent with previous studies,22 LPS increased the retention of leukocytes at the injection sites, as shown by the enhanced fluorescent intensities (Figure 2B and C), and high mortality 12 h after injection (Figure 2D). LCC-09 did not affect the recruitment of neutrophils under basal conditions but significantly inhibited LPS-induced neutrophil retention in the yolk (Figure 2B and C). Consequently, LCC-09 increased the survival rates from 0% to 68% at 130 h after LPS injection (Figure 2D). Furthermore, we intravenously injected LPS through caudal veins to validate the effect of LCC-09 on systemic inflammatory response (Figure 2E). Systemic LPS induces vascular inflammatory response, increased vascular permeability and tissue damage, all of which lead to the extravasation of leukocytes into extravascular compartments.33 As shown in Figure 2F and G, 6 h after injection, the tail fin showed increased neutrophil retention after LPS injection but treatment of LCC-09 significantly inhibited LPS-induced neutrophil accumulation. Taken together, these results collectively support that LCC-09 inhibits inflammatory response in vivo.
LCC-09 Did Not Elicit Significant Toxicity in Cultured Endothelial Cells or Zebrafish Embryos
Organ toxicity is a major limiting factor in drug development and is commonly observed in the applications of small molecule kinase inhibitors.35 Thus, we examined the cytotoxic effect of LCC-09 on cultured endothelial cells. MTS assay was first performed and showed that LCC-09 at 5 μM and 10 μM decreased the MTS absorbance (Supplemental Figure 3), indicating that LCC-09 reduces the cell proliferation or viability. Annexin V-PI staining was further performed and showed that LCC-09 did not induce cell death (Figure 5A), suggesting LCC-09 does not induce death but inhibits the proliferation of ECs. Furthermore, the effect of LCC-09 on cardiac toxicity was examined in zebrafish models, which have close resemblance of the genetic cascade governing heart development, physiological and functional regulation to that of human heart.36 Wild-type zebrafish embryos (Danio rerio) were incubated with LCC-09 or DMSO as control up to 48 and 72 h, and then heart chamber size and heart rates were examined, respectively, to evaluate the effect of LCC-09 on cardiac morphology and function. As shown in Figure 5B and C, no significant growth delay or developmental defect was observed after the treatment of LCC-09 for 48 and 72 h. Meanwhile, LCC-09 did not significantly affect the heart chamber size or basal-level heart rates (Figure 5D and E). These results collectively support that LCC-09 application has no significant cardiac toxicity in vivo.
