")
Back to Journals » Cancer Management and Research » Volume 12
LACTB Regulates PIK3R3 to Promote Autophagy and Inhibit EMT and Proliferation Through the PI3K/AKT/mTOR Signaling Pathway in Colorectal Cancer
Authors Xu W, Yu M, Qin J, Luo Y, Zhong M
Received 20 February 2020
Accepted for publication 30 May 2020
Published 30 June 2020 Volume 2020:12 Pages 5181—5200
DOI https://doi.org/10.2147/CMAR.S250661
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Wei Xu, Minhao Yu, Jun Qin, Yang Luo, Ming Zhong
Department of Gastrointestinal Surgery, Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai 200127, People’s Republic of China
Correspondence: Ming Zhong Email [email protected]
Background: Colorectal cancer (CRC) is one of the most common aggressive malignancies. LACTB functions as a tumor suppressor, and previous findings have demonstrated that LACTB can inhibit epithelial-to-mesenchymal transition (EMT) and proliferation of breast cancer and CRC cells. However, few studies have investigated the roles of LACTB in autophagy and proliferation in CRC. The current study aimed to identify the roles of LACTB in EMT and proliferation associated with autophagy in CRC and to elucidate the probable molecular mechanisms through which LACTB are involved in these processes.
Materials and Methods: Transwell invasion, MTT, transmission electron microscopy, RNA-seq, immunoprecipitation, immunohistochemistry and Western blotting assays were performed to evaluate the migratory, invasive, proliferative and autophagic abilities of CRC cells, and the levels of active molecules involved in PI3K/AKT signaling were examined through Western blotting analysis. In addition, the in vivo function of LACTB was assessed using a tumor xenograft model.
Results: Weaker LACTB expression was found in CRC tissue samples than in nonmalignant tissue samples, and LACTB inhibited cell invasion, migration, and proliferation by promoting autophagy in vitro. Furthermore, the regulatory effects of LACTB on autophagy and EMT were partially attributed to the PI3K/AKT signaling pathway. The in vivo results also showed that LACTB modulated CRC tumorigenesis.
Conclusion: LACTB can regulate the activity of PIK3R3 to influence the level of PI3K, and it also promotes autophagy and inhibits EMT and proliferation in part through the PI3K/AKT/mTOR signaling pathway.
Keywords: colorectal cancer, LACTB, PIK3R3, EMT, proliferation, autophagy
Introduction
Colorectal cancer (CRC) is one of the most lethal and common malignancies diagnosed and is currently the second leading cause of cancer-related death worldwide.1 The published research studies suggest that the incidence of CRC continues to increase, which might be related to the increasing obesity rate.2 Despite the development of early diagnostic methods and molecular targeted therapy, the mortality rate remains significant.3 In addition, distant metastases play an increasingly important role in impairing the overall survival of patients with CRC. The molecular mechanisms underlying the invasive behaviors of CRC urgently need to be elucidated.
Weinberg’s study was inspired by the different tumor incidence rates in different tissues, such as the extreme rarity of heart cancer and skeletal muscle cancer and the high incidence of cancer in the lungs and colon, and both gene expression profiles of different tissues and molecular experimental results suggest that beta-lactamase-like (LACTB) might be a new factor that could be used for the diagnosis and treatment of cancer.4 LACTB, which is mainly located in the mitochondria, might influence the membrane organization and microcompartmentalization progression of mitochondria, which is related with regulating oxidative phosphorylation and alterations in mitochondrial lipid metabolism.5 This factor, a type of mammalian active-site serine protein expressed in most mammalian tissues, is related to bacterial penicillin-binding B-lactamase proteins, which are endowed with a novel biochemical function in cell progression.5,6 Recent studies have shown that LACTB can play a role as a tumor suppressor in breast cancer, glioma and CRC, and these previous studies focused on the inhibition of CRC invasion and migration by LACTB and revealed that these effects are mediated by regulating the stability of the p53 tumor suppressor pathway.3,6-8 And recent studies showed that autophagy was associated with the mitochondria under metabolic stress conditions and could regulate cancer development.9,10 So, we want to investigate the relationship between the LACTB and autophagy and what the underlying mechanisms are, such as whether autophagy is induced by LACTB and whether LACTB-induced autophagy modulates EMT and proliferation in CRC cells.
In this study, a microarray analysis suggests that LACTB overexpression can influence cell proliferation and autophagy induction in CRC. The cell cycle is a process with highly regulated, orchestrated steps, and impaired function of its critical gatekeepers will induce abnormally persistent cell proliferation, namely, cancer development.9 Autophagy, which is a type of homeostatic mechanism and a self-degradative process, plays a significant role in the recycling of cellular proteins, organelles and cytoplasmic components for balancing sources of energy and performing quality control in the presence of an insufficient energy supply.9,10 At an appropriate level, autophagy can maintain basic cellular activities and is treated as a defense mechanism that is upregulated when cancer cells are subjected to adverse environmental stimuli during anticancer treatments or under nutrient-deficient conditions.11,12 Because autophagy is linked to mitochondria under hypoxic or metabolic stress conditions, mitophagy can recycle intracellular components to sustain the metabolic demands,13,14 but the function of autophagy in cancer cells remains complex and controversial.15 Numerous studies have revealed that autophagy can decrease the level of destruction and promote apoptosis in cancerous cells to suppress the development of malignant tumors.16,17 However, this process can also provide energy to cancer cells by activating the metabolic stress responses to promote tumor growth, and the levels of autophagic flux in distant metastases differ from those in primary tumors.18–20 The transformation of autophagy between carcinogenic and suppressor states is manifested in the modulation of metabolism and cytokines,21–23 but the role of autophagy in regulating the migration and invasion of CRC cells remains controversial. The Rho family of small GTPases and downstream signaling molecules, which are key factors of membrane protrusion and cytoskeletal remodeling that influence different patterns of migration, could be controlled by autophagy.27 Autophagy can also regulate EMT by influencing the levels of individual factors, such as ATP, AMKP and phosphatidylinositol-3-kinase (PI3K).23 The studies conducted by Li and Zhu have shown that the energy provided by autophagy can promote the invasion of cancer cells by inducing EMT in hepatocellular carcinoma and pancreatic cancer.24,25
In the present study, we investigated the capacity of LACTB to induce autophagy in CRC and explored the effects of LACTB on CRC cell autophagy, EMT and proliferation both in vitro and in vivo. We further identified whether LACTB modulates EMT and proliferation of CRC cells by regulating autophagy and elucidated the mechanism underlying the LACTB-mediated induction of autophagy by mainly focusing on the PI3K/AKT/mTOR signaling pathway.
Materials and Methods
Patients and Tissue Samples
In our study, 80 paired CRC and adjacent nontumor tissue samples were acquired from CRC patients who underwent surgery between January 2017 and July 2019 at our hospital. These patients included 48 males and 32 females with a mean age of 55.3±8.8 years.The detail clinical data of patients were described in Table 1. None of the patients had received standard chemotherapy prior to surgery, and all the patient tissue samples were pathologically confirmed to be CRC tissue. The study was approved by the Ethics Committee of Renji Hospital, School of Medicine, Shanghai Jiaotong University, and all the samples were obtained with informed consent provided by the patients prior to their participation.
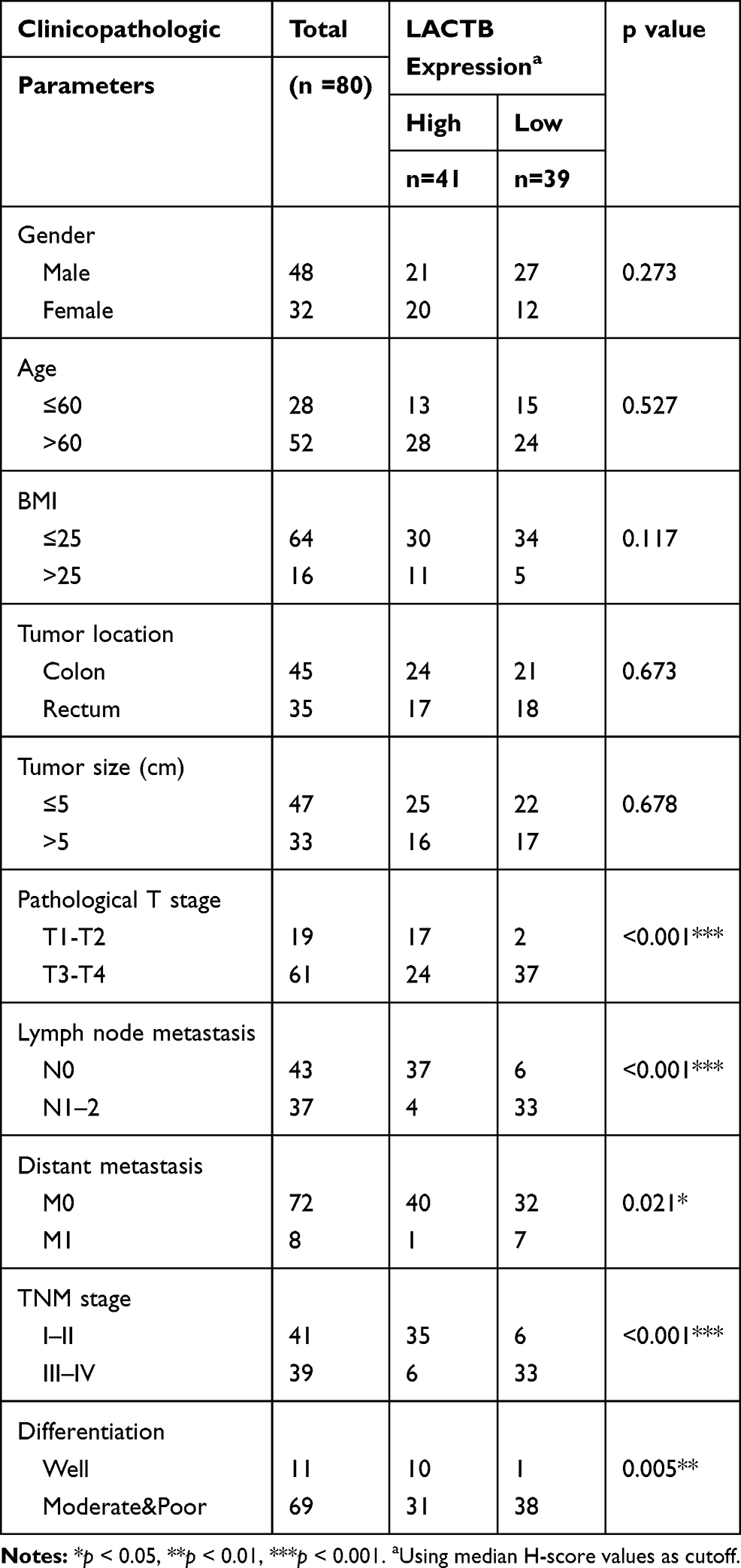 |
Table 1 The Correlation of LACTB Expression and Clinical Characteristics in CRC Patients (n = 80) |
Analyses of LACTB Expression Based on TCGA Databases
A total of 438 cases of colon cancer and 159 cases of rectal cancer were provided by TCGA project. Based on the expression value of LACTB, the cohort obtained after merging the colon and rectal cancer cases was classified into a high-expression group and a low-expression group (cut-off = 50%). Box plots were generated to compare the LACTB expression level between the tumor and normal tissues of patients with CRC and to identify the features of LACTB expression at different pathological stages. A tool named The Human Protein Atlas, which is an interactive web server for analyzing the RNA sequencing expression data from TCGA projects, was used for batch processing and visualization of TCGA data in this study.
