")
Back to Journals » The Application of Clinical Genetics » Volume 14
Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum
Authors Seixas S , Marques PI
Received 11 January 2021
Accepted for publication 24 February 2021
Published 22 March 2021 Volume 2021:14 Pages 173—194
DOI https://doi.org/10.2147/TACG.S257511
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Susana Seixas,1,2 Patricia Isabel Marques1,2
1i3S - Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Porto, Portugal; 2Institute of Molecular Pathology and Immunology of the University of Porto (IPATIMUP), Porto, Portugal
Correspondence: Susana Seixas
i3S, Rua Alfredo Allen, 208, Porto, 4200-135, Portugal
Tel +351 220408806
Email [email protected]
Abstract: Alpha-1-Antitrypsin deficiency (AATD), caused by SERPINA1 mutations, is one of the most prevalent Mendelian disorders among individuals of European descend. However, this condition, which is characterized by reduced serum levels of alpha-1-antitrypsin (AAT) and associated with increased risks of pulmonary emphysema and liver disease in both children and adults, remains frequently underdiagnosed. AATD clinical manifestations are often correlated with two pathogenic variants, the Z allele (p.Glu342Lys) and the S allele (p.Glu264Val), which can be combined in severe ZZ or moderate SZ risk genotypes. Yet, screenings of AATD cases and large sequencing efforts carried out in both control and disease populations are disclosing outstanding numbers of rare SERPINA1 variants (> 500), including many pathogenic and other likely deleterious mutations. Generally speaking, pathogenic variants can be subdivided into either loss- or gain-of-function according to their pathophysiological effects. In AATD, the loss-of-function is correlated with an uncontrolled activity of elastase by its natural inhibitor, the AAT. This phenomenon can result from the absence of circulating AAT (null alleles), poor AAT secretion from hepatocytes (deficiency alleles) or even from a modified inhibitory activity (dysfunctional alleles). On the other hand, the gain-of-function is connected with the formation of AAT polymers and their switching on of cellular stress and inflammatory responses (deficiency alleles). Less frequently, the gain-of-function is related to a modified protease affinity (dysfunctional alleles). Here, we revisit SERPINA1 mutation spectrum, its origins and population history with a greater emphasis on variants fitting the aforementioned processes of AATD pathogenesis. Those were selected based on their clinical significance and wider geographic distribution. Moreover, we also provide some directions for future studies of AATD clinically heterogeneity and comprehensive diagnosis.
Keywords: SERPINA1 variants, Z allele, S allele, MMalton allele and QOurém allele
Introduction
Alpha-1-antitrypsin deficiency (AATD) is an autosomal codominant disorder caused by multiple mutations affecting SERPINA1 gene. Although in most cases these are translated into a functional protein present in the bloodstream at lower concentrations, some mutations can also lead to dysfunctional molecules or null variants.1–3 The major clinical implications of AATD are an increased risk of chronic obstructive pulmonary disease (COPD) and emphysema in adults, and hepatic illness during childhood or later in adult life. Nonetheless, AATD has been associated with disorders affecting not only the respiratory system but also other organs, such as bronchiectasis, anti-neutrophil cytoplasmic antibody (ANCA) – associated vasculitis, and panniculitis.1,4–7 AATD has been typically correlated with European populations, where its prevalence can reach up to 1:2000 subjects.8,9 However, this genetic condition can be virtually detected anywhere due to a worldwide dispersal of two more prevalent deficiency alleles, the S and Z alleles, and a large spectrum of rare pathogenic mutations, which are typically more geographically confined.2,10–13
The AAT Molecule and Pathophysiology of AATD
Alpha-1-Antitrypsin (AAT) is the major protease inhibitor in the serum and the archetypical member of the SERerine Protease INhibitors (SERPIN) superfamily. Therefore, it is often referred to as SERPINA1 – SERPIN clade A member.14,15 AAT is a monomeric glycoprotein that once in the bloodstream weighs approximately 52 KDa and is composed of 394 amino acid residues. However, prior to its secretion, the AAT molecule is post-translationally modified by the removal of its signal peptide, composed of 24 amino acids, and by the linkage of three carbohydrate side chains to asparagine residues located at positions 46, 83 and 247.5,7,16
AAT is primarily synthetized at high levels by hepatocytes, but also to a lower extent by many other tissues, such as whole blood innate immunity cells (neutrophils and monocytes), epithelial lung cells, the kidney and the skin (Figure 1A).4,17–19 At the structural level, AAT is a globular protein packed into three β-sheets (A-C) and nine α-helices (A-I), showing a solvent-exposed reactive center loop (RCL; Figure 1B) that determines its inhibitory affinity towards distinct proteases.3,20 Precisely, it is the Met358-Ser359 bond (P1-P1ʹ; Figure 1B) located within the RCL that by mimicking a protease substrate, works like a specific bite in a kind of molecular mouse-trap.3,20 This remarkable feature of AAT can only be accomplished by the ability of SERPINs to undergo major conformational rearrangements upon RCL cleavage, in which proteases are moved into the opposite pole of the SERPIN molecule. Thereafter, the structure of the protease is disturbed and permanently inactivated in a stable complex with its inhibitor (Figure 1C).3,20 Although AAT is mainly recognized as a potent inhibitor of neutrophil elastase, which is released in the lung during infectious and inflammatory states, it is also capable of regulating other proteases, including proteinase 3, kallikrein 7, matriptase, and caspase-3.4,21 Importantly, AAT is also an acute phase molecule with multiple immunomodulatory and anti-inflammatory functions, such as in the control and release of cytokines, the recruitment of innate immunity cells and in responses to diverse pathogens.4,21,22
