")
Back to Journals » OncoTargets and Therapy » Volume 12
LAPTM4B knockdown increases the radiosensitivity of EGFR-overexpressing radioresistant nasopharyngeal cancer cells by inhibiting autophagy
Authors Chu C , Niu X, Ou X, Hu C
Received 22 March 2019
Accepted for publication 11 June 2019
Published 15 July 2019 Volume 2019:12 Pages 5661—5677
DOI https://doi.org/10.2147/OTT.S207810
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gaetano Romano
Chu Chu, Xiaoshuang Niu, Xiaomin Ou, Chaosu Hu
Department of Radiation Oncology, Fudan University Shanghai Cancer Center, Shanghai 200032, People’s Republic of China
Purpose: Nasopharyngeal carcinoma (NPC) is a malignant tumor that commonly occurs in southern China and Southeast Asia. Radiation therapy is the main treatment for patients with NPC, and the radioresistance of NPC is an unresolved clinical problem. This study focuses on the mechanism of NPC radioresistance and explores therapeutic targets and research directions for increasing the radiosensitivity of radioresistant cells.
Methods: We used a gradient dose model to establish radioresistant strains of 6-10B and CNE-2 human NPC cells. Plate colony formation assays were used to verify the radioresistance of the cells. We evaluated the expression of epidermal growth factor receptor (EGFR), lysosome-associated transmembrane protein 4β (LAPTM4B), Beclin1 and the autophagy-related proteins p62, LC3I, and LC3II by Western blot and observed GFP-LC3 puncta by confocal microscopy. The interaction between proteins was verified by immunofluorescence and coimmunoprecipitation analyses. Flow cytometry was performed to detect differences related to the apoptosis of radioresistant strains.
Results: The EGFR and LAPTM4B expression levels and autophagic flux were higher in radioresistant cells than in nonradioresistant cells, suggesting that EGFR and LAPTM4B are associated with autophagy levels. We observed that EGFR and LAPTM4B interact and stabilize each other in endosomes by confocal microscopy. LAPTM4B knockdown decreased the survival fraction of radioresistant cells and increased apoptosis after exposure to radiation. Coimmunoprecipitation experiments demonstrated that LAPTM4B interacts with Beclin1, which in turn promotes the initiation of autophagy.
Conclusion: This study illustrates a relationship among EGFR, LAPTM4B and autophagy in radioresistant NPC cell lines. LAPTM4B interacts with EGFR and Beclin 1, which promotes autophagy. LAPTM4B knockdown decreases radioresistance by inhibiting autophagy. This study proposes a possible mechanism for NPC radioresistance and provides a new research direction and theoretical basis for addressing the radioresistance of NPC.
Keywords: radioresistance, autophagy, lysosome-associated transmembrane protein 4β, epidermal growth factor receptor, nasopharyngeal cancer
Introduction
Nasopharyngeal carcinoma (NPC) is a malignant tumor that commonly occurs in southern China and Southeast Asia. Hereditary factors, the environment, Epstein-Barr (EB) virus infection and pathogenic factors contribute to the occurrence of NPC. Radiation therapy is currently the first-line treatment for nasopharyngeal cancer.1,2 Although most NPCs are sensitive to radiation, some patients still exhibit radioresistance. Radioresistance of cancer cells leads to recurrence and metastasis shortly after radiation therapy. These patients often have a worse prognosis than those who are sensitive to radiotherapy.3 Thus, elucidating the mechanism of radioresistance in NPC is key for enhancing treatment. Understanding radioresistance can help improve the therapeutic effect for patients with radioresistance and prolong their life.
The radioresistance of cancer leads to the survival and proliferation of cancer cells after radiation exposure, and survival and proliferation are closely related to cell survival signaling pathways, growth factors and their receptors. The role of epidermal growth factor receptor (EGFR) is of great concern. EGFR is expressed in most human epithelial cancers, and high EGFR expression in tumors is associated with more invasive phenotypes, more significant therapeutic resistance and worse prognosis.4–6 Studies have confirmed that a large proportion of patients with NPC express EGFR, and EGFR plays a critical role in the proliferation, invasion and metastasis of NPC cells.7,8 Lysosome-associated transmembrane protein 4β (LAPTM4B) is a lysosome-targeted protein that acts to stabilize the lysosomal membrane and promotes the proliferation and migration of tumors.9 LAPTM4B is reportedly overexpressed in some cancers and associated with prognosis,10 and high LAPTM4B expression indicates a high risk of tumor metastasis.11,12 The roles of EGFR and LAPTM4B in nasopharyngeal cancer need further study.
Autophagy is an important lysosome-mediated pathway for the degradation of intracellular substances and maintains the internal stability of cells by removing damaged organelles and proteins.13 Autophagy flux starts with double-membrane autophagosomes, which in turn encapsulate the intracellular components that need to be degraded. Then, autophagosomes fuse with lysosomes to form autophagosomes and degrade the contents.14 In addition to its homeostatic functions, autophagy is also involved in a variety of human diseases, such as metabolic diseases, neurodegenerative diseases, cardiovascular diseases and cancer.15,16 The roles of autophagy in tumorigenesis and progression are complex and contradictory. On the one hand, autophagy inhibits the occurrence of tumors by eliminating misfolded proteins in cells. On the other hand, in the late stage of tumor progression, autophagy promotes cell survival by providing energy and eliminating proteins that have been damaged by drugs and radiation.13 Thus, autophagy plays a dual role in tumor survival.However, compared to the number of studies reporting a tumor inhibitory role for autophagy, more studies have reported that autophagy plays a major role in radiation and that drug resistance is higher in autophagic cells.
Relationships among the EGFR pathway, tumor radiosensitivity and autophagy have been reported in glioma, lung cancer and other tumors.17,18 EGFR is thought to regulate the autophagy signaling pathway and radioresistance.19–21 However, few studies have reported on NPC. This study explored the relationship between the EGFR-regulated autophagy signaling pathway and radioresistance in NPC cell lines.
Materials and methods
Cell lines and agents
The human nasopharyngeal cancer cell line 6-10B was ordered from Icellbioscience (Shanghai, China), and CNE-2 was kindly given by Zhongshan University. RPMI-1640 was purchased from HyClone (Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Gibco (Waltham, MA, USA). Radioimmunoprecipitation (RIPA) lysis buffer was purchased from Beyotime Biotechnology (Beijing, China). The BCA Protein Assay Kit was purchased from Pierce (Rockford, IL, USA). PVDF membrane was purchased from Merck Millipore (Billerica, MA, USA). Chloroquine (CQ) was purchased from MedChemExpress (Monmouth, NJ, USA). Lipofectamine 2000 was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Cell apoptosis staining kits were purchased from BD Biosciences (Franklin Lakes, NJ, USA). The electrochemiluminescence kit was purchased from Yeasen Biotech (Shanghai, China). The anti-EGFR rabbit monoclonal antibody and anti-Beclin1 mouse monoclonal antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). The anti-EGFR mouse polyclonal antibody and anti-LAPTM4B rabbit polyclonal antibody were purchased from Abcam (Cambridge, MA, USA). The anti-p62 rabbit polyclonal antibody, anti-Beclin1 rabbit polyclonal antibody, anti-β-actin rabbit monoclonal antibody and horseradish peroxidase (HRP)-conjugated secondary anti-rabbit and anti-mouse antibodies were purchased from Proteintech (Chicago, IL, USA). The anti-LC3 B rabbit monoclonal antibody was purchased from Novus Biologicals (Littleton, CO, USA). The Institutional Review Board of Fudan University Shanghai Cancer Center approved this study protocol and usage of the CNE-2 cell line.
Cell culture and establishment of radioresistant cell lines
Cells were cultured in RPMI-1640 medium supplemented with 10% FBS, 5% penicillin (100 units/mL) and 5% streptomycin (100 units/mL). The cells were incubated at 37 °C in an incubator (5% CO2, YCP-50S, Changsha Huaxi Electronic Technology Co., Ltid., China). Parental 6-10B and CNE-2 cells were irradiated with X-rays at 3.845 Gy/min on the Small Animal Radiation Research Platform (SARRP, GulmayMedical, ND, USA) upon reaching 60–70% confluence. Cells were digested and seeded in new T25 flasks when the cell confluence reached 80%. Then, the cells were serially irradiated with 2, 4, 6, 8, and 10 Gy for 2 cycles. We chose 1 clone from each cell line and named them 6-10BR and CNE-2R. The chosen clones were irradiated with 10 Gy for 2 additional cycles.
