")
Back to Journals » Cancer Management and Research » Volume 10
L61H46 shows potent efficacy against human pancreatic cancer through inhibiting STAT3 pathway
Authors Bai E, Yang L, Xiang Y, Hu W, Li C, Lin J, Dai X, Liang G, Jin R , Zhao C
Received 6 December 2017
Accepted for publication 25 January 2018
Published 23 March 2018 Volume 2018:10 Pages 565—581
DOI https://doi.org/10.2147/CMAR.S159090
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leylah Drusbosky
Encheng Bai,1,2,* Lehe Yang,1,* Youqun Xiang,2,* Wanle Hu,3 Caleb Li,4 Jiayuh Lin,5 Xuanxuan Dai,2 Guang Liang,1 Rong Jin,2 Chengguang Zhao1
1Chemical Biology Research Center, School of Pharmaceutical Sciences, 2Department of Epidemiology, First Affiliated Hospital, 3Department of Coloproctology, The Second Affiliated Hospital and Yuying Children’s Hospital, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 4Dublin Coffman High School, Dublin, OH, 5Department of Biochemistry and Molecular Biology, University of Maryland Greenebaum Comprehensive Cancer Center, University of Maryland School of Medicine, Baltimore, MD, USA
*These authors contributed equally to this work
Background: Pancreatic cancer is the fourth leading cause of cancer-related death worldwide. The poor prognosis of this disease highlights the urgent need to develop more effective therapies. Activation of the STAT3 represents a potential drug target for pancreatic cancer therapy. Currently, clinically available small-molecule inhibitors targeting STAT3 are lacking.
Methods: Through bioassay screening and molecular docking, we identified a small molecule L61H46 that can potently target constitutive STAT3 signaling and kill human pancreatic cancer cells in vitro and in vivo.
Results: L61H46 effectively reduced colony formation and the viability of pancreatic cancer cells in a dose-dependent manner with half-maximal inhibitory concentration (IC50) values in the range between 0.86 and 2.83 µM. L61H46 significantly inhibited STAT3 phosphorylation (Tyr705) and the subsequent nucleus translocation but did not downregulate STAT1 phosphorylation. Moreover, L61H46 demonstrated a potent activity in suppressing pancreatic tumor growth in BXPC-3 xenograft model in vivo. Furthermore, L61H46 showed no signs of adverse effects on liver, heart, and kidney cells in vivo.
Conclusion: Collectively, our results suggest that L61H46 could be further optimized into a highly potent STAT3 inhibitor for the treatment of pancreatic cancer.
Keywords: L61H46, STAT3, cancer therapy, interleukin-6, pancreatic cancer
Erratum for this paper has been published
Introduction
Pancreatic cancer is the fourth leading cause of cancer-related death worldwide with a 5-year survival rate of ~5% and a median survival of 6 months.1,2 Only 5–10% of the diagnosed patients have localized tumors suitable for operation, of whom only 10–20% of the patients survive for >5 years afterwards.3 A number of conventional therapies for pancreatic cancer include gemcitabine, cisplatin, fluoropyrimidine, irinotecan, and taxanes.4 However, these treatments develop inevitable drug resistance and cause serious side effects such as leukopenia, thrombocytopenia, and gastrointestinal irritation.5,6 Therefore, an urgent need arises for an effective and safer therapeutic approach to treat pancreatic cancer.7
STAT3 is a transcription factor and oncogenic driver, which can promote malignant tumors.8 Persistent activation of STAT3 has been detected in many human tumors, including pancreatic and colon cancer cells.6,9 In the cytoplasm, phosphorylated STAT3 proteins form homodimers and translocate to the nucleus where they regulate the expression of various target genes involved in cell proliferation and survival.10 Constitutive activation of STAT3 plays a vital role in regulating cell proliferation, apoptosis, invasion, angiogenesis, and immune suppression.9,11 Studies show that STAT3 inhibition can promote the apoptosis of human pancreatic cancer cells.12–14 Evidence has been accumulated that STAT3 can serve as a viable therapeutic target of the major oncogenic pathways activated in pancreatic cancer.13,14
Curcumin (Figure 1A), an active compound of a medicinal herb Curcuma longa L, has been found to have anticarcinogenic effects with pharmacological safety both in vivo and in vitro.15 Recently, a report has reviewed the pharmacological properties of curcumin and shown evidence that curcumin is an unstable, reactive, and nonbioavailable compound.16 The compound has been tested safe even at high doses in animal and clinical trial studies but exhibits poor stability and bioavailability.17 To improve stability and bioavailability, curcumin derivatives were designed and synthesized to improve their pharmacokinetics potency and bioavailability, while retaining the same safety profile.18 Several curcumin analogs, namely FLLL31, FLLL32, FLLL62, and GO-030, have been shown to bind selectively to STAT3 SH2 and inhibit STAT3 phosphorylation and dimerization.19–22 These curcumin analogs selectively inhibited STAT3 phosphorylation without suppressing the expression of STAT1 phosphorylation and induced apoptosis in various cancer cells both in vitro and in vivo.23,24 Previously, our laboratory designed and synthesized a series of mono-carbonyl analogs of curcumin (MACs) via deletion of β-diketone moiety. These MACs showed a significantly improved chemical stability in vitro and a good pharmacokinetic profile and bioavailability in vivo.18,25 We hypothesize that some of these MACs compounds could induce apoptosis in pancreatic cell lines by inhibiting the STAT3 pathway activation. Molecular docking screenings were carried out to predict possible MAC binders for the STAT3 SH2 domain using the AutoDock Vina program.26 By methylthiazolyldiphenyl-tetrazolium bromide (MTT) bioassay screening of the predicted MAC binders against human pancreatic cancer cells, we identified L61H46 (Figure 1A)as a potent inhibitor of human pancreatic cancer cells. The underlying mechanisms were also examined.
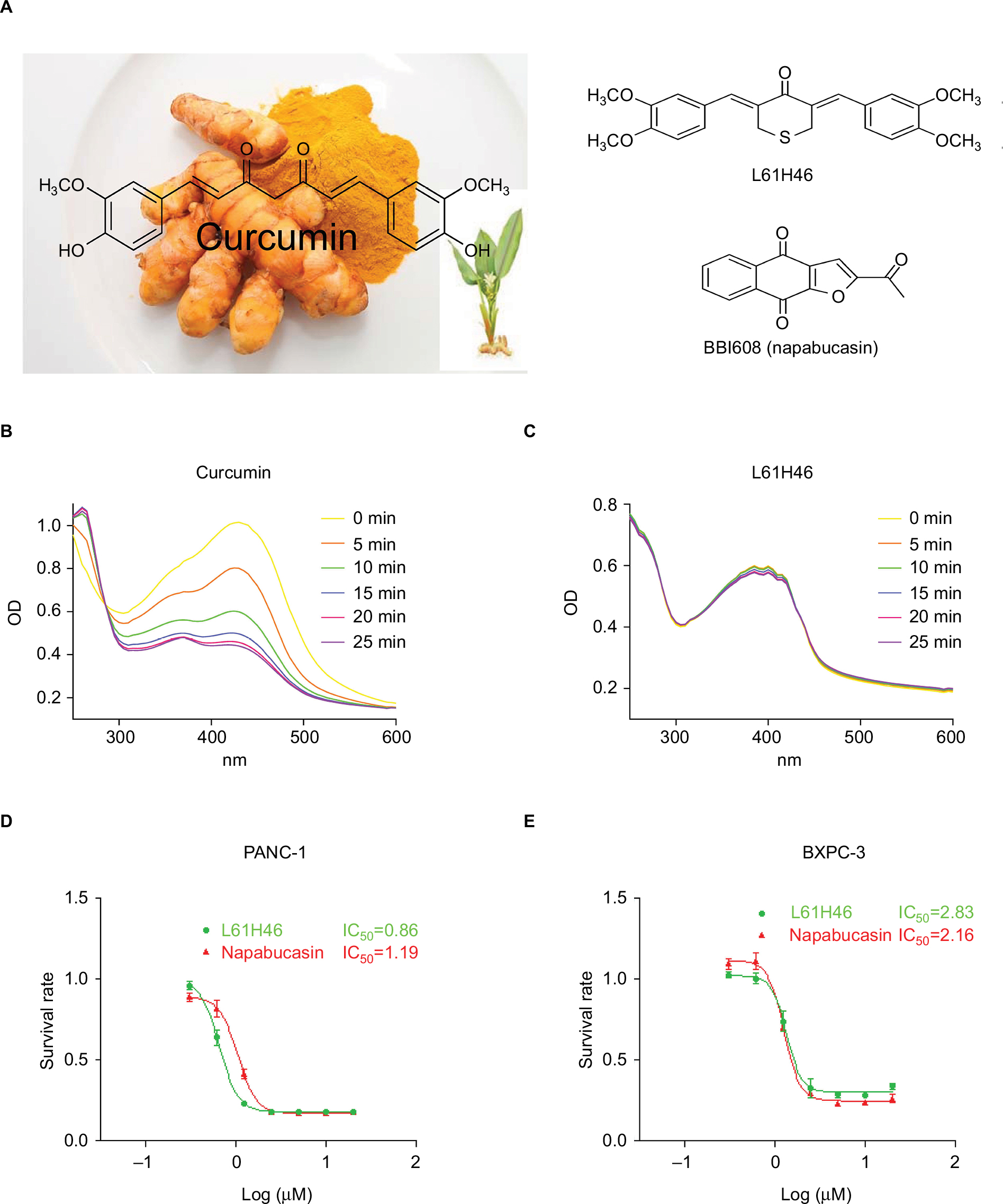 | Figure 1 L61H46 shows superior stability than curcumin and effectively suppressed cell viability. Notes: (A) Chemical structure of curcumin, L61H46, and napabucasin. UV–visible absorption spectrum of curcumin (B) and L61H46 (C) in phosphate buffer (pH 7.4). The effects of L61H46 on the proliferation of human pancreatic cancer cell lines PANC-1 (D) and BXPC-3 cells (E) were incubated with increasing doses of L61H46 and napabucasin (0.3–10 μM) for 48 h, respectively. Cell viability was determined by MTT assay. Data represent similar results from at least three independent experiments. Abbreviations: IC50, half-maximal inhibitory concentration; MTT, methylthiazolyldiphenyl-tetrazolium bromide; OD, optical density; UV, ultraviolet. |
Our results demonstrate that L61H46 could effectively induce apoptosis in pancreatic cancer cells via inhibiting the STAT3 pathway and downregulating the downstream target genes. The blockade of the STAT3 signaling inhibited cell proliferation, migration, and colony formation. L61H46 also exhibited potent anticancer effects in a mouse xenograft model in vivo. These data suggest that lead compound L61H46 could be further developed into a promising candidate for the treatment of pancreatic cancer.
