")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 9
Knockout Of BIRC5 Gene By CRISPR/Cas9 Induces Apoptosis And Inhibits Cell Proliferation In Leukemic Cell Lines, HL60 And KG1
Authors Narimani M, Sharifi M , Jalili A
Received 25 September 2019
Accepted for publication 2 November 2019
Published 27 November 2019 Volume 2019:9 Pages 53—61
DOI https://doi.org/10.2147/BLCTT.S230383
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor David Dingli
Manizheh Narimani,1 Mohammadreza Sharifi,2 Ali Jalili1
1Cancer and Immunology Research Center, Institute of Research for Health Development, Kurdistan University of Medical Sciences, Sanandaj, Iran; 2Department of Genetics and Molecular Biology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
Correspondence: Ali Jalili
Cancer and Immunology Research Center, Institute of Research for Health Development, Kurdistan University of Medical Sciences, Sanandaj, Iran
Tel +98-9183771862
Email [email protected]
Introduction: Human Baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5) which encodes survivin exhibits multiple biological activities, such as cell proliferation and apoptosis. Survivin is overexpressed in numerous malignant diseases including acute myeloid leukemia (AML). Recent studies have shown that the CRISPR/Cas9 nuclease-mediated gene-editing systems are suitable approach’s for editing or knocking out various genes including oncogenes.
Methods and materials: We used CRISPR-Cas9 to knockout the BIRC5 in the human leukemic cell line, HL60, and KG1, and these cell lines were transfected with either the Cas9- and three sgRNAs expressing plasmids or negative control (scramble) using Lipofectamine 3000. The efficacy of the transfection was determined by quantitative reverse transcription-polymerase chain (RT-qPCR) and surveyor mutation assays. Cell proliferation and apoptosis were measured by MTT assay and flow cytometry, respectively.
Results: We have successfully knocked out the BIRC5 gene in these leukemic cells and observed that the BIRC5-knocked out cells by CRISPR/Cas9 showed a significant decrease (30 folds) of survivin at mRNA levels. Moreover, cell death and apoptosis were significantly induced in BIRC5-CRISPR/Cas9-transfected cells compared to the scramble vector.
Conclusion: We demonstrated for the first time that targeting BIRC5 by CRISPR/Cas9 technology is a suitable approach for the induction of apoptosis in leukemic cells. However, further studies targeting this gene in primary leukemic cells are required.
Keywords: BIRC5, survivin, CRISPR/Cas9 nuclease, AML, KG1 cells, HL60 cell
Introduction
Acute myeloid leukemia (AML) is a genetically and clinically heterogeneous disorder defined by undifferentiated malignant clonal myeloid cell proliferation which is more common in older adults.1–3 Accumulating evidence indicates that a number of mutations and aberrant expression of many genes are involved in the pathogenesis of AML.4 Although various therapeutic strategies including chemotherapy and bone marrow stem cells transplantation are now available for the treatment of patients with AML, the rate of mortality is still high in these patients.5–7 Therefore, there is an urgent need to precisely dissect fundamental molecular mechanisms regulating leukemia and empower personalized medicine strategy via technologically advanced tools.8 Accordingly, concurrent targeting multiple components of survival, proliferation, and apoptotic pathways in leukemic cells seems to be suitable approaches for future therapeutic strategies.6
Human Baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5), belongs to the inhibitor of apoptosis (IAP) family, encodes survivin protein as a bifunctional regulator of apoptosis inhibition and cell cycle progression. An increasing number of studies have revealed that survivin preferentially upregulated in leukemia and fetal cells, but it does not exist in normal differentiated adult tissues.9–12 Since a strong correlation between the elevated survivin expressions and clinicopathological features of a number of cancers has been reported, many Researchers paid attention to target survivin by employing various kinds of molecular and cellular technologies such as antisense oligonucleotides targeting survivin mRNA,13 induction of specific cytotoxic T cells and generation of antibody against it;14 however, most of these preclinical studies were not clinically successful.
Through technological breakthroughs in the field of biotechnology and molecular medicine over the recent years, now we have come in the “gene editing era”. High-throughput genome engineering tools would break new ground by preventing and/or treating the most devastating genetic disorders. In the last decades or so, targeted-specific nucleases, such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the “clustered regularly interspaced short palindromic repeats” (CRISPR)/“CRISPR-associated genes 9” (Cas9), have extremely been used in genetic modification industry.15–17 However, CRISPR/Cas9 has been demonstrated as an easy-handle, highly specific and efficient approach for editing the eukaryotic genome.18 As survivin plays vital roles in inhibition of apoptosis and cell death in leukemic cells, here we set out to explore the disruption of its gene, BIRC5 by CRISPR/Cas9. Herein, we demonstrated that knockout of BIRC5 by CRISPR/Cas9 in leukemic cells is technically feasible and efficient, also this gene-editing leads to induction of apoptosis and inhibition of cell growth. Moreover, our data show the therapeutic application of CRISPR/Cas9 for disruption of oncogenic BIRC5 in cancers including leukemia.