Colony formation assay
Cells were collected and seeded into 6-well plates (Corning, NY, USA) at 200, 300, 400, 800, 1500, and 2000 cells per well and then exposed to 0, 1, 2, 4, 6, and 8 Gy doses of radiation. After incubation for 14 d, the cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. The clones were photographed and counted using a clone imager (Thermo Fisher Scientific).
Apoptosis detection
Cell apoptosis was detected by flow cytometry using a cell apoptosis staining kit (BD Biosciences, NJ, USA). The cells and supernatant were collected and centrifuged at 1500 rpm for 5 min. Then, the cells were washed with PBS, resuspended in binding buffer and mixed with Annexin V-PE and 7AAD. The cells were transferred into flow tubes and incubated at room temperature for 20 min in the dark. The samples were analyzed by flow cytometry (FC500 MPL, Beckman Coulter).
Western blotting
Cellular proteins were extracted by lysis with RIPA buffer for 1 h on ice followed by centrifugation at 12,000× g for 15 min at 4 °C. The total protein concentration was measured using the BCA Protein Assay Kit. Suitable amounts of protein were separated by SDS-PAGE electrophoresis and transferred to a PVDF membrane. The membranes were blocked with 5% nonfat milk and then sequentially probed with primary antibodies and HRP-conjugated secondary antibodies. The samples were imaged using the Chemioscope Mini system (ChemiQ 4800, Bioshine, Shanghai, China)
Immunofluorescence
The cells were seeded into 24-well plates on slides and incubated for 24 h. The cells on the slides were fixed with 4% paraformaldehyde, washed with PBS, and treated with 0.1% Triton for 15 min. The cells were incubated with primary antibodies overnight and then probed with fluorophore-conjugated secondary antibodies and DAPI. The cellular proteins were detected by laser scanning confocal microscopy (Eclipse Ti-S, Nikon, Japan).
Cell transfection
The cells were collected and plated in 6-well plates 24 h before transfection at a proper cell concentration. Lipofectamine 2000 was combined with small interfering RNAs (siRNAs) or plasmids and added to the cells. The specific siRNAs were purchased from Biotend Biotechnology Company (Shanghai, China). The plasmids were purchased from Vigene Bioscience (Shandong, China). The shRNA was purchased from Genechem (Shanghai, China).
Autophagy detection
A GFP-LC3 lentivirus was used to infect cells to help detect autophagy. The green fluorescent protein GFP was observed by confocal microscopy (Eclipse Ti-S, Nikon, Japan), which showed the number and location of autophagosomes and autolysosomes.
Statistical analysis
The experimental data were statistically analyzed using SPSS 24.0 software, and images were drawn using GraphPad Prism 7.0 software. All data are presented as the mean ± standard deviation. Experimental data from multiple groups were analyzed by two-way ANOVA, and multiple comparisons between groups were made using the LSD-t test. Data comparison between two groups was performed using Student’s t-test. Differences were statistically significant at p<0.05.
Results
Radioresistant nasopharyngeal cancer cells exhibit increased autophagy
We established radioresistant cell lines (RR) derived from the human nasopharyngeal cancer cell lines 6-10B and CNE-2. The radioresistant cell sublines were termed 6-10BR and CNE-2R. To evaluate the radioresistance of the radioresistant cells compared with that of parental cells, we performed colony formation assays and calculated the fractions of surviving cells. Figures 1A and S1A show that the 6-10BR and CNE-2R cells demonstrated increased radioresistance compared with that of the parental cell lines. The cell survival curve was analyzed with a linear quadratic model (Figure 1B).
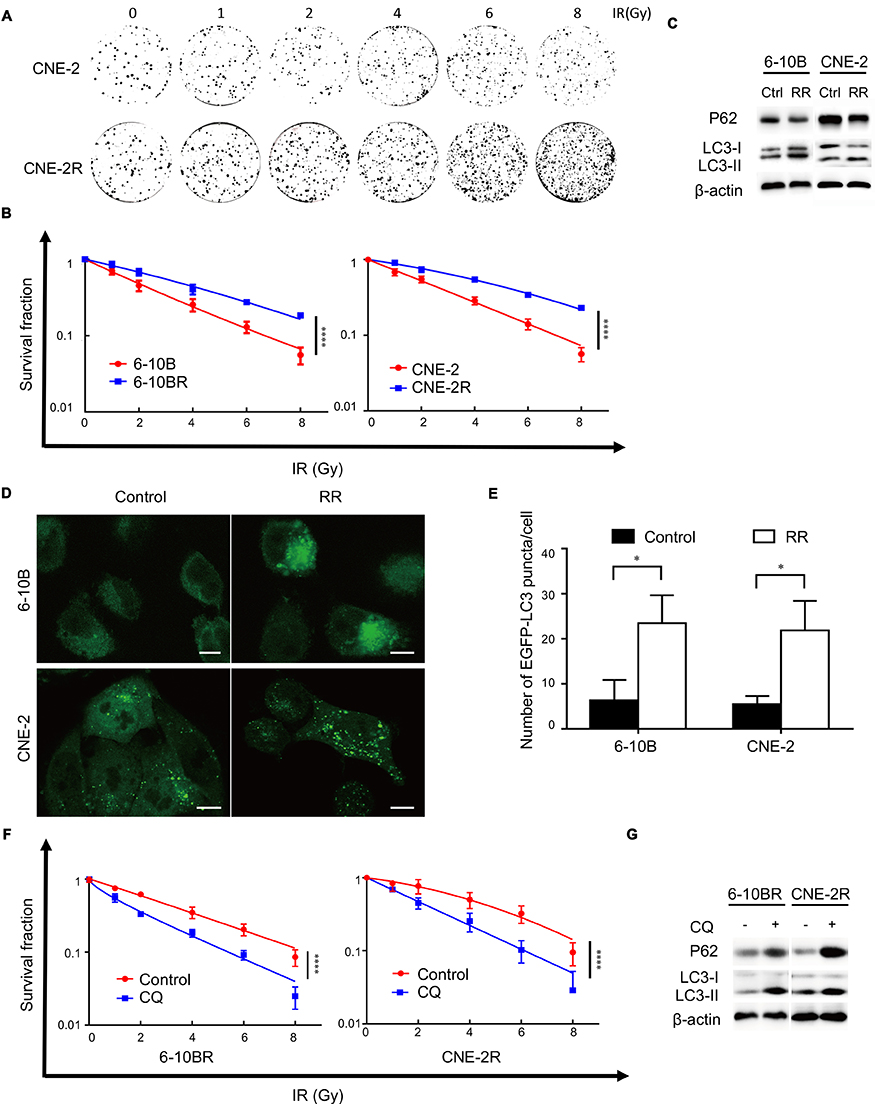 |
Figure 1 Radioresistant nasopharyngeal cancer cells exhibit increased autophagy. Notes: (A) Representative crystal violet staining of the colonies formed by CNE2 and CNE2R cells 14 d after irradiation. (B) The survival fractions of established RR cells and their parental cell lines after irradiation. ****P<0.0001 (C) Expression of the p62, LC3 I and LC3 II proteins in RR cells and their parental cell lines. (D) Representative images of EGFP-LC3 puncta in RR cells and their parental cells. Scale bars, 10 μm. (E) Quantification of the numbers of EGFP-LC3 puncta in (D); mean + SD, n=3, *P<0.05. (F) The survival fraction of RR cells treated with CQ 14 d after irradiation. ****P<0.0001 (G) Expression of p62, LC3 I and LC3 II proteins in RR cells treated with and without CQ.Abbreviations: IR, irradiation; Ctrl, control; RR, radioresistant; CQ, chloroquine. |
Radiation-induced autophagy is one factor contributing to radioresistance. Autophagy facilitates the survival of cells after radiation by reducing cellular damage and inhibiting radiation-induced apoptosis. Therefore, we assumed that radioresistant cells may exhibit increased autophagy. We found increased LC3-II expression and decreased p62 expression in radioresistant cells by Western blot analysis (Figure 1C). Then, we established cells stably expressing the GFP-LC3 fusion protein. We found more GFP-LC3 puncta in the radioresistant cells than in the parental cells, suggesting that more autophagic vacuoles were formed (Figure 1D and E). To evaluate whether the level of autophagy flux influenced the radioresistance of the cell lines, we treated the radioresistant cell lines 6-10BR and CNE-2R with 10 μM CQ, an autophagy inhibitor, for 24 h. Western blotting was performed to detect autophagy level variation after the addition of CQ. As shown in Figure 1G, the expression of both LC3-II and p62 was increased, indicated the blockage of autophagy flux. As shown in Figure 1F and S1B, radioresistant cells treated with CQ exhibited decreased radioresistance, while treatment with CQ had no effect on cell proliferation. Therefore, we confirmed that inhibiting autophagy with CQ might resensitize nasopharyngeal cancer cells to radiation. Finally, our results indicate that radioresistance in nasopharyngeal cancer cells is associated with increased autophagic flux.