Cell Culture
The human CRC cell lines LOVO, SW480 and HCT116 were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All the cells were cultured in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 100 U/mL penicillin/streptomycin (HyClone, Shanghai, China) under standard conditions at 37°C in an atmosphere containing 5% CO2. The cells were used in the experiments once they reached the logarithmic phase of growth. For the induction and inhibition of autophagy, the cells were treated with 250 nM Torin 1 (Sigma-Aldrich, MO, USA) and 2 μM MHY1485 (Sigma-Aldrich, Missouri, USA), respectively, and to regulate PI3K activity, the cells were treated with 150 nM wortmannin (Sigma-Aldrich, MO, USA) and 50 μg/mL 740Y-P (Cayman, MI, USA).
Immunohistochemistry (IHC)
Tissue samples embedded in paraffin were cut into 5-µm sections, and the sections were dewaxed in Bioclear (Bio-Optica, Milan, Italy) and rehydrated in decreasing concentrations of ethanol. The paraffin-embedded sections were pretreated in 0.01 M citrate buffer in a microwave oven. Normal horse serum was used as a blocking agent. The sections were incubated with a primary antibody against LACTB (1:200, CST, USA) overnight at 4°C, washed three times, exposed to the appropriate secondary antibody for 30 min at 20°C and visualized with DAB/H2O2 (DAKO, Shanghai, China). The sections were subsequently counterstained with hematoxylin and washed. The degree of antigen expression was scored based on the staining intensity (0, no staining; 1, weak staining; 2, moderate staining; and 3, strong staining) and proportion (0, no cells stained; 1+, <10% cells showing positive staining; 2+, 10–50% cells showing positive staining; and 3+, >50% cells showing positive staining). The final scores for the IHC images were graded using a four-point scale, which was defined as follows: no positive cells, <10% positive cells, 10–50% positive cells and >50% positive cells. The IHC images were examined by two experienced pathologists who were blinded to clinicopathological data, and the final score was evaluated twice.
Quantitative PCR
Total RNA was isolated from tissues and cells using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) on the basis of the manufacturer’s protocol. Treating with quantitative PCR, total RNA was reverse-transcribed using a Transcriptor First-Strand cDNA Synthesis Kit (Roche Diagnostics). And then the reaction system was operated in 96-well plates and analyzed with a 7500 Real-Time PCR System and 7500 software. The specific LACTB PCR primers were following: 5ʹ-GTGGTTGGAGTTTCTGTAGATGGAA-3ʹ (forward), 5ʹ-AGTAATCTTGTTGTGACAGAAACCT-3ʹ (reverse). All of PCR reactions have duplicated the third time. Significant differences were indicated by a P value <0.05.
Western Blotting
In our study, total protein was extracted with lysis buffer (50 mM Tris-HCl, pH 6.8, 150 mM NaCl, 0.5% sodium deoxycholate, and 1% NP-40), and the supernatant was then collected by centrifugation. Equal amounts of protein were separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% nonfat milk in TBS containing 0.05% Tween-20 (TBST), incubated with primary antibodies overnight at 4°C, washed with TBST, and incubated with the secondary antibody (DyLight 488-conjugated mouse anti-rabbit IgG; 1:5000). Protein expression was then detected using an ECL detection reagent (Proteintech, Hubei, China). In this analysis, the following primary antibodies were used: anti-LACTB, anti-E-cadherin, anti-vimentin, anti-N-cadherin, anti-Twist1, anti-PI3K, anti-PIK3R3, anti-AKT, anti-p-AKT, anti-mTOR, anti-4E-BP1, anti-LC3, anti-P62, anti-C-Myc, anti-cyclinD1, and anti-ULK1 (Cell Signaling Technology, USA). An anti-β-actin antibody (Santa Cruz Biotechnology, USA) was used as a loading control.
Coimmunoprecipitation
CRC cells were lysed in lysis buffer for coimmunoprecipitation assays, and immunoprecipitation was performed using lysates incubated with the indicated antibodies overnight at 4°C and pulled down with magnetic protein A/G beads (Thermo Scientific, USA). After extensive washes in accordance to the manufacturer’s instructions, the antibody-bound proteins were eluted in loading buffer, and the pulled-down protein lysates were analyzed by Western blotting.26 The following primary antibodies were used: anti-LACTB (1:100, CST, USA) and anti-PIK3R3 (1:100, CST, USA).
Synthetic Lentiviral Transfection
The LACTB-shRNA vector sequence was 5ʹ-GCTTTGTATAAAGTGGAGATT-3ʹ, whereas a control shRNA was synthesized using a scrambled sequence. LACTB (LV-LACTB-shRNA-puromycin) knockdown using shRNA and LACTB (LV-LACTB-puromycin) overexpression by a lentiviral carrier were performed using reagents obtained from Shanghai GeneChem Co., Ltd. (China). The PIK3R3-shRNA vector sequence was 5ʹ-GGGAGGAGGTAAATGACAAAT-3ʹ, whereas a control shRNA was synthesized using a scrambled sequence. PIK3R3 (LV- PIK3R3-shRNA-puromycin) knockdown using shRNA and PIK3R3 (LV- PIK3R3-puromycin) overexpression by a lentiviral carrier were performed with reagents obtained from Shanghai GeneChem Co., Ltd. (China). Three cell lines were seeded in six-well plates for 24 h prior to lentiviral transduction and cultured until they reached 70–80% subconfluency. LV-LACTB-shRNA-puromycin was used for the transduction of LOVO cells at a multiplicity of infection (MOI) of 20 using polybrene (5 µg/mL), which enhanced the infection efficiency (GeneChem, China). In contrast, HCT116 and SW480 cells were transfected with LV-LACTB-puromycin at an MOI of 10 (HCT116) and 30 (SW480). The negative control puromycin-LV (GeneChem, China) was then used to identify the underlying influence of the viral vector. After 12 to 24 h of incubation, the medium was removed, and fresh medium was added. After 48 h of incubation, the appropriate concentrations of puromycin were added to the cell cultures to select cells stably over-expression or knockdown LACTB after the cell transfection with approximately 85% efficiency. We then utilized these selected cells for protein and mRNA analyses and other assays.
Cell Proliferation, Invasion and Migration Assays
First, CRC cells were seeded in 96-well plates at a density of 20,000 cells per well. After transfection as previously described and 0, 24, 36, 48, 72 or 96 h after treatment, 20 μL of MTT dye solution (5 mg/mL, Sigma, NY, USA) was added to each well. After incubation for another 4 h, 150 μL of DMSO was added to each well. The optical density (OD) was measured at 490 nm. For the invasion and migration assays, 1 × 105 cells were cultured in the upper chamber of a 24-well plate in serum-free medium, whereas the bottom chamber was supplemented with complete medium. After 24 h of culture, the migrated cells were washed, fixed, and stained with 0.1% crystal violet for 10 min. Representative images were captured under a light microscope. In addition, cell invasion was similarly detected based on the addition of inserts precoated with Matrigel (BD Biosciences, USA), which can mimic invasion-triggering circumstances. All of these assays were performed using at least three independent experiments.
Colony Formation Assay
Initially, the cells were seeded in 10-cm Petri dishes (500 cells/well) and cultured for 2 weeks. After this time, the colonies were washed, fixed in 4% formaldehyde for 15 min and then stained with 0.1% crystal violet for 15 min. The violet-stained colonies were counted and captured. This experiment was repeated at least three times for each cell line.
Cell Immunofluorescence and Confocal Microscopy
In brief, the cells were homogeneously cultured on six-well coverslips to 50%-80% confluency, immobilized with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 (Beyotime, Zhenjiang, Jiangsu, China) and then blocked with 10% goat serum at 37°C for 30 min. Primary anti-LC3 (1:300 dilution; Cell Signaling Technology, USA) and anti-E-catenin (1:300 dilution; Cell Signaling Technology, USA) antibodies were added to the coverslips, and the coverslips were then incubated overnight at 4°C. The coverslips were washed in PBS, and the corresponding secondary antibody (SA00009-2Cy3-goat anti-rabbit IgG for LC3 and E-catenin, Proteintech) was incubated with the coverslips for 2 h in the dark. The nuclei were stained with DAPI for 10 min, which resulted in the manifestation of a blue color, and a confocal microscope was used to acquire images (400×).
RNA-Seq
RNA was extracted using the Pure Link RNA Mini Kit (Ambion; Foster City, CA, USA) according to the manufacturer’s instructions, and its quality was assessed based on the minimum concentration and size range. The cDNA libraries were established using the TruSeq Total Stranded RNA with RiboZero Kit (Ambion, Set-A) and quantified using the Qubit System (Invitrogen; Carlsbad, CA, USA), and the quality and size were assessed using the Experion DNA 1 K Chip (Bio-Rad; Hercules, CA, USA). All the samples surpassed the quality control standards of minimum concentration and RNA quality indicator > 9, as demonstrated by discrete 18S and 28S bands. The library of n = 6 pooled samples was sequenced using the NextSeq 500 High Output Kit (300 cycles, paired-end 100 bp) with the Illumina NextSeq 500 platform (Illumina; San Diego, CA, USA). The sequenced reads and Fastq sequence files were used to align reads to the reference genome USCS-GRCm38/mm10 in the RNA-Seq Alignment Application using STAR aligner with the Illumina Cloud Computing Platform. The fragments per kilobase of transcript per million mapped reads (FPKM) values of the reference genes and transcripts were generated using Cufflinks 2.
Transmission Electron Microscopy (TEM)
According to standard protocols, the cells were exposed to RF-EMFs (1, 2, or 4 W/kg) for 24 h, harvested, washed with PBS and then fixed at room temperature with glutaraldehyde followed by osmium tetroxide. The cells were then washed, dehydrated, embedded in paraffin and cut into 70-nm-thick sections using an Ultrathin microtome (Leica EM UC6; Leica Microsystems, Wetzlar and Mannheim, Germany), and the sections were stained with uranyl acetate-lead citrate. The stained sections were prepared for examination with an electron microscope (JEM1230, JEOL Ltd., Tokyo, Japan) to detect autophagic vacuoles.
Tumor Xenograft Study
Animal studies were performed in line with the rules for the care and use of laboratory animals from the National Academy of Sciences. Animal experiments were approved by Institutional Animal Care and Use Committee of Shanghai jiaotong University (Approval No. 2019040112). For in vivo tumorigenesis assays, 0.5 mL of basal medium containing approximately 1 × 106 CRC cells that were equivalently transfected with either a lentivirus encoding LACTB-shRNA or a lentivirus encoding Control-shRNA was injected subcutaneously into the left flank of 4-week-old immunodeficient BALB/c male nude mice. During the tumor-formation period, the tumor volume was measured every week and calculated using the formula V (mm3) = length×width2/2. The mice were then anesthetized and ethically euthanized. All the mouse experiments were performed in accordance with internationally recognized guidelines.
Statistical Analyses
Prism 5 (GraphPad Software, San Diego, CA, USA) was used to analyze the data, and all the results are presented as the mean ± standard error of the mean (SEM). A two-tailed unpaired t-test was used to compare two groups. Unless otherwise indicated, p<0.05 and p<0.01, which are denoted by single (*) and double (**) asterisks, respectively, were considered statistically significant. Statistical analysis was performed with SPSS version 17.0. Survival curves were plotted according to the Kaplan–Meier method, and the Log rank test was used to compare the overall survival (OS).