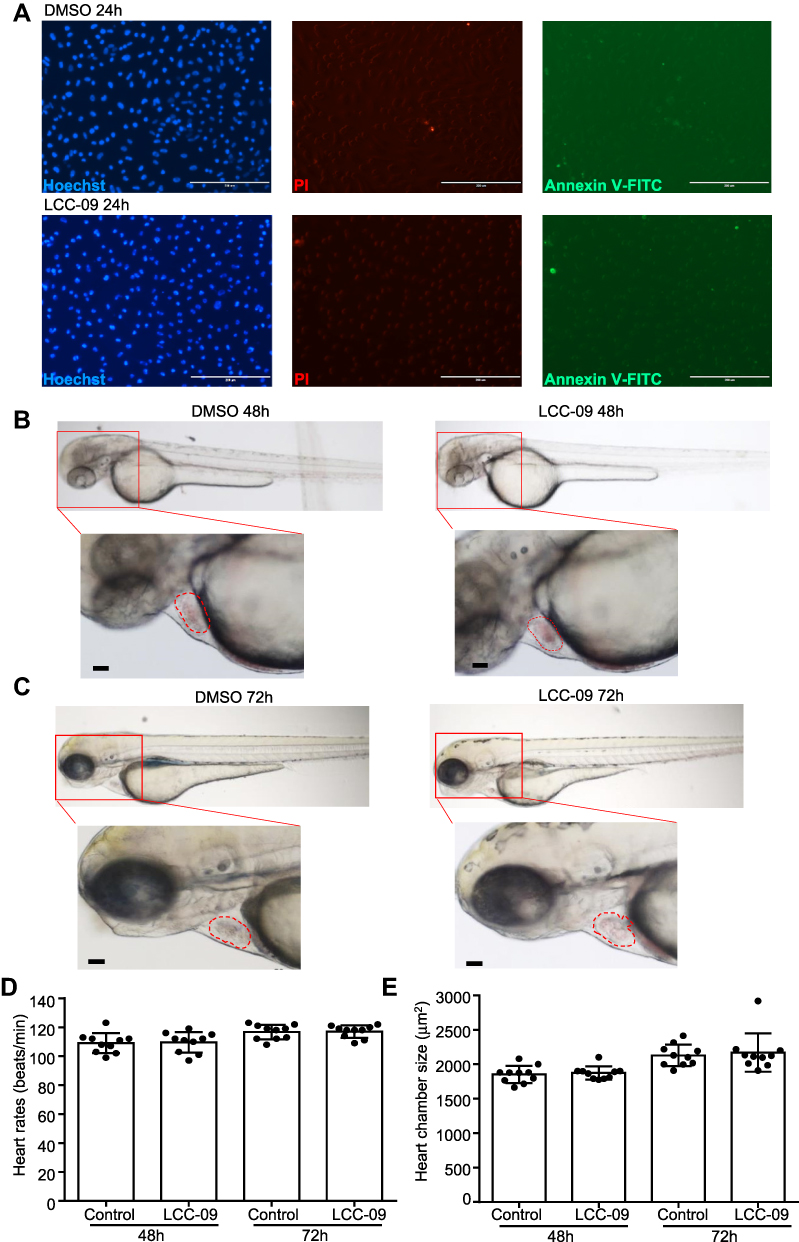 |
Figure 5 LCC-09 does not induce toxicity in cultured endothelial cells or zebrafish embryos. (A) HUVECs were cultured with LCC (10 μM) or DMSO control for 24 h and then stained with Hoechst, PI and annexin V-FITC. Images are representative of 3 independent experiments. (B–E) Zebrafish embryos at 6 hpf were incubated with LCC-09 (10 μM) or DMSO as control for 48 and 72 h, respectively. Heart rates and chamber sizes were measured and compared. Scale bar: 200 μM (A), 50 μM (B and C). |
Discussion
Leukocyte recruitment to the vessel wall in inflammation is a multistep process which involves tethering, rolling, activation, arrest, spreading, and crawling of leukocytes on the endothelium, and finally transmigration across the endothelium.37 Induction of surface adhesion molecules in ECs is a rate-limiting step to mediate adhesive interactions between leukocytes and ECs.38 Adhesion molecules recognize and bind to ligands expressed by leukocytes such as glycan epitopes and α4β1-integrin, etc., to mediate efficient leukocyte rolling and transmigration.39,40 CXCL10 is a chemokine which facilitates recruitment of monocytes, and it is shown to be involved in sustained monocyte influx upon chronic TNFα stimulation.41 Our results indicate LCC-09 reduced TNFα-induced expression of E-selectin, CXCL10 (Figure 1), and VCAM-1 (Figure 1), resulting in decreased leukocyte adhesion to cultured ECs and demonstrating that LCC-09 attenuates inflammation through regulation of multiple steps involved in leukocyte recruitment. The effect of E-selectin antibody on leukocyte adhesion has been confirmed by several previous studies,39,42–45 in which E-selectin antibody consistently blocked leukocyte adhesion in vitro and in vivo. Interestingly, LCC-09 did not significantly affect the levels of monocyte chemoattractant protein-1 or intercellular adhesion molecule 1 (data not shown). Given that LCC-09 effectively reduced the leukocyte adhesion to endothelial cells (Figure 1E and F), it is thus likely that the reduced expression of E-selectin and VCAM-1 contributes to the decreased leukocyte-endothelial cell adhesion.