EGFR expression is associated with autophagy flux
EGFR overexpression is commonly observed in nasopharyngeal cancer cells. We found that EGFR was upregulated in radioresistant cells, and this change was accompanied by increased autophagy (Figure 2A). Therefore, we hypothesized that EGFR is necessary for autophagic flux. To test this hypothesis, we knocked down EGFR with two different siRNAs. Figure 2B suggests that EGFR knockdown inhibited autophagic flux, as shown by the decreased LC3-II levels, increased p62 levels and reduced LC3 puncta numbers (Figure 2C and D). Then, we overexpressed EGFR with a plasmid and found autophagy upregulation (Figure 2E–G). Overall, we concluded that EGFR expression is correlated with autophagy and plays a role in autophagy formation.
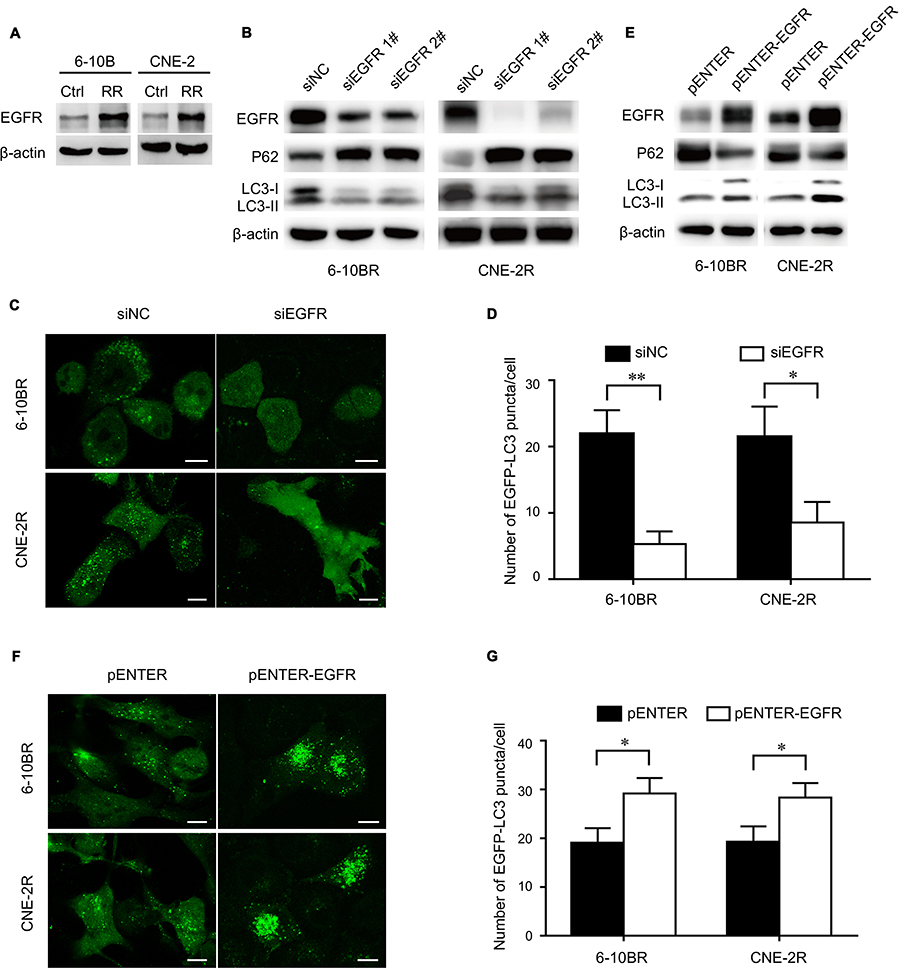 |
Figure 2 The expression of EGFR is associated with autophagy flux. Notes: (A) Expression of the EGFR protein in RR cells and their parental cell lines. (B) Expression of the EGFR, p62, LC3 I and LC3 II proteins in RR cells treated with control or EGFR siRNAs. (C) Representative images of EGFP-LC3 puncta in RR cells treated with control or EGFR siRNAs. Scale bars, 10 μm. (D) Quantification of the numbers of EGFP-LC3 puncta in (C); mean + SD, n=3, *P<0.05, **P<0.01. (E) Expression of the EGFR, p62, LC3 I and LC3 II proteins in RR cells treated with control or pENTER-EGFR. (F) Representative images of EGFP-LC3 puncta in RR cells treated with control or pENTER-EGFR. Scale bars, 10 μm. (G) Quantification of the numbers of EGFP-LC3 puncta in (F); mean + SD, n=3, *P<0.05.Abbreviations: Ctrl, control; RR, radioresistant; EGFR, epidermal growth factor receptor; NC, negative control. |
LAPTM4B promotes autophagy and interacts with EGFR
The LAPTM4B protein localizes in lysosomes, increasing membrane permeability and promoting the maturation of autophagosomes in later stages of autophagy. To determine whether LAPTM4B plays a role in increasing the autophagy-induced radioresistance of NPC, the expression of EGFR and LAPTM4B in radioresistant cells was measured. LAPTM4B was upregulated in radioresistant cells (Figure 3A), and this change was accompanied by increased EGFR levels. Figure 3B shows that knocking down LAPTM4B with two different siRNAs inhibited the autophagic flux. Consistently, GFP-LC3 puncta were decreased upon the loss of LAPTM4B (Figure S2A and B). Likely, LAPTM4B overexpression resulted in increased autophagic flux and GFP-LC3 puncta formation (Figure 3C, Figure S2C and D). This finding demonstrated that LAPTM4B is required for autophagy and may contribute to the radioresistance of nasopharyngeal cancer cells.
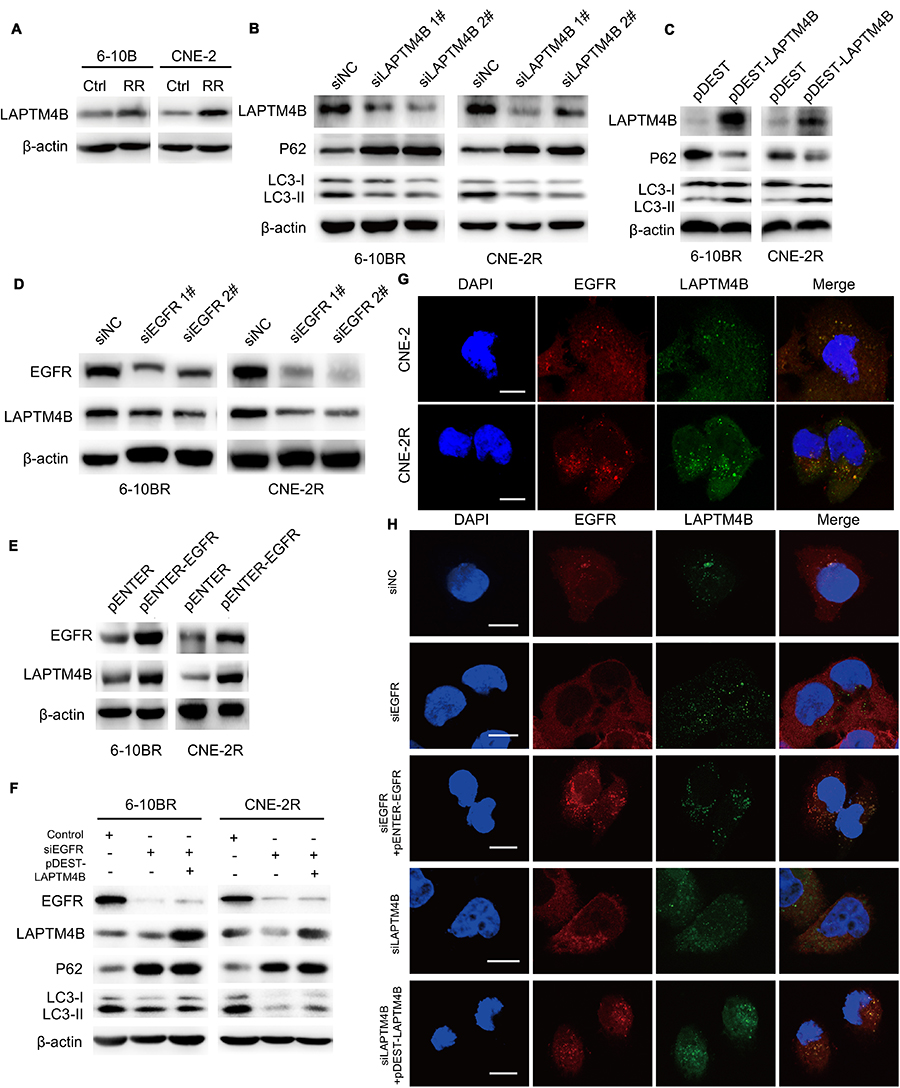 |
Figure 3 LAPTM4B promotes autophagy and interacts with EGFR. Notes: (A) Expression of the LAPTM4B protein in RR cells and their parental cell lines. (B) Expression of the LAPTM4B, p62, LC3 I and LC3 II proteins in RR cells treated with control or LAPTM4B siRNAs. (C) Expression of the LAPTM4B, p62, LC3 I and LC3 II proteins in RR cells treated with control or pDEST-LAPTM4B. (D) Expression of the EGFR and LAPTM4B proteins in RR cells treated with control or EGFR siRNAs. (E) Expression of the EGFR and LAPTM4B proteins in RR cells treated with control or pENTER-EGFR. (F) Expression of the EGFR, LAPTM4B, p62, LC3 I and LC3 II proteins in RR cells treated with control, EGFR siRNA or pDEST-LAPTM4B. (G) Representative images of the costaining of EGFR (red) and LAPTM4B (green) in CNE2 and CNE2R cells. Scale bars, 10 μm. (H) Representative images of the costaining of EGFR (red) and LAPTM4B (green) in RR cells treated with control, EGFR siRNA, or EGFR siRNA followed by pENTER-EGFR or LAPTM4B siRNA and then by pDEST-LAPTM4B. Scale bars, 10 μm.Abbreviations: Ctrl, control; RR, radioresistant; LAPTM4B, lysosome-associated transmembrane protein 4β; EGFR, epidermal growth factor receptor; NC, negative control. |
To confirm that EGFR regulated autophagic flux along with LAPTM4B, EGFR was knocked down or overexpressed, and we detected a decrease or increase in LAPTM4B expression (Figure 3D and E). As shown in Figure 3F, Figure S2E and F, ectopic LAPTM4B expression partially rescued autophagy in EGFR knockdown cells. These results thus indicated that LAPTM4B might act as a cofactor of EGFR in the initiation of autophagic flux.