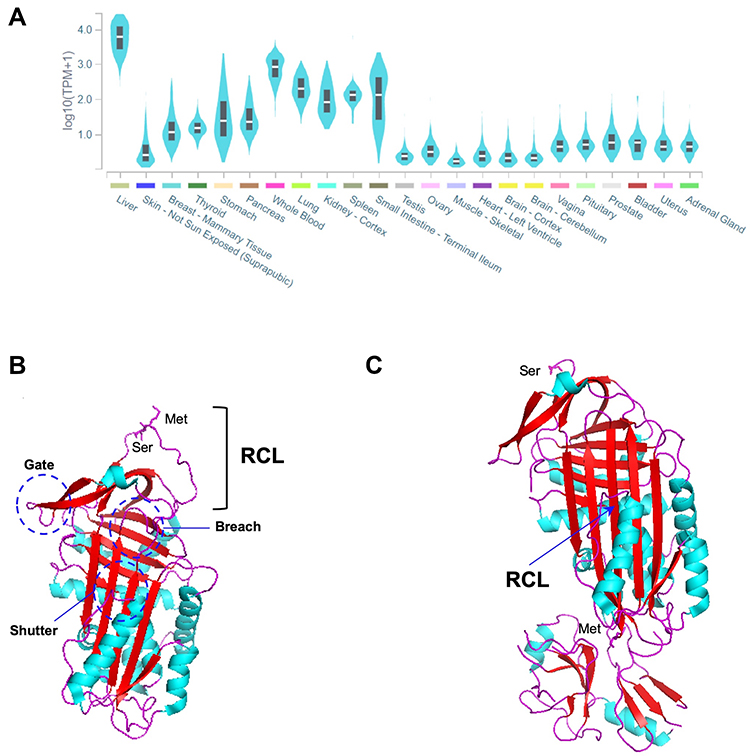 |
Figure 1 (A) SERPINA1 expression across different tissues. The graph was built in GTEx portal (https://gtexportal.org/home/) using the RNAseq data from multiple individuals generated by the Genotype-Tissue Expression (GTEx) project. (B) Three-dimensional structure of an active AAT molecule (PDB code: 1QLP). The breach, shutter and gate domains are highlighted by dark blue circles. The reactive center loop (RCL) and Met-Ser bond (P1-P1ʹ) therein are indicated. (C) Three-dimensional structure of an AAT molecule bond to trypsin in a protease-inhibitor complex (PDB code: 1EXZ). The inserted β-strand of the RCL and the Met358 and Ser359 residues from the cleaved P1-P1ʹ are indicated. α-helixes are shown in light blue, β-strands in red and loops in magenta. The three-dimensional images were generated using PyMOL.112 |
Broadly speaking, the clinical manifestations of the AATD condition are partially attributed to the low levels of AAT and an unbalanced activity of proteases, which can lead to tissue degradation through the cleavage of extracellular matrix proteins as described for pulmonary emphysema.9,19 In general, a serum concentration of 11 uM (57 mg/dl) is considered as the minimum protective threshold against the lung proteolytic attack.1,5 Nevertheless, some authors already questioned its universal application given that some AATD patients with values above the proposed limit may still exhibit respiratory symptoms and benefit from specific clinical interventions.23,24 On the other hand, AATD complications can be also correlated with the AAT conformational flexibility and its propensity to polymerize when misfolded. The toxic effects of AAT polymers inside the endoplasmic reticulum (ER) have long been recognized as a primary cause of the hepatic disease associated with AATD. More recently, the role of AAT polymers in the pathophysiology of pulmonary emphysema was also acknowledged after their detection in the lungs.9,19 The underlying mechanisms of panniculitis and ANCA vasculitis linked to AATD are not yet fully elucidated, but those are probably correlated with both processes of unbalanced proteolysis and polymer accumulation.1,6
The SERPINA1 Gene and Its Variants
AAT is encoded by SERPINA1 (SERPIN family A member 1) gene, which is located at chromosomal region 14q32.13, and spans for about 13.9 kb (GRCh38.p13/hg38 genomic coordinates chr14:94,376,747–94,390,654). SERPINA1 is organized into seven exons of which the first three are untranslated regions (UTR), often labeled as IA, IB and IC, and the remaining four comprise the coding region and the terminal UTR (exons II–V). The 5ʹ UTR contains distinct transcription initiation sites being IA and IB exons reported as alternatively used by monocytes, and IC specifically employed in the gene expression by hepatocytes.6,7,25
So far, more than 120 SERPINA1 mutations have been reported in the literature, of which near 40% are described to cause some type of AATD.2 Despite the vast majority of pathogenic mutations is localized within the coding region (II–V exon) and engages either single nucleotide substitutions or small insertion and deletions (1 to 3 nucleotides), there are also a few reports of AATD causing variants affecting splice sites, as well as large deletions eliminating partially or entirely the sequence of SERPINA1 gene (Table 1).2
 |  |  | 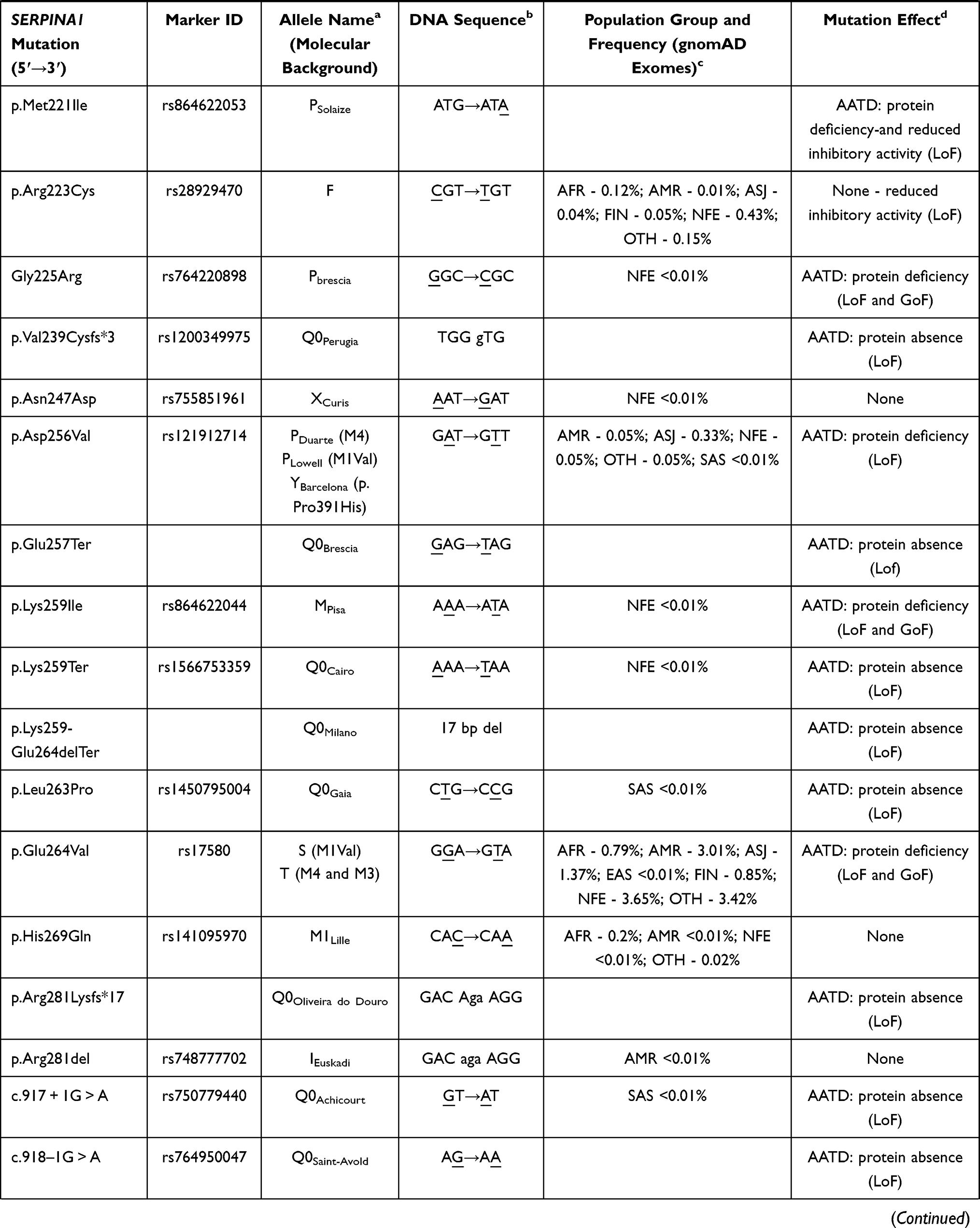 | 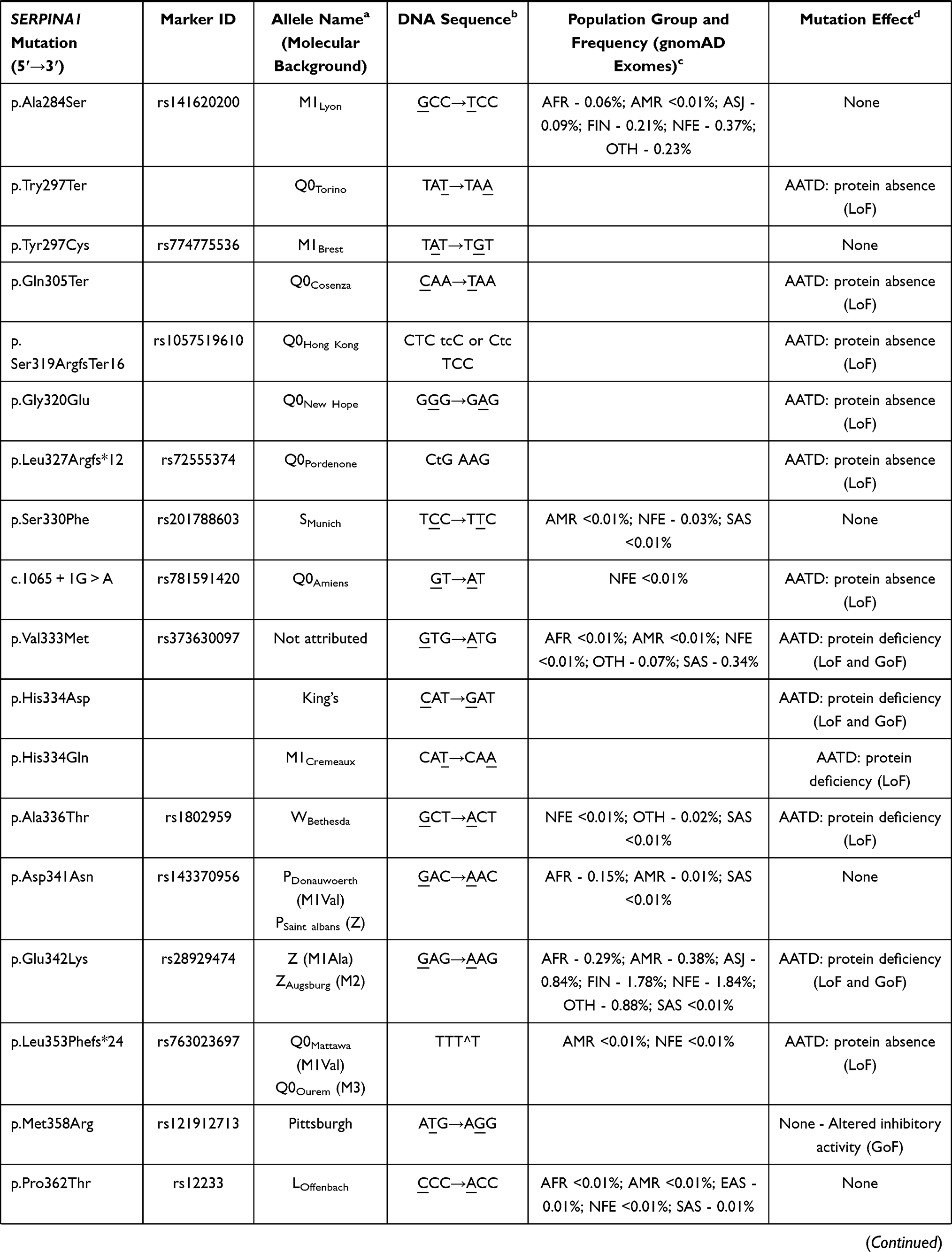 | 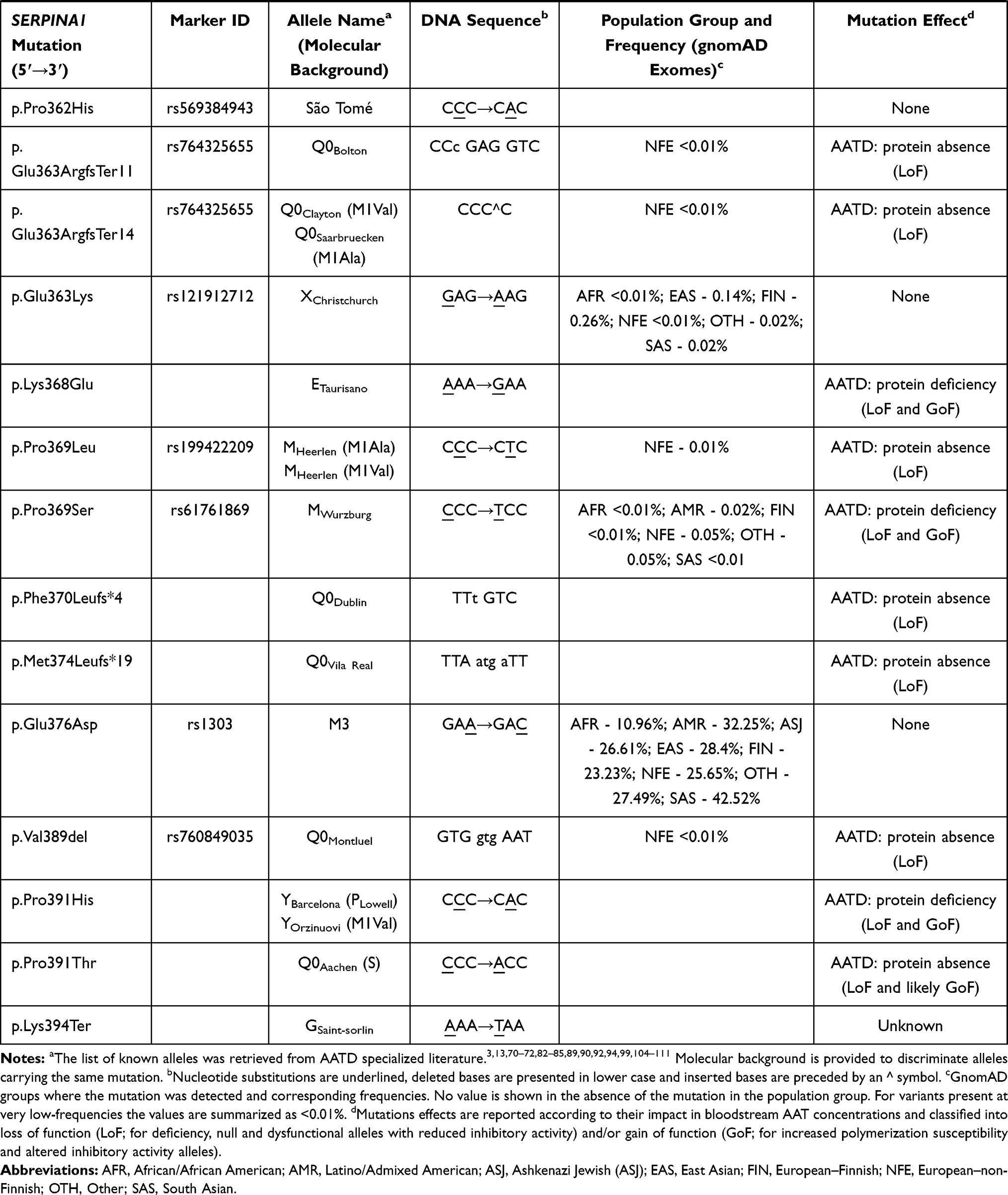 |
Table 1 List of Known SERPINA1 Mutations Defining Different AAT Protein Alleles. Mutations are Listed According to Their Order of Appearance in Gene (Exon IA to Exon V) or Protein Sequence (Amino Acid −24 to 394) |
To date, sequencing efforts are targeting the exome/genome screening of thousands of individuals from ethnically diverse healthy (controls; eg, gnomAD – The Genome Aggregation Database) and diseased populations (eg, TOPMed – The Trans-Omics for Precision Medicine). These are uncovering an unprecedented number of very low-frequency variants (<0.01%), many of them with predicted negative effects in AAT function, if considering distinct bioinformatics pathogenicity scores (Supplementary Table 1).2,12 This indicates that we are only aware of the tip of the iceberg regarding AATD causing mutations. In this respect and as discussed in the following sections, whereas polymorphic variants (>1%) and several low-frequency variants tend to be dispersed worldwide, or at least found across wider geographical regions, other rarer mutations are more likely to be restricted to specific populations. While low-frequency variants will probably be detected in control populations of genomic databases, population-specific pathogenic variants are likely to be absent from those datasets and identified mainly through AATD clinical cases, or among related disease groups such as COPD subjects included in TOPMED. From a clinical standpoint, this highlights the importance of not neglecting SERPINA1 rare variants while performing AATD genetic testing, especially when subjects show symptoms and reduced AAT serum levels.