Results
The Level of LACTB Is Lower in CRC Tissue Than in Nonmalignant Tissue
To investigate the functional roles of LACTB in CRC, we analyze the LACTB expression levels from TCGA data showed that the LACTB mRNA levels were lower in the CRC tissue samples than in the nonmalignant tissue samples (Figure 1A). The qRT-PCR analysis showed that LACTB expression was lower in CRC tissues compared with nonmalignant tissues (Figure 1B), and the analysis of the LACTB protein levels in CRC samples detected by Western blotting yielded the same results (Figure 1C). Statistical analysis revealed that decreased LACTB expression was significantly associated with T stage, local lymph node metastasis, distant metastasis, advanced clinical stage and worse differentiation in CRC using our q-PCR data (p < 0.05) (Table 1). In addition, we screened the LACTB expression levels in 80 randomly selected CRC patients, and our IHC results revealed that LACTB expression in the cytoplasm was lower in CRC tissue sections than in nonmalignant tissue sections (Figure 1D). To study the biological function of LACTB in CRC, we compared the LACTB expression levels among three CRC cell lines, namely, HCT116, SW480 and LOVO cells, which were used as our cellular models. The results revealed that LOVO cells exhibited high LACTB expression, whereas the other two cell lines showed relatively low expression of LACTB (Figure 1E). A Kaplan–Meier analysis of CRC patients was also conducted, and TCGA data revealed that the LACTB-high group exhibited significantly improved OS (Figure 1F) (p < 0.05).
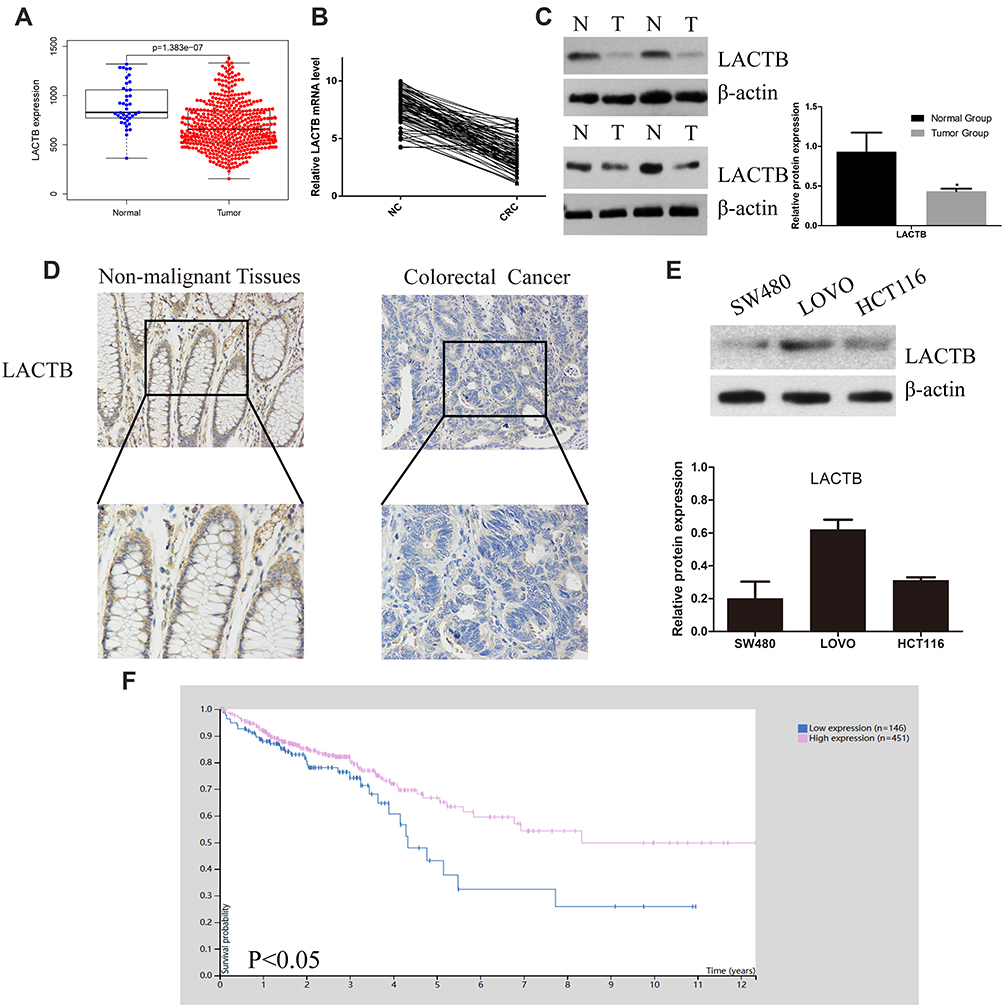 |
Figure 1 LACTB expression in CRC tissues is low and could promote the overall survival of CRC patients. (A) The analysis of the LACTB mRNA levels in CRC tissues and paired noncancerous tissues from TCGA revealed that these levels were significantly decreased in CRC tissues, as demonstrated by the Mann–Whitney U-test. (B) The LACTB mRNA expression levels in 80 CRC tissues and paired noncancerous tissues were assessed by qRT-PCR, which showed that LACTB mRNA expression was significantly decreased in CRC tissues, as demonstrated using the 2-ΔΔct method and determined by the Mann–Whitney U-test. (C) LACTB protein expression in representative samples of CRC tissues and paired adjacent noncancerous tissues. The level of LACTB was downregulated in CRC tissues (*P<0.05), as determined by Log rank test. (D) Immunohistochemical staining of LACTB in CRC tissues and normal tissues. The level of LACTB was weaker in CRC tissues than in nonmalignant tissues. (E) Levels of LACTB protein expression in three different CRC cell lines. The HCT116 and SW480 cells showed inconspicuous LACTB expression, and strong expression of LACTB was detected in LOVO cells. (F) The analysis of TCGA data showed that the CRC patients with high LACTB expression exhibited significantly improved OS as determined by Student’s t-test (P<0.05). |
LACTB Inhibits CRC Cell Proliferation and Colony Formation in vitro
To investigate whether LACTB alters the biological characteristics of CRC cells, LOVO, SW480 and HCT116 cells stably expressing ectopic LACTB from a lentiviral vector were established, and the transduced and negative control cells were cultured for 14 days. As expected, the colonies formed by the LACTB-knockdown cells were denser than those formed by the negative control cells, which suggested that LACTB upregulation weakened the ability of cancer cells to form colonies (Figure 2A–C) (p < 0.05). Moreover, the proliferation of these cells was examined through MTT assays. The results showed that the proliferative capacity of LOVO cells with suppressed LACTB expression was higher than that of control LOVO cells; however, overexpression of LACTB in HCT116 or SW480 cells led to a decline in the proliferative ability of these cells (Figure 2D–F) (p < 0.05).
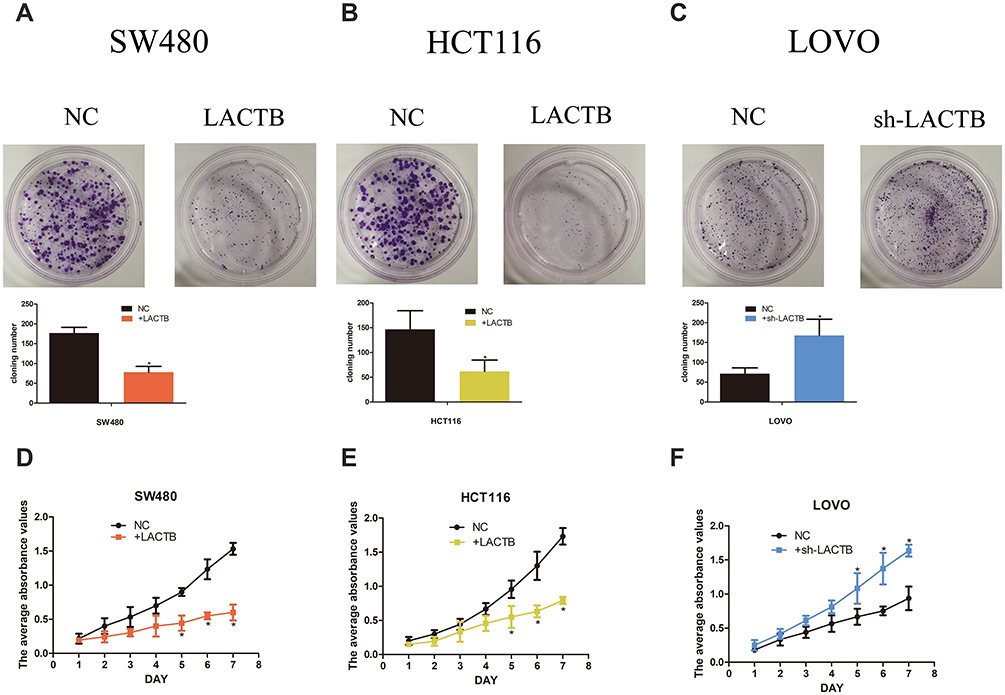 |
Figure 2 LACTB inhibits CRC cell proliferation and colony formation in vitro. (A–C) The colonies formed by the LACTB-knockdown cells were denser than those formed by the negative control cells, which suggests that LACTB upregulation weakens the ability of cancer cells to form colonies (*P<0.05), as determined by Student’s t-test. (D–F) The MTT assay showed that the proliferative capacity of LOVO cells with suppressed LACTB expression was higher than that of control LOVO cells; however, the overexpression of LACTB in HCT116 or SW480 cells led to a decline in the proliferative ability of these cells (*P<0.05), as determined by Student’s t-test. |
LACTB Inhibits EMT and Promotes Autophagy in CRC
The SW480 and HCT116 cell lines manifested relatively low endogenous LACTB expression, and the treatment of both cell lines with a LACTB-overexpressing lentivirus inhibited EMT. Cancer cells undergoing the EMT process are characterized by changes in the expression of classic EMT markers, including a decrease in the level of the cell-cell contact factor E-cadherin and increases in the expression of the transcription factor Twist1 and the mesenchymal markers vimentin and N-cadherin.5 Immunofluorescence and Western blotting assays revealed that E-cadherin expression was enhanced in LACTB-overexpressing SW480 and HCT116 cells and dysregulated in LACTB-knockdown LOVO cells compared with that in control cells transfected with a nonspecific lentivirus. In contrast, the opposite trend was obtained for N-cadherin, vimentin, C-Myc, CyclinD1 and Twist1 expression in these groups of cells (Figure 3A–C, Figure 4A–C). These factors exhibited upregulated expression in LACTB-silenced LOVO cells, which indicates that LACTB plays a significant role in mediating EMT in CRC.