To further explore the interaction between EGFR and LAPTM4B, double immunofluorescence staining and colocalization experiments were utilized to observe their colocalization in plasma. Figure 3G shows that EGFR and LAPTM4B colocalized in radioresistant cells but colocalized much less in parental cells. This result demonstrated that endosomal EGFR accumulates and colocalizes with LAPTM4B in radioresistant cells, which might contribute to increased autophagic flux. EGFR knockdown with siRNA led to reduced endosomal LAPTM4B levels, which were rescued by re-expressing EGFR. Similarly, EGFR endosomes were markedly decreased after LAPTM4B knockdown, and this change was rescued by re-expression of LAPTM4B (Figure 3H). These data indicated that EGFR and LAPTM4B colocalize in the cytoplasm, which might lead to autophagy initiation. EGFR stabilizes LAPTM4B in the cytoplasm. Likewise, LAPTM4B mediates the endosomal accumulation of EGFR.
To verify the effect of the EGFR and LAPTM4B interaction on cell radiosensitivity, we performed colony formation assays. As shown in Figure S3, cells in which EGFR was knocked down showed the lowest survival fraction, while cells in which EGFR was knocked down and LAPTM4B was overexpressed showed a higher survival fraction. Both groups showed less radioresistance than the control group. Therefore, we considered that the EGFR-LAPTM4B interaction also affects cell radiosensitivity.
LAPTM4B induces radioresistance and inhibits radiation-induced apoptosis
To further investigate whether LAPTM4B has a potential role in the radioresistance of NPC, we knocked down LAPTM4B in the 6-10BR and CNE-2R cell lines with shRNA and termed the resulting cells 6-10BR-shLAPTM4B and CNE-2R-shLAPTM4B. When exposed to different doses of radiation, the 6-10BR-shLAPTM4B and CNE-2R-shLAPTM4B cells had lower survival fractions in colony formation assays than the 6-10BR and CNE-2R cells (Figures 4A and B and S4A). This finding indicated that LAPTM4B might induce radioresistance in nasopharyngeal cancer cells. When LAPTM4B knockdown was combined with 0.5 mM cetuximab, the survival fraction dramatically decreased, suggesting that blockage of both EGFR and LAPTM4B may further inhibit radioresistance.
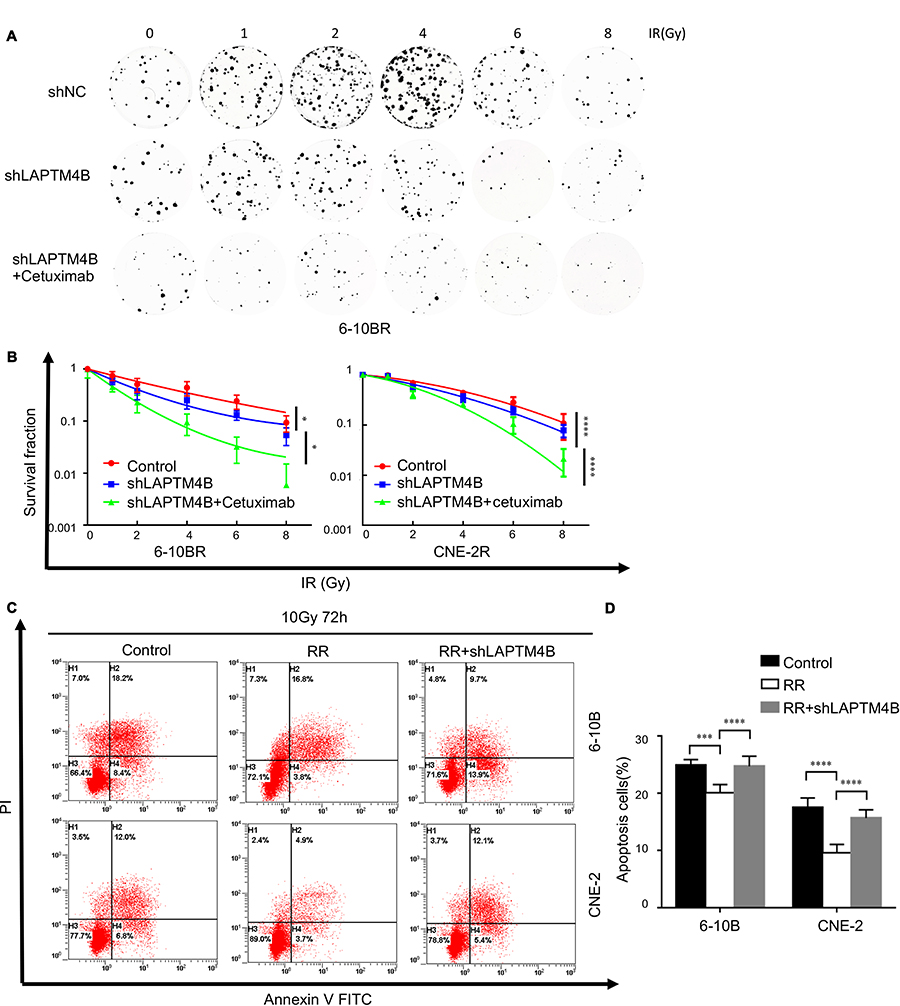 |
Figure 4 LAPTM4B induces radioresistance and inhibits radiation-induced apoptosis. Notes: (A) Representative crystal violet staining of the colonies formed by 6-10BR cells treated with control, LAPTM4B shRNA and LAPTM4B shRNA combined with cetuximab 14 d after irradiation. (B) The survival fractions of RR cells treated with control, LAPTM4B shRNA and LAPTM4B shRNA combined with cetuximab 14 d after irradiation. *P<0.05, ****P<0.0001 (C) Apoptotic cells treated with 10 Gy were stained with Annexin V-PE/7AAD and analyzed by flow cytometry 72 h after irradiation. (D) The percentage of apoptotic cells. Data are presented as the mean ± standard deviation. Control, parental cells treated with 10 Gy; RR, RR cells treated with 10 Gy; RR+shLAPTM4B, RR cells treated with LAPTM4B shRNA and 10 Gy. ***P<0.001, ****P<0.0001.Abbreviations: RR, radioresistant; LAPTM4B, lysosome-associated transmembrane protein 4β; NC, negative control; PI, propidium iodide; FITC, fluorescein isothiocyanate. |
Apoptosis is an essential response contributing to radiation-induced cell death. To determine whether LAPTM4B induces radioresistance by inhibiting apoptosis, we examined the apoptosis of the cells. When exposed to no radiation, the apoptosis rates of all of the cell lines did not differ (Figure S4B and C). Figure 4C and D show that compared with that of parental cells, radioresistant cells exhibited significantly less apoptosis after exposure to 10 Gy radiation over 72 h. Consistently, the apoptosis rates of 6-10BR-shLAPTM4B and CNE-2R-shLAPTM4B cells were markedly increased after radiation. These data indicated that LAPTM4B induced radioresistance partly via inhibiting radiation-induced apoptosis.