Materials and methods
Chemistry
Napabucasin (BBI608) was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Erlotinib was purchased from Selleck Chemicals (Houston, TX, USA). L61H46 (Figure 1A), a MAC, was synthesized in our laboratory. The synthesis procedure was the same as our previous reports.25 The compound L61H46 was recrystallized from CH2Cl2/CH3CH2OH, with a purity of 98.85% as determined by high performance liquid chromatography.
Cell culture
Human pancreatic cancer cell lines (BXPC-3 and PANC-1) human umbilical vein endothelial cells (HUVEC), and normal human bronchial epithelium cells (BEAS-2B), were obtained from Shanghai Institute of Biosciences and Cell Resources Center (Chinese Academy of Sciences, Shanghai, China). PANC-1 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA). BXPC-3, HUVEC, and BEAS-2B cells were routinely cultured in Roswell Park Memorial Institute (RPMI)-1640 media (Thermo Fisher Scientific). The culture medium was a mixture of RPMI-1640 or DMEM, 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific), 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells were cultured in a humidified cell incubator with an atmosphere of 5% CO2 at 37°C.
Antibodies and reagents
Antibodies for anticleaved poly(ADP-ribose) polymerase (PARP), anti-BCL-2, anti-Caspase 3, anticleaved Caspase 3, and secondary antibodies goat antimouse IgG-horseradish peroxidase and donkey antirabbit IgG-horseradish peroxidase were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Antibodies including anti-p-STAT3, anti-STAT3, anti-p-STAT1, and anti-STAT1 were purchased from Cell Signal Technology (Danvers, MA, USA). MTT and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich Co. Fluorescein isothiocyanate Annexin V Apoptosis Detection Kit, propidium iodide (PI), and basement membrane Matrigel were obtained from BD (Franklin Lakes, NJ, USA). Crystal violet staining solution was purchased from Beyotime Institute of Biotechnology (Shanghai, China). L61H46 was dissolved in DMSO before being used in experiments.
Docking of L61H46 and napabucasin to the STAT3 SH2
The STAT3 SH2 crystal structure was obtained from the Protein Data Bank (protein data bank code: 1BG1) and used as the protein target for docking simulation.27 AutoDockTools (ADT) 1.5.6 was employed for the preparation of input PDBQT files of ligands and protein receptor STAT3 SH2 as described in the instruction.28 Docking simulations were performed using AutoDock Vina 1.0.2.26 The docking grid was a 3D 24 Å ×16 Å ×24 Å box, which encompassed the entire binding region. The Vina docking parameters were as follows: a receptor binding center of x=103, y=72.5, and z=64.1, an energy range of 5 kcal/mol, an exhaustiveness of 25, and an output number of five binding modes ranked by highest binding affinity. The binding modes were viewed and analyzed using the ADT tool, and the top binding modes with best binding affinity (most negative binding energy) were selected.
Stability evaluation of L61H46 and curcumin in phosphate buffer
Absorbance readings were taken from 250 to 600 nm using a spectrum Max M5 (Molecular Devices LLC, Sunnyvale, CA, USA). A stock solution of 1 mM curcumin or L61H46 was prepared and diluted by phosphate buffer (pH 7.4) to a final concentration of 20 μM. In the experiments where degradation of curcumin was recorded, the absorption spectra were collected for >25 min at 5 min intervals. The ultraviolet–visible absorbance spectrum was measured at 25°C at varying time intervals in a 1 cm path-length quartz cuvette.
Cell viability assay
The cytotoxicity of L61H46 against pancreatic cancer cells was determined by the MTT assay. BXPC-3 and PANC-1 cell lines were incubated at 3×103 cells/well in 96-well plates with RPMI-1640 or DMEM and treated with a dose escalation of L61H46 (0.3125-10 μM) for 48 h. An MTT solution was added at 20 μL/well and incubated for 4 h at 37°C. The crystals were dissolved with DMSO at 150 μL/well, and the absorbance of each well was measured at 490 nm using microplate reader. The cell viability was calculated according to the following formula: viability = (average optical density values of treatment wells/average optical density values of vehicle control wells) ×100%. And, half-maximal inhibitory concentration (IC50) values were determined by GraphPad Pro Prism 5.0.
Cell apoptosis analysis
BXPC-3 and PANC-1 cells were seeded on six-well plates overnight and then treated with L61H46 (5 and 10 μM) or napabucasin (10 μM) for 24 h. Cells were then harvested, washed with phosphate-buffered saline (PBS), and stained with Annexin V for 10 min. Afterward, PI was added to stained cells for 5 min. Flow cytometric analysis was performed to evaluate the apoptosis using FACSCalibur (BD).
Western blot analysis
Cells or tissues were homogenized in protein lysate buffer, with sample loading buffer denatured with boiling water bath added. Thereafter, protein samples were electrophoresed by 10 or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene fluoride (PVDF) membrane. The membranes were blocked with fresh 5% nonfat milk for 1.5 h at room temperature in Tris-buffered saline and Tween 20 (TBST) and then incubated with specific primary antibody in TBST at 4°C overnight. After washing with TBST for three times, the membranes were incubated with secondary antibodies for 1 h. The bands were visualized using enhanced chemiluminescence detection kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). The density of all immunoreactive bands was analyzed using the ImageJ software (National Institute of Health, Bethesda, MD, USA). Cartograms of them are listed in the Supplementary materials.
Cell transfections for gene overexpression
The human STAT3 transcript expression vector coding STAT3 protein was obtained from Sino Biological Inc (Catalog: HG10034-CF; Beijing, China). The STAT3 plasmid was transfected into pancreatic cancer cell line (PANC-1) using the Lipofectamine 2000 reagent (Thermo Fisher Scientific) according to the manufacturer’s instruction. After 48 h post-transfection, STAT3 expression in PANC-1 cells was confirmed by Western blotting.
Immunohistochemistry (IHC) staining and hematoxylin and eosin (H&E) staining
Tumor tissues were fixed in 10% paraformaldehyde at room temperature and embedded in paraffin. Paraffin-embedded tissues were sectioned (5 μm thick). Then, the specimens were incubated with indicated antibodies overnight at 4°C. The signal was detected by corresponding secondary antibodies. Thereafter, these slides were stained with diaminobenzidine and counterstained with hematoxylin. Quantity assay of the immunochemistry data was assessed by Image-Pro Plus 6.0 (Media Cybernetics, Bethesda, MD, USA). For histological analysis, the heart, kidney, and liver tissues of the control group and the L61H46 group were fixed in 4% formaldehyde and embedded in paraffin. The paraffin tumor tissue sections (5 μM) were deparaffinized and rehydrated and then stained with eosin and hematoxylin. The images were captured using a light microscope (200× amplification, Eclipse Ti; Nikon Corporation, Tokyo, Japan).
Immunofluorescent staining
Cells were plated on sterilized coverslips in a six-well plate and allowed to grow overnight. For nuclear translocation experiment, BXPC-3 cells were grown in serum-free RPMI-1640 overnight. After that, cells were treated with L61H46 (2.5 μM) for 12 h, followed with the stimulation of interleukin-6 (IL-6) or interferon (IFN)-γ (50 ng/mL) for 30 min. After the treatments, the cells were washed with ice-cold PBS buffer and fixed with 4% paraformaldehyde for 15 min. The cells were then washed with PBS for twice, permeabilized with 0.3% Triton X-100, and blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. All cell samples were incubated with the primary antibodies anti-p-STAT3 (1:200 dilution) overnight at 4°C and, then, the cells were incubated with polyethylene-conjugated goat antirabbit secondary antibody (1:400 dilution) for 1 h at room temperature. Afterward, 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) was used to stain the nuclei. Pictures were captured by confocal microscopy (800× amplification, Eclipse Ti).
Clonogenic assay
The BXPC-3 and PANC-1 cells were seeded at 1000 cells/well on six-well plates. Cells were treated with L61H46 (1 and 2.5 μM) or napabucasin (2.5 μM) for 24 h. After being replaced with fresh RPMI-1640 medium, cells were allowed to grow for 18 days until the colonies were visible. Colonies were stained with crystal violet staining solution and photographed after 7 days.
Hoechst 33258 staining
After treated with L61H46 (5 or 10 μM) or napabucasin (10 μM) for 24 h, cells were fixed with 4% formaldehyde solution, washed twice with PBS, and stained with Hoechst 33258 staining kit according to the manufacturer’s instructions (Beyotime Institute of Biotechnology). Apoptotic features of cell death were determined by the morphology of cell nuclei using the fluorescence microscope (Nikon Corporation) with 400× amplification. Five microscopic fields were randomly selected to observe in each group. The apoptotic cells were defined as those with nuclei brighter and smaller than normal cells.
Matrigel invasion assay
Matrigel invasion assay was performed to evaluate the invasion abilities of tumor cells in transwell insert chambers (8.0 μm; Corning Incorporated, Corning, NY, USA) according to the manufacturer’s instructions. A total of 500 μL of culture medium containing 10% FBS was added to the lower chamber, and then, cells together with L61H46 (1 and 2.5 μM) or napabucasin (2.5 μM) were seeded on the upper chamber (precoated with diluted matrigel) at 10,000 cells/well in 200 μL of serum-free medium. After incubation for 24 h at 37°C, the cells on the lower surface of chamber were fixed and stained with crystal violet, while the cells on the upper chamber were removed completely with a cotton swab. The migration cells were captured with light microscopy (Nikon Corporation) with 200× amplification.
Wound-healing assay
BXPC-3 cells’ migration was determined using the wound-healing assay. When cells grew into 100% confluent in six-well plate, the monolayer cells were scratched in same width using yellow pipette tip and washed twice with PBS to remove nonadherent cells. After washing, cells were treated with L61H46 (5 and 10 μM), napabucasin (10 μM) or DMSO. After 12 h, growth medium was replaced with a fresh complete RPMI-1640 medium with 10% FBS. Cells were allowed to grow for 24–36 h, and the images were captured with a light microscopy (100× amplification). The percentage of migration was calculated by the following formula: 100%− (final gap area/initial gap area ×100%).