The trans-migration of leukocyte determines the levels of leukocyte recruitment to inflamed tissues and is regulated by several molecules including junctional adhesion molecules, clusters of VCAM-1 and ICAM-1, etc.46,47 The inhibitive effect of LCC-09 on VCAM-1 expression suggests that it may reduce the transmigration of leukocytes. Furthermore, the trans-migration of leukocytes was examined in zebrafish model in vivo (Figure 2), in which the extravasation and accumulation of neutrophils in yolk sac and tail fin were quantified after local and systemic LPS injection, respectively. Although the molecular details through which LCC-09 controls the trans-migration process require further investigation, our current results support that LCC-09 inhibits the recruitment and extravasation of leukocytes in vivo. The anti-inflammatory effect of LCC-09 is supported by results from cultured endothelial cells in vitro and zebrafish models in vivo. Specifically, endothelial cells pre-treated with LCC-09 exhibited reduced expression of adhesion molecules, inflammatory cytokines, and decreased leukocyte adhesion upon TNFα stimulation. Zebrafish embryos treated with LCC-09 also showed reduced leukocyte extravasation when challenged with LPS. Given that NFκB and STAT1 pathways, and ROS generation are also required for myeloid cell activation, it is possible that LCC-09 has dual effects on both myeloid cells and endothelial cells, which collectively contributes to the reduced neutrophil retention at the site of LPS injection. In our in vitro experiments, monocytes were co-cultured with HUVECs (Figure 1), while neutrophil accumulation was examined in the zebrafish model in vivo (Figure 2). Although monocytes and neutrophil are distinct subsets of leukocyte populations, the molecular interactions mediating leukocyte-endothelial cell interactions are similar. Thus, reduced expression of adhesion molecules and chemokines in endothelial cells can lead to decreased recruitment of both monocytes and neutrophil. Given that endothelial cells and zebrafish embryos exhibited reduced monocyte adhesion and neutrophil extravasation, respectively, these results collectively indicate that LCC-09 is an anti-inflammatory compound. Interestingly, post-treatment of LCC-09 showed a profound inhibitive effect on the expression of E-selectin and CXCL10 (Supplemental Figure 1). TNFα is known to induce the peak activation of its downstream signaling pathways including NFκB in a few minutes, however, oscillations of NFκB activation happens over time48,49 and sustained activation of STAT1 is required for continuous expression of inflammatory cytokines.50 It is thus likely that the inhibitive effect of LCC-09 on NFκB and STAT1 contributes to reduced expression of E-selectin and CXCL10 post-TNFα stimulation. Nevertheless, this result suggests that LCC-09 is a promising anti-inflammatory compound for the treatment of inflammatory disease.
We used LPS-induced zebrafish inflammation model to evaluate the anti-inflammatory role of LCC-09 in this study. Zebrafish has been widely used in the studies of inflammatory responses because of their conservation with mammals in immune cell lineages and advantages of rapid development, high fecundity, and optical transparency. Even though the zebrafish TLR4 paralogs do not bind to LPS, they still have a significant inflammatory response after LPS injection.33,51 LPS-induced downstream signaling pathways, including induction of TNFα, activation of NFκB, and enhanced production of ROS, have shown to be conserved between zebrafish and mammals.33 Indeed, the anti-inflammatory effect of LCC-09 in the zebrafish model is further supported by results from cultured human endothelial cells (Figure 1) and a recent study using murine rheumatoid arthritis models.11 Taken together, our results support that LPS-induced zebrafish inflammation model is an excellent tool for screening new anti-inflammatory drugs.
One recent study has shown that administration of LCC-09 for 4 weeks in a murine glioblastoma model did not elicit any significant toxicity or side effect.9 Another study reported that administration of LCC-09 over a 12-week period attenuated synovial inflammation and macrophage infiltration in rheumatoid arthritis mouse models with no observed side effects.11 Taken together with our results in cultured ECs and zebrafish embryos, these data support that LCC-09 reduces inflammation and has mild toxicity.