AATD and AAT Variation as a Classical Polymorphism
AATD was firstly discovered by Laurell and Eriksson more than 50 years ago, in 1963, when they noticed the absence of a protein band corresponding to AAT in the electrophoresis strips of plasma samples from several pulmonary emphysema cases.26,27 Other separating systems with improved band resolution were later developed, and used in multiple studies to assess the genetic polymorphism of blood proteins, including AAT. In general, the alleles of those proteins were labeled as F (fast) and S (slow) according to their electrophoretic mobility in starch, agarose or polyacrylamide gels.28,29 For AAT in particular, this classic nomenclature of protein variants is still maintained nowadays. Precisely, it relies on the band patterns achieved upon an electrophoretic separation in an isoelectric focusing (IEF) gel with a narrow pH gradient (pH 4–5). There, alleles are classified from “A – Z”, being “A” the faster ones (closer to the anode – the positive end of the IEF gel), “M” those found at the middle region, and “Z” the slowest migrating alleles (Figure 2).28,30
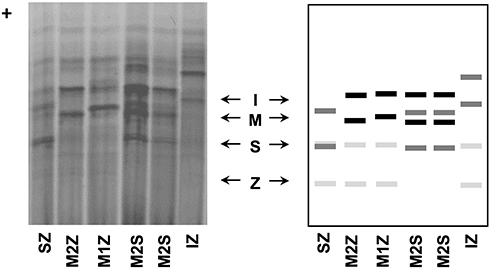 |
Figure 2 AAT phenotypes as obtained by IEF in polyacrylamide gels after Coomassie blue staining (left side) and schematic representation of major diagnostic bands of AAT alleles. |
Another typical feature of this terminology is correlated with the “Q0” usage to denote null alleles: variants in which no band can be detected in IEF gels. Furthermore, this nomenclature can also admit a linked city name to discriminate alleles with similar electrophoretic mobility, but characterized by distinct mutations (eg, MMalton, ZAugsburg or Q0Ourém). This is more often employed in a rare variant classification where the city name corresponds to the birth-place of the oldest carrier with the identified mutation.28 Currently, the most updated genomic databases follow the Human Genome Variation Society (HGVS) guidelines,31 in which the first amino acid is always the initiation methionine. Still, in the AATD field, it is a common rule to employ the residue numbering according to the mature protein (ie, without the first 24 residues of the signal peptide). Thus, for consistency with the former literature, we will use this same labeling of AAT residues, and whenever available we will provide their genomic reference codes.
Common and Rare AAT Variants
Like other genetic variations, AAT alleles can be divided not only according to their association with the AATD condition but also depending on their frequencies across distinct human populations. If using 1% as the threshold for common variants (former assumption of polymorphism), the spectrum of AAT mutations is quite limited, comprising only five variable positions in which just two are linked to AATD. Precisely, these comprise p.Arg101His (rs709932), p.Ala213Val (rs6647), p.Glu264Val (rs17580), p.Glu342Lys (rs28929474) and p.Glu376Asp (rs1303) amino acid replacements that altogether define seven prevalent protein alleles: M1 (Ala or Val), M2, M3, M4, S and Z (Table 2).28,32 Whereas M1 to M4 are all considered normal alleles placed in the central area of IEF gels; S and Z, are both deficiency alleles associated with fainter bands, located closer to the cathode (Figure 2). Here, the Z allele defined by the p.Glu342Lys (rs28929474) mutation, which replaces a negatively charged residue with a positive one, displays the largest separation from the M zone in IEF gels and also the fainter protein bands (Figure 2). On the other hand, the S allele characterized by a p.Glu264Val (rs17580) mutation changes a negatively charged residue by a neutral one, which results in a protein band with intermediate intensity focusing between M and Z zones (Figure 2).
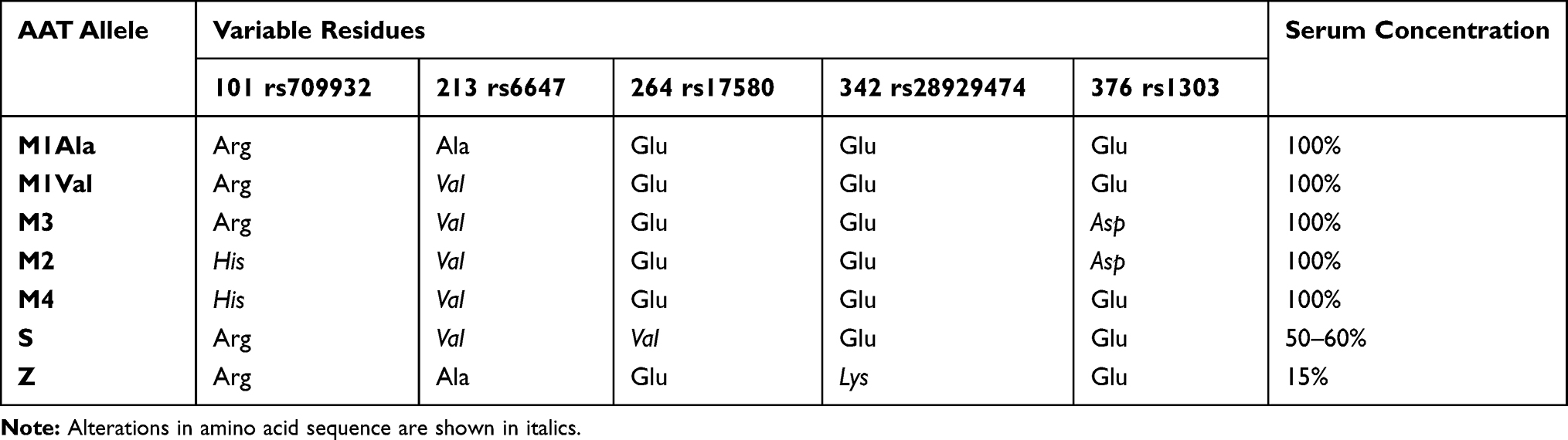 |
Table 2 Common SERPINA1 Variants Defining the Most Prevalent AAT Protein Alleles |
Along with these major protein alleles, there are a large plethora of other variants segregating in human populations. In general, those are found at much lower frequencies (<1%) and thus often referred to as rare AAT alleles, which comprise variants associated with regular AAT activity and serum concentrations, as well as many pathogenic alleles (Table 1).
In overview, common and rare pathogenic variants (or mutations) can be subdivided into two non-mutually exclusively categories according to their negative effects in AAT function and/or activity:
- Deficiency alleles: This category includes all alleles causing a significant reduction in AAT serum levels. Some authors suggested a 20 uM threshold to consider an allele as causing AATD.5 Null alleles (Q0), showing only residual AAT concentrations and no detectable protein band in IEF gels, can also be included in this category. In other words, this group comprises all alleles or mutations that can be connected with a loss-of-function classification.8,9,19
- Dysfunctional alleles: In a strict sense, this category includes only those alleles described to affect the inhibitory activity of AAT, in a qualitative or quantitative manner. Nonetheless, in a broader interpretation of dysfunction and fitting the currently used gain-of-function definition, variants increasing AAT susceptibility to polymerization may also fall within this group.8,9,19
In the following sections, we will revisit some of the collected data about the most relevant pathogenic mutations, by considering not only their clinical importance and functional effects in AAT but also the prevalence among studied AATD cohorts and control populations from the gnomAD database.33
The Z Allele and p.Glu342Lys Mutation
The Z allele is characterized by a p.Glu342Lys (rs28929474) mutation in an M1Ala background allele (Table 2).28,32,34 This replacement of a glutamic acid by a lysine residue at a key region of the SERPINs structure – the breach (upper extremity of the shutter domain; Figure 1B and C; Figure 3) – has been experimentally demonstrated to change the conformational properties of AAT. Briefly, the p.Glu342Lys mutation generates the synthesis of unstable monomers, in which the breach and the shutter are opened. This structural modification predisposes the Z allele to dimerize in a process that can be explained by three alternative mechanisms: 1) the loop-sheet rearrangement, where the RCL alone occupies the shutter of another AAT molecule; 2) the β-hairpin configuration, where the RCL together with a contiguous β-strand forms a β-hairpin structure that is inserted into the core of an acceptor AAT molecule; and 3) the C-terminal swap, where the donor AAT undergoes an RCL self-insertion coupled with an abnormal folding of the C-terminal region, which in turn is incorporated in the following AAT molecule. Indeed, it is the steady state of this dimerization process that when carried in a sequential manner leads to the production of the elongated AAT polymers.9,18,19,35,36 According to a recent study by Faull et al (2020), it is the latter model of the C-terminal swap that best suits the structural information and high-resolution images of ex vivo polymers obtained from several ZZ liver explants. In addition, when compared with the other mechanisms, the C-terminal swap also predicates for an increased stability and irreversibility of Z allele conformational changes.35
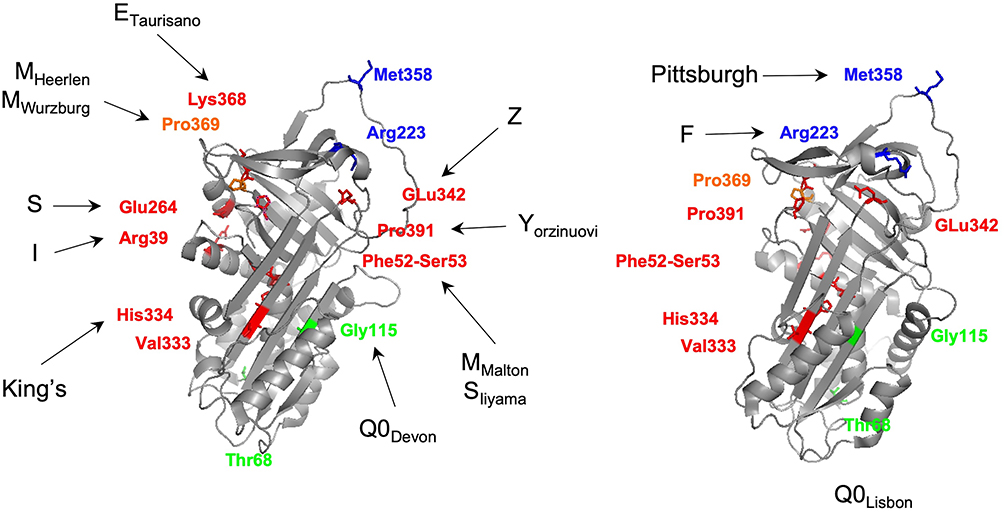 |
Figure 3 Three-dimensional structure of an active AAT molecule (PDB code: 1QLP) displaying the mutated residues in selected pathogenic AAT alleles. Amino acids affected in variants with increased polymerization susceptibility are shown in red, in dysfunctional alleles are highlighted in blue, in null alleles are displayed in green and combining polymerogenic and null alleles are presented in orange. The three-dimensional images were generated using PyMOL.112 |
Notably, although Z polymers are described to aggregate primarily in the ER of hepatocytes, they are suggested to also accumulate in other cell types, such as pneumocytes.8,9,19,37 Furthermore, Z polymers were found extracellular as well, in the lung interstitium where these are supposed to have a pro-inflammatory activity with repercussions in the progression of emphysema.9,18,19
This increased susceptibility of the Z allele to polymerize underlies the dual feature of p.Glu342Lys as a loss- and gain-of-function mutation. If, on one hand, it reduces the secretion of AAT into the bloodstream by 85% thus, enhancing the risk of pulmonary disease due to the unbalanced activity of proteases (loss-of-function); on the other hand, the formation of AAT granules that escape the intracellular pathways of protein degradation induces a stress response that may culminate in cell death and fibrosis (gain-of-function).8,9,19,37 Notably, this represents a critical step not only in liver disease among ZZ individuals9,38 but also in alveolar cell loss in emphysema, where extracellular polymers found in the interstitium were correlated with another gain-of-function effect that amplifies the damage to lung parenchyma.19 It is through a chemotactic action that the extracellular polymers stimulate the recruitment and degranulation of neutrophils in the alveolar membranes, maximizing proteolytic activity and spreading the focus of inflammation in the lower lung.8,9,19,37 On top of this major mechanism of AATD pathophysiology, the p.Glu342Lys mutation per se is also known to reduce the inhibitory capacity of the AAT molecule further enhancing its loss-of-function effects.3,39
From an epidemiological perspective, the Z allele is more prevalent in Scandinavia and Baltic countries, where its frequency is estimated to vary between 2% and 4%. Accordingly, the proportion of severe AATD cases caused by the ZZ genotype is also relatively high considering their population sizes – about 2000 to 4500 individuals.10,11,40 The elevated prevalence of the Z allele in Northern Europe, together with its wide distribution across other regions, namely in Southern Europe and Mediterranean areas, is on the genesis of a Viking migration (800–1100 A.D) theory for its dispersal.28,41 Additionally, hypotheses for a selective advantage of the Z allele were also advanced. For instance, taking into account that Z polymers magnify the inflammatory response of an individual, it could thus have been beneficial in fighting pathogens during a pre-antibiotic era.41 Another scenario was a possible increase in the reproductive success of ZZ subjects. Although an AAT function in the regulation of acrosin proteolytic activity and sperm-egg fertilization might appear as a valid argument for an improved fitness of the Z allele, this selective hypothesis was later rejected by family-based transmission studies.