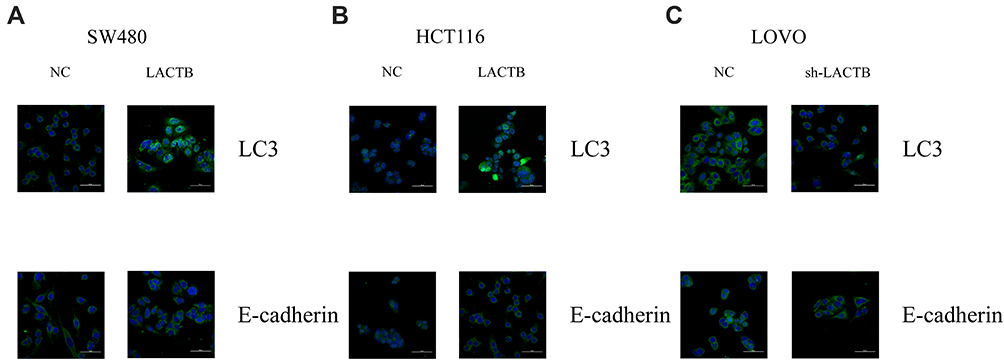 |
Figure 3 LACTB inhibits EMT and promotes autophagy in CRC, as demonstrated by immunofluorescence. (A–C) Immunofluorescence results revealed that E-cadherin expression was enhanced in LACTB-overexpressing SW480 and HCT116 cell lines and dysregulated in LACTB-knockdown LOVO cells compared with control cells transfected with a nonspecific lentivirus. The upregulation of LACTB expression in SW480 and HCT116 cells significantly elevated the LC3-II/LC3-I ratio, and the knockdown of LACTB expression in LOVO cells decreased the LC3-II/LC3-I ratio and the E-cadherin level. |
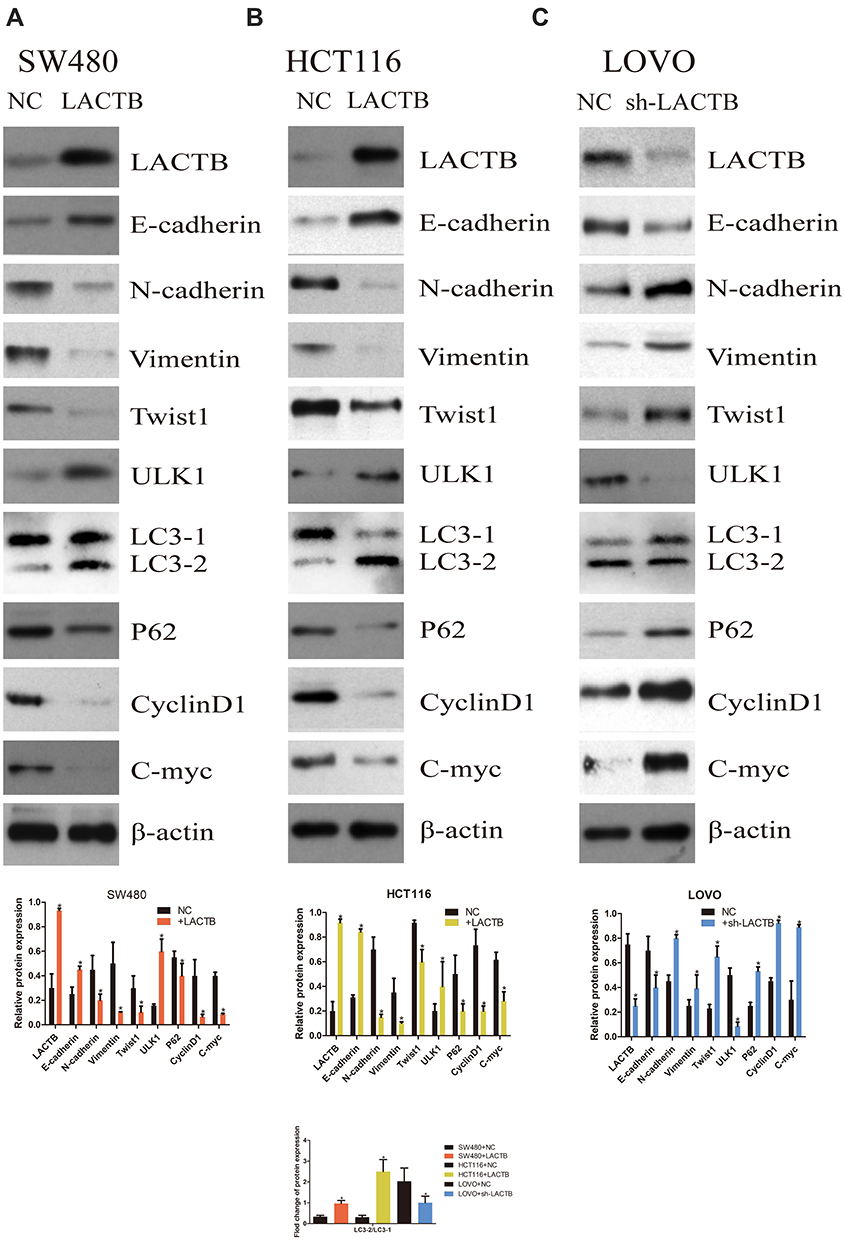 |
Figure 4 LACTB inhibits EMT and promotes autophagy in CRC, as demonstrated by Western blotting. (A–C) Western blotting assays revealed that E-cadherin expression was enhanced in LACTB-overexpressing SW480 and HCT116 cell lines and dysregulated in LACTB-knockdown LOVO cells compared with control cells transfected with a nonspecific lentivirus. In contrast, the opposite trend was obtained for N-cadherin, vimentin, C-Myc, CyclinD1 and Twist1 expression in these groups of cells. The upregulation of LACTB expression in SW480 and HCT116 cells significantly elevated the LC3-II/LC3-I ratio and the ULK1 level and decreased the P62 expression level. In contrast, the knockdown of LACTB expression in LOVO cells decreased the LC3-II/LC3-I ratio and the ULK1 level and promoted the expression of P62 (*P<0.05), as determined by Student’s t-test. |
Furthermore, a recent study showed that autophagy can promote the survival of isolated dormant cells. However, autophagy can decrease EMT by inhibiting necrosis and the infiltration of prometastatic inflammatory cells, which results in the promotion of dormancy in disseminated cancer cells for extended periods of time. To investigate this phenomenon, we further explored the role of LACTB in CRC cell autophagy. Immunofluorescence and Western blotting assays revealed that the upregulation of LACTB expression in SW480 and HCT116 cells significantly elevated the LC3-II/LC3-I ratio and the Unc51-like autophagy activating kinase-1 (ULK1) level and decreased P62 expression. In contrast, the knockdown of LACTB expression in LOVO cells decreased both the LC3-II/LC3-I ratio and the ULK1 level and promoted the expression of P62 (Figure 3A–C, Figure 4A–C). The opposite results were found for E-cadherin expression in the transduced cells: high expression in SW480 and HCT116 cells with upregulated LACTB expression and low expression in LACTB-knockdown LOVO cells (Figure 3A–C). Additionally, as observed by TEM, autophagosome formation was significantly induced in the SW480 and HCT116 cells transduced with the LACTB-overexpressing constructs, whereas the inhibition of LACTB expression in LOVO cells resulted in decreased formation of autophagosomes (Figure 5A–C). In summary, these findings demonstrate that LACTB plays significant roles in EMT and autophagy in CRC.
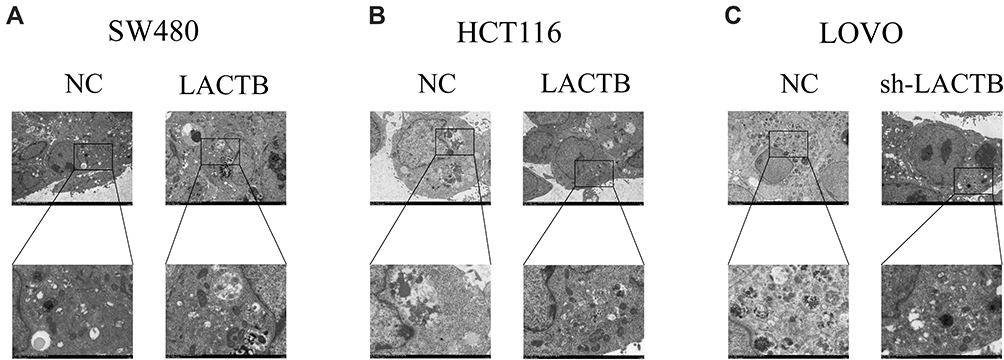 |
Figure 5 Effects of LACTB on autophagosomes in CRC cells. (A–B) Autophagosomes were observed in CRC cells by TEM. Additionally, in LACTB-overexpressing HCT116 and SW480 cells exhibited higher levels of autophagosomes compared with the control cells. (C) The inhibition of LACTB expression in LOVO cells decreased the numbers of autophagosomes. Magnification, 1500× and 5000×. |
The Regulatory Effects of LACTB on Autophagy and EMT are Partially Due to the PI3K/AKT Signaling Pathway
To understand the underlying molecular mechanisms involved in the inhibition of migration and invasion and the promotion of autophagy by LACTB, we subsequently investigated the differentially expressed genes in LACTB-overexpressing HCT116 cells by a transcriptomic analysis based on RNA‐seq. Compared with the control HCT116 cells, LACTB-overexpressing cells exhibited 2178 differentially expressed genes (p < 0.05), which included 1464 (67.2%) upregulated and 714 (32.8%) downregulated genes, respectively. In addition, many of the differentially expressed genes were involved in autophagy, and PIK3R3 expression was significantly downregulated in the LACTB-overexpressing cells (Figure 6A). Coimmunoprecipitation assays showed that LACTB can directly regulate the level of PIK3R3 (Figure 6B), which is a regulatory subunit of the PI3K complex associated with activation of the PI3K complex and a novel oncogene in many cancers. The PI3K/Akt/mTOR signaling pathway is considered an important mechanism in the regulation of autophagy.22 Based on the above-described results, we hypothesized that LACTB mediates autophagy to affect the tumor characteristics of CRC cells and that this activity partially inhibits the activation of the PI3K/Akt/mTOR signaling pathway. We evaluated the expression of PIK3R3 and PI3K in the three cell lines, and Western blotting results indicated that PIK3R3 and PI3K expression was significantly decreased in the two LACTB-overexpressing cell lines compared with the LACTB-knockdown LOVO cells (Figure 7A–C). Simultaneously, LACTB overexpression decreased the levels of p-AKT and mTOR and increased the expression of 4E-BP1, and LACTB-silenced LOVO cells showed increased levels of these proteins but low 4E-BP1 expression (Figure 7A–C).