In vivo antitumor study
All animal experiments were followed according to the Wenzhou Medical University’s Policy on the Care and Use of Laboratory Animals. Operations for animal experiments were approved by the Wenzhou Medical University Animal Policy and Welfare Committee. For analysis of tumorigenicity, BXPC-3 cells (5×106 cells/mice) were mixed with 100 μL of PBS and were injected subcutaneously into the flank of 6-week-old athymic nude mice (18–22 g). Animals were housed under barrier conditions and fed with a standard rodent diet and water. After 1 week, tumor volume reached 50 mm3 and mice were randomly divided into three treatment groups (n=6): 1) PBS as vehicle control, 2) 10 mg/kg of L61H46 (dissolved in 10% castor oil and 90% PBS), and 3) 10 mg/kg of erlotinib (dissolved in 10% castor oil and 90% PBS). Vehicle and L61H46 were administered via intraperitoneal injection every other day for 36 days. The tumor volumes were determined by measuring length (l) and width (w) with a caliper. And, tumor volume was calculated according to the following formula: volume = (ϖ/6) × L × W2. At the end of treatment, tumors were removed from euthanized mice, snap-frozen in liquid nitrogen, and stored at −80°C. A portion of tumor tissues was fixed with formalin and embedded in paraffin for IHC staining. Tumor tissue homogenates were lysed and separated by SDS-PAGE to examine the expression of p-STAT3, BCL-2, and cleaved Caspase 3. BCL-2 was also examined by IHC staining.
Statistical analysis
All of the data were assayed in triplicate (n=3). Data are expressed as mean ± standard error of mean. All statistical analyses were performed using GraphPad Pro. Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Student’s t-test and one-way analysis of variance were used to analyze the differences between sets of data. A P-value of <0.05 was considered statistically significant.
Results
L61H46 shows superior stability than curcumin and effectively suppressed cell viability
The structure of curcumin and L61H46 is shown in Figure 1A. We first study the stability of L61H46 in PBS by ultraviolet–visible absorption spectrum. The result showed that the absorption intensity of curcumin spectra declined significantly in phosphate buffer (pH 7.4) with time. The intensity of curcumin dropped to ~40% after incubation in the phosphate buffer for 25 min, while L61H46 showed no degradation under equal condition (Figure 1B and C). To evaluate the antitumor activity of the lead compound L61H46, we performed MTT assays to examine the effects of L61H46 on the cell viability of human pancreatic cancer cell lines, PANC-1 and BXPC-3. Napabucasin (BBI608, Figure 1A), an orally potent inhibitor of STAT3 pathway in cancer stemness, was used as a reference control.29 As shown in Figure 1D and E, L61H46 significantly induced cell death in a dose-dependent manner in both human pancreatic cancer cell lines, with IC50 values 0.86 and 2.83 μM respectively. For comparison, the IC50 values of napabucasin in these two cell lines were 1.19 and 2.16 μM, respectively. L61H46 showed a similar anticancer potency in human pancreatic cancer cell lines, comparing with the reported STAT3 inhibitor napabucasin.
L61H46 induced apoptosis and decreased colony formation
We also examined the apoptosis-inducing effects of L61H46 using Annexin V/PI staining assay. Both pancreatic cancer cell lines had a dose-dependent apoptosis after the treatment with L61H46 for 24 h (Figure 2A and B). L61H46 at 10 μM induced significant apoptosis, while napabucasin (10 μM) at the same concentration induced less apoptosis. We further determined the expression levels of apoptosis-related proteins in BXPC-3 cells treated with L61H46. Consistent with the results obtained by Annexin V/PI staining assay, we found that treatment with L61H46 for 24 h dose-dependently increased the level of cleaved PARP and decreased the levels of pro-Caspase 3 and BCL-2 (Figures 2C and S1A). In addition, cell morphological changes were examined to determine cell apoptosis using DAPI staining assay in BXPC-3 cells. It was found that L61H46 dose-dependently increased the number of apoptotic cells (Figures 2D and S1B). L61H46 displayed more intense apoptotic effects than the known STAT3 inhibitor napabucasin. Furthermore, L61H46 remarkably inhibited the colony formation capacity in both PANC-1 and BXPC-3 pancreatic cancer cell lines (Figure 2E).
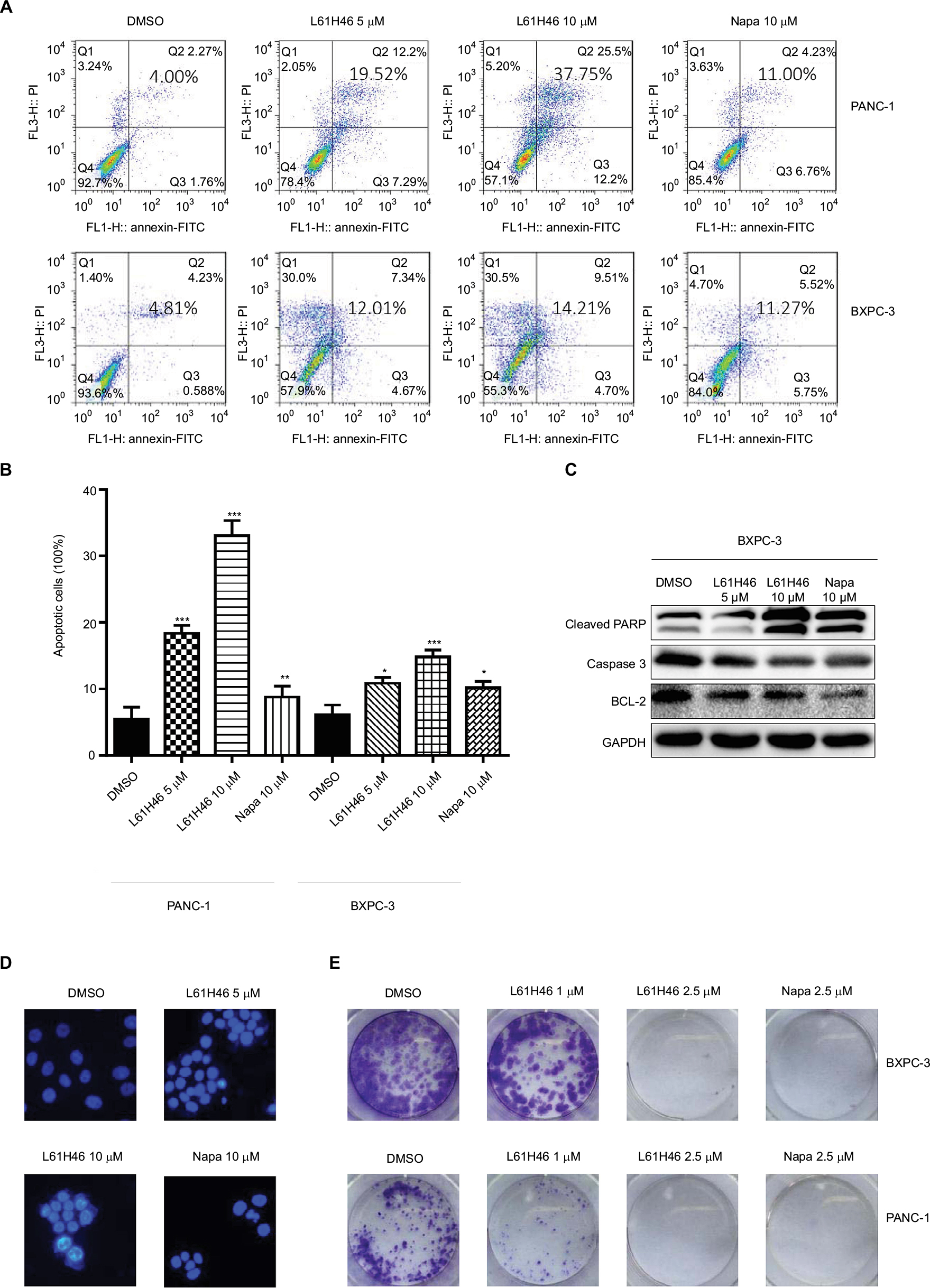 | Figure 2 L61H46 induced apoptosis and decreased colony formation. Notes: (A) Induction of apoptosis in human pancreatic cancer cells was determined by flow cytometry after treatment with L61H46 (5 or 10 μM) and napabucasin (10 μM) for 24 h. (B) The percentage of apoptotic cells in the treatment groups was calculated. (C) BXPC-3 cells were treated with L61H46 (5 or 10 μM) or napabucasin (10 μM) for 24 h. Western blotting was employed to assess the expression of cell apoptosis-related proteins. GAPDH was used as an internal control. (D) The L61H46 treatment induced increased apoptotic morphology in BXPC-3 cells compared with napabucasin. BXPC-3 cells were treated with L61H46 (5 or 10 μM) or napabucasin (10 μM) for 24 h. Cell morphology was observed by an inverted microscope after Hoechst 33258 staining. The magnification is 400×. (E) Representative images of colony-formation assay with L61H46 (1 or 2.5 μM) or napabucasin (2.5 μM). All images are representative of three independent experiments. Data are mean ± standard error of mean of three independent experiments. *P<0.05, **P<0.01, ***P<0.001. Abbreviations: DMSO, dimethyl sulfoxide; FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Napa, napabucasin; PARP, poly(ADP-ribose) polymerase; PI, propidium iodide. |
L61H46 inhibited cell invasion and migration
The invasion and migration ability is critical for cancer progression.30 We performed the migration transwell assay and wound-scratch assay to evaluate the inhibitory effects of L61H46 on cell invasion and migration in BXPC-3 cells. Our results showed that treatment with L61H46 resulted in a dose-dependent decrease in cell invasion ability (Figure 3A and B). Similar results were obtained in wound-scratch assays (Figure 3C and D). The inhibitory rate of migration was ~66 and 81% at concentrations of 5 and 10 μM, respectively (Figure 3C).