More recently, the Z allele was proposed to have been positively selected by height and/or weight adaptive traits to colder climates or conversely, to represent an example of a balanced polymorphism correlated with better lung function values (FEV1: Forced Expiratory Volume in 1 s and FVC: Forced Vital Capacity) in heterozygotes.42 Yet, the evidence of extended Z haplotypes collected by North et al 2016, as well as the former arguments for a selective advantage of the p.Glu342Lys, may be questioned by similar findings obtained for other genetics variants. For example, the 32 bp deletion in C-C chemokine receptor 5 gene (CCR5-del32) that confers an effective resistance to HIV infection and is present in North-Europeans at a 16% frequency was proposed to have been selected during the Middle ages, due to a protective effect against the bubonic plague and/or smallpox.43,44 Nevertheless, CCR5-del32 long-range haplotypes did not differ from other variants found in the same frequency bins, suggesting a likely neutral evolution for this variant.45–47
Interestingly, the age estimates of CCR5-del32 mutation fall within a 3000–5000 yrs. old range,45–47 which closely overlaps with the origins of Z allele calculated to be around 2000 to 6000 yrs. ago.2,34,48,49 For the Z allele, this age variability has been correlated with a large haplotype diversity described in Southern European regions compared to Northern ones.49 Indeed, this south-north cline of genetic diversity is not restricted to SERPINA1 or CCR5 mutations, but rather a global European trend as recently shown by genome-wide data. To date, it is well accepted that the genetic variability within Europe mirrors the local geography, and the observed gradients are likely to result from several population movements towards the north, like the ones associated with the expansion of agriculturalists during Neolithic times.50 Furthermore, the larger sustained population sizes in Southern European regions, because of their improved long-term survival conditions, are likely to have contributed as well to their larger genetic diversity.50 In summary, the updated knowledge of human genetic variation discards, not only a Scandinavian origin of the Z allele but also any linked benefit to its carries. Taking into account the limited mortality of ZZ subjects in their early life, and that major clinical complications occur after post-reproductive ages (>40 yrs. old), often connected with cigarette smoking, AATD is likely to represent a modern challenge to human health.
The S Allele and the p.Glu264Val Mutation
The S allele is defined by a p.Glu264Val (rs17580) mutation in an M1Val background allele (Table 2).16,28,32 The pathogenic consequences of this allele result from the loss of a hydrogen bond linking the glutamic acid to a tyrosine residue located in the opposed 38 position (Figure 3). This alteration was found to disturb the shutter domain in its periphery affecting the stability of the AAT molecule. An increased turnover of the S protein was initially advanced as the source for its reduced secretion at 50–60% of the normal level.51 However, the p.Glu264Val mutation has been shown to turn AAT susceptible to polymerization, and to cause its retention in the ER, although at a lesser extent than the Z allele.51–53 This finding provides an explanation for the S allele being less frequently associated with detectable granules in hepatocytes, a condition that may be shifted in SZ subjects, given that the two variants are capable of forming heteropolymers.51,53,54 According to recent co-expression assays of Z and S alleles, the first molecules slow down and reduce the intracellular degradation of the misfolded S ones without compromising their secretion.53,54 Indeed, this phenomenon of heteropolymerization affords a better understanding of the incidence of hepatic disease among SZ subjects, particularly when there is a second hit caused by other risk factors, such as infection, fat liver or alcoholism.53,55
Oddly, the risk of lung disease among SZ individuals is not yet fully elucidated. This results from some contradictory studies concerning the association of the SZ genotype with pulmonary emphysema and COPD, but also from the small numbers of individuals analyzed so far. Notwithstanding, in smokers, this same genotype seems to increase the risk of COPD, although the disease itself does not differ from what is observed in non-AATD cases.55–59 On the other hand, the proportion of SZ patients showing serum levels below the protective threshold (11 uM) is limited to about 16%23,60 and to our knowledge, there are no data concerning the heteropolymerization in the lung, either intra- or extracellularly. At this point, it may be difficult to accurately address the loss- and gain-of-function effects of p.Glu264Val mutation in COPD and emphysema development, and if a second “hit” is required in SZ subjects similar to what happens in hepatic disease. Importantly, this genotype is several times more prevalent worldwide than the severe risk ZZ genotype, especially within Europe where numbers are estimated to vary within the following ranges: 600,000–700,000 for SZ and 80,000–125,000 for ZZ individuals.10,40,61 Considering that AATD persists as an underdiagnosed condition, SZ subjects could still represent a vulnerable group to respiratory illnesses.
The S allele is more common in Iberia, where frequencies as high as 15% were described in both North and West Iberian regions.49,62 Previous works also report an increased prevalence of the S allele in Madeira island (Portugal) with an 18% frequency, and in a few African countries, like Angola and Namibia with a prevalence of 18.8% and 14.6%, respectively.10,11,63 Still, those reports are likely to be correlated with the past demography of the studied groups and their recognized Iberian heritage.64,65 In this respect, the most updated and unbiased surveys of human genetic variation carried out in African and African-American populations show only minor frequencies of the S allele (<1%; Table 1).
Similar to the Z allele, the p.Glu264Val mutation was proposed to have arisen in the region where the S allele is more common to date – in the Iberian Peninsula. In this case, the 8000–14,000 yrs. age estimates place the origin of the S allele in the Late Paleolithic after the last glacial maximum, in a period when modern humans are thought to have left their refugees in Southern Mediterranean regions.49,50 Later, Neolithic migrations might have facilitated a first dispersal of the S allele throughout Eurasia and North Africa. More recently, the Iberian diaspora initiated in the XV century with the maritime expansion could have contributed to the spread of the S allele in sub-Saharan Africa and in Central and South America.50,64,66
The MMalton, MPalermo, MNichinan, Q0LaPalma and SIiyama Alleles
The five variants MMalton, MPalermo, MNichinan, Q0LaPalma and SIiyama are connected by their alterations of the contiguous 51–53 residues, where the Phe-Phe-Ser sequence is fundamental to maintain a stable conformation of the shutter domain (Figure 3; Table 1).67–72 The deletion of a phenylalanine p.Phe52del (rs775982338) found in MMalton, MPalermo or MNichinan and the replacement of serine by a phenylalanine p.Ser53Phe (rs55819880) as observed in SIiyama, cause in a similar way to the Z allele the opening of the breach/shutter, enhancing the susceptibility of these variants to polymerize. However, whereas the SIiyama allele is an extremely rare variant identified in a few families from Japan,67,68,73 the other alleles characterized by the p.Phe52del are widely distributed across different human groups as indicated by the genomics studies of large control populations, where this mutation was found at low or very frequencies (<0.01–0.04%) (Table 1). The discrimination of MMalton, MPalermo or MNichinan alleles is only achieved by the analysis of their molecular background. The MMalton has a p.Phe52del mutation linked to an M2 allele and the MPalermo to an M1Val instead.69–71 The MNichinan allele, on the other hand, has a p.Phe52del in an M1Val background together with another rare mutation that does not lead to AATD, the p.Gly148Arg (V allele – rs112030253).68 Finally, in Q0LaPalma allele, the p.Phe52del is in phase with the p.Glu264Val mutation, which typically defines the S variant. Interestingly, this combination of pathogenic mutations appears to not only impair AAT secretion, as well as its polymerization. No evidence of hepatic dysfunction was detected in Q0LaPalma carriers.72
Whereas MMalton and MPalermo alleles have been identified in several AATD cases across different European and North African countries,13,74,75 the MNichinan was only reported in AATD subjects from Japan.73 So far, only a few studies performed a clinical evaluation of MMalton carriers, homozygous and heterozygous, confirming the association of p.Phe52del with both pulmonary and hepatic diseases.76–78 Interestingly, one of those studies reports a case of end-stage liver disease in the presence of a non-deficiency allele, in an M3MMalton genotype, suggesting a high pathogenicity and heteropolymerization potential of p.Phe52del variant.53,79 According to a recent work, MMalton granules found in the liver of AATD patients contain calcium and are mineralized. These modifications were hypothesized to upregulate ER chaperones fostering cell necrosis in a distinct molecular process from one of the Z and SIiyama alleles.80
The haplotype analysis of MMalton and MPalermo alleles disclosed their independent origin, indicating that the p.Phe52del mutation occurred at least twice.2 In addition, such data were used to estimate their time to the most common recent ancestor, which in both instances is close to 5000 years old.2 This date is consistent with a European and Mediterranean distribution, as well as, a spread associated with Neolithic migrations.50 Although the molecular base linked to p.Phe52del is rarely acknowledged, thus not allowing the discrimination of MMalton and MPalermo alleles, their association with an M2 or M1Val is not expected to further modify protein structure or disease risk.