LAPTM4B interacts with Beclin1 in autophagy initiation
Beclin1 is a protein recognized as being necessary for the initiation of autophagy. We hypothesize that LAPTM4B might mediate the initiation of autophagy by interacting with Beclin1. To test this hypothesis, LAPTM4B was knocked down with siRNAs, and Beclin1 levels were decreased. Consistently, overexpression of LAPTM4B resulted in increased Beclin1 levels (Figure 5A and B). Coimmunoprecipitation revealed that endogenous LAPTM4B interacted with Beclin1 in radioresistant nasopharyngeal cancer cells (Figure 5C and D). Double immunofluorescence staining was adopted and confirmed that LAPTM4B colocalized with Beclin1 (Figure 5E). In conclusion, we speculate that LAPTM4B interacts with Beclin1 in nasopharyngeal cancer cells and that LAPTM4B may promote the initiation of autophagy by promoting Beclin1 aggregation.
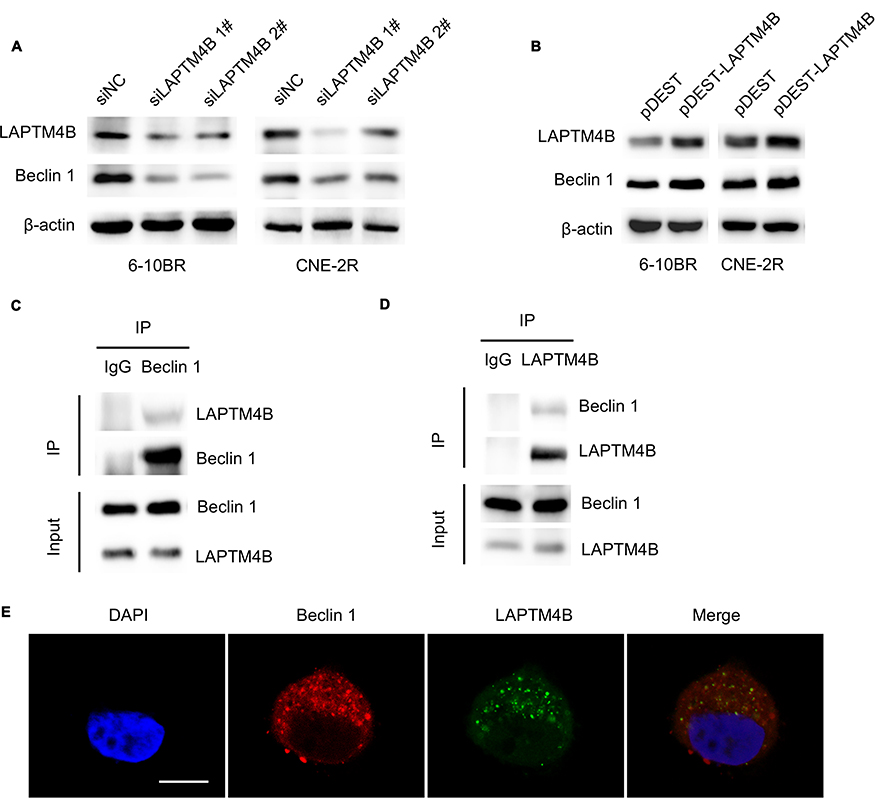 |
Figure 5 LAPTM4B interacts with Beclin1 in autophagy initiation. Notes: (A) Expression of the LAPTM4B and Beclin1 proteins in RR cells treated with control or LAPTM4B siRNAs. (B) Expression of the LAPTM4B and Beclin1 proteins in RR cells treated with control or pDEST-LAPTM4B. (C) Endogenous Beclin1 protein complexes were immunoprecipitated from CNE-2R cell lysates using the anti-Beclin1 antibody, and LAPTM4B was coprecipitated with Beclin1. (D) Endogenous LAPTM4B protein complexes were immunoprecipitated from CNE-2R cell lysates using the anti-LAPTM4B antibody, and Beclin1 was coprecipitated with LAPTM4B. (E) Colocalization analysis of LAPTM4B and Beclin1 by confocal microscopy. Representative images of the costaining of Beclin1 (red) and LAPTM4B (green) in RR cells. Scale bars, 10 μm.Abbreviations: LAPTM4B, lysosome-associated transmembrane protein 4β; IP, immunoprecipitation. |
Discussion
In this study, we identified a connection among autophagy and EGFR and LAPTM4B levels in radioresistant cells. Then, we showed that LAPTM4B interacts with EGFR and Beclin1, which promotes autophagy initiation. Finally, we confirmed that LAPTM4B knockdown sensitizes nasopharyngeal cancer cells to irradiation. Thus, we show that LAPTM4B is a critical regulator of autophagy and enhances the radioresistance of nasopharyngeal cancer cells by promoting autophagy.
Autophagy is a conserved mechanism of cell self-protection induced by metabolic stress, starvation and hypoxia. Studies have shown that autophagy plays a key role in tumor initiation and progression.22 In the process of tumorigenesis, autophagy suppresses tumorigenesis by sustaining the stability of intracellular homeostasis, regulating the cell cycle and limiting necrosis. Conversely, autophagy also protects tumor cells from anticancer therapies by resisting stress, providing energy and maintaining homeostasis.23–25 Autophagy also affects the responses of tumor cells to anticancer therapies.26–28 Increasingly, studies have confirmed that in the context of stress caused by radiotherapy and chemotherapy, autophagy acts as a cell survival mechanism. Increased autophagy enhances the radioresistance of tumors, while tumor tissues with defective autophagy signaling pathways become more sensitive to radiotherapy.29 We herein established radioresistant nasopharyngeal cancer cells and found that their level of autophagy flux was higher than that of their parental cells. CQ treatment inhibits autophagy and reduces the cell survival fraction after radiation. Therefore, we speculate that the level of autophagy is related to radioresistance. Lower autophagy levels can sensitize tumor cells to radiation.
EGFR, also known as ErbB-1 and HER1, is a transmembrane receptor that can be activated by specific ligands, ionizing radiation, hypoxia, and oxidative stress. EGFR promotes malignant transformation and cell growth by inducing several signal transduction pathways. Then, activated transcription factors downstream of EGFR further regulate cell proliferation, apoptosis, invasion and other functions.30 EGFR is overexpressed in many types of tumor cells. Data show that nasopharyngeal cancer cell lines and approximately 85% of nasopharyngeal cancer patients have moderate to high EGFR expression and that patients with high EGFR expression tend to have a worse prognosis.6 In this study, we verified that the expression of EGFR was higher in radioresistant nasopharyngeal cancer cells than in parental cells. When knocking down and overexpressing EGFR, the level of autophagy decreased and increased, respectively. Thus, we conclude that EGFR expression is associated with autophagy levels in cells.
LAPTM4B is an essential protein for lysosomal homeostasis, acidification, and maintenance of normal function and thus allow tumor cells to resist the lysosome-mediated cell death triggered by stress.31 LAPTM4B promotes tumor cell survival and proliferation. Depletion of LAPTM4B increases lysosomal membrane permeability and promotes increased pH, cathepsin release, and apoptosis.9,11 LAPTM4B deletion can block the maturation of autophagosomes, thereby inhibiting late autophagy. Studies have reported that LAPTM4B-deficient cells are more sensitive to hypoxia or nutrient deficiencies,32 whereas LAPTM4B overexpression promotes autophagic flow and cell survival and stimulates tumor growth.33 Tan et al reported that LAPTM4B promotes the initiation of autophagy induced by serum starvation by binding to nonactivated EGFR.34 We herein found that LAPTM4B expression was upregulated in radioresistant nasopharyngeal cancer cells and related to autophagy formation. Further observation of immunofluorescence by confocal microscopy revealed that EGFR and LAPTM4B colocalized in the cytoplasm and formed stable endosomes in the radioresistant cells. When one of the two proteins was knocked down, the levels of the other protein and colocalized endosomes were significantly reduced, which were restored by transfection of an exogenous plasmid. Because LAPTM4B localizes to the lysosomal membrane, we hypothesize that LAPTM4B stabilizes EGFR on endosomes and promotes its accumulation and activation. The formation of EGFR-LAPTM4B colocalized endosomes may facilitate the initiation of autophagy. The survival rate of LAPTM4B knockdown radioresistant cells was decreased, and apoptosis was increased after irradiation, suggesting that LAPTM4B may affect irradiation-induced apoptosis and promote radioresistance in cells. LAPTM4B knockdown combined with cetuximab significantly reduces the survival rate of radioresistant cells, indicating that simultaneous blockage of EGFR and LAPTM4B dramatically sensitizes radioresistant cells to radiation.