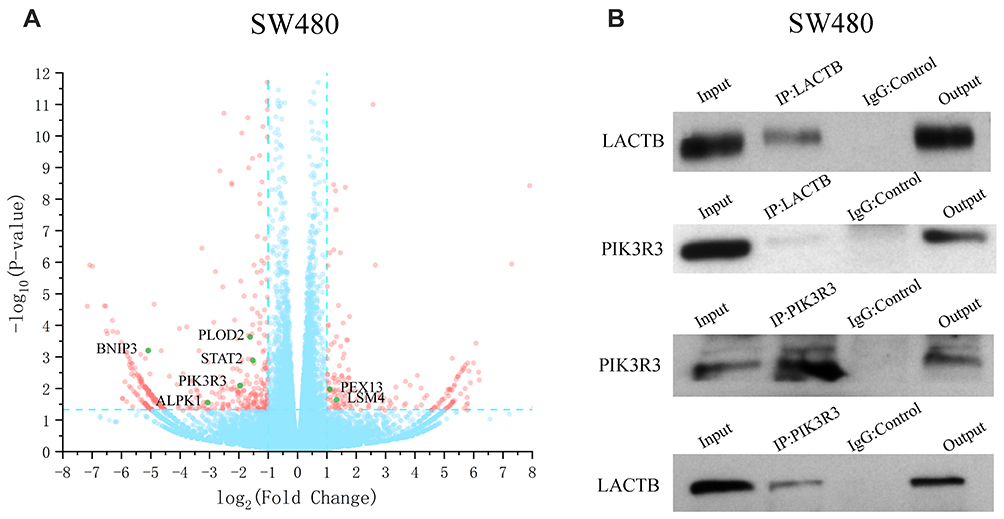 |
Figure 6 LACTB can regulate the level of PIK3R3 to influence cancer development. (A) The RNA‐seq-based analysis showed that the LACTB-overexpressing SW480 cells exhibited 2178 differentially expressed genes (p < 0.05), including 1464 (67.2%) upregulated and 714 (32.8%) downregulated cells, compared with the control SW480 cells. (B) Coimmunoprecipitation assays indicated that LACTB can directly regulate the level of PIK3R3. |
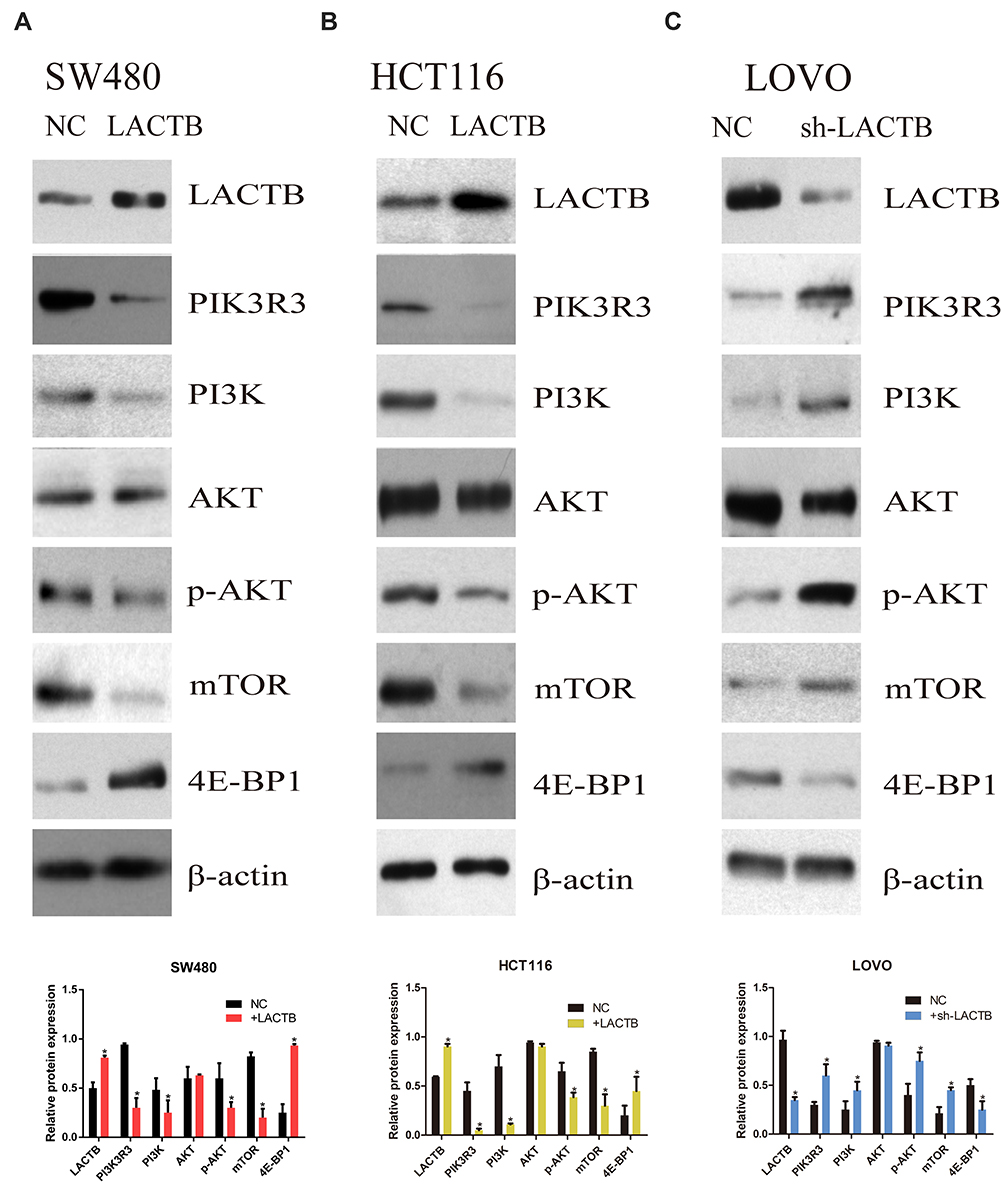 |
Figure 7 The regulatory effects of LACTB on autophagy and EMT are partially due to the PI3K/AKT signaling pathway. (A–C) Western blotting showed that PIK3R3 and PI3K expression was clearly decreased in the two LACTB-overexpressing cell lines compared with the control cell lines, whereas the LACTB-knockdown LOVO cells showed high PIK3R3 and PI3K expression. Simultaneously, the levels of p-AKT and mTOR were decreased after LACTB overexpression, and the LACTB-silenced LOVO cells showed increased levels of these proteins. In addition, 4E-BP1 exhibited adverse outcomes (*P<0.05), as determined by Student’s t-test. |
Although the tumor suppressor function of LACTB is specific, further investigation is urgently needed to elucidate the mechanism. As expected, LACTB-shRNA stimulated a cascade of events that led to the upregulation of PIK3R3 expression, which induced the activation of PI3K. Thus, PI3K expression was increased and blocked in stable PIK3R3-overexpressing and PIK3R3-knockdown cell lines, respectively, and these changes affected PI3K, AKT, p-AKT and mTOR expression, as demonstrated by Western blotting (Figure 8A–C). The results showed that LACTB-overexpressing SW480 and HCT116 cells exhibited increased levels of PI3K, p-AKT and mTOR after the induction of PIK3R3 overexpression, but these levels were reduced in LACTB-knockdown LOVO cells after PIK3R3 silencing (Figure 8A–C). In addition, the LC3-II/LC3-I expression ratio was decreased in LACTB-overexpressing SW480 and HCT116 cells overexpressing PIK3R3, but the levels of P62, C-Myc and cyclinD1 were increased in these cells (Figure 8A–C). Additionally, PIK3R3 silencing inhibited the PI3K/AKT/mTOR signaling pathway, leading to reduced expression of PI3K, p-AKT and mTOR. In contrast, the LC3-II/LC3-I expression ratio was augmented after PIK3R3 silencing, which resulted in suppression of the PI3K/AKT/mTOR signaling pathway and thereby the induction of autophagy, as demonstrated by a decreased level of the negative autophagy marker P62. In addition, the inhibition of proliferation decreased the levels of C-Myc and cyclinD1 (Figure 8A–C). As mentioned above, we conclude that the regulatory effects of LACTB on autophagy can be partially attributed to the PI3K/AKT/mTOR signaling pathway via regulation of the level of PIK3R3.
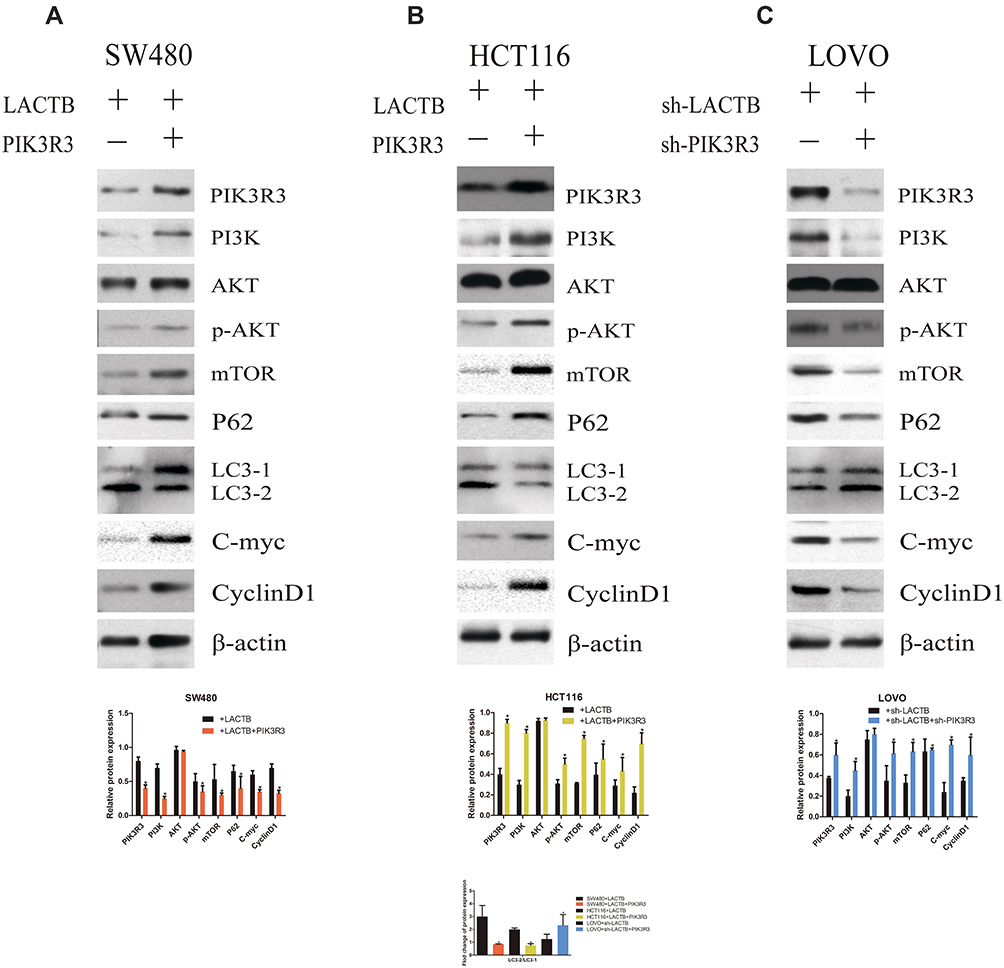 |
Figure 8 The regulatory effects of LACTB on autophagy can be partially attributed to the PI3K/AKT/mTOR signaling pathway via regulation of the level of PIK3R3. (A–C) PI3K expression was increased and blocked in stable PIK3R3-overexpressing and PIK3R3-knockdown cell lines, respectively, and these changes affected the expression of PI3K, AKT, p-AKT and mTOR, as observed by Western blotting. Additionally, PIK3R3 silencing inhibited the PI3K/AKT/mTOR signaling pathway, which lead to reduced expression of PI3K, p-AKT and mTOR. In contrast, the LC3-II/LC3-I expression ratio was augmented after PIK3R3 silencing, and this enhancement resulted in suppression of the PI3K/AKT/mTOR signaling pathway and thereby the induction of autophagy, as demonstrated by a decreased level of the negative autophagy marker P62. In addition, decreased levels of C-Myc and cyclinD1 resulted in the inhibition of proliferation (*P<0.05), as determined by Student’s t-test. |
LACTB Can Mediate EMT and Proliferation of CRC Cells by Regulating Autophagy
To confirm the inhibitory function of autophagy in cancer cell development, we analyzed the migration and invasion of CRC cells transduced with LACTB-overexpression or LACTB-knockdown constructs. Our previous results demonstrated that LACTB can promote autophagy in CRC cells via the PI3K/AKT/mTOR signaling pathway. Additionally, we questioned whether LACTB can suppress EMT by promoting autophagic activity, and whether LACTB can regulate autophagy to influence EMT in CRC remains to be evaluated. Thus, we applied MHY1485, an autophagy inhibitor (mTOR promoter), and the activator Torin 1 (mTOR inhibitor) to cells exhibiting stable LACTB overexpression or knockdown and evaluated the expression of LC3, P62 and Twist1. The results showed that LACTB-overexpressing SW480 and HCT116 cells exhibited increased expression levels of P62 and Twist1 after treatment with the autophagy inhibitor MHY1485, but these levels were reduced in LACTB-knockdown LOVO cells treated with the autophagy activator Torin 1 (Figure 9A–C). In addition, the LC3-II/LC3-I expression ratio was decreased in LACTB-overexpressing SW480 and HCT116 cells following MHY1485 treatment, but this ratio was increased in LACTB-knockdown LOVO cells treated with Torin 1 (Figure 9A–C). The inhibition of autophagy is associated with an increased level of P62, which prevents the degradation of Twist1. Transwell assays revealed that cell migration and invasion were promoted by the attenuation of autophagy in LACTB-overexpressing SW480 and HCT116 cells treated with the autophagy inhibitor MHY1485, whereas LOVO-knockdown cells treated with the autophagy activator Torin 1 showed poor cell migration and invasion abilities (Figure 9D). These findings demonstrate that autophagy can partially inhibit the migration and invasion of CRC cells, and this effect can be partly attributed to the PI3K/AKT/mTOR signaling pathway.