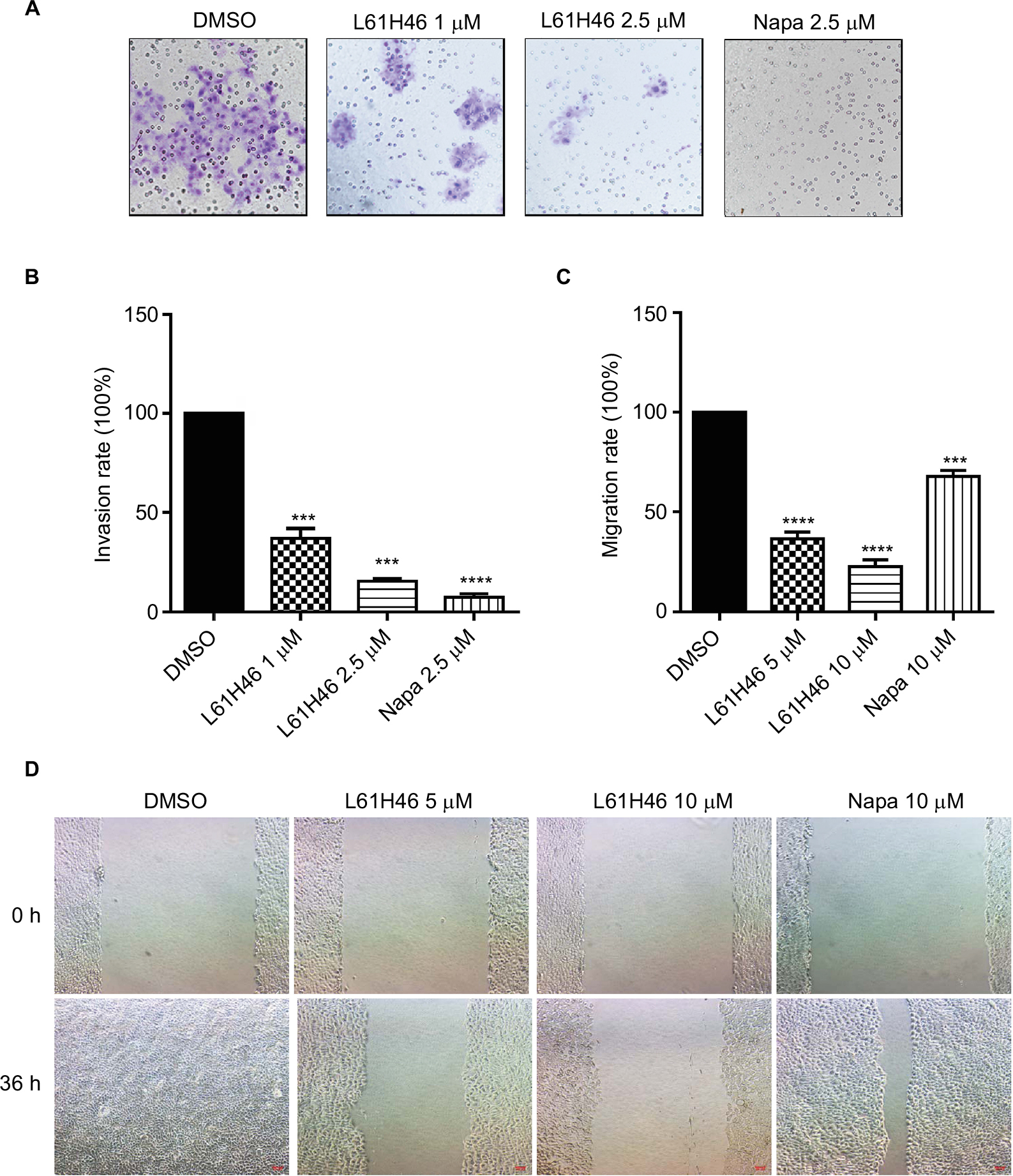 | Figure 3 L61H46 inhibited cell invasion and migration. Notes: (A) BXPC-3 cells (10,000/well) and L61H46 (1 or 2.5 μM) or napabucasin (2.5 μM) were added at the same time to the upper chamber. After incubation for 24 h at 37°C, invasive cells stuck to the lower chamber were fixed and stained with crystal violet and their number was quantified with microscopy. The magnification is 200×. (B) The invasive cells on the surface of the lower chamber were calculated and represented as the percent of control. (C) The inhibition of cell migration was assessed by the ImageJ software. (D) Wound healing assay was conducted for migration in BXPC-3 cells treated with L61H46 (5 or 10 μM) or napabucasin (10 μM). After 12 h, growth medium was replaced with a fresh complete RPMI-1640 medium with 10% FBS. The magnification is 100×. All images are representative of three independent experiments. Data are mean ± standard error of mean of three independent experiments. ***P<0.001, ****P<0.0001. Abbreviations: DMSO, dimethyl sulfoxide; FBS, fetal bovine serum; Napa, napabucasin; RPMI, Roswell Park Memorial Institute. |
L61H46 effectively inhibited constitutive STAT3 phosphorylation in human pancreatic cancer cells
We tested the inhibiting ability of napabucasin on STAT3 Tyr705 phosphorylation (p-STAT3 at Y705) in pancreatic cancer cells. The results showed that napabucasin inhibited STAT3 phosphorylation dose-dependently (Figure S2A). To confirm whether L61H46 could also suppress STAT3 Tyr705 phosphorylation, PANC-1 cells were treated with L61H46 for a series of time periods. The Western blotting results showed that L61H46 time-dependently inhibited the persistent STAT3 phosphorylation. The phosphorylation of STAT3 at Tyr705 was reduced significantly at ~24 h after the treatment (Figure 4A). We also found that L61H46 induced a dose-dependent reduction of STAT3 phosphorylation in both PANC-1 (Figure 4B) and BXPC-3 (Figure 4C) cell lines. These results show that L61H46 is a potent inhibitor of STAT3 phosphorylation (p-STAT3) at a low concentration in multiple pancreatic cancer cell lines.
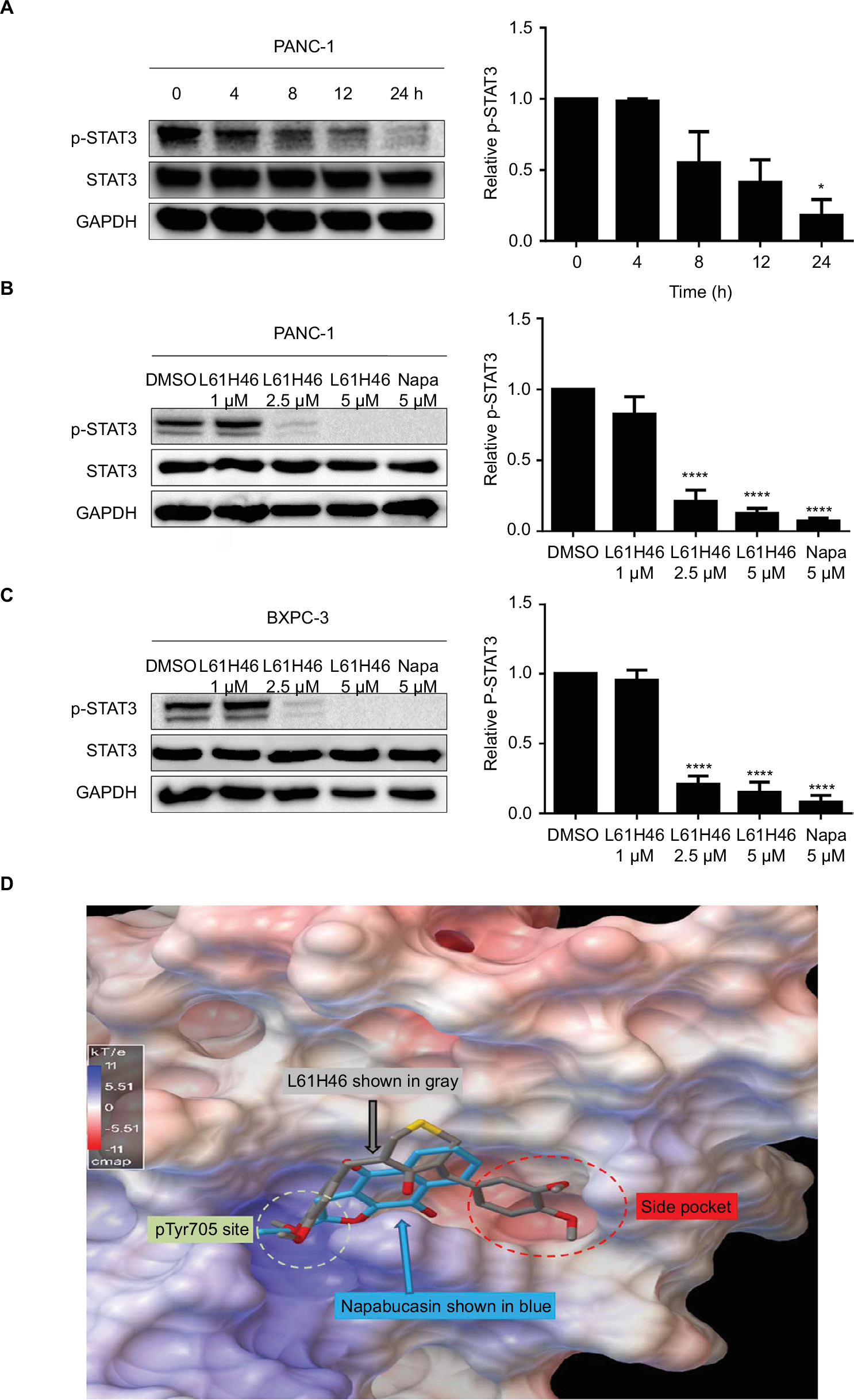 | Figure 4 L61H46 inhibited constitutive phosphorylation of STAT3 in pancreatic cancer cells. Notes: (A) L61H46 (5 μM) time dependently (0, 4, 8, 12, and 24 h) inhibited STAT3 phosphorylation in PANC-1 cells. L61H46 dose dependently (1.0, 2.5, and 5 μM) inhibited STAT3 phosphorylation in PANC-1 (B) and BXPC-3 (C) human pancreatic cancer cells for 24 h; napabucasin is 5 μM. The column figure was the normalized optical density as a percentage of the relevant total STAT3 protein. (D) The docking modeling of L61H46 to the binding hot spots of STAT3 SH2 (protein code: 1BG1). STAT3 SH2 is in electrostatic potential surface representation. L61H46 is rendered in a gray stick-ball, and napabucasin is rendered in a blue stick-ball. L61H46 (gray stick-ball) occupied the main pTyr705 hot spot and the side pocket with a binding energy of −6.5 kcal/mol, which could effectively disrupt the native pTyr705 binding to prevent STAT3 SH2 dimerization. As a comparison, the known inhibitor napabucasin (blue stick-ball) had similar binding modes and energy, binding to the pTyr705 site and partially to the side pocket with a binding energy of −6.1 kcal/mol. Data represent similar results from at least three independent experiments. *P<0.05, ****P<0.0001. Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Napa, napabucasin. |
To further explore the relationship between inhibition on STAT3 phosphorylation and cell viability inhibition, we tested L61H46 in two normal cells. The results showed that L61H46 significantly inhibited HUVEC cells with IC50 values 1.23 μM, while the IC50 values of the compound in BEAS-2B cells were >10 μM (Figure S2B and C). We next detected the STAT3 phosphorylation levels of two pancreatic cancer cells and two normal cells. Western blotting analysis indicated that BEAS-2B cells lacked STAT3 phosphorylation, while other three cells express high levels of STAT3 phosphorylation (Figure S2D). This may explain huge difference on IC50 values between HUVEC and BEAS-2B cells.