Beclin1, which plays a key role in autophagy and apoptotic signaling pathways, is the major determinant of autophagy initiation, and upregulation of Beclin1 promotes autophagy.35,36 Beclin1 forms a complex with class III PI3K, which regulates the localization of other autophagy target gene (ATG) proteins in the autophagy precursor structure, thereby regulating autophagy activity.37,38 In this study, we found a correlation between LAPTM4B and Beclin1 expression, which has been indicatedin other cell lines.39 We revealed that LAPTM4B and Beclin1 colocalize in the cytoplasm and interact with each other. Therefore, we speculate that LAPTM4B and EGFR form stable endosomes in radioresistant cells and that LAPTM4B interacts with Beclin1 to promote the initiation of autophagy flux, possibly by promoting the formation of the class III PI3K complex with Beclin1. However, the specific mechanism by which LAPTM4B affects tumor radioresistance in vitro and initiates autophagy by interacting with Beclin1 needs to be further studied.
Conclusion
In this study, we demonstrated that EGFR and LAPTM4B play roles in autophagy formation in radioresistant nasopharyngeal cancer cells. LAPTM4B interacts with EGFR and Beclin 1, which promotes autophagy. LAPTM4B knockdown increases the radiosensitivity of radioresistant cells by inhibiting autophagy.
Acknowledgments
This study was supported by the CSCO-Merck Serono Oncology Research Fund, the National Natural Science Foundation of China(Grant No.81602372) and the National Key Research Project on Prevention and Control of Chronic Non-communicable Diseases. We acknowledge the support of Professor Musheng Zeng from Zhongshan University. The Institutional Review Board of Fudan University Shanghai Cancer Center approved the study protocol and usage of the CNE-2 cell line. The views expressed in this publication are those of the authors.
Disclosure
The authors report that no conflicts of interest are associated with this work.
References
1. Zhang MX, Li J, Shen GP, et al. Intensity-modulated radiotherapy prolongs the survival of patients with nasopharyngeal carcinoma compared with conventional two-dimensional radiotherapy: a 10-year experience with a large cohort and long follow-up. Eur J Cancer. 2015;51(17):2587–2595. doi:10.1016/j.ejca.2015.08.006
2. Kong L, Hu C, Niu X, et al. Neoadjuvant chemotherapy followed by concurrent chemoradiation for locoregionally advanced nasopharyngeal carcinoma: interim results from 2 prospective phase 2 clinical trials. Cancer. 2013;119(23):4111–4118. doi:10.1002/cncr.28324
3. Kong F, Zhou J, Du C, et al. Long-term survival and late complications of intensity-modulated radiotherapy for recurrent nasopharyngeal carcinoma. BMC Cancer. 2018;18(1):1139. doi:10.1186/s12885-018-4242-8
4. Ruan L, Li XH, Wan XX, et al. Analysis of EGFR signaling pathway in nasopharyngeal carcinoma cells by quantitative phosphoproteomics. Proteome Sci. 2011;9:35. doi:10.1186/1477-5956-9-35
5. Ma X, Huang J, Wu X, et al. Epidermal growth factor receptor could play a prognostic role to predict the outcome of nasopharyngeal carcinoma: a meta-analysis. Cancer Biomark Sect A Dis Markers. 2014;14(4):267–277. doi:10.3233/CBM-140401
6. Lin DC, Meng X, Hazawa M, et al. The genomic landscape of nasopharyngeal carcinoma. Nat Genet. 2014;46(8):866–871. doi:10.1038/ng.3006
7. Zheng LS, Yang JP, Cao Y, et al. SPINK6 promotes metastasis of nasopharyngeal carcinoma via binding and activation of epithelial growth factor receptor. Cancer Res. 2016;77(2):579–589. doi:10.1158/0008-5472.CAN-16-1281.
8. Meng DF, Xie P, Peng LX, et al. CDC42-interacting protein 4 promotes metastasis of nasopharyngeal carcinoma by mediating invadopodia formation and activating EGFR signaling. J Exp Clin Cancer Res. 2017;36(1):21. doi:10.1186/s13046-016-0483-z
9. Li Y, Zhang Q, Tian R, et al. Lysosomal transmembrane protein LAPTM4B promotes autophagy and tolerance to metabolic stress in cancer cells. Cancer Res. 2011;71(24):7481–7489. doi:10.1158/0008-5472.CAN-11-0940
10. Maki Y, Fujimoto J, Lang W, et al. LAPTM4B is associated with poor prognosis in NSCLC and promotes the NRF2-mediated stress response pathway in lung cancer cells. Sci Rep. 2015;5:13846. doi:10.1038/srep13846
11. Xiao M, Yang S, Meng F, et al. LAPTM4B predicts axillary lymph node metastasis in breast cancer and promotes breast cancer cell aggressiveness in vitro. Cell Physiol Biochem. 2017;41(3):1072–1082. doi:10.1159/000464115
12. Dong X, Tamura K, Kobayashi D, Ando N, Sumita K, Maehara T. LAPTM4B-35 is a novel prognostic factor for glioblastoma. J Neurooncol. 2017;132(2):295–303. doi:10.1007/s11060-017-2369-0
13. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi:10.1016/j.ccr.2006.06.001
14. Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
15. Ren J, Zhang Y. Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol Sci. 2018;39(12):1064–1076. doi:10.1016/j.tips.2018.10.005
16. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017. doi:10.1038/nrc.2017.53
17. Han X, Xue X, Zhou H, Zhang G. A molecular view of the radioresistance of gliomas. Oncotarget. 2017;8(59):100931–100941. doi:10.18632/oncotarget.21753
18. Bokobza SM, Jiang Y, Weber AM, Devery AM, Ryan AJ. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget. 2014;5(13):4765–4778. doi:10.18632/oncotarget.2017
19. Sooro MA, Zhang N, Zhang P. Targeting EGFR-mediated autophagy as a potential strategy for cancer therapy. Int J Cancer. 2018. doi:10.1002/ijc.31398
20. Jutten B, Rouschop KM. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle. 2014;13(1):42–51. doi:10.4161/cc.27518
21. Henson E, Chen Y, Gibson S. EGFR family members’ regulation of autophagy is at a crossroads of cell survival and death in cancer. Cancers. 2017;9:4. doi:10.3390/cancers9040027
22. Hait WN, Jin S, Yang JM. A matter of life or death (or both): understanding autophagy in cancer. Clin Cancer Res. 2006;12(7 Pt 1):1961–1965. doi:10.1158/1078-0432.CCR-06-0011
23. Zhou S, Zhao L, Kuang M, et al. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde? Cancer Lett. 2012;323(2):115–127. doi:10.1016/j.canlet.2012.02.017
24. Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. doi:10.1016/j.cell.2009.03.048
25. Singh SS, Vats S, Chia AY, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37(9):1142–1158. doi:10.1038/s41388-017-0046-6
26. Bhat P, Kriel J, Shubha Priya B, Basappa SNS, Loos B. Modulating autophagy in cancer therapy: advancements and challenges for cancer cell death sensitization. Biochem Pharmacol. 2018;147:170–182. doi:10.1016/j.bcp.2017.11.021
27. Zou SH, Du X, Sun FD, Wang PC, Li M. Cisplatin suppresses tumor proliferation by inhibiting autophagy in ovarian cancer via long non-coding RNA RP11-135L22.1. Eur Rev Med Pharmacol Sci. 2018;22(4):928–935. doi:10.26355/eurrev_201802_14372
28. Chen X, Wang P, Guo F, et al. Autophagy enhanced the radioresistance of non-small cell lung cancer by regulating ROS level under hypoxia condition. Int J Radiat Biol. 2017;93(8):764–770. doi:10.1080/09553002.2017.1325025
29. Wang F, Tang J, Li P, et al. Chloroquine enhances the radiosensitivity of bladder cancer cells by inhibiting autophagy and activating apoptosis. Cell Physiol Biochem. 2018;45(1):54–66. doi:10.1159/000486222
30. Kovacs E, Zorn JA, Huang Y, Barros T, Kuriyan J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu Rev Biochem. 2015;84:739–764. doi:10.1146/annurev-biochem-060614-034402
31. Li S, Wang L, Meng Y, Chang Y, Xu J, Zhang Q. Increased levels of LAPTM4B, VEGF and survivin are correlated with tumor progression and poor prognosis in breast cancer patients. Oncotarget. 2017;8(25):41282–41293. doi:10.18632/oncotarget.17176
32. Meng Y, Wang L, Chen D, et al. LAPTM4B: an oncogene in various solid tumors and its functions. Oncogene. 2016;35(50):6359–6365. doi:10.1038/onc.2016.189
33. Blom T, Li S, Dichlberger A, et al. LAPTM4B facilitates late endosomal ceramide export to control cell death pathways. Nat Chem Biol. 2015;11(10):799–806. doi:10.1038/nchembio.1889
34. Tan X, Thapa N, Sun Y, Anderson RA. A kinase-independent role for EGF receptor in autophagy initiation. Cell. 2015;160(1–2):145–160. doi:10.1016/j.cell.2014.12.006
35. Li Z, Li Q, Lv W, et al. The interaction of Atg4B and Bcl-2 plays an important role in Cd-induced crosstalk between apoptosis and autophagy through disassociation of Bcl-2-Beclin1 in A549 cells. Free Radic Biol Med. 2018;130:576–591. doi:10.1016/j.freeradbiomed.2018.11.020.