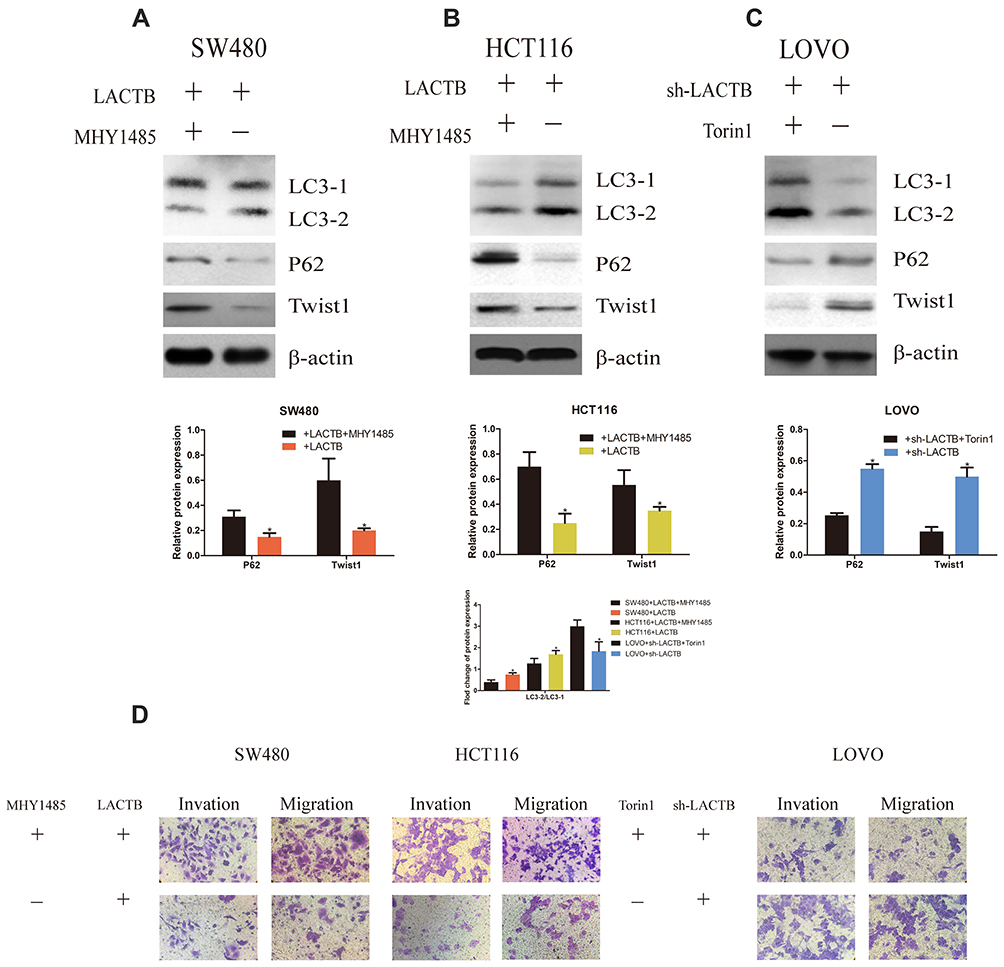 |
Figure 9 LACTB can mediate EMT and proliferation of CRC cells by regulating autophagy. (A–C) The results showed that LACTB-overexpressing SW480 and HCT116 cells exhibited increased expression levels of P62 and Twist1 after treatment with the autophagy inhibitor MHY1485, but these levels were reduced in LACTB-knockdown LOVO cells treated with the autophagy activator Torin1. In addition, the LC3-II/LC3-I expression ratio was decreased in LACTB-overexpressing SW480 and HCT116 cells following MHY1485 treatment, but this ratio was increased in LACTB-knockdown LOVO cells treated with Torin1. The inhibition of autophagy is associated with an increased level of P62, which prevents the degradation of Twist1 (*P<0.05), as determined by Student’s t-test. (D) Transwell assays revealed that cell migration and invasion were promoted after the attenuation of autophagy in LACTB-overexpressing SW480 and HCT116 cells treated with the autophagy inhibitor MHY1485, whereas LOVO-knockdown cells treated with the autophagy activator Torin1 showed poor cell migration and invasion abilities. |
LACTB Modulates CRC Tumorigenesis in vivo
To explore the effects of the tumor suppressor role of LACTB on tumor growth in vivo, we constructed a tumor xenograft model. Untreated control LOVO, SW480 or HCT116 cells or those transduced with LACTB-shRNA or the LACTB-overexpression vectors (1 × 106) were injected into the left flank of nude mice. The tumor size was measured weekly using calipers. At 8 weeks postinoculation, the mice were euthanized. The sizes of the tumors generated from the corresponding untransduced control cells were enhanced compared with those of the tumors derived from LACTB-overexpressing cells, whereas the tumors generated from LACTB-silenced cells exhibited a considerable increase in tumor volume compared with those generated from the corresponding control cells. Additionally, the trend obtained for the tumor weight was similar to that found for the tumor volume (Figure 10). In conclusion, these data are consistent with the results from the in vitro cell proliferation assay and indicate that LACTB is associated with CRC progression and tumorigenesis in vivo (p < 0.05 or p < 0.01).
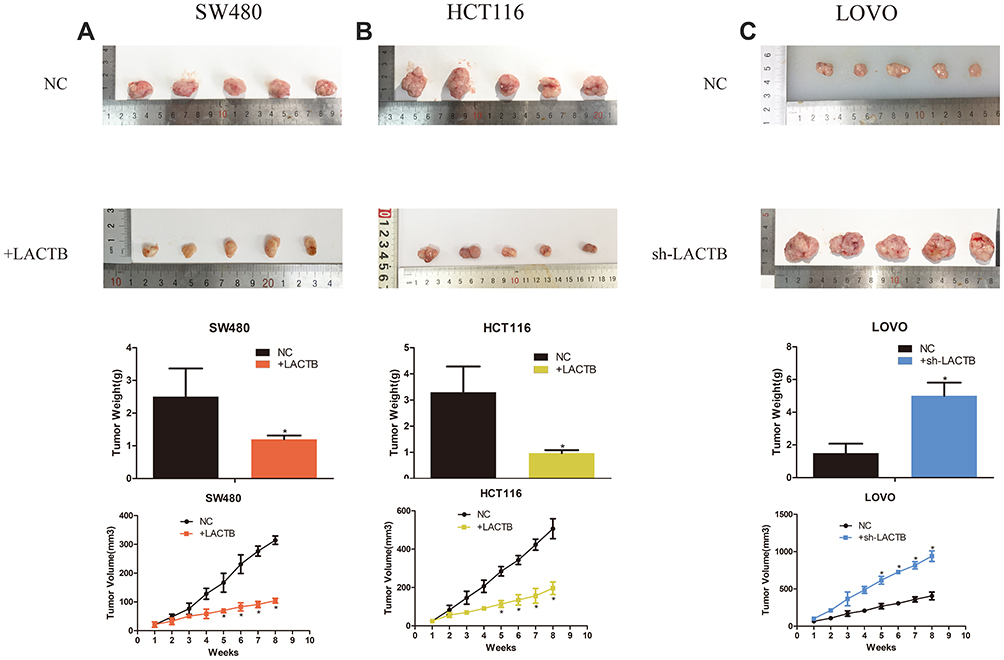 |
Figure 10 LACTB modulates CRC tumorigenesis in vivo. (A–C) We constructed a tumor xenograft model. Untreated control LOVO, SW480 or HCT116 cells or those transduced with LACTB-shRNA or the LACTB-overexpression vectors (1 × 106) were injected into the left flank of nude mice. The sizes of the tumors generated from the corresponding untransduced control cells were enhanced compared with those of the tumors derived from LACTB-overexpressing cells, whereas the tumors generated from LACTB-silenced cells exhibited a considerable increase in volume compared with those from the corresponding control cells. Furthermore, as a consequence of these effects in the tumor volume, a similar trend was obtained for the tumor weight (*P<0.05), as determined by Student’s t-test. |
Discussion
CRC is a common aggressive malignancy, and novel sensitive and specific diagnostic molecular biomarkers and therapeutic targets are urgently needed.27 LACTB reportedly exerts strong tumor-suppressive effects and can decrease CRC progression by attenuating the ubiquitination of MDM2 and p53.28,29 LACTB and autophagy were revealed to play a significant role during tumour development and energy metabolism, we want to investigate the relationship between the LACTB and autophagy and what the underlying mechanisms are. In this study, we provide the first demonstration that autophagy was induced in CRC cells by LACTB and that autophagy positively contributed to the inhibition of cell EMT and proliferation both in vitro and in vivo. In addition, the induction of autophagy by LACTB was dependent on the activation of the PI3K/AKT/mTOR signaling pathway via regulation of the PIK3R3 level. These results demonstrate that autophagy induction plays significant roles in cell EMT and the inhibitory effect of LACTB on CRC cell proliferation, and these findings might provide a novel point of view for understanding the mechanism through which LACTB suppresses cancer development.