We further determined whether overexpression of STAT3 would dampen the cytotoxic effect of L61H46. We performed transfections to increase STAT3 expression in PANC-1 cells (Figure S2E). Overexpression of STAT3 reduced L61H46-induced inhibition of growth (Figure S2F). These findings indicate that the anticancer activity of L61H46 is, at least partly, mediated by targeting STAT3.
Molecular docking of L61H46 to the binding hot spots of STAT3 SH2
To predict the binding modes and affinity of compound L61H46 to the binding hot spots of STAT3 SH2 domain, we performed molecular docking simulations of L61H46 to the STAT3 SH2. Figure 4D shows the predicted top binding mode of L61H46 to the STAT3 SH2 domain (protein code: 1BG1). L61H46 (gray stick-ball), with a binding energy ΔE of −6.5 kcal/mol, occupied both the main pTyr705 hot spot and the side pocket, which could effectively disrupt the native pTyr peptide (pYLK) binding to prevent STAT3 SH2 activation and dimerization. Interestingly, the docking simulations show that the known STAT3 inhibitor napabucasin (blue color) bound similarly to the pTyr705 binding sub-pocket and partially to the side pocket with a binding energy ΔE of −6.1 kcal/mol.
L61H46 inhibited IL-6-induced STAT3 phosphorylation and STAT3 nuclear translocation
IL-6 is known to stimulate STAT3 phosphorylation on Tyr705 in many cancer cells.31 Our results showed that L61H46 inhibited the STAT3 phosphorylation (p-STAT3) induced by IL-6 in a dose-dependent manner. No significant changes were observed in the total amount of STAT3 (Figure 5A). To examine if L61H46 selectively inhibits the phosphorylation of STAT3 (p-STAT3), not STAT1 (p-STAT1), we further investigated whether L61H46 could inhibit STAT1 phosphorylation mediated by IFN-γ. We found that L61H46 had no obvious effect on p-STAT1 level (Figure 5B). The data showed that the lead compound suppressed the IL-6 induced STAT3 phosphorylation but not the IFN-γ stimulated STAT1 phosphorylation.
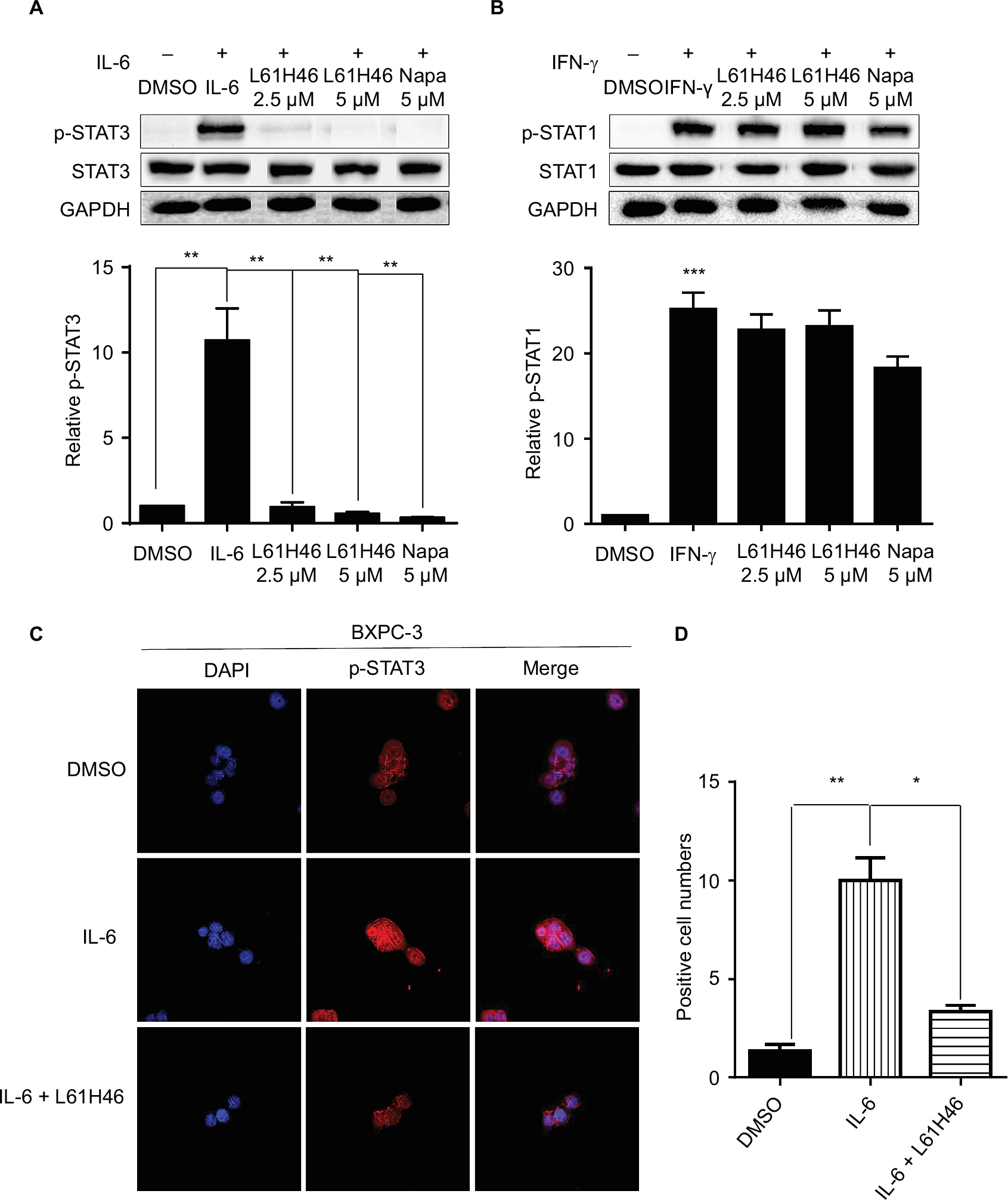 | Figure 5 L61H46 inhibited STAT3 phosphorylation and p-STAT3 nuclear translocation induced by IL-6. Notes: (A) L61H46 (2.5–5 μM) inhibited STAT3 phosphorylation induced by IL-6 in BXPC-3 human pancreatic cancer cells. After treating with L61H46 (2.5–5 μM) for 24 h, cells were stimulated by IL-6 (50 ng/mL) or (B) IFN-γ (50 ng/mL) for 30 min; napabucasin is 5 μM. The cells were harvested and analyzed for STAT3 and STAT1 phosphorylation by Western blotting as described in the “Western blot analysis” section. The density of p-STAT3 and p-STAT1 was analyzed by the ImageJ software. (C) After a serum-free overnight, BXPC-3 pancreatic cancer cells were pretreated with L61H46 (2.5 μM) for 24 h, followed by IL-6 (50 ng/mL) for 30 min, and then detected for STAT3 nuclear translocation by immunofluorescence staining. (D) The percentage of positive cells per field in each group was calculated. All images are representative of three independent experiments. The level of significance is indicated by *P<0.05, **P<0.01, and ***P<0.001. Abbreviations: DAPI, 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IFN, interferon; IL, interleukin; Napa, napabucasin. |
We next detected that once STAT3 was activated by IL-6, the phosphorylated STAT3 (p-STAT3) would translocate into the nucleus of BXPC-3 cancer cells. We examined whether L61H46 could suppress the nuclear translocation of STAT3 induced by IL-6. The result indicated that L61H46 blocked the IL-6 induced STAT3 phosphorylation (p-STAT3) and the subsequent nucleus translocation (Figure 5C and D). Interestingly, once phosphorylated p-STAT3 consistently localized in the nucleus even without IL-6 treatment. Hence, L61H46 prevented the nucleus translocation most likely through inhibition of p-STAT3.
L61H46 suppressed tumor growth of pancreatic cancer cells in vivo
To examine the in vivo antitumor effects of L61H46 on BXPC-3 pancreatic cancer cells, we used a nude mice xenograft model. Erlotinib, the epidermal growth factor receptor-targeted therapy drug for pancreatic cancer, was used as a positive control.32 As shown in Figure 6A–C, treatment with L61H46 at 10 mg/kg for 36 days resulted in significant reduction in both tumor volume and weight in BXPC-3-xenografted mice (P<0.05) as compared to the vehicle-treated controls and erlotinib-treated positive controls. In addition, no significant difference was found in body weight between the vehicle group and the L61H46-treated group, suggesting that L61H46 showed no significant toxicity on nude mice within the 36 days treatment (Figure 6D). We further measured the expressions of various cancer-related genes in tumor tissues by Western blotting. The result showed that L61H46 inhibited STAT3 phosphorylation, increased the level of cleaved Caspase 3, and decreased the level of BCL-2 (Figures 6E and S3). L61H46 also inhibited the level of Bcl-2 as shown by IHC staining (Figure 6F). The toxicity of L61H46 was further evaluated by H&E staining analysis in mouse kidneys, livers, and hearts. No noticeable difference was found in morphology between the vehicle group and the L61H46-treated group (Figure 6G), indicating no signs of adverse effects on liver, heart, and kidney cells in vivo. These results showed that L61H46 had a potent antitumor efficacy in vivo using human pancreatic cancer xenograft model.