Activation of IKK/NFκB pathway and JAK/STAT is well recognized to play essential roles in TNFα-induced EC inflammation.52 The essential roles of NFκB and STAT1 in TNFα-induced inflammatory response are supported by several previous studies52–55 which reported that inhibitors/siRNAs of NFκB and STAT1 decreased TNFα-induced expression of pro-inflammatory adhesion molecules and leukocyte adhesion. Our results indicate that LCC-09 inhibited TNFα-induced IκBα degradation and NFκB p65 phosphorylation, as well as the binding of NFκB to the human E-selectin promoter, collectively suggests LCC-09 inhibits NFκB pathway. Interestingly, the molecular docking model suggests that LCC-09 preferentially binds to NEMO but not IKKβ, the upstream kinase of IκBα. NEMO is a noncatalytic component in the activation of NFκB pathway and exerts its function by recruiting IKK to form the IKK complex.56 An extensive-binding interface is present between NEMO amino residues 44–111 and IKKβ residues 701–745,57 thus, previous studies have developed small peptide inhibitors to prevent the interaction between NEMO and IKKβ to inhibit NFκB activation in inflammation.56,58 Our molecular docking experiments suggest that LCC-09 potentially binds to NEMO Glu-89 to reduce the interaction between NEMO and IKKβ, leading to decreased IκBα phosphorylation and degradation.
The JAK-STAT pathway is essential for a wide range of cytokines and growth factors in both development and diseases.59,60 STAT1 was originally identified to be a downstream signaling component of TNF receptor (TNFR)-1 and shown to be required for TNFα-induced TNFR2-interferon regulatory factor-1 (IRF1)- interferon-β autocrine loop in monocyte recruitment to endothelial cells,41,61 suggesting the essential role of STATs in TNFα-induced inflammation. Although our molecular docking model suggests that LCC-09 potentially binds to JAK1, JAK2 and JAK3 (Figure 4), LCC-09 was shown to selectively inhibit the phosphorylation of STAT1 but not STAT3 (Figure 3). Due to the strict homolog and distinct function between STAT1 and STAT3,62 successful development of a specific STAT inhibitor has been challenging. Our results raise the possibility that LCC-09 can be used in conditions when blockage of STAT1 but not STAT3 activation is required. However, how LCC-09 specifically inhibits STAT1 activation remains to be investigated and is out of the scope of the current study. Notably, LCC-09 was shown to inhibit the STAT1 phosphorylation at basal level, suggesting that it interferes with some endogenous mechanism to block STAT1 activation. It is also possible that some JAK-independent mechanism of STAT1 activation is involved.60,63,64 Meanwhile, our current results do not exclude the possibility that other NFκB- and STAT1-independent signaling pathways are involved in the anti-inflammatory effect of LCC-09.
Salicylates have a long history in the clinical practice of treating pain and inflammation. Previous studies have reported that sodium salicylate inhibits EC inflammatory response, TNFα-induced IκBα degradation, and adhesion molecule expression through both cyclooxygenase- dependent and independent mechanisms.65–68 However, salsalate, the prodrug dimer of salicylates, did not provide additional benefits in overweight or obese patients with coronary artery diseases receiving statins.69 Furthermore, high-dose disalicylate therapy was shown to impair endothelium-dependent vasodilation,70 thus raising concerns about the clinical use of salicylates in cardiovascular diseases and highlighting the need to develop novel anti-inflammatory agents. Salicylanilide derivatives have been known for a wide range of biological activities including anti-infection, anti-tumor and anti-oxidation, etc.71,72 Our results show that LCC-09, a derivative of salicylanilide consisting of the functional fragment of magnolol, 2,4-difluorophenyl, and paeonol moiety of salicylate, inhibits EC inflammatory response in both cultured ECs and zebrafish models but does not induce significant cardiac toxicity. Additionally, our results show that LCC-09 inhibited expression of adhesion molecules and NFκB activation at the dose of 5 μM, suggesting it is more potent than sodium salicylate and aspirin, which were shown to reduce NFκB activation at 10 mM previously.65–67 Previous studies reported that LCC-09 reduced the monocyte accumulation in the rodent rheumatoid arthritis models,11 and our results suggest LCC-09-mediated inhibition of the EC inflammatory response is likely involved in the beneficial role of LCC-09 in this disease model.