Autophagy, which is a lysosomal degradation pathway that leads to the degradation and recycling of intracellular proteins and organelles to provide hydrolytic enzymes, occurs when cells cannot obtain sufficient nutrients from the extracellular milieu to sustain ATP production and biosynthesis.30,31 Recent studies have shown that autophagy can not only inhibit primary tumor growth but also protect cancer cells at advanced stages of the disease against chemotherapy or radiotherapy.32,33 The upregulation of LACTB expression observed in treated CRC cells could increase the levels of LC3 and ULK1 and reduce the accumulation of P62/SQSTM1, as demonstrated by our Western blotting results, which indicates that LACTB might promote autophagy in CRC. As a specific autophagosomal biomarker for autophagy induction, LC3 plays a significant role in autophagosome biosynthesis in mammals.34 LC3 is found in two forms in cells, LC3-I and LC3-II: LC3-I is cytosolic and binds to PE to generate LC3-II, which is used to monitor autophagic activity.35 P62 can take part in meditating the transport of protein aggregates and damaged organelles,36 and it can also interact with LC3 and localize to the outer surface of autophagosomes; as a result, P62 can be treated as a significant marker of autophagic proteins and autophagolysosomal activity.37 Furthermore, P62 can induce increased oxidative stress and mitochondrial damage, and insufficient P62 degradation is accompanied by defective autophagy.38 As a serine/threonine protein kinase, ULK1 can form a complex with both FIP200 and Atg13, and this complex plays a critical role in inducing autophagy and functions as a downstream target of mTOR because mTOR phosphorylates ULK1 at Ser 757 to inhibit ULK1 activation to mediate autophagy.39
EMT is widely considered a significant physiological process that regulates the initial steps in the invasion and migration of cancer cells.40 This process can be followed by monitoring the levels of some EMT-associated markers, and among these, Snail1, Snail2 and Twist1 are known to repress E-cadherin expression in epithelial cells undergoing EMT and increase the levels of N-cadherin and vimentin.41,42 Recent studies have shown that an intricate relationship exists between autophagy and EMT in cancer.41 Autophagy is thought to protect tumor initiation by limiting genome instability and decreasing the accumulation of damaged organelles and proteins.43 In addition, cancer cells can utilize autophagy to promote their metastasis in a hypoxic and stressful microenvironment; specifically, these cells exploit autophagy to provide energy to cancer cells that exhibit activated metabolic stress responses and to thus allow their escape from the physiological responses to cancer and therapy.44 On the one hand, autophagy can increase metastasis via anoikis resistance and defends the survival of isolated dormant cells, and on the other hand, autophagy can decrease EMT by inhibiting necrosis and the infiltration of prometastatic inflammatory cells, which promotes the dormancy of disseminated cancer cells for extended periods of time.45 Recent studies have indicated that increasing the levels of the autophagic machinery inhibits the EMT phenotype by degrading key EMT molecular markers.46 In this study, we explored the molecular and cellular functions of LACTB in regulating EMT and autophagy, and the results showed that the level of Twist1 was upregulated in CRC cells by the silencing of LACTB and was decreased by LACTB overexpression. The level of Twist1 could be upregulated by treatment with the mTOR promoter MHY1485, and this treatment inhibited cell autophagy.46 The mTOR inhibitor Torin1 can promote autophagy but decrease the level of Twist1, which can increase the level of E-cadherin.47 The results related to the Twist1 and autophagy levels revealed that LACTB might inhibit EMT by promoting autophagy in CRC cells. Under autophagy-deficient conditions, accumulation of the selective autophagy receptor P62 is directly linked to tumorigenesis and the subsequent ubiquitination of Twist1, which promotes Twist1 stabilization.48 Twist1 can be removed through both autophagy and proteasomal degradation.49 However, in autophagy-deficient cells, the degradation of Twist1 by the autophagosome is inhibited by its steady protection mediated by the accumulation of P62,50 which supports an important role for autophagy in EMT repression.
The p110 protein, which is a significant subunit of PI3K, is a heterodimeric protein with a regulatory subunit and a catalytic subunit.51 The regulatory subunit is regulated by a variety of genes, such as PIK3R2 and PIK3R3, and our results demonstrated that LACTB can inhibit the expression of PIK3R3, which can bind to the p110 catalytic subunit through the iSH2 domain, and its unique NH2 terminus can directly bind to key proteins in cell cycle regulation.52,53 Phosphorylation mediated by PI3K transforms PIP2 into PIP3, and PIP3 can promote the phosphorylation of Ser308 and 473 of AKT.54 In addition, phosphorylated AKT (p-Akt) can inhibit TSC2 and TSC to activate mTOR,55 and mTOR can induce mRNA translation by phosphorylating S6K and then p70S6K to increase the adhesion of ribosomes to the endoplasmic reticulum and thereby inhibit both the formation of autophagic membranes and autophagy activity.56,57 The corresponding results from our study showed that LACTB might inhibit the level of PIK3R3 to reduce the levels of PI3K, p-AKT and mTOR and thereby promote autophagy.
To clarify the role of LACTB in regulating CRC proliferation, we tested the levels of 4E-BP1, C-Myc and cyclinD1, and the results showed that LACTB upregulation decreased the cyclinD1 level but increased the 4E-BP1 level. CyclinD1 plays a crucial role in regulating the cell cycle, and it can combine with CDK4 or CDK6 and translocate from the cytoplasm to the nucleus, which promotes progression from the G1 phase to the S phase to accelerate cell proliferation.58,59 A previous study demonstrated that AKT knockdown or inhibition could clearly reduce NF-κB phosphorylation and promote the activation of GSK-3 to decrease the level of CyclinD1.60 4E-BP1, a downstream molecule, plays a negative but pivotal role in protein translation in cancer cells.61 This protein can be activated by dephosphorylation and binds to eIF4E, and this binding can stop the formation of the cap‐dependent translation initiation complex.62 Increased 4E-BP1 expression is associated with the inhibition of cellular proliferation and elevated cyclin D1 and C-Myc mRNA levels.63 In addition, the functions of 4E-BP1 and eIF4E can be inhibited by increasing mTOR expression.64 Our Western blotting, MTT and colony formation assays showed that LACTB upregulation inhibits CRC proliferation and that LACTB downregulation promotes the opposite outcome. Therefore, LACTB might regulate cell proliferation through the PI3K/AKT/mTOR signaling pathway. Furthermore, the downregulation and upregulation of LACTB contribute to the enhancement and attenuation of CRC growth, respectively, in BALB/c nude mice.
Conclusions
Taken together, our data demonstrate that LACTB can regulate proliferation and autophagy to suppress the development of CRC. Moreover, LACTB silencing inhibits CRC cell autophagy and stimulates the proliferation, invasion, migration and EMT of these cells, whereas the overexpression of LACTB yields the opposite results and enhances autophagy. The stimulatory effects of LACTB on autophagy occur through the upregulation of LC3 expression, the downregulation of P62 expression, and the effects of LACTB on proliferation, as evidenced by the detected changes in the expression levels of 4E-BP1, C-Myc and cyclinD1 through the PI3K/AKT/mTOR signaling pathway. Moreover, LACTB can induce epithelial polarity and cell junction formation and inhibit CRC cell EMT by promoting autophagy. Furthermore, the upregulation of LACTB expression leads to a reduction in xenograft tumor growth in vivo, whereas LACTB downregulation yields the opposite outcome. Therefore, we conclude that LACTB regulates the PIK3R3 level to promote autophagy and inhibit EMT and proliferation, and these effects are partly achieved through the PI3K/AKT/mTOR signaling pathway (Figure 11). We anticipate that this study will provide a basis for establishing new strategic approaches for the development of effective CRC therapies.
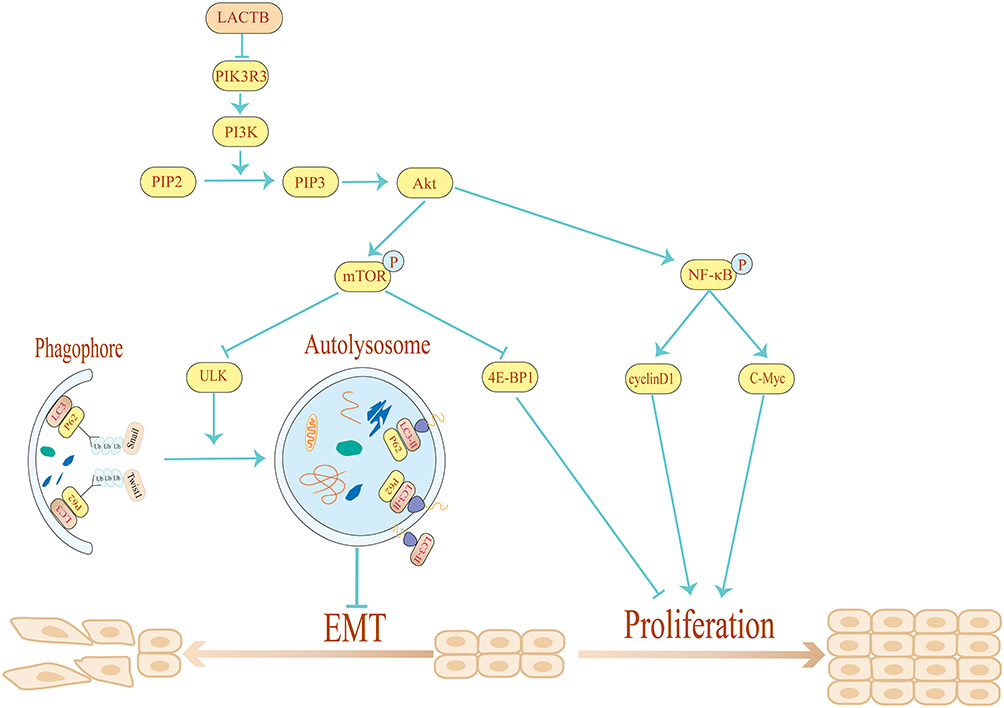 |
Figure 11 Schematic diagram of the mechanism through which LACTB inhibits EMT and promotes autophagy in CRC via the PI3K/AKT/mTOR signaling pathway. By overexpressing and silencing LACTB and PIK3R3, we demonstrated that LACTB might negatively regulate PIK3R3 to inhibit cancer development. In addition, LACTB can inhibit EMT by promoting autophagy and decreasing the level of Twist1. |
Abbreviations
CRC, colorectal cancer; LACTB, beta-lactamase-like; DAB, diaminobenzidine; ECL, enhanced chemiluminescence; EMT, epithelial-to-mesenchymal transition; FBS, fetal bovine serum; IHC, immunohistochemistry; PBS, phosphate buffer solution; q-PCR, quantitative real-time PCR; SD, standard deviation; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; shRNA, short hairpin RNA; ATP, adenosine triphosphate; OD, optical density; OS, overall survival; BD, Matrigel; BCA, BCA protein assay kit; MOI, multiplicity of infection; LV, lentiviral; TEM, transmission electron microscopy; PIK3R3, phosphatidylinositol 3-kinase regulatory subunit gamma; PI3K, phosphoinositide 3-kinase; 4E-BP1, eukaryotic translation factor 4E-binding protein 1; p-AKT, phosphorylated AKT; ULK1, unc-51-like kinase 1.
Consent for Publication
We declare that the paper is being submitted for consideration for publication in the Cancer Management and Research and that the content has not been published or submitted for publication elsewhere. All the authors have made significant contributions and agree with the content of the manuscript. Ming Zhong made the greatest contribution to this study and is the corresponding author.
Data Sharing Statement
The datasets generated from the patients during the current study are not publicly available in accordance with local health research ethics protocols but might be available from the corresponding author.
Ethical Approval and Consent to Participate
The study was highly supported by Department of Gastrointestinal Surgery, Renji Hospital, School of Medicine, Shanghai Jiaotong University. And prior to patients’ participation, we have informed all patients about the use of human specimens in the study and obtained consent from each patient. All the patients comprehend it and signed paper informed consent.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Wingo PA, Cardinez CJ, Landis SH, et al. Long-term trends in cancer mortality in the United States, 1930–1998. Cancer. 2003;97(S12):3133–3275. doi:10.1002/cncr.11380
3. Kahi CJ, Boland CR, Dominitz JA, et al. United states multi-society task force on colorectal, colonoscopy surveillance after colorectal cancer resection: recommendations of the us multi-society task force on colorectal cancer. Gastroenterology. 2016;150(3):758–768.e711. doi:10.1053/j.gastro.2016.01.001
4. Keckesova Z, Donaher JL, De Cock J, et al. LACTB is a tumour suppressor that modulates lipid metabolism and cell state. Nature. 2017;543(7647):681–686. doi:10.1038/nature21408
5. Zeng K, Chen X, Hu X, et al. LACTB, a novel epigenetic silenced tumor suppressor, inhibits colorectal cancer progression by attenuating MDM2-mediated p53 ubiquitination and degradation. Oncogene. 2018;37(41):5534–5551. doi:10.1038/s41388-018-0352-7
6. Xue C, He Y, Zhu W, et al. Low expression of LACTB promotes tumor progression and predicts poor prognosis in hepatocellular carcinoma. Am J Transl Res. 2018;10(12):4152–4162.