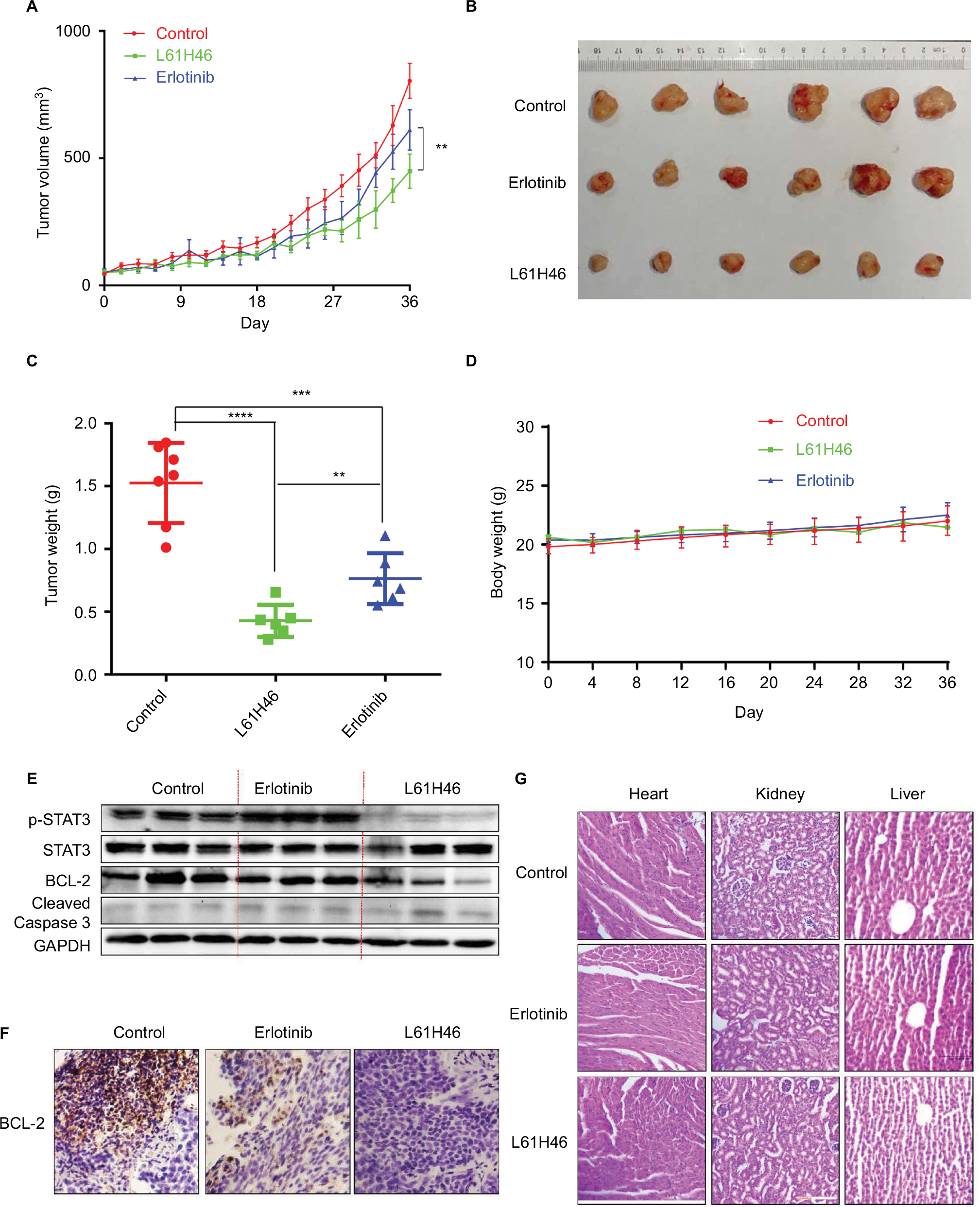 | Figure 6 L61H46 suppressed tumor growth of pancreatic cancer cells in vivo. Notes: Tumor volume (A and B) and tumor weight (C) in BXPC-3 human pancreatic cancer xenografts in nude mice. (D) Body weight of mice. (E) L61H46 inhibited STAT3 phosphorylation, increased the level of cleaved Caspase 3, and decreased BCl-2 in mouse xenografts in vivo. (F) L61H46 decreased the expression of BCL-2 as shown by IHC staining. (G) No histological abnormalities were observed in kidneys, livers, and hearts in the L61H46 group. Hearts, kidneys, and livers from two groups were sectioned at 5 μm, and the slides were stained with H&E (n=5 in each group). All images were obtained by microscope with 200× magnification. All images are representative of three independent experiments. Data are mean ± standard error of mean of three independent experiments. The level of significance is indicated by **P<0.01, ***P<0.001, and ****P<0.0001. Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H&E, hematoxylin and eosin; IHC, immunohistochemistry. |
Discussion
The crucial role of STAT3 in the cancer signaling makes it a promising therapeutic target for cancer treatments.6,19 Here, we identified a small molecule L61H46 as a novel STAT3 inhibitor using bioassay screening and molecular docking. We found that L61H46 could significantly reduce the viability of pancreatic cancer cells in a dose-dependent manner with IC50 values in the range between 0.86 and 2.83 µM. Moreover, our results confirmed that L61H46 can potently suppress pancreatic tumor growth in BXPC-3 xenograft model in vivo (Figure 6).
In addition to its proapoptotic function, L61H46 showed antiproliferation and antimigration activities (Figures 2 and 3). Our data further confirmed that L61H46 inhibited the STAT3 pathway at very low concentration (2.5 μM) in PANC-1 and BXPC-3 cancer cell lines (Figure 4B and C). L61H46 blocked the IL-6-induced STAT3 phosphorylation in a dose-dependent manner but had no noticeable effect on the STAT1 phosphorylation in BXPC-3 cells, suggesting that it selectively inhibits STAT3 phosphorylation (Figure 5). L61H46 was slightly more potent in downregulating STAT3 phosphorylation in BXPC-3 cells than the reported STAT3 inhibitor napabucasin, an orphan drug that targets STAT3 in the treatment of pancreatic cancer and gastric cancer. The binding hot spots of STAT3 SH2 consist of the following three sub-pockets: the main pTyr705 site, a Pro704 side pocket, and a Leu706 site.33 These binding sites are critical for STAT3 dimerization and biological activity. Previous docking simulations have shown that the majority of the reported direct STAT3 inhibitors bind to the main pTyr705-binding site and the side pocket of STAT SH2.34,35 Our results of docking simulation showed that L61H46 (gray stick-ball in Figure 4D) occupied the main pTyr705-binding site and the side pocket (ΔE: −6.5/kcal/mol). The results agree with the binding modes of the previously reported STAT3 inhibitors.
In our study, we compared the antitumor effect of L61H46 with napabucasin, an orphan drug that targets STAT3. The results showed that L61H46 could significantly reduce the viability of pancreatic cancer cells with a similar IC50 value compared to napabucasin. At the same concentration of 10 µM, L61H46 promoted more cells’ apoptosis than napabucasin, consistent with the result of apoptosis-related proteins. The inhibitory rate of cell invasion of L61H46 was slightly weaker than napabucasin, while L61H46 inhibited cell migration more effectively than the positive control drug. Furthermore, L61H46 inhibited the STAT3 pathway at very low concentration (2.5 μM) in PANC-1 and BXPC-3 cancer cell lines and blocked the IL-6-induced STAT3 phosphorylation in a dose-dependent manner. The inhibition effects of p-STAT3 on BXPC-3 cancer cell lines of these two drugs were very close. Finally, we performed the docking modeling of L61H46 and napabucasin to the binding hot spots of STAT3 SH2. For a comparison, the known inhibitor napabucasin (blue stick-ball) had a similar binding mode and energy (ΔE: −6.1/kcal/mol), binding to the pTyr705 site and partially to the side pocket. Of note, L61H46 binding encompassed the entire side pocket, while napabucasin only covered part of the sub-pocket. A fully covered side pocket binding could favor L61H46’s specificity to inhibit the STAT3 activation and help effectively compete with the native pTyr705 binding to block STAT3 phosphorylation and dimerization.
Drug safety is a vital factor to consider in drug discovery and development. Curcumin, a natural food spice, was reported to have anti-cancer activity. Despite the evidence of its excellent safety, no clinical trials of curcumin have been successful due to its instability and nonbioavailability.16 L61H46 is an optimized MAC. Previous reports showed that the mono-carbonyl structure significantly improved the stability and bioavailability while retaining superior safety.25 We also found that L61H46 has no signs of adverse effects on liver, heart, and kidney cells in vivo (Figure 6G). In the future, it is worth while to further evaluate the pharmacological kinetics and bioavailability of L61H46.
Various chemotherapy drugs including gemcitabine, cisplatin, irinotecan, and taxanes have been used to treat pancreatic cancer.36 Gemcitabine and erlotinib have shown a survival benefit in the first-line setting in metastatic pancreatic cancers. Unfortunately, these anticancer agents have shown limited benefits because of drug resistance.37 Emerging evidence has shown that STAT3 activation is implicated in the treatment failure of targeted therapies or chemotherapy.6 Zhang et al38 reported that certain doses of gemcitabine promoted the binding of the phosphorylated STAT3 to the promoter of Bmi1, Nanog, and Sox2 genes. Furthermore, inhibition of STAT3 partially reversed gemcitabine-induced sphere formation, migration, chemo-resistance, and tumor relapse. These findings suggest that STAT3 could be a potential target to sensitize pancreatic cancer cells to chemotherapy. A clinical trial has been designed to examine the effects of napabucasin paired with paclitaxel on pancreatic cancer patients in comparison to the effects of a placebo paired with paclitaxel.39,40 Bao et al41 found that a natural product andrographolide caused apoptosis via inactivation of STAT3 and potentiated antitumor activity of gemcitabine in pancreatic cancer. In the future, we would investigate the synergistic effects of combination of L61H46 and chemotherapy drugs on pancreatic cancer.
Conclusion
Our data confirm that L61H46 is a selective STAT3 inhibitor and exhibits highly potent antitumor activity against pancreatic cancer cells both in vitro and in vivo (Figure 7). These results suggest that L61H46 could be further developed as a promising STAT3 inhibitor for the treatment of pancreatic cancer.