Other Rare Variants Leading to AAT Polymerization
There are at least 10 other variants with polymerogenic potential, if taking into account their association with hepatic disease and/or evidence from experimental studies.12,18 Among those only King’s (p.His334Asp),81 ETaurisano (p.Lys368Glu)82 and YOrzinuovi (p.Pro391His)83 are rare variants absent from public databases (Table 1). Conversely, the I (p.Arg39Cys; rs28931570)71 and MWurzburg (p.Pro369Ser; rs61761869)84 alleles are the most prevalent of those low-frequency variants within Europe (0.2% - 0.05%, respectively); the p.Val333Met (rs373630097)12 is another mutation observed mainly in South Asians at 0.3% (Table 1). Importantly, to the best of our knowledge, only the I and MWurzburg alleles have their polymerogenic properties carefully and experimentally validated.53,54 Whereas the I variant is a functional equivalent of the S allele (the residue 39 faces the 264 one, Figure 3) interfering as well in the periphery of the shutter domain, the MWurzburg is closely located to the C-terminal region of the AAT molecule, and described to affect the stability of another key SERPIN domain – the gate.2,54,83 Moreover, the amino acid replacements of I and MWurzburg may also contribute to their ER accumulation by increasing AAT hydrophilicity, although at a lesser extent than Z or MMalton, given that their association with hepatic disease is dependent on a stronger polymerogenic allele.53,54,83 Although the low prevalence of these alleles may prevent an accurate evaluation of their impact in AATD manifestations, their detection among symptomatic cases supports the hypothesis of some associated risk of liver and lung diseases.
Dysfunctional AAT Alleles
The best example of a dysfunctional allele is illustrated by the Pittsburgh, which is characterized by a p.Met358Arg substitution (rs121912713), altering the P1 residue of the reactive bond located within the RCL (Figure 1B and Figure 3).85 This change causes the AAT molecule to shift its protease affinity towards thrombin instead of its regular anti-elastase inhibitory activity. Interestingly, the clinical outcomes associated with the Pittsburgh variant are not connected with the loss of AAT function, but rather with an abnormal control of coagulation cascades. Indeed, this mutation was first described in a young boy who died of a fatal bleeding disorder, in which the AAT properties as an acute-phase reactant after trauma were proposed to increase its new anti-coagulation potential, consequently causing the patient severe hemorrhages.85 This variant is extremely rare and it has been reported in few isolated cases and in a single family, where it always arose as a de novo mutation.86 Unsurprisingly, this variant is also absent from control populations currently available through large genomic databases (Table 1).
The F allele is another well-known example of a dysfunctional variant. In this case, if compared with a normal M allele, the F variant displays a considerably reduced inhibitory activity towards neutrophil elastase but also proteinase 3.39,87 This functional impaired variant results from a p.Arg223Cys (rs28929470) substitution that in spite of being located outside of the RCL is believed to alter the architecture of P1-P1ʹ region.39,88 This allele has been proposed to increase the risk of emphysema; however, this may only apply to FZ heterozygous, given that no evidence of elastase-induced damage was detected in an FF homozygous patient.39 Importantly, the F allele is a relatively uncommon variant that is found mainly in Europeans, reaching its highest frequency in Sweden (0.9% in gnomAD). Considering that there is also some heterogeneity in the clinical manifestations of AATD cases, the study of additional patients would be fundamental to clarify whether FF subjects are susceptible to lung disease, or not. Still, in no circumstance, the respiratory manifestations would be a consequence of the lower AAT concentrations given that the secretion of F allele is within normal ranges.39,88 There are recent reports of additional dysfunctional variants but their rarity has prevented so far, a detailed evaluation of their association with AATD disease.89
Null Alleles
In most instances, null alleles (Q0) are caused by the premature termination of protein sequence, due to either a nonsense mutation, or the introduction of small insertions and deletions altering the open reading frame. Nonetheless, in a few instances, AAT null alleles can result from standard amino acid substitutions where a misfolded protein is synthetized and rapidly degraded by the ER-associated machinery.2,90 The MHeerlen (p.Pro369Leu; rs199422209) and Q0Lisbon (p.Thr68Ile; rs1490133295) alleles are two such examples, in which a full protein is produced but not detected intracellularly as granules and only poorly secreted from cells, thus being nearly absent from the serum.71,84,91 In addition, these are likely to be the most prevalent of the null alleles, in populations of European descent and native American admixed (approximately 0.01% and 0.02%, respectively; Table 1), as suggested also by studies of AATD patients.2,13,74,92 In South Asians, a similar situation occurs with the p.Gly115Ser (rs11558261) replacement, which defines the Q0Devon and Q0Newport alleles (0.07%, Table 1).
Splice site changes are another source of null alleles. Interestingly, some of the few cases reported in the literature engage exon IC donor site (Table 1) and an impairment of SERPINA1 expression restricted to hepatocytes.2,93,94 Given that other cell types use upstream transcription initiation sites located in exons IA and IB, some AAT will be synthetized, but those molecules are far from reestablishing the normal serum levels secreted by the liver. The Q0Porto (rs775786225), Q0Faro and Q0Madrid alleles belong to this category of splice site mutations and were proposed to produce abnormal transcripts targeted by mechanisms of nonsense-mediated mRNA decay (NMD).2,93,94 The same NMD phenomenon is likely to also be involved in the processing of most truncated variants preventing the accumulation of unusually long or short transcripts within the cells.2,90,95
Independently of the underlying molecular mechanisms of null alleles, those are by default clinically connected with the loss-of-function effects, and subsequently, with AATD pulmonary manifestations. Here, it is important to strengthen that they will be strictly correlated with the unopposed activity of neutrophil elastase and other proteases. Once there is no formation of AAT polymers, the progression of pulmonary emphysema cannot be exacerbated by a polymer amplified pro-inflammatory response.
The Example of Q0Ourém, Q0Mattawa and the p.Leu353Phefs*24 Mutation
The Q0Ourém, which is characterized by an insertion of a thymine at 353 codon (p.Leu353Phefs*24; rs763023697) in an M3 background allele, is perhaps one of the most prevalent truncated variants of SERPINA1 (Table 1).93,96 Although it has been identified in only three individuals from control populations (2 Europeans and 1 Latino American), in the Iberian Peninsula it has been associated with several unrelated cases of severe AATD, including several homozygous patients.74,96–98 The haplotype analysis of Q0Ourém alleles identified in Portuguese patients confirmed not only a single origin of p.Leu353Phefs*24 mutation but also allowed to estimate its age in 650 yrs.96 Remarkably, this timeframe concurs with the major settlements of Ourém village, in central Portugal, which took place during the XIV-XV centuries. Considering that most cases cluster in Ourém and other neighboring areas, a founder effect offers a plausible explanation for its spread in Portugal.96
Notably, the p.Leu353Phefs*24 mutation was originally identified in an M1Val background and named as Q0Mattawa according to the birthplace of the index case, in Ontario, Canada.99 This suggested that the p.Leu353Phefs*24 mutation could have occurred at least twice. However, there is recent evidence for the segregation of Q0Ourém allele in two unrelated subjects from Quebec city population (Quebec, Canada).97 Although an independent origin of the two alleles may now be questionable considering the proximity of Mattawa and Quebec cities (700 km) in Canada, the hypothesis may only be ruled out by an updated characterization of Q0Mattawa haplotypes.
A Comprehensive Analysis of AATD Causing Mutations
The mutational spectrum of SERPINA1 was proposed to be composed of a limited repertoire of genetic variants, in which specific sites were more prone to accumulate events of homoplasy.2,100 Although this conjecture may still hold true for some pathogenic variants (eg, p.Phe52del, p.Glu264Val and p.Glu342Lys; Table 1), the improvements achieved over the last years in AATD testing, as well as the efforts made in the exome/genome sequencing of thousands of individuals, uncovered unprecedented numbers of SERPINA1 variants present in different populations at low and very low frequencies (<1% and <0.01%).2,12 In the absence of clinical or experimental data addressing the functional impact of those variants, there are currently multiple predictive scores of variant pathogenicity that offer an alternative bioinformatics approach to evaluate disease-causing mutations.101 In the context of AATD, a recent study has already analyzed the accuracy of those predictive scores to infer the pathogenicity of known and novel AAT mutations.12 The authors confirmed not only the scores feasibility in the classification of recognized AATD causing variants, as well as, in the detection of new ones later validated by functional assays.12 Currently, the compiling of variants identified mainly through the screening of AATD patients (Table 1) with those reported in the gnomAD database33 linked to their pathogenicity scores for a series of predictive algorithms (Supplementary Table 1), shows that SERPINA1 has a vast number of mutations spreading throughout its coding region and splice sites, many of them with expected negative consequences. In overview, SERPINA1 variants exceed the 500, in which more than a third likely have implications for AATD according to clinical data and/or computational predictive tools.2,12,89
Still and as mentioned above, there are some variants fitting the concept of homoplasy,2,100 for which the existence of haplotype data is fundamental to address their underlying molecular mechanism. The MMalton and MPalermo (p.Phe52del in M2 and M1Val, respectively) alleles occurring in a short repeat sequence (Table 1), are one of the few examples with sustained evidence of mutational recurrence.2 The T allele is another known case of homoplasy (p.Glu264Val in an M471 or M3 background2 instead of the common M1Val) possibly resulting from either crossover or mutation recurrence events.2 The ZAugsburg (p.Glu342Lys in M2 instead of M1Ala), the MHeerlen (p.Pro369Leu in M1Ala or M1Val), the MNichinan (p.Phe52del in a V allele) among other alleles (Table 1) represent additional examples, in which the molecular events behind their origin were not yet elucidated.2,68,91,102
Although the AATD condition is excepted to be mainly caused by pathogenic mutations, we cannot rule out the influence of their extended genomic background in symptoms variability and clinical outcomes. In this respect, it was demonstrated that the p.Ala213Val variant modifies AAT interactions with neutrophil elastase and lipoproteins,103 and on the other hand, several cis-quantitative trait loci (QTL) were shown to affect SERPINA1 expression (eQTL) and splicing (sQTL; Figure 4).