36. Wei Y, Zou Z, Becker N, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154(6):1269–1284. doi:10.1016/j.cell.2013.08.015
37. Tian M, Chen Y, Tian D, Qiao X, Ma Z, Li J. Beclin1 antagonizes LAPTM4B-mediated EGFR overactivation in gastric cancer cells. Gene. 2017. doi:10.1016/j.gene.2017.05.006
38. Chang SH, Minai-Tehrani A, Shin JY, et al. Beclin1-induced autophagy abrogates radioresistance of lung cancer cells by suppressing osteopontin. J Radiat Res. 2012;53(3):422–432. doi:10.1269/jrr.11148
39. Tian M, Chen Y, Tian D, Qiao X, Ma Z, Li J. Beclin1 antagonizes LAPTM4B-mediated EGFR overactivation in gastric cancer cells. Gene. 2017;626:48–53. doi:10.1016/j.gene.2017.05.006
Supplementary materials
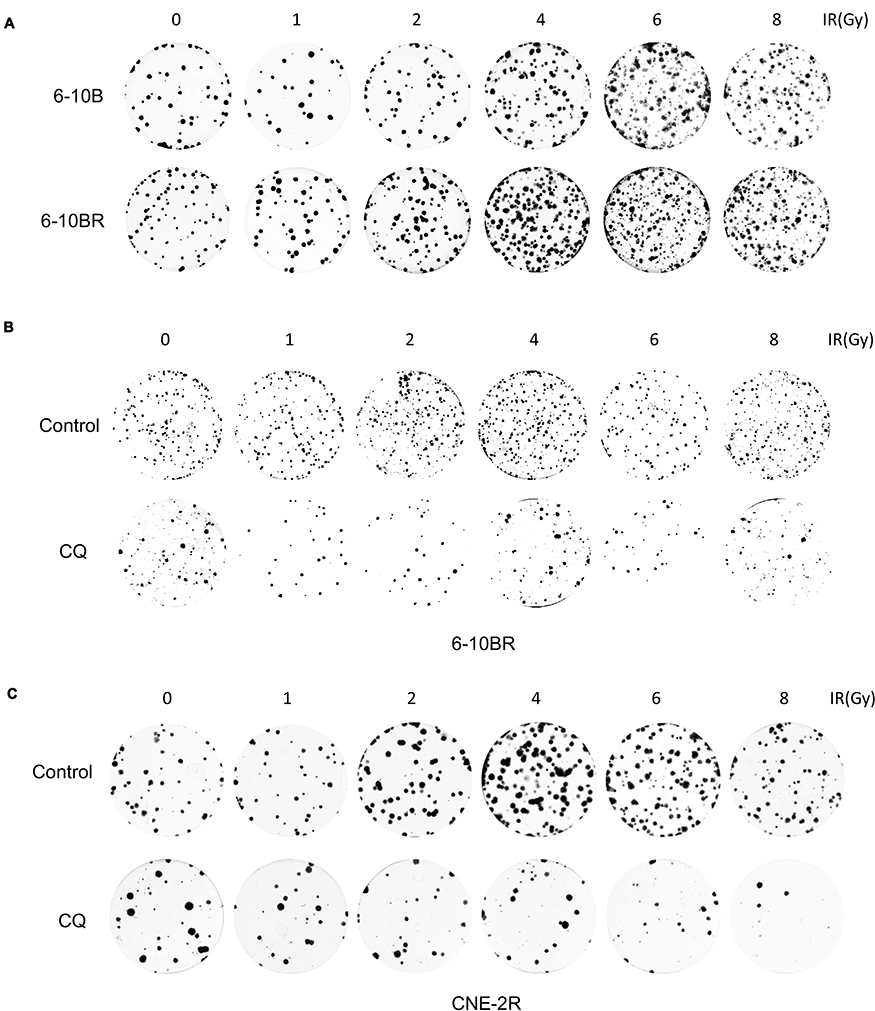 |
Figure S1 (A) Representative crystal violet staining of the colonies formed by 6-10B and 6-10BR cells 14 d after irradiation. (B) Representative crystal violet staining of the colonies formed by 6-10BR and CNE-2R cells treated with control or CQ 14 d after irradiation.Abbreviations: IR, irradiation; Ctrl, control; RR, radioresistant; CQ, chloroquine. |
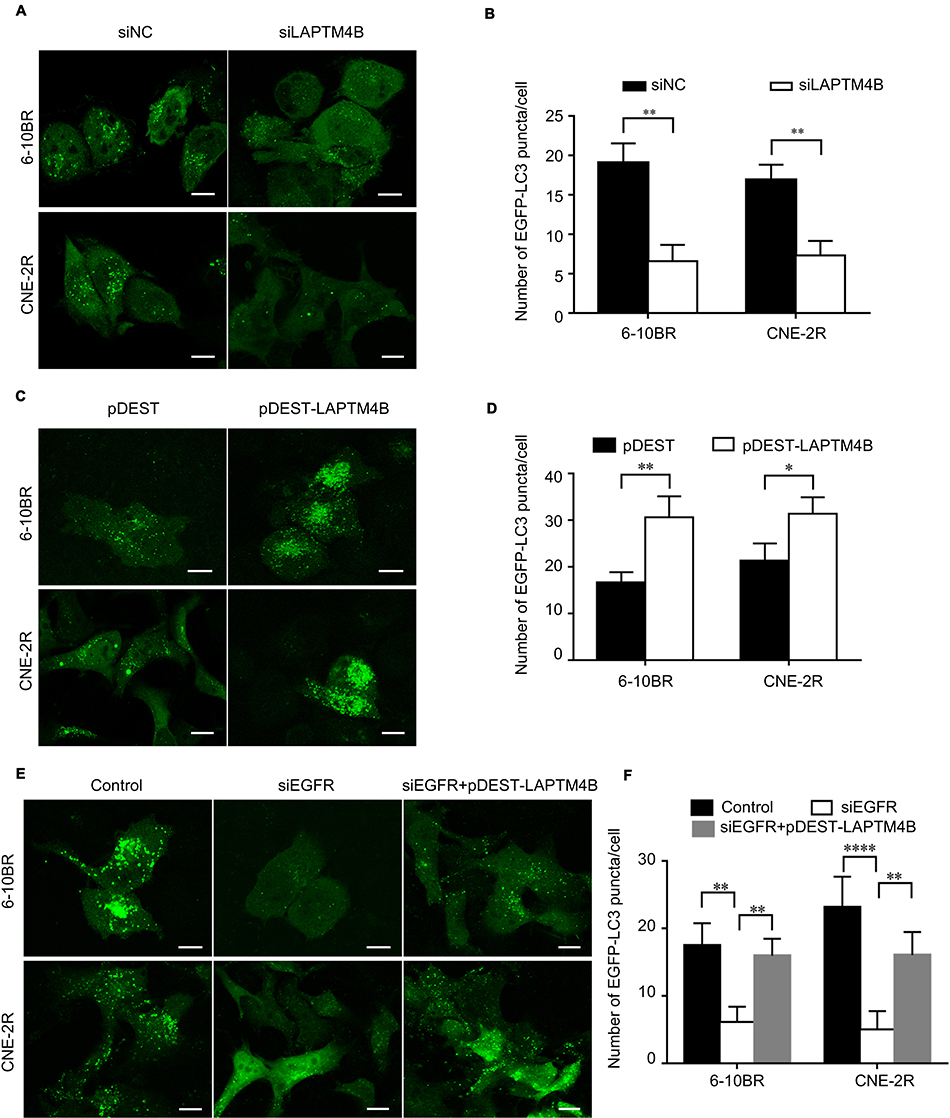 |
Figure S2 (A) Representative images of EGFP-LC3 puncta in RR cells treated with control or LAPTM4B siRNAs. Scale bars, 10 μm. (B) Quantification of the numbers of EGFP-LC3 puncta in (A); mean + SD, n=3, **P<0.01. (C) Representative images of EGFP-LC3 puncta in RR cells treated with control or pDEST-LAPTM4B. Scale bars, 10 μm. (D) Quantification of the numbers of EGFP-LC3 puncta in (C); mean + SD, n=3, *P<0.05, **p<0.01. (E) Representative images of EGFP-LC3 puncta in RR cells treated with control, EGFR siRNA or pDEST-LAPTM4B. Scale bars, 10 μm. (F) Quantification of the numbers of EGFP-LC3 puncta in Figure 3G; mean + SD, n=3, **p<0.01, ****P<0.0001.Abbreviations: LAPTM4B, lysosome-associated transmembrane protein 4β; EGFR, epidermal growth factor receptor; NC, negative control. |