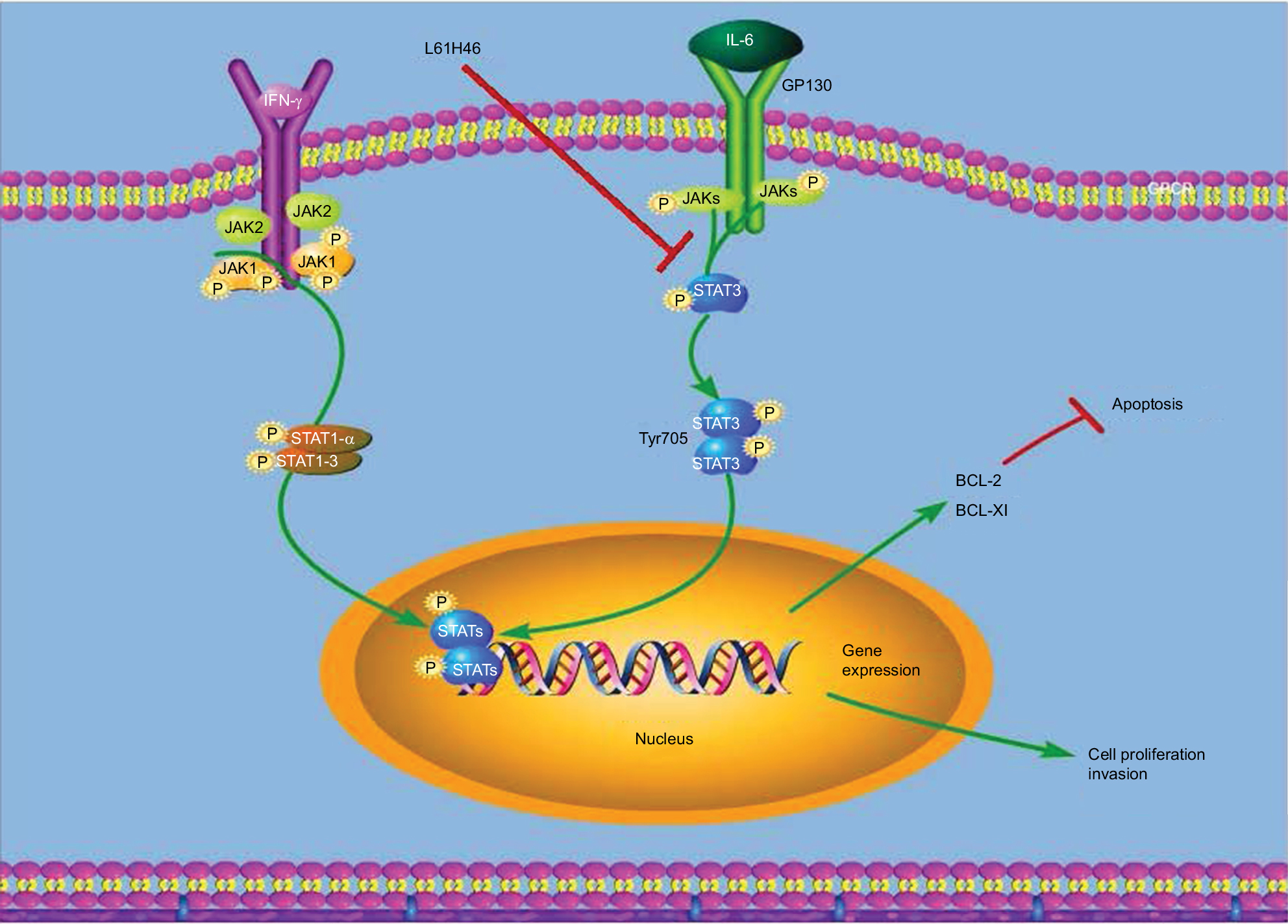 | Figure 7 Schematic illustration of the underlying mechanism of L61H46’s anticancer activity. Abbreviations: IFN, interferon; IL, interleukin. |
Acknowledgments
This research was supported by the Zhejiang Provincial Natural Science Fund (LY18H160047, LY16H160052, and LY17H160055), Medical Scientific Research Fund of Zhejiang Province (2017192276), and Wenzhou Science and Technology Project (Y20170176, Y20170280, and Y20170174). We also thank the Chinese Society of Clinical Oncology (CSCO Y-MX2016-058) and Zhejiang Anticancer Association.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Xing L, Shi Q, Zheng K, et al. Ultrasound-mediated microbubble destruction (UMMD) facilitates the delivery of CA19-9 targeted and paclitaxel loaded mPEG-PLGA-PLL nanoparticles in pancreatic cancer. Theranostics. 2016;6(10):1573–1587. | ||
Mahajan UM, Teller S, Sendler M, et al. Tumour-specific delivery of siRNA-coupled superparamagnetic iron oxide nanoparticles, targeted against PLK1, stops progression of pancreatic cancer. Gut. 2016;65(11):1838–1849. | ||
Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26(2):207–221. | ||
Zhao C, Li H, Lin HJ, Yang S, Lin J, Liang G. Feedback activation of STAT3 as a cancer drug-resistance mechanism. Trends Pharmacol Sci. 2016;37(1):47–61. | ||
Yin F, Hu K, Chen Y, et al. SiRNA delivery with PEGylated graphene oxide nanosheets for combined photothermal and genetherapy for pancreatic cancer. Theranostics. 2017;7(5):1133–1148. | ||
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. | ||
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746. | ||
Herrmann A, Priceman SJ, Swiderski P, et al. CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J Clin Invest. 2015;125(6):2547. | ||
Jung KH, Yoo W, Stevenson HL, et al. Multi-functional effects of a small-molecule STAT3 inhibitor on NASH and HCC in mice. Clin Cancer Res. 2017;23(18):5537–5546. | ||
Sahu N, Chan E, Chu F, et al. Co-targeting of MEK and PDGFR/STAT3 pathways to treat pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2017;16(9):1729–1738. | ||
Long KB, Tooker G, Tooker E, et al. IL-6 receptor blockade enhances chemotherapy efficacy in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2017;16(9):1898–1908. | ||
Zhang Z, Wang F, Du C, et al. BRM/SMARCA2 promotes the proliferation and chemoresistance of pancreatic cancer cells by targeting JAK2/STAT3 signaling. Cancer Lett. 2017;402:213–224. | ||
Huminiecki L, Horbanczuk J, Atanasov AG. The functional genomic studies of curcumin. Semin Cancer Biol. 2017;46:107–118. | ||
Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA. The essential medicinal chemistry of curcumin. J Med Chem. 2017;60(5):1620–1637. | ||
Baker M. Deceptive curcumin offers cautionary tale for chemists. Nature. 2017;541(7636):144–145. | ||
Zhao C, Liu Z, Liang G. Promising curcumin-based drug design: mono-carbonyl analogues of curcumin (MACs). Curr Pharm Des. 2013;19(11):2114–2135. | ||
Lin L, Hutzen B, Zuo M, et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010;70(6):2445–2454. | ||
Wu J, Patmore DM, Jousma E, et al. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene. 2014;33(2):173–180. | ||
Mace TA, Ameen Z, Collins A, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73(10):3007–3018. | ||
Bill MA, Nicholas C, Mace TA, et al. Structurally modified curcumin analogs inhibit STAT3 phosphorylation and promote apoptosis of human renal cell carcinoma and melanoma cell lines. PLoS One. 2012;7(8):e40724. | ||
Cen L, Hutzen B, Ball S, et al. New structural analogues of curcumin exhibit potent growth suppressive activity in human colorectal carcinoma cells. BMC Cancer. 2009;9:99. | ||
Rath KS, Naidu SK, Lata P, et al. HO-3867, a safe STAT3 inhibitor, is selectively cytotoxic to ovarian cancer. Cancer Res. 2014;74(8):2316–2327. | ||
Liang G, Shao L, Wang Y, et al. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg Med Chem. 2009;17(6):2623–2631. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394(6689):145–151. | ||
Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57–61. | ||
Hubbard JM, Grothey A. Napabucasin: an update on the first-in-class cancer stemness inhibitor. Drugs. 2017;77(10):1091–1103. | ||
Agrawal P, Fontanals-Cirera B, Sokolova E, et al. A systems biology approach identifies FUT8 as a driver of melanoma metastasis. Cancer Cell. 2017;31(6):804.e–819.e. | ||