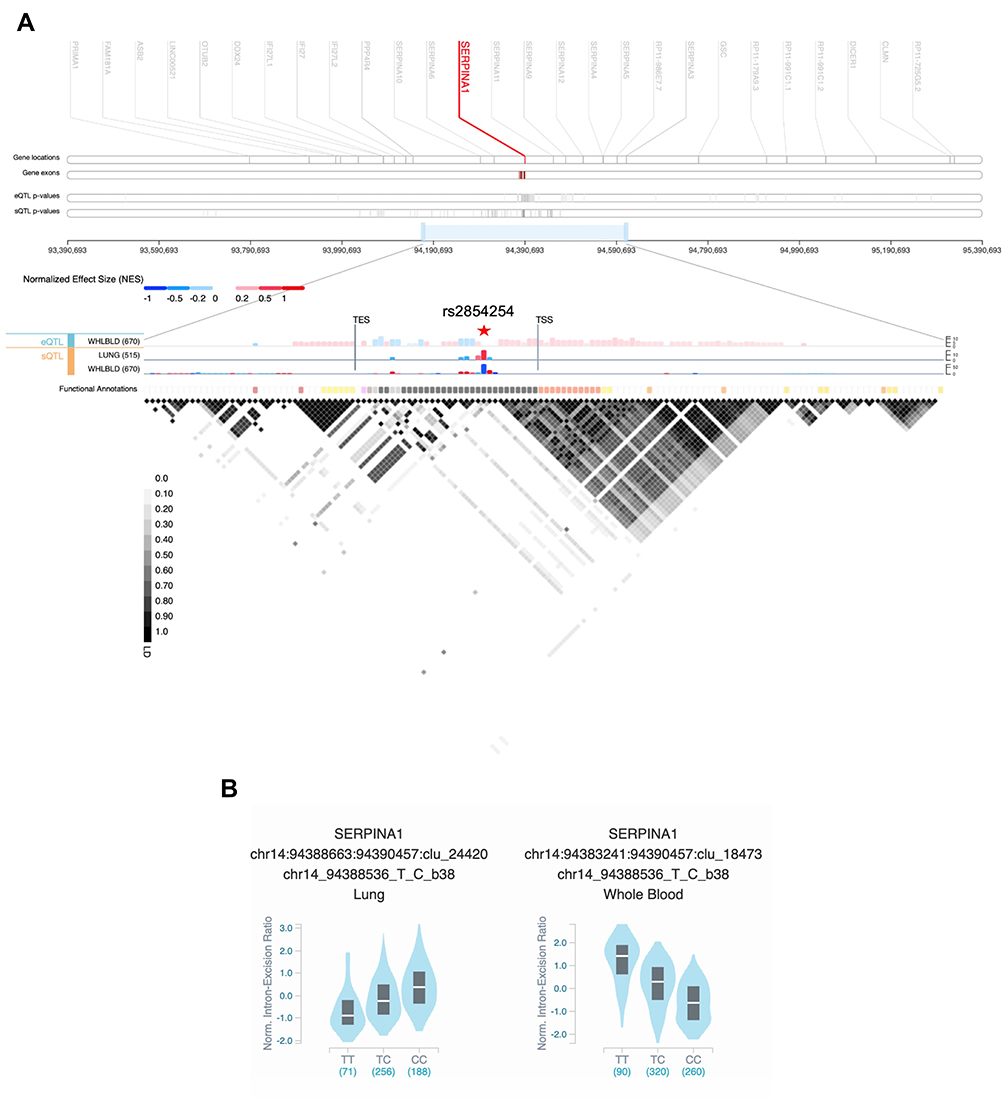 |
Figure 4 (A) SERPINA1 cis-quantitative trait loci (QTL) view in Genotype-Tissue Expression (GTEx) Locus Browser (https://gtexportal.org/home/). The top section shows the genomic region 1Mb up- and downstream of SERPINA1. In the middle, expression QTLs (eQTL) and splicing QTLs (sQTL) of SERPINA1 are portrayed as bar charts in lung and whole blood with the –log10(p-value) represented in the y-axis. The liver tissue was not available for QTLs analysis. The variant with the most significant p-value – rs2854254 – is highlighted by a red star symbol. The last row shows the functional annotation of the variants according to Ensembl’s Variant Predictor (VEP), Loss-Of-Function Transcript Effect (LOFTEE) and Ensembl Regulatory Build. At the bottom, the plot depicts the pairwise linkage disequilibrium (LD) values (R2) of the QTL variants. TSS, transcription start site; TES, transcription end site. (B) Violin plots of allele-specific cis-sQTL effects of rs2854254 genotypes on SERPINA1 in human lung and whole blood (GTEx database – GTEx Analysis Release v8). |
Conclusion
The repertoire of AATD causing mutations has expanded over the last years, and to date, it is acknowledged that along with the major disease risk genotypes, ZZ and SZ, there are many others resulting from different combinations with rare alleles. Indeed, previous studies have shown that rare mutations can be detected in up to 17% of clinically cases74,75 and like for Z and S alleles, several low-frequency pathogenic variants have been segregating in human populations for hundreds or thousands of years. Therefore, their geographical dispersal could have been facilitated by past human migrations, as well as, by the occurrence of some founders’ effects. Besides these, there are other even rarer variants, which probably arisen recently in time, and thus, are familial or regional confined. Altogether, this data highlights the importance of AATD genetic diagnosis to contemplate more inclusive screenings of SERPINA1 variants, and not only p.Glu264Val and p.Glu342Lys mutations. This goal may be achieved by the traditional AATD laboratorial algorithms combining serum concentration, phenotyping, genotyping and SERPINA1 sequencing, or conversely by the implementation of novel state-of-the-art genotyping/sequencing approaches.
Moreover, it is fundamental to continue unravelling the mechanisms of AATD pathophysiology, for both common and rare pathogenic variants and to define their effects as loss- and/or gain-of-function mutations. Namely, it is imperative to discriminate processes and variants purely associated with an unopposed proteolytic activity, from those stimulating pathways of ER stress and inflammation related to homo- and hetero-polymerization of misfolded alleles. Finally, in a personalized medicine era, it is urgent to solve controversies regarding the disease risk associated with different genotypes (or groups of genotypes for rare variants), and to address the impact in the AATD clinical outcomes of different genetic/genomic modifiers and other factors to which an individual might be exposed (eg smoking, alcoholism, infection, air pollution, obesity).
Acknowledgments
IPATIMUP integrates the i3S Research Unit, which is partially supported by the Portuguese Foundation for Science and Technology (FCT). PIM is supported by the FCT post-doctoral fellowship (SFRH/BPD/120777/2016), financed from the Portuguese State Budget of the Ministry for Science, Technology and High Education and from the European Social Fund, available through the Programa Operacional do Capital Humano.
Disclosure
The authors have no competing interests to declare.
References
1. Lopes AP, Mineiro MA, Costa F, et al. Portuguese consensus document for the management of alpha-1-antitrypsin deficiency. Pulmonology. 2018;24(Suppl 1):1–21. doi:10.1016/j.pulmoe.2018.09.004
2. Silva D, Oliveira MJ, Guimarães M, Lima R, Gomes S, Seixas S. Alpha-1-antitrypsin (SERPINA1) mutation spectrum: three novel variants and haplotype characterization of rare deficiency alleles identified in Portugal. Respir Med. 2016;116:8–18. doi:10.1016/j.rmed.2016.05.002
3. Stein PE, Carrell RW. What do dysfunctional serpins tell us about molecular mobility and disease? Nat Struct Biol. 1995;2(2):96–113. doi:10.1038/nsb0295-96
4. Janciauskiene SM, Bals R, Koczulla R, Vogelmeier C, Kohnlein T, Welte T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir Med. 2011;105(8):1129–1139. doi:10.1016/j.rmed.2011.02.002
5. ATS/ERS. American thoracic society/European respiratory society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. doi:10.1164/rccm.168.7.818.
6. Greene CM, Marciniak SJ, Teckman J, et al. Alpha1-antitrypsin deficiency. Nat Rev Dis Primers. 2016;2(1):16051. doi:10.1038/nrdp.2016.51
7. WHO. Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397–415.
8. Ghouse R, Chu A, Wang Y, Perlmutter DH. Mysteries of 1-antitrypsin deficiency: emerging therapeutic strategies for a challenging disease. Dis Model Mech. 2014;7(4):411–419. doi:10.1242/dmm.014092
9. Lomas DA, Hurst JR, Gooptu B. Update on alpha-1 antitrypsin deficiency: new therapies. J Hepatol. 2016;65(2):413–424. doi:10.1016/j.jhep.2016.03.010
10. de Serres FJ, Blanco I. Prevalence of alpha1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review. Ther Adv Respir Dis. 2012;6(5):277–295. doi:10.1177/1753465812457113
11. Blanco I, de Serres FJ, Carcaba V, Lara B, Fernandez-Bustillo E. Alpha-1 antitrypsin deficiency PI*Z and PI*S gene frequency distribution using on maps of the world by an inverse distance weighting (IDW) multivariate interpolation method. Hepat Mon. 2012;12(10HCC):e7434. doi:10.5812/hepatmon.7434
12. Giacopuzzi E, Laffranchi M, Berardelli R, et al. Real-world clinical applicability of pathogenicity predictors assessed on SERPINA1 mutations in alpha-1-antitrypsin deficiency. Hum Mutat. 2018;39(9):1203–1213. doi:10.1002/humu.23562
13. Rodriguez-Frias F, Miravitlles M, Vidal R, Camos S, Jardi R. Rare alpha-1-antitrypsin variants: are they really so rare? Ther Adv Respir Dis. 2012;6(2):79–85. doi:10.1177/1753465811434320
14. Irving JA, Cabrita LD, Kaiserman D, Worrall MM, Whisstock JC. Evolution and classification of the serpin superfamily. In: Silverman GA, Lomas DA, editors. Molecular and Cellular Aspects of the Serpinopathies and Disorders in Serpin Activity. World Scientific Publishing;2007:1–33.chap 1.
15. Seixas S. The human SERPIN repertoire and the evolution of 14q32.1 and 18q21.3 gene clusters. In: Geiger M, Furtmüller M, Wahlmüller F, editors. The Serpin Family Proteins with Multiple Functions in Health and Disease. Springer International Publishing; 2015:1–14.