Acknowledgment
We thank Dr. Laura Lewis-Tuffin at Mayo Clinic, Jacksonville for assisting the zebrafish imaging. We thank Dr. Stephen C Ekker at Mayo Clinic, Rochester, for providing the (MPO):GFP zebrafish line. We thank Drs. Si Pham, Baoan Ji, Krishnendu Pal, Santanu Bhattacharya, Pritam Das, Tanmay A Kulkarni at Mayo Clinic, Jacksonville, for helpful suggestions. We thank Ms. Sarah Chau at Mayo Clinic, Rochester, for assisting the revision of this manuscript. Victoria Pham was a scholar of the Mayo Clinic Science Program for the Advancement of Research Knowledge (SPARK) program and Gabriel D Perez Cordero was supported by Mayo Clinic-University of Puerto Rico medical student NIH CCATS summer research scholarship.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, agreed to the submitted journal, and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Institutes of Health [HL140411 and CA78383-20 to D. Mukhopadhyay, HL148339 to Y. Wang], Florida Department of Health Cancer Research Chair’s Fund Florida [grant number#3J-02 to D. Mukhopadhyay], American Heart Association [19CDA34700013 to Y. Wang] and Mayo Clinic Center for Clinical and Translational Science (CCaTS) [Ted and Loretta Rogers Cardiovascular Career Development Award Honoring Hugh C. Smith to Y. Wang]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196:430–443. doi:10.1002/jcp.10333
2. Liberale L, Montecucco F, Schwarz L, Luscher TF, Camici GG. Inflammation and cardiovascular diseases: lessons from seminal clinical trials. Cardiovasc Res. 2021;117:411–422.
3. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi:10.1038/nri2171
4. Dinarello CA. Anti-inflammatory agents: present and future. Cell. 2010;140:935–950. doi:10.1016/j.cell.2010.02.043
5. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi:10.1056/NEJMoa1707914
6. Samuel M, Tardif JC, Khairy P, et al. Cost-effectiveness of low-dose colchicine after myocardial infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur Heart J Qual Care Clin Outcomes. 2020;qcaa045. doi:10.1093/ehjqcco/qcaa045
7. Diamantis E, Kyriakos G, Quiles-Sanchez LV, Farmaki P, Troupis T. The anti-inflammatory effects of statins on coronary artery disease: an updated review of the literature. Curr Cardiol Rev. 2017;13:209–216. doi:10.2174/1573403X13666170426104611
8. Kosmas CE, Silverio D, Sourlas A, Montan PD, Guzman E, Garcia MJ. Anti-inflammatory therapy for cardiovascular disease. Ann Transl Med. 2019;7:147. doi:10.21037/atm.2019.02.34
9. Wen YT, Wu AT, Bamodu OA, et al. A novel multi-target small molecule, LCC-09, inhibits stemness and therapy-resistant phenotypes of glioblastoma cells by increasing miR-34a and deregulating the DRD4/Akt/mTOR signaling axis. Cancers. 2019;11:1442. doi:10.3390/cancers11101442
10. Lee CC, Liu Fl, Chen CL, Chen TC, Chang DM, Huang HS. Discovery of 5-(2ʹ,4ʹ-difluorophenyl)-salicylanilides as new inhibitors of receptor activator of NF-kappaB ligand (RANKL)-induced osteoclastogenesis. Eur J Med Chem. 2015;98:115–126. doi:10.1016/j.ejmech.2015.05.015
11. Dai SP, Hsieh WS, Chen CH, et al. TDAG8 deficiency reduces satellite glial number and pro-inflammatory macrophage number to relieve rheumatoid arthritis disease severity and chronic pain. J Neuroinflammation. 2020;17:170. doi:10.1186/s12974-020-01851-z
12. Tousoulis D, Oikonomou E, Economou EK, Crea F, Kaski JC. Inflammatory cytokines in atherosclerosis: current therapeutic approaches. Eur Heart J. 2016;37:1723–1732. doi:10.1093/eurheartj/ehv759
13. Zhang H, Park Y, Wu J, et al. Role of TNF-α in vascular dysfunction. Clin Sci (Lond). 2009;116:219–230. doi:10.1042/CS20080196
14. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65.