Liu Y, Li PK, Li C, Lin J. Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J Biol Chem. 2010;285(35):27429–27439. | ||
Wu Z, Gabrielson A, Hwang JJ, et al. Phase II study of lapatinib and capecitabine in second-line treatment for metastatic pancreatic cancer. Cancer Chemother Pharmacol. 2015;76(6):1309–1314. | ||
Li H, Liu A, Zhao Z, et al. Fragment-based drug design and drug repositioning using multiple ligand simultaneous docking (MLSD): identifying celecoxib and template compounds as novel inhibitors of signal transducer and activator of transcription 3 (STAT3). J Med Chem. 2011;54(15):5592–5596. | ||
Li H. Multiple Ligand Simultaneous Docking (MLSD) and Its Applications to Fragment Based Drug Design and Drug Repositioning [Electronic Thesis or Dissertation] Ohio State University; 2012. Available from: https://etd.ohiolink.edu/. Accessed February 6, 2018 | ||
Yu W, Li C, Zhang W, et al. Discovery of an orally selective inhibitor of signal transducer and activator of transcription 3 using advanced multiple ligand simultaneous docking. J Med Chem. 2017;60(7):2718–2731. | ||
Vennin C, Chin VT, Warren SC, et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci Transl Med. 2017;9(384): eaai8504. | ||
Zeng H, Qu J, Jin N, et al. Feedback activation of leukemia inhibitory factor receptor limits response to histone deacetylase inhibitors in breast cancer. Cancer Cell. 2016;30(3):459–473. | ||
Zhang Z, Duan Q, Zhao H, et al. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-kappaB/STAT3 signaling cascade. Cancer Lett. 2016;382(1):53–63. | ||
Jonker DJ, Nott L, Yoshino T, et al. A randomized phase III study of napabucasin [BBI608] (NAPA) vs placebo (PBO) in patients (pts) with pretreated advanced colorectal cancer (ACRC): the CCTG/AGITG CO.23 trial. Ann Oncol. 2016;27:454O. | ||
Grothey A, Tebbutt N, Van Cutsem E, et al. CanStem303C trial: a phase III study of BBI-608 (napabucasin) in combination with 5-fluorouracil (5-FU), leucovorin, irinotecan (FOLFIRI) in adult patients with previously treated metastatic colorectal cancer (mCRC). Ann Oncol. 2016;35:27. | ||
Bao GQ, Shen BY, Pan CP, Zhang YJ, Shi MM, Peng CH. Andrographolide causes apoptosis via inactivation of STAT3 and Akt and potentiates antitumor activity of gemcitabine in pancreatic cancer. Toxicol Lett. 2013;222(1):23–35. |
Supplementary materials
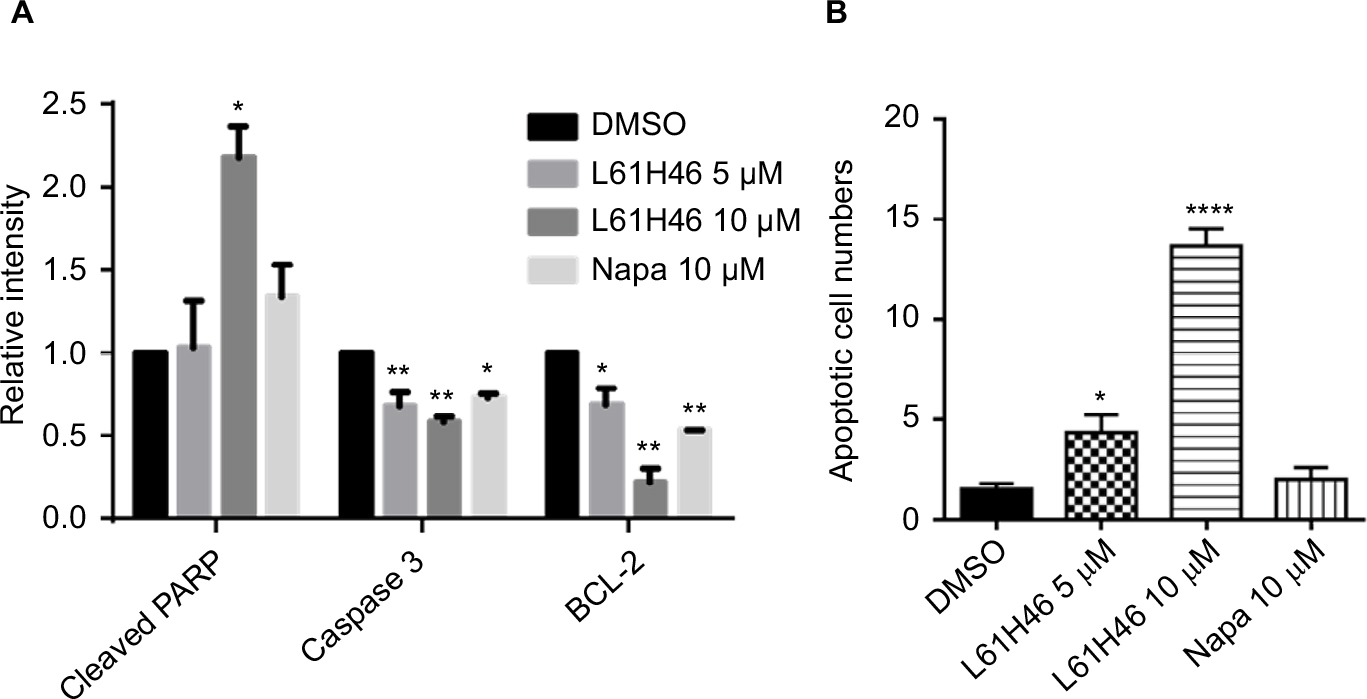 | Figure S1 (A) The relative intensity of various apoptosis-related proteins. (B) The number of apoptotic cells per field in each group was calculated. Notes: Data represent similar results from at least three independent experiments. *P<0.05; **P<0.01; ****P<0.0001. Abbreviations: DMSO, dimethyl sulfoxide; Napa, napabucasin; PARP, poly(ADP-ribose) polymerase. |
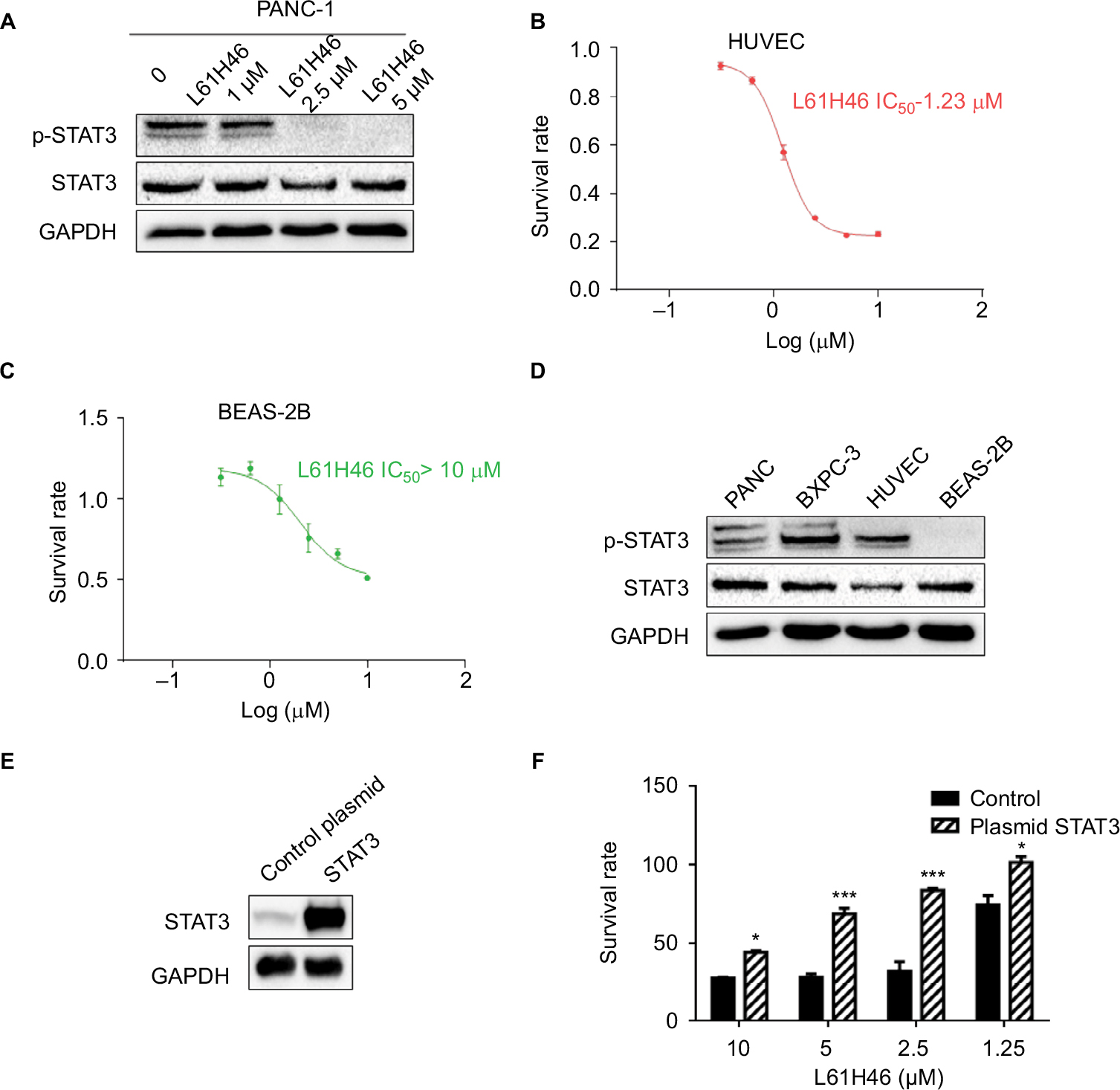 | Figure S2 L61H46 inhibited constitutive phosphorylation of STAT3 in pancreatic cancer cells. Notes: (A) Napabucasin dose dependently (1.0, 2.5, and 5 μM) inhibited STAT3 phosphorylation in PANC-1 human pancreatic cancer cells. (B and C) The effects of L61H46 on the viability of two normal cells. HUVEC and BEAS-2B cells were incubated with increasing doses of L61H46 (0.3–10 μM) for 48 h. Cell viability was determined by MTT assay. (D) The STAT3 phosphorylation levels of two pancreatic cancer cells and two normal cells. (E) Western blotting analysis of stable overexpression of STAT3 protein in PANC-1 cells after STAT3 plasmid transfection (control plasmid = control vehicle vector, STAT3 = STAT3 plasmid transfection). (F) PANC-1 cells with control vehicle vector or STAT3 plasmid transfection were incubated with increasing doses of L61H46 (1.25–10 μM) for 48 h. Cell viability was determined by MTT assay. Data represent similar results from at least three independent experiments. *P<0.05; ***P<0.001. Abbreviations: BEAS-2B, human bronchial epithelial cells; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HUVEC, human umbilical vein endothelial cells; IC50, half-maximal inhibitory concentration; MTT, methylthiazolyldiphenyl-tetrazolium bromide. |
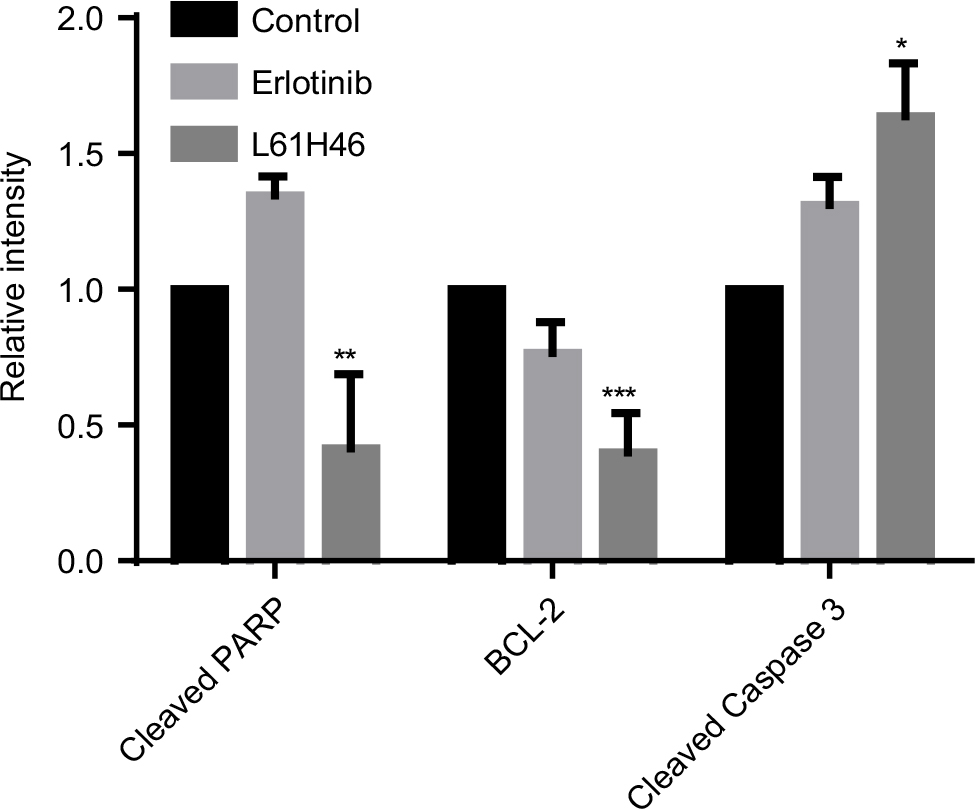 | Figure S3 The relative intensity of various proteins expressed in mouse xenografts in vivo was calculated. Notes: Data represent similar results from at least three independent experiments. *P<0.05; **P<0.01; ***P<0.001.
|
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.