16. Long GL, Chandra T, Woo SL, Davie EW, Kurachi K. Complete sequence of the cDNA for human alpha 1-antitrypsin and the gene for the S variant. Biochemistry. 1984;23(21):4828–4837. doi:10.1021/bi00316a003
17. Bergin DA, Reeves EP, Meleady P, et al. Alpha-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120(12):4236–4250. doi:10.1172/JCI41196
18. Gooptu B, Dickens JA, Lomas DA. The molecular and cellular pathology of alpha(1)-antitrypsin deficiency. Trends Mol Med. 2014;20(2):116–127. doi:10.1016/j.molmed.2013.10.007
19. Gooptu B, Ekeowa UI, Lomas DA. Mechanisms of emphysema in 1-antitrypsin deficiency: molecular and cellular insights. Eur Respir J. 2009;34(2):475–488. doi:10.1183/09031936.00096508
20. Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 2000;10(12):1845–1864. doi:10.1101/gr.GR-1478R
21. Bergin DA, Hurley K, McElvaney NG, Reeves EP. Alpha-1 antitrypsin: a potent anti-inflammatory and potential novel therapeutic agent. Arch Immunol Ther Exp (Warsz). 2012;60(2):81–97. doi:10.1007/s00005-012-0162-5
22. Jonigk D, Al-Omari M, Maegel L, et al. Anti-inflammatory and immunomodulatory properties of alpha1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci U S A. 2013;110(37):15007–15012. doi:10.1073/pnas.1309648110
23. Ferrarotti I, Thun GA, Zorzetto M, et al. Serum levels and genotype distribution of alpha1-antitrypsin in the general population. Thorax. 2012;67(8):669–674. doi:10.1136/thoraxjnl-2011-201321
24. Tonelli AR, Brantly ML. Augmentation therapy in alpha-1 antitrypsin deficiency: advances and controversies. Ther Adv Respir Dis. 2010;4(5):289–312. doi:10.1177/1753465810373911
25. Marsden MD, Fournier RE. Organization and expression of the human serpin gene cluster at 14q32.1. Front Biosci. 2005;10(1–3):1768–1778. doi:10.2741/1660
26. Laurell CB, Eriksson S. The electrophoretic alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. Copd. 2013;10(Suppl 1):3–8. doi:10.3109/15412555.2013.771956
27. Laurell C-B, Eriksson S. The electrophoretic α;1-globulin pattern of serum in α;1-antitrypsin deficiency. Scand J Clin Lab Invest. 1963;15(2):132–140. doi:10.1080/00365516309051324
28. Luisetti M, Seersholm N. Alpha1-antitrypsin deficiency. 1: epidemiology of alpha1-antitrypsin deficiency. Thorax. 2004;59(2):164–169. doi:10.1136/thorax.2003.006494
29. Cavalli-Sforza LL, Menozzi P, Piazza A. The History and Geography of Human Genes Abridged Paperback Edition. Princeton University Press; 1994.
30. Rachelefsky G, Hogarth DK. Issues in the diagnosis of alpha 1-antitrypsin deficiency. J Allergy Clin Immunol. 2008;121(4):833–838. doi:10.1016/j.jaci.2007.12.1183
31. den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37(6):564–569. doi:10.1002/humu.22981
32. Brantly M, Nukiwa T, Crystal RG. Molecular basis of alpha-1-antitrypsin deficiency. Am J Med. 1988;84(6A):13–31. doi:10.1016/0002-9343(88)90154-4
33. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi:10.1038/s41586-020-2308-7
34. Cox DW, Woo SL, Mansfield T. DNA restriction fragments associated with alpha 1-antitrypsin indicate a single origin for deficiency allele PI Z. Nature. 1985;316(6023):79–81. doi:10.1038/316079a0
35. Faull SV, Elliston ELK, Gooptu B, et al. The structural basis for Z α 1-antitrypsin polymerization in the liver. Sci Adv. 2020;6(43):eabc1370. doi:10.1126/sciadv.abc1370
36. Yamasaki M, Sendall TJ, Pearce MC, Whisstock JC, Huntington JA. Molecular basis of alpha1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep. 2011;12(10):1011–1017. doi:10.1038/embor.2011.171
37. Patel D, Teckman JH. Alpha-1-antitrypsin deficiency liver disease. Clin Liver Dis. 2018;22(4):643–655. doi:10.1016/j.cld.2018.06.010
38. Mayhew D, Devos N, Lambert C, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax. 2018. doi:10.1136/thoraxjnl-2017-210408
39. Sinden NJ, Koura F, Stockley RA. The significance of the F variant of alpha-1-antitrypsin and unique case report of a PiFF homozygote. BMC Pulm Med. 2014;14(1):132. doi:10.1186/1471-2466-14-132
40. Blanco I, de Serres FJ, Fernandez-Bustillo E, Lara B, Miravitlles M. Estimated numbers and prevalence of PI*S and PI*Z alleles of alpha1-antitrypsin deficiency in European countries. Eur Respir J. 2006;27(1):77–84. doi:10.1183/09031936.06.00062305
41. Lomas DA. The selective advantage of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2006;173(10):1072–1077. doi:10.1164/rccm.200511-1797PP
42. North TL, Ben-Shlomo Y, Cooper C, et al. A study of common Mendelian disease carriers across ageing British cohorts: meta-analyses reveal heterozygosity for alpha 1-antitrypsin deficiency increases respiratory capacity and height. J Med Genet. 2016;53(4):280–288. doi:10.1136/jmedgenet-2015-103342
43. Stephens JC, Reich DE, Goldstein DB, et al. Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes. Am J Hum Genet. 1998;62(6):1507–1515. doi:10.1086/301867
44. Galvani AP, Slatkin M. Evaluating plague and smallpox as historical selective pressures for the CCR5-delta 32 HIV-resistance allele. Proc Natl Acad Sci U S A. 2003;100(25):15276–15279. doi:10.1073/pnas.2435085100
45. Sabeti PC, Walsh E, Schaffner SF, et al. The case for selection at CCR5-delta32. PLoS Biol. 2005;3(11):e378. doi:10.1371/journal.pbio.0030378
46. Novembre J, Han E. Human population structure and the adaptive response to pathogen-induced selection pressures. Philos Trans R Soc Lond B Biol Sci. 2012;367(1590):878–886. doi:10.1098/rstb.2011.0305
47. Novembre J, Galvani AP, Slatkin M. The geographic spread of the CCR5 delta32 HIV-resistance allele. PLoS Biol. 2005;3(11):e339. doi:10.1371/journal.pbio.0030339
48. Lace B, Sveger T, Krams A, Cernevska G, Krumina A. Age of SERPINA1 gene PI Z mutation: Swedish and Latvian population analysis. Ann Hum Genet. 2008;72(Pt3):300–304. doi:10.1111/j.1469-1809.2008.00431.x
49. Seixas S, Garcia O, Trovoada MJ, Santos MT, Amorim A, Rocha J. Patterns of haplotype diversity within the serpin gene cluster at 14q32.1: insights into the natural history of the alpha1-antitrypsin polymorphism. Hum Genet. 2001;108(1):20–30. doi:10.1007/s004390000434
50. Veeramah KR, Novembre J. Demographic events and evolutionary forces shaping European genetic diversity. Cold Spring Harb Perspect Biol. 2014;6(9):a008516. doi:10.1101/cshperspect.a008516
51. Elliott PR, Stein PE, Bilton D, Carrell RW, Lomas DA. Structural explanation for the deficiency of S alpha 1-antitrypsin. Nat Struct Biol. 1996;3(11):910–911. doi:10.1038/nsb1196-910
52. Teckman JH, Perlmutter DH. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J Biol Chem. 1996;271(22):13215–13220. doi:10.1074/jbc.271.22.13215
53. Laffranchi M, Berardelli R, Ronzoni R, Lomas DA, Fra A. Heteropolymerization of alpha-1-antitrypsin mutants in cell models mimicking heterozygosity. Hum Mol Genet. 2018;27(10):1785–1793. doi:10.1093/hmg/ddy090
54. Mahadeva R, Chang WS, Dafforn TR, et al. Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. J Clin Invest. 1999;103(7):999–1006. doi:10.1172/JCI4874
55. McElvaney GN, Sandhaus RA, Miravitlles M, et al. Clinical considerations in individuals with α1-antitrypsin PI*SZ genotype. Eur Respir J. 2020;55(6):1902410. doi:10.1183/13993003.02410-2019
56. Green CE, Vayalapra S, Hampson JA, Mukherjee D, Stockley RA, Turner AM. PiSZ alpha-1 antitrypsin deficiency (AATD): pulmonary phenotype and prognosis relative to PiZZ AATD and PiMM COPD. Thorax. 2015;70(10):939–945. doi:10.1136/thoraxjnl-2015-206906
57. Choate R, Mannino DM, Holm KE, Sandhaus RA. Comparing patients with ZZ versus SZ alpha-1 antitrypsin deficiency: findings from AlphaNet’s disease management program. Chronic Obstr Pulm Dis. 2018;6(1):29–39. doi:10.15326/jcopdf.6.1.2018.0134
58. Piras B, Ferrarotti I, Lara B, et al. Clinical phenotypes of Italian and Spanish patients with alpha1-antitrypsin deficiency. Eur Respir J. 2013;42(1):54–64. doi:10.1183/09031936.00104712
59. Nakanishi T, Forgetta V, Handa T, et al. The undiagnosed disease burden associated with alpha-1 antitrypsin deficiency genotypes. Eur Respir J. 2020;56(6):2001441. doi:10.1183/13993003.01441-2020
60. Bornhorst JA, Greene DN, Ashwood ER, Grenache DG. Alpha1-antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest. 2013;143(4):1000–1008. doi:10.1378/chest.12-0564
61. Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*SZ genotype: estimated prevalence and number of SZ subjects worldwide. Int J Chron Obstruct Pulmon Dis. 2017;12:1683–1694. doi:10.2147/COPD.S137852
62. Hutchison DC. Alpha 1-antitrypsin deficiency in Europe: geographical distribution of Pi types S and Z. Respir Med. 1998;92(3):367–377. doi:10.1016/s0954-6111(98)90278-5
63. Spinola C, Bruges-Armas J, Pereira C, Brehm A, Spinola H. Alpha-1-antitrypsin deficiency in Madeira (Portugal): the highest prevalence in the world. Respir Med. 2009;103(10):1498–1502. doi:10.1016/j.rmed.2009.04.012
64. Fortes-Lima C, Verdu P. Anthropological genetics perspectives on the transatlantic slave trade. Hum Mol Genet. 2020. doi:10.1093/hmg/ddaa271
65. Tomas G, Seco L, Seixas S, Faustino P, Lavinha J, Rocha J. The peopling of Sao Tome (Gulf of Guinea): origins of slave settlers and admixture with the Portuguese. Hum Biol. 2002;74(3):397–411. doi:10.1353/hub.2002.0036
66. Kehdy FS, Gouveia MH, Machado M, et al. Origin and dynamics of admixture in Brazilians and its effect on the pattern of deleterious mutations. Proc Natl Acad Sci U S A. 2015;112(28):8696–8701. doi:10.1073/pnas.1504447112
67. Seyama K, Nukiwa T, Takabe K, Takahashi H, Miyake K, Kira S. Siiyama (serine 53 (TCC) to phenylalanine 53 (TTC)). A new alpha 1-antitrypsin-deficient variant with mutation on a predicted conserved residue of the serpin backbone. J Biol Chem. 1991;266(19):12627–12632. doi:10.1016/S0021-9258(18)98945-3
68. Matsunaga E, Shiokawa S, Nakamura H, Maruyama T, Tsuda K, Fukumaki Y. Molecular analysis of the gene of the alpha 1-antitrypsin deficiency variant, Mnichinan. Am J Hum Genet. 1990;46(3):602–612.
69. Curiel DT, Holmes MD, Okayama H, et al. Molecular basis of the liver and lung disease associated with the alpha 1-antitrypsin deficiency allele Mmalton. J Biol Chem. 1989;264(23):13938–13945. doi:10.1016/S0021-9258(18)80090-4
70. Graham A, Kalsheker NA, Newton CR, Bamforth FJ, Powell SJ, Markham AF. Molecular characterisation of three alpha-1-antitrypsin deficiency variants: proteinase inhibitor (Pi) nullcardiff (Asp256—-Val); PiMmalton (Phe51—-deletion) and PiI (Arg39—-Cys). Hum Genet. 1989;84(1):55–58. doi:10.1007/BF00210671
71. Faber JP, Poller W, Weidinger S, et al. Identification and DNA sequence analysis of 15 new alpha 1-antitrypsin variants, including two PI*Q0 alleles and one deficient PI*M allele. Am J Hum Genet. 1994;55(6):1113–1121.