15. Sedger LM, McDermott MF. TNF and TNF-receptors: from mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev. 2014;25:453–472.
16. Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103:398–406. doi:10.1007/s00395-008-0733-0
17. Cordero-Maldonado ML, Siverio-Mota D, Vicet-Muro L, et al. Optimization and pharmacological validation of a leukocyte migration assay in zebrafish larvae for the rapid in vivo bioactivity analysis of anti-inflammatory secondary metabolites. PLoS One. 2013;8:e75404. doi:10.1371/journal.pone.0075404
18. Renshaw SA, Trede NS. A model 450 million years in the making: zebrafish and vertebrate immunity. Dis Model Mech. 2012;5:38–47. doi:10.1242/dmm.007138
19. Ji B, Gaiser S, Chen X, Ernst SA, Logsdon CD. Intracellular trypsin induces pancreatic acinar cell death but not NF-κB activation. J Biol Chem. 2009;284(26):17488–17498. doi:10.1074/jbc.M109.005520
20. Westerfield M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio Rerio). University of Oregon press; 1993.
21. Renshaw SA, Loynes CA, Trushell DM, Elworthy S, Ingham PW, Whyte MK. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108:3976–3978. doi:10.1182/blood-2006-05-024075
22. Yang LL, Wang GQ, Yang LM, Huang ZB, Zhang WQ, Yu LZ. Endotoxin molecule lipopolysaccharide-induced zebrafish inflammation model: a novel screening method for anti-inflammatory drugs. Molecules. 2014;19:2390–2409. doi:10.3390/molecules19022390
23. Burns CG, Milan DJ, Grande EJ, Rottbauer W, MacRae CA, Fishman MC. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat Chem Biol. 2005;1:263–264. doi:10.1038/nchembio732
24. Pober JS. Endothelial activation: intracellular signaling pathways. Arthritis Res. 2002;4(Suppl 3):S109–S116. doi:10.1186/ar576
25. Dore M, Sirois J. Regulation of P-selectin expression by inflammatory mediators in canine jugular endothelial cells. Vet Pathol. 1996;33:662–671. doi:10.1177/030098589603300605
26. Mako V, Czucz J, Weiszhar Z, et al. Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL-1beta, TNF-alpha, and LPS. Cytometry A. 2010;77:962–970. doi:10.1002/cyto.a.20952
27. Mehta NN, Teague HL, Swindell WR, et al. IFN-γ and TNF-α synergism may provide a link between psoriasis and inflammatory atherogenesis. Sci Rep. 2017;7:13831. doi:10.1038/s41598-017-14365-1
28. Schindler U, Baichwal VR. Three NF-kappa B binding sites in the human E-selectin gene required for maximal tumor necrosis factor alpha-induced expression. Mol Cell Biol. 1994;14:5820–5831.
29. Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–2301. doi:10.1161/ATVBAHA.107.149179
30. Smith CW. Endothelial adhesion molecules and their role in inflammation. Can J Physiol Pharmacol. 1993;71:76–87. doi:10.1139/y93-012
31. Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. 2007;101:234–247. doi:10.1161/CIRCRESAHA.107.151860b
32. Chen X, Andresen BT, Hill M, Zhang J, Booth F, Zhang C. Role of reactive oxygen species in tumor necrosis factor-alpha induced endothelial dysfunction. Curr Hypertens Rev. 2008;4:245–255. doi:10.2174/157340208786241336
33. Philip AM, Wang Y, Mauro A, et al. Development of a zebrafish sepsis model for high-throughput drug discovery. Mol Med. 2017;23:134–148. doi:10.2119/molmed.2016.00188
34. Forn-Cuni G, Varela M, Pereiro P, Novoa B, Figueras A. Conserved gene regulation during acute inflammation between zebrafish and mammals. Sci Rep. 2017;7:41905. doi:10.1038/srep41905
35. Jin Y, Xu Z, Yan H, He Q, Yang X, Luo P. A comprehensive review of clinical cardiotoxicity incidence of FDA-approved small-molecule kinase inhibitors. Front Pharmacol. 2020;11:891. doi:10.3389/fphar.2020.00891
36. Zakaria ZZ, Benslimane FM, Nasrallah GK, et al. Using zebrafish for investigating the molecular mechanisms of drug-induced cardiotoxicity. Biomed Res Int. 2018;2018:1642684. doi:10.1155/2018/1642684
37. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi:10.1038/nri3399
38. Granger DN, Vowinkel T, Petnehazy T. Modulation of the inflammatory response in cardiovascular disease. Hypertension. 2004;43:924–931. doi:10.1161/01.HYP.0000123070.31763.55
39. Ley K, Allietta M, Bullard DC, Morgan S. Importance of E-selectin for firm leukocyte adhesion in vivo. Circ Res. 1998;83:287–294. doi:10.1161/01.RES.83.3.287
40. Silva M, Videira PA, Sackstein R. E-selectin ligands in the human mononuclear phagocyte system: implications for infection, inflammation, and immunotherapy. Front Immunol. 2017;8:1878. doi:10.3389/fimmu.2017.01878
41. Venkatesh D, Ernandez T, Rosetti F, et al. Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-β autocrine signaling to promote monocyte recruitment. Immunity. 2013;38(5):1025–1037. doi:10.1016/j.immuni.2013.01.012
42. Kunkel EJ, Ley K. Distinct phenotype of E-selectin-deficient mice: E-selectin is required for slow leukocyte rolling in vivo. Circ Res. 1996;79:1196–1204. doi:10.1161/01.RES.79.6.1196
43. Su Y, Lei X, Wu L, Liu L. The role of endothelial cell adhesion molecules P-selectin, E-selectin and intercellular adhesion molecule-1 in leucocyte recruitment induced by exogenous methylglyoxal. Immunology. 2012;137:65–79. doi:10.1111/j.1365-2567.2012.03608.x
44. Yu Y, Moulton KS, Khan MK, et al. E-selectin is required for the antiangiogenic activity of endostatin. Proc Natl Acad Sci U S A. 2004;101:8005–8010. doi:10.1073/pnas.0402551101
45. Rodriguez-Romero A, Almog O, Tordova M, Randhawa Z, Gilliland GL. Primary and tertiary structures of the Fab fragment of a monoclonal anti-E-selectin 7A9 antibody that inhibits neutrophil attachment to endothelial cells. J Biol Chem. 1998;273:11770–11775. doi:10.1074/jbc.273.19.11770
46. Muller WA. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol. 2014;184:886–896. doi:10.1016/j.ajpath.2013.12.033
47. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. 2015;15:692–704. doi:10.1038/nri3908
48. Nelson DE, Ihekwaba AE, Elliott M, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–708. doi:10.1126/science.1099962
49. Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi:10.1126/science.1071914
50. Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9:378–387. doi:10.1038/ni1576