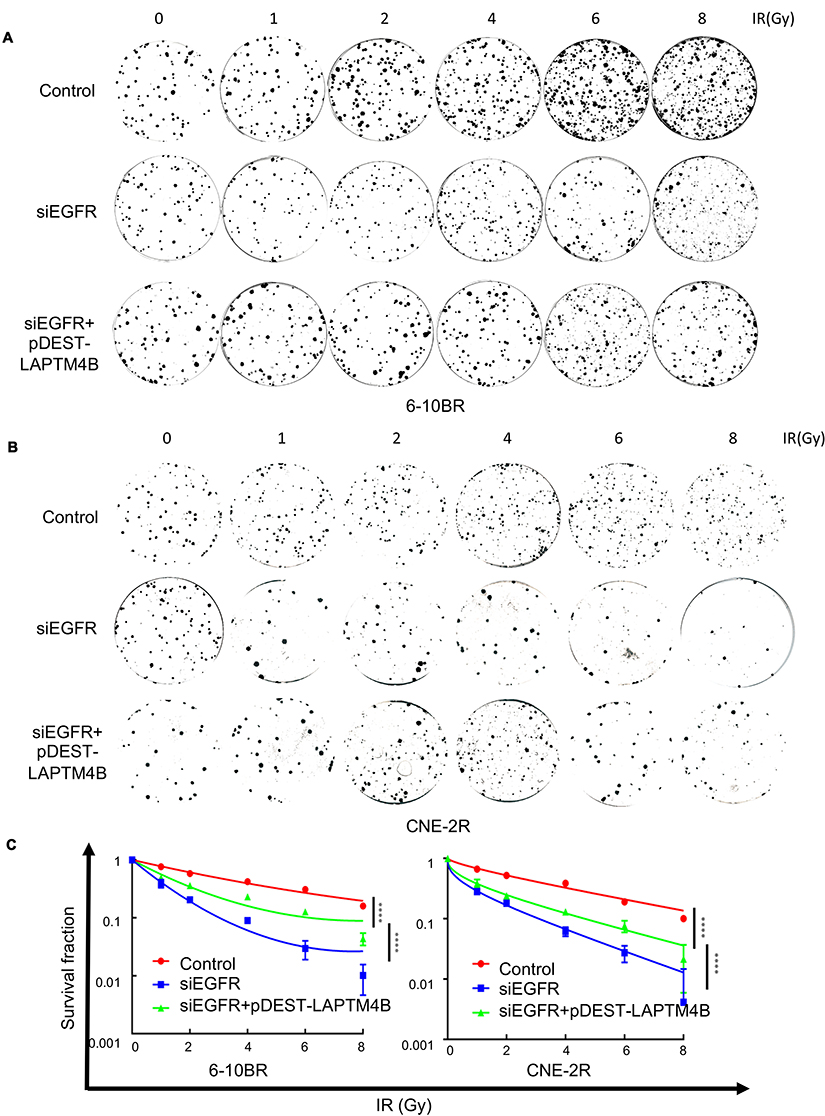 |
Figure S3 (A) Representative crystal violet staining of the colonies formed by 6-10BR cells treated with control, siEGFR, or siEGFR combined with pDEST-LAPTM4B 14 d after irradiation. (B) Representative crystal violet staining of the colonies formed by CNE-2R cells treated with control, siEGFR, or siEGFR combined with pDEST-LAPTM4B 14 d after irradiation. (C) The survival fractions of RR cells treated with control, siEGFR, or siEGFR combined with pDEST-LAPTM4B 14 d after irradiation. ****P<0.0001.Abbreviations: IR, irradiation; RR, radioresistant. |
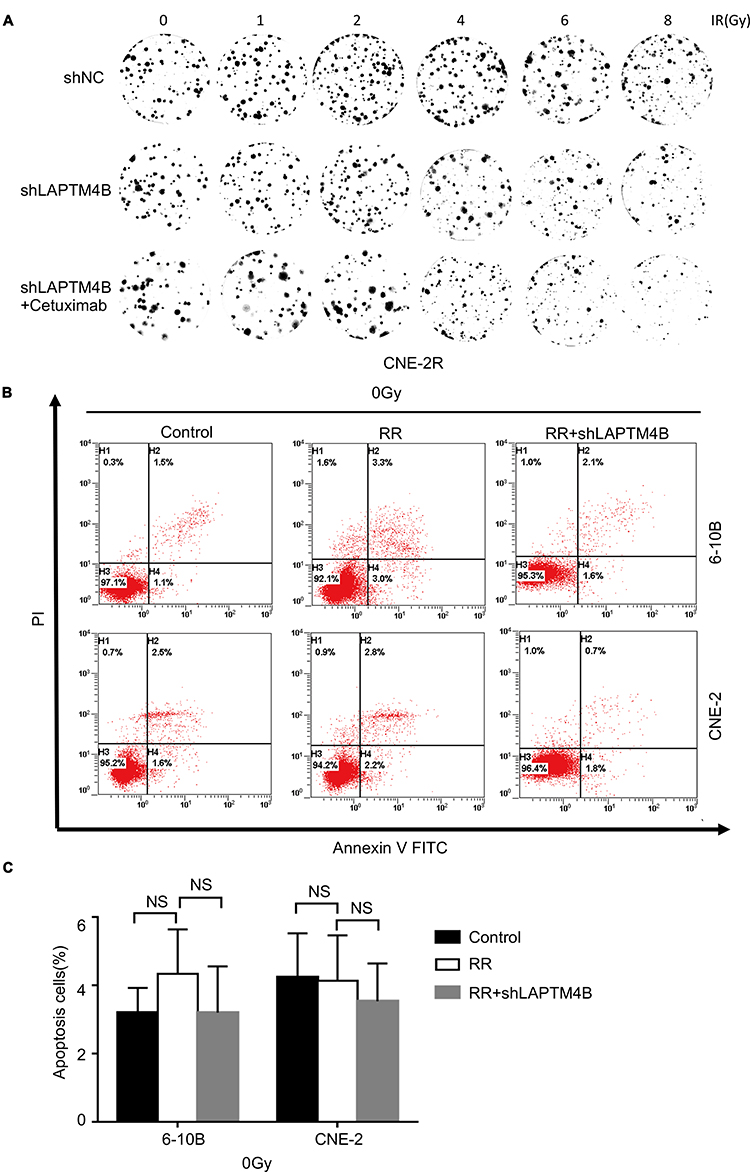 |
Figure S4 (A) Representative crystal violet staining of the colonies formed by CNE-2 and CNE-2R cells treated with control, LAPTM4B shRNA or LAPTM4B shRNA combined with cetuximab 14 d after irradiation. (B) Apoptotic cells treated with the control were stained with Annexin V-FITC/PI and analyzed by flow cytometry. (C) The percentage of apoptotic cells. Data are presented as the mean ± standard deviation. Control, parental cells without treatment; RR, RR cells without treatment; RR+shLAPTM4B, RR cells treated with LAPTM4B shRNA. NS, not significant.Abbreviations: RR, radioresistant; LAPTM4B, lysosome-associated transmembrane protein 4β; NC, negative control; PI, propidium iodide; FITC, fluorescein isothiocyanate; NS, not significant. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.