72. Hernandez-Perez JM, Ramos-Diaz R, Fumero-Garcia S, Perez JA. Molecular characterization of PI*Q0la palma, a new alpha-1-antitrypsin null allele that combines two defective genetic variants. Clin Genet. 2017;91(6):927–928. doi:10.1111/cge.12889
73. Seyama K, Nukiwa T, Souma S, Shimizu K, Kira S. Alpha 1-antitrypsin-deficient variant Siiyama (Ser53[TCC] to Phe53[TTC]) is prevalent in Japan. Status of alpha 1-antitrypsin deficiency in Japan. Am J Respir Crit Care Med. 1995;152(6 Pt 1):2119–2126. doi:10.1164/ajrccm.152.6.8520784
74. Meira L, Boaventura R, Seixas S, Sucena M. Alpha-1 antitrypsin deficiency detection in a Portuguese population. COPD. 2018;1–6. doi:10.1080/15412555.2017.1414779
75. Ferrarotti I. Prevalence and phenotype of subjects carrying rare variants in the Italian registry for alpha1-antitrypsin deficiency. J Med Genet. 2005;42(3):282–287. doi:10.1136/jmg.2004.023903
76. Canva V, Piotte S, Aubert JP, et al. Heterozygous M3Mmalton alpha1-antitrypsin deficiency associated with end-stage liver disease: case report and review. Clin Chem. 2001;47(8):1490–1496. doi:10.1093/clinchem/47.8.1490
77. Joly P, Guillaud O, Hervieu V, Francina A, Mornex JF, Chapuis-Cellier C. Clinical heterogeneity and potential high pathogenicity of the Mmalton Alpha 1 antitrypsin allele at the homozygous, compound heterozygous and heterozygous states. Orphanet J Rare Dis. 2015;10(1):130. doi:10.1186/s13023-015-0350-6
78. Figueira Goncalves JM, Martinez Bugallo F, Diaz perez D, Martin Martinez MD, Garcia-Talavera I, Pitti Perez R. Clinical manifestations of the Mmalton alpha-1 antitrypsin deficiency variant. Pulmonology. 2017. doi:10.1016/j.rppnen.2017.11.004
79. Lomas DA, Elliott PR, Sidhar SK, et al. Alpha 1-antitrypsin mmalton (Phe52-deleted) forms loop-sheet polymers in vivo. Evidence for the C sheet mechanism of polymerization. J Biol Chem. 1995;270(28):16864–16870. doi:10.1074/jbc.270.28.16864
80. Callea F, Giovannoni I, Francalanci P, et al. Mineralization of alpha-1-antitrypsin inclusion bodies in Mmalton alpha-1-antitrypsin deficiency. Orphanet J Rare Dis. 2018;13(1):79. doi:10.1186/s13023-018-0821-7
81. Miranda E, Perez J, Ekeowa UI, et al. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha1-antitrypsin deficiency. Hepatology. 2010;52(3):1078–1088. doi:10.1002/hep.23760
82. Medicina D, Montani N, Fra AM, et al. Molecular characterization of the new defective P(brescia) alpha1-antitrypsin allele. Hum Mutat. 2009;30(8):E771–81. doi:10.1002/humu.21043
83. Fra AM, Gooptu B, Ferrarotti I, et al. Three new alpha1-antitrypsin deficiency variants help to define a C-terminal region regulating conformational change and polymerization. PLoS One. 2012;7(6):e38405. doi:10.1371/journal.pone.0038405
84. Poller W, Merklein F, Schneider-Rasp S, et al. Molecular characterisation of the defective alpha 1-antitrypsin alleles PI Mwurzburg (Pro369Ser), Mheerlen (Pro369Leu), and Q0lisbon (Thr68Ile). Eur J Hum Genet. 1999;7(3):321–331. doi:10.1038/sj.ejhg.5200304
85. Owen MC, Brennan SO, Lewis JH, Carrell RW. Mutation of antitrypsin to antithrombin. alpha 1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N Engl J Med. 1983;309(12):694–698. doi:10.1056/NEJM198309223091203
86. Hua B, Fan L, Liang Y, Zhao Y, Tuddenham EGD. 1-antitrypsin Pittsburgh in a family with bleeding tendency. Haematologica. 2009;94(6):881–884. doi:10.3324/haematol.2008.004739
87. Cook L, Burdon JGW, Brenton S, et al. Kinetic characterisation of alpha-1-antitrypsin F as an inhibitor of human neutrophil elastase. Pathology. 1996;28(3):242–247. doi:10.1080/00313029600169074
88. Okayama H, Brantly M, Holmes M, Crystal RG. Characterization of the molecular basis of the alpha 1-antitrypsin F allele. Am J Hum Genet. 1991;48(6):1154–1158.
89. Renoux C, Odou MF, Tosato G, et al. Description of 22 new alpha-1 antitrypsin genetic variants. Orphanet J Rare Dis. 2018;13(1):161. doi:10.1186/s13023-018-0897-0
90. Lee JH, Brantly M. Molecular mechanisms of alpha1-antitrypsin null alleles. Respir Med. 2000;94(Suppl C):S7–11. doi:10.1053/rmed.2000.0851
91. Hofker MH, Nukiwa T, van Paassen HM, et al. A Pro—-Leu substitution in codon 369 of the alpha-1-antitrypsin deficiency variant PI MHeerlen. Hum Genet. 1989;81(3):264–268. doi:10.1007/BF00279001
92. Fregonese L, Stolk J, Frants RR, Veldhuisen B. Alpha-1 antitrypsin null mutations and severity of emphysema. Respir Med. 2008;102(6):876–884. doi:10.1016/j.rmed.2008.01.009
93. Seixas S, Mendonca C, Costa F, Rocha J. Alpha1-antitrypsin null alleles: evidence for the recurrence of the L353fsX376 mutation and a novel G–>A transition in position +1 of intron IC affecting normal mRNA splicing. Clin Genet. 2002;62(2):175–180. doi:10.1034/j.1399-0004.2002.620212.x
94. Lara B, Martinez MT, Blanco I, et al. Severe alpha-1 antitrypsin deficiency in composite heterozygotes inheriting a new splicing mutation QOMadrid. Respir Res. 2014;15:125. doi:10.1186/s12931-014-0125-y
95. Nguyen LS, Wilkinson MF, Gecz J. Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci Biobehav Rev. 2014;46(Pt 2):175–186. doi:10.1016/j.neubiorev.2013.10.016
96. Vaz Rodrigues L, Costa F, Marques P, Mendonca C, Rocha J, Seixas S. Severe alpha-1 antitrypsin deficiency caused by Q0(Ourem) allele: clinical features, haplotype characterization and history. Clin Genet. 2012;81(5):462–469. doi:10.1111/j.1399-0004.2011.01670.x
97. Bellemare J, Gaudreault N, Valette K, et al. The clinical utility of determining the allelic background of mutations causing alpha-1 antitrypsin deficiency: the case with the null variant Q0(Mattawa)/Q0(ourem). Chronic Obstr Pulm Dis. 2020. doi:10.15326/jcopdf.8.1.2020.0168
98. Hernández Pérez JM, Ramos Díaz R, Fumero García S, Pérez Pérez JA. Description of alpha-1-antitrypsin deficiency associated with PI*Q0ourém allele in La Palma Island (Spain) and a genotyping assay for detection. Arch Bronconeumol. 2015;51(1):e1–e3. doi:10.1016/j.arbr.2014.11.019
99. Curiel D, Brantly M, Curiel E, Stier L, Crystal RG. Alpha 1-antitrypsin deficiency caused by the alpha 1-antitrypsin Nullmattawa gene. An insertion mutation rendering the alpha 1-antitrypsin gene incapable of producing alpha 1-antitrypsin. J Clin Invest. 1989;83(4):1144–1152. doi:10.1172/JCI113994
100. Hildesheim J, Kinsley G, Bissell M, Pierce J, Brantly M. Genetic diversity from a limited repertoire of mutations on different common allelic backgrounds: alpha 1-antitrypsin deficiency variant Pduarte. Hum Mutat. 1993;2(3):221–228. doi:10.1002/humu.1380020311
101. Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37(3):235–241. doi:10.1002/humu.22932
102. Faber JP, Weidinger S, Olek K. Sequence data of the rare deficient alpha 1-antitrypsin variant PI Zaugsburg. Am J Hum Genet. 1990;46(6):1158–1162.
103. Malik R, Dau T, Gonik M, et al. Common coding variant in SERPINA1 increases the risk for large artery stroke. Proc Natl Acad Sci U S A. 2017;114(14):3613–3618. doi:10.1073/pnas.1616301114
104. Dickens JA, Lomas DA. Why has it been so difficult to prove the efficacy of alpha-1-antitrypsin replacement therapy? Insights from the study of disease pathogenesis. Drug Des Devel Ther. 2011;5:391–405. doi:10.2147/DDDT.S14018
105. Ferrarotti I, Carroll TP, Ottaviani S, et al. Identification and characterisation of eight novel SERPINA1 Null mutations. Orphanet J Rare Dis. 2014;9(1):172. doi:10.1186/s13023-014-0172-y
106. Seixas S, Garcia O, Amorim A, Rocha J. A novel alpha-1-antitrypsin r281del variant found in a population sample from the Basque country. Hum Mutat. 2000;15(1):121–122. doi:10.1002/(SICI)1098-1004(200001)15:1<121::AID-HUMU37>3.0.CO;2-U
107. Jardi R, Rodriguez F, Miravitlles M, et al. Identification and molecular characterization of the new alpha-1-antitrypsin deficient allele PI Y Barcelona (Asp256–>Val and Pro391–>His). Mutations in brief no. 174. online. Hum Mutat. 1998;12(3):213.
108. Seixas S, Trovoada M, Santos M, Rocha J. A novel alpha-1-antitrypsin P362H variant found in a population sample from São Tomé e Príncipe (Gulf of Guinea, West Africa). Hum Mutat. 1999;13(5):414.
109. Matamala N, Gomez-Mariano G, Perez JA, et al. New cis-acting variants in pi*s background produce null phenotypes causing alpha-1 antitrypsin deficiency. Am J Respir Cell Mol Biol. 2020;63(4):444–451. doi:10.1165/rcmb.2020-0021OC
110. Matamala N, Lara B, Gomez-Mariano G, et al. Characterization of novel missense variants of SERPINA1 gene causing alpha-1 antitrypsin deficiency. Am J Respir Cell Mol Biol. 2018;58(6):706–716. doi:10.1165/rcmb.2017-0179OC
111. Crystal RG. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest. 1990;85(5):1343–1352. doi:10.1172/JCI114578
112. DeLano WL. The PyMOL molecular graphics system. DeLano Scientific; 2002. Available from: http://www.pymol.org.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.