7. Cucchi D, Mauro C. LACTB-mediated tumour suppression by increased mitochondrial lipid metabolism. Cell Death Differ. 2017;24(7):1137–1139. doi:10.1038/cdd.2017.60
8. Zhang J, He Y, Yu Y, et al. Upregulation of miR-374a promotes tumor metastasis and progression by downregulating LACTB and predicts unfavorable prognosis in breast cancer. Cancer Med. 2018;7(7):3351–3362. doi:10.1002/cam4.1576
9. Fischer-Huchzermeyer S, Dombrowski A, Hagel C, Mautner VF, Schittenhelm J, Harder A. The cellular retinoic acid binding protein 2 promotes survival of malignant peripheral nerve sheath tumor cells. Am J Pathol. 2017;187(7):1623–1632. doi:10.1016/j.ajpath.2017.02.021
10. Zhang X, Cheng Q, Yin H, Yang G. Regulation of autophagy and EMT by the interplay between p53 and RAS during cancer progression (Review). Int J Oncol. 2017;51(1):18–24. doi:10.3892/ijo.2017.4025
11. Gugnoni M, Sancisi V, Manzotti G, Gandolfi G, Ciarrocchi A. Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell Death Dis. 2016;7(12):e2520–e2520. doi:10.1038/cddis.2016.415
12. Colella B, Faienza F, Di Bartolomeo S. EMT regulation by autophagy: a new perspective in glioblastoma biology. Cancers (Basel). 2019;11(3):312. doi:10.3390/cancers11030312
13. Feng H, Zhao X, Guo Q, et al. Autophagy resists EMT process to maintain retinal pigment epithelium homeostasis. Int J Biol Sci. 2019;15(3):507–521. doi:10.7150/ijbs.30575
14. Li H, Li J, Chen L, et al. HERC3-mediated SMAD7 ubiquitination degradation promotes autophagy-induced EMT and chemoresistance in glioblastoma. Clin Cancer Res. 2019;25(12):3602–3616. doi:10.1158/1078-0432.CCR-18-3791
15. Baek A, Yoon S, Kim J, et al. Autophagy and KRT8/keratin 8 protect degeneration of retinal pigment epithelium under oxidative stress. Autophagy. 2017;13(2):248–263. doi:10.1080/15548627.2016.1256932
16. Jiang Y, Woosley AN, Sivalingam N, Natarajan S, Howe PH. Cathepsin-B-mediated cleavage of Disabled-2 regulates TGF-β-induced autophagy. Nat Cell Biol. 2016;18(8):851–863. doi:10.1038/ncb3388
17. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–1930. doi:10.1101/gad.287524.116
18. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14(1):48. doi:10.1186/s12943-015-0321-5
19. Fulda S, Kögel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene. 2015;34(40):5105–5113. doi:10.1038/onc.2014.458
20. Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2017;14(4):247–258. doi:10.1038/nrclinonc.2016.183
21. Chen P, Cescon M, Bonaldo P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy. 2014;10(2):192–200. doi:10.4161/auto.26927
22. Cicchini M, Chakrabarti R, Kongara S, et al. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy. 2014;10(11):2036–2052. doi:10.4161/auto.34398
23. Akalay I, Janji B, Hasmim M, et al. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy. 2013;9(7):1104–1106. doi:10.4161/auto.24728
24. Peng Y-F, Shi Y-H, Ding Z-B, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy. 2013;9(12):2056–2068. doi:10.4161/auto.26398
25. Heldring N, Nyman U, Lönnerberg P, et al. NCoR controls glioblastoma tumor cell characteristics. Neuro Oncol. 2014;16(2):241–249. doi:10.1093/neuonc/not214
26. Colwill K, Gräslund S. Renewable Protein Binder Working, S. Gräslund, A roadmap to generate renewable protein binders to the human proteome. Nat Methods. 2011;8(7):551–558. doi:10.1038/nmeth.1607
27. Brody H. Colorectal cancer. Nature. 2015;521(7551):S1–S1. doi:10.1038/521S1a
28. Torrano V, Carracedo A. Quiescence-like metabolism to push cancer out of the race. Cell Metab. 2017;25(5):997–999. doi:10.1016/j.cmet.2017.04.027
29. Smith TS, Southan C, Ellington K, Campbell D, Tew DG, Debouck C. Identification, genomic organization, and mRNA expression of LACTB, encoding a serine beta-lactamase-like protein with an amino-terminal transmembrane domain. Genomics. 2001;78(1–2):12–14. doi:10.1006/geno.2001.6643
30. Xiong Y, Yepuri G, Forbiteh M, et al. ARG2 impairs endothelial autophagy through regulation of MTOR and PRKAA/AMPK signaling in advanced atherosclerosis. Autophagy. 2014;10(12):2223–2238. doi:10.4161/15548627.2014.981789
31. Di BS, Nazio F, Cecconi F. The role of autophagy during development in higher eukaryotes. Traffic. 2010;11(10):1280. doi:10.1111/j.1600-0854.2010.01103.x
32. Qiang L, Zhao B, Ming M, et al. Regulation of cell proliferation and migration by P62 through stabilization of Twist1. Autophagy. 2014;111:9241–9246.
33. Qiang L, He YY. Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy. 2014;10(10):1864–1865. doi:10.4161/auto.32171
34. Saiki S, Sasazawa Y, Imamichi Y, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011;7(2):176–187. doi:10.4161/auto.7.2.14074
35. Bodine SC, Stitt TN, Gonzalez M, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3(11):1014–1019. doi:10.1038/ncb1101-1014
36. Gao N, Flynn DC, Zhang Z, et al. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am J Physiol Cell Physiol. 2004;287(2):C281. doi:10.1152/ajpcell.00422.2003
37. Qian Y, Corum L, Meng Q, et al. PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration. Am J Physiol Cell Physiol. 2004;286(1):C153. doi:10.1152/ajpcell.00142.2003
38. Jian X, Xiao-yan Z, Bin H. MiR-204 regulate cardiomyocyte autophagy induced by hypoxia-reoxygenation through LC3-II. Int J Cardiol. 2011;148(1):110–112. doi:10.1016/j.ijcard.2011.01.029
39. Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate P62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. doi:10.1038/ncb2021
40. Vucicevic L, Misirkic-Marjanovic M, Paunovic V, et al. Autophagy inhibition uncovers the neurotoxic action of the antipsychotic drug olanzapine. Autophagy. 2014;10(12):2362–2378. doi:10.4161/15548627.2014.984270
41. Li J, Yang B, Zhou Q, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis. 2013;34(6):1343–1351. doi:10.1093/carcin/bgt063
42. Zhu H, Wang D, Zhang L, et al. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol Rep. 2014;32(3):935–942. doi:10.3892/or.2014.3298
43. Durrant LG, Metheringham RL, Brentville VA. Autophagy, citrullination and cancer. Autophagy. 2016;12(6):1055–1056. doi:10.1080/15548627.2016.1166326
44. Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–946. doi:10.1038/nature09076
45. Liu H, He Z, Simon H-U. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin Cancer Biol. 2013;23(5):352–360. doi:10.1016/j.semcancer.2013.06.008
46. Lu Y, Xiao L, Liu Y, et al. MIR517C inhibits autophagy and the epithelial-to-mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy. 2015;11(12):2213–2232. doi:10.1080/15548627.2015.1108507
47. Zi D, Zhou Z-W, Yang Y-J, et al. Danusertib induces apoptosis, cell cycle arrest, and autophagy but inhibits epithelial to mesenchymal transition involving PI3K/Akt/mTOR signaling pathway in human ovarian cancer cells. Int J Mol Sci. 2015;16(11):27228–27251. doi:10.3390/ijms161126018
48. Qiang L, He -Y-Y. Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy. 2014;10(10):1864–1865.
49. Nam HY, Han MW, Chang HW, Kim SY, Kim SW. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy. 2013;9(10):1631–1632. doi:10.4161/auto.25879
50. Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8(9):528–539. doi:10.1038/nrclinonc.2011.71
51. Yang W, Zhang Y, Li Y, Wu Z, Zhu D. Myostatin induces cyclin D1 degradation to cause cell cycle arrest through a phosphatidylinositol 3-kinase/AKT/GSK-3 beta pathway and is antagonized by insulin-like growth factor 1. J Biol Chem. 2007;282(6):3799. doi:10.1074/jbc.M610185200
52. Xu W, Wang Z, Zhang W, et al. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/β-catenin signaling pathway. Cancer Lett. 2015;356(2):613–627. doi:10.1016/j.canlet.2014.10.008
53. Haraguchi K, Ohsugi M, Abe Y, Semba K, Akiyama T, Yamamoto T. Ajuba negatively regulates the Wnt signaling pathway by promoting GSK-3β-mediated phosphorylation of β-catenin. Oncogene. 2007;27(3):274. doi:10.1038/sj.onc.1210644
54. Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK‐3β and β‐catenin and promotes GSK‐3β‐dependent phosphorylation of β‐catenin. EMBO J. 1998;17(5):1371–1384. doi:10.1093/emboj/17.5.1371
55. Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition (EMT) gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res off J Am Assoc Cancer Res. 2013;19(1):279–290. doi:10.1158/1078-0432.CCR-12-1558
56. Qiying X, Zelin S, Meifang C, Qiaoqing Z, Tianlun Y, Jun Y. IL-8 up-regulates proliferative angiogenesis in ischemic myocardium in rabbits through phosphorylation of Akt/GSK-3β(ser9) dependent pathways. Int J Clin Exp Med. 2015;8:12498.
57. Zhang L, Huang J, Yang N, et al. Integrative genomic analysis of phosphatidylinositol 3ʹ-kinase family identifies PIK3R3 as a potential therapeutic target in epithelial ovarian cancer. Clin Cancer Res. 2007;13(18):5314–5321. doi:10.1158/1078-0432.CCR-06-2660
58. Lu Q-B, Wan M-Y, Wang P-Y, et al. Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFκB/mTOR/P70S6K signaling cascade. Redox Biol. 2018;14:656–668. doi:10.1016/j.redox.2017.11.012
59. Zhao T, Du H, Ding X, Walls K, Yan C. Activation of mTOR pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal(-/-) mice. Oncogene. 2015;34(15):1938–1948. doi:10.1038/onc.2014.143
60. Deeb D, Gao X, Liu Y, et al. The inhibition of cell proliferation and induction of apoptosis in pancreatic ductal adenocarcinoma cells by verrucarin A, a macrocyclic trichothecene, is associated with the inhibition of Akt/NF-кB/mTOR prosurvival signaling. Int J Oncol. 2016;49(3):1139–1147. doi:10.3892/ijo.2016.3587
61. Chen X, Wang Y, Tao J, et al. mTORC1 up-regulates GP73 to promote proliferation and migration of hepatocellular carcinoma cells and growth of xenograft tumors in mice. Gastroenterology. 2015;149(3):741–752.e714. doi:10.1053/j.gastro.2015.05.005
62. Deng F, Ma Y-X, Liang L, Zhang P, Feng J. The pro-apoptosis effect of sinomenine in renal carcinoma via inducing autophagy through inactivating PI3K/AKT/mTOR pathway. Biomed Pharmacother. 2018;97:1269–1274. doi:10.1016/j.biopha.2017.11.064
63. Guan L, Song K, Pysz MA, et al. Protein kinase C-mediated down-regulation of cyclin D1 involves activation of the translational repressor 4E-BP1 via a phosphoinositide 3-kinase/Akt-independent, protein phosphatase 2A-dependent mechanism in intestinal epithelial cells. J Biol Chem. 2007;282(19):14213–14225. doi:10.1074/jbc.M610513200
64. Murooka TT, Rahbar R, Platanias LC, Fish EN. CCL5-mediated T-cell chemotaxis involves the initiation of mRNA translation through mTOR/4E-BP1. Blood. 2008;111(10):4892–4901. doi:10.1182/blood-2007-11-125039
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.