51. Sepulcre MP, Alcaraz-Perez F, Lopez-Munoz A, et al. Evolution of lipopolysaccharide (LPS) recognition and signaling: fish TLR4 does not recognize LPS and negatively regulates NF-κB activation. J Immunol. 2009;182:1836–1845. doi:10.4049/jimmunol.0801755
52. Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33:5308–5319. doi:10.1093/nar/gki836
53. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12:49–62. doi:10.1038/nrrheum.2015.169
54. Laug R, Fehrholz M, Schutze N, et al. IFN-γ and TNF-α synergize to inhibit CTGF expression in human lung endothelial cells. PLoS One. 2012;7:e45430. doi:10.1371/journal.pone.0045430
55. Yarilina A, Xu K, Chan C, Ivashkiv LB. Regulation of inflammatory responses in tumor necrosis factor-activated and rheumatoid arthritis synovial macrophages by JAK inhibitors. Arthritis Rheum. 2012;64:3856–3866. doi:10.1002/art.37691
56. Barczewski AH, Ragusa MJ, Mierke DF, Pellegrini M. The IKK-binding domain of NEMO is an irregular coiled coil with a dynamic binding interface. Sci Rep. 2019;9:2950. doi:10.1038/s41598-019-39588-2
57. Rushe M, Silvian L, Bixler S, et al. Structure of a NEMO/IKK-associating domain reveals architecture of the interaction site. Structure. 2008;16:798–808. doi:10.1016/j.str.2008.02.012
58. Rhodes CA, Dougherty PG, Cooper JK, et al. Cell-permeable bicyclic peptidyl inhibitors against NEMO-IκB kinase interaction directly from a combinatorial library. J Am Chem Soc. 2018;140:12102–12110. doi:10.1021/jacs.8b06738
59. Harrison DA. The Jak/STAT pathway. Cold Spring Harb Perspect Biol. 2012;4:a011205. doi:10.1101/cshperspect.a011205
60. Bousoik E, Montazeri Aliabadi H. “Do we know jack” about JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. 2018;8:287.
61. Guo D, Dunbar JD, Yang CH, Pfeffer LM, Donner DB. Induction of Jak/STAT signaling by activation of the type 1 TNF receptor. J Immunol. 1998;160:2742–2750.
62. Ghoreschi K, Jesson MI, Li X, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. 2011;186:4234–4243. doi:10.4049/jimmunol.1003668
63. Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6:587–595. doi:10.1038/nrclinonc.2009.129
64. Vignais ML, Sadowski HB, Watling D, Rogers NC, Gilman M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol Cell Biol. 1996;16:1759–1769. doi:10.1128/MCB.16.4.1759
65. Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T. Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996;156:3961–3969.
66. Gerli R, Paolucci C, Gresele P, et al. Salicylates inhibit adhesion and transmigration of T lymphocytes by preventing integrin activation induced by contact with endothelial cells. Blood. 1998;92:2389–2398. doi:10.1182/blood.V92.7.2389
67. Shirakawa K, Wang L, Man N, et al. Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity. Elife. 2016;5:e11156. doi:10.7554/eLife.11156
68. Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956–959. doi:10.1126/science.8052854
69. Hauser TH, Salastekar N, Schaefer EJ, et al. Effect of targeting inflammation with salsalate: the TINSAL-CVD randomized clinical trial on progression of coronary plaque in overweight and obese patients using statins. JAMA Cardiol. 2016;1:413–423. doi:10.1001/jamacardio.2016.0605
70. Nohria A, Kinlay S, Buck JS, et al. The effect of salsalate therapy on endothelial function in a broad range of subjects. J Am Heart Assoc. 2014;3:e000609. doi:10.1161/JAHA.113.000609
71. Vinsova J, Imramovsky A, Buchta V, et al. Salicylanilide acetates: synthesis and antibacterial evaluation. Molecules. 2007;12:1–12. doi:10.3390/12010001
72. Hu M, Ye W, Li J, Zhou P, Chu Z, Huang W. The salicylanilide derivatives inhibit signal transducer and activator of transcription 3 pathways in A549 lung cancer cells. Anticancer Drugs. 2016;27:41–47. doi:10.1097/CAD.0000000000000303
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.