Materials And Methods
Cell Culture
The human erythroleukemia KG1 cell line and human promyelocytic leukemia HL60 were obtained from Pasteur Institute of Iran. The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, USA) containing 10% heat-inactivated (30 min, 56°C) fetal bovine serum (FBS) (Gibco, Langley, OK), 1% Glutamax (Gibco), 1% penicillin/streptomycin at 37°C and 5% CO2 in fully humidified incubator.
Vector Construction And Expression
CRISPR/Cas9 system was purchased from GeneCopoeia (Rockville, MD). To drive the expression of the Cas9 protein, Cas9 nuclease expression clone was specifically designed to have CMV promoter, a mammalian antibiotic resistance gene as a stable selection marker (Neo), and a reporter gene, mCherry fluorescent protein. Also, the negative control (Scramble) vector for pCRISPR-SG01 was constructed as the same as the sgRNA plasmid backbone which did not contain a sgRNA sequence. Three different sgRNAs (sgRNAa, sgRNAb, and sgRNAc) were then designed to construct the CRISPR/Cas9 to target human BIRC5. Briefly, the sequences of sgRNAa, sgRNAb, and sgRNAc are 5ˊ CCAGGCAGGGGGCAACGTCG 3ˊ, 5ˊ ACTTACATGGGGTCGTCATC 3ˊ, and 5ˊ GGGCAGTCTCACCCGCTCCG 3ˊ, respectively, which targeted exon 1 except for sgRNAb that generated an Indel mutation in exon 2 of the gene. These sequences were separately inserted into the sgRNA expression cassettes of pCRISPR-SG01 vector containing the U6 promoter. All of the vectors were introduced by chemical transformation into the competent E.coli DH5α for cloning purposes using a selectable marker of Ampicillin. These strains were cultured on LB agar plates supplemented with 1 mg/mL Ampicillin at 37°C and subsequently in LB broth, liquid culture, containing Ampicillin in the 37°C shaking incubator at 180 rpm/min speed. Then, all of the plasmids were purified with EndoFree Plasmid Maxi Kit (Qiagen, Germantown, MD) according to the manufacturer’s protocol.
Cell Transfection
8 × 105 KG1 and HL60 cells were dispensed in 6-well plastic tissue-culture plates containing 2 mL of fresh RPMI 1640. The cells were then transfected with either the Cas9- and three sgRNAs expressing plasmids or scramble DNA using Lipofectamine 3000 (Invitrogen Waltham, MA). For a single well of a 6-well dish, 2500 ng plasmid DNA, 5 µL of P3000 Reagent, 6 µL of Lipofectamine 3000 reagent, and 250 µL of OptiMEM were used, according to the supplier’s protocol. After 6 hrs, supplemented media consisting of 10% FBS and 1% penicillin/streptomycin was added to each well. To investigate co-transfection efficiency, cultures were visualized through fluorescence microscopy 48 hrs’ post-transfection by monitoring the expression of the reporter gene, mCherry.DNA Extraction and PCR Amplification Assay Genomic DNA (gDNA) was extracted using the PrimePrepTM Genomic DNA Isolation Kit (GeNet Bio, Daejeon, Korea) 48 hours after transfection, and quantification of gDNA was performed by NanoDrop spectrophotometer (WPA Biowave II, Cambridge, UK). The DNA region encompassing the CRISPR target site in BIRC5 was amplified with Pfu high fidelity DNA Polymerase (Vivantis Technologies, Kuala Lumper, Malaysia) using the sense 5ʹ- GACTACAACTCCCGGCACAC −3ʹ and antisense 5ʹ– AAGGCATCAGGCATCTTACG −3ʹ primers. PCR was performed under the standard following conditions: 1 cycle initial denaturation of 3 mins at 95°C, followed by 35 cycles of 1 min at 95°C, 1 min at 59°C, and 1 min at 72°C, with a final 10 mins at 72°C for post extension. Amplified PCR products were simply subjected to electrophoresis on 1.5% agarose gel prestained with DNA Green Viewer (Parstous, Iran) at 80 volts for 30–45 mins. The 868 bp fragment was then excised from the gel and purified using the AccuPrep™ Gel Purification Kit (Bioneer, Daejeon, Korea).
Surveyor Mutation Detection Assay
BIRC5 oncogene knockout was examined by the SURVEYOR Mutation Detection Kit (Integrated DNA technologies, USA) according to the producer’s instructions. Briefly, 400 ng of equal amounts of the test (transfected) and reference (untransfected) purified PCR products were mixed in a microtube for DNA duplex formation. Then, to promote heteroduplex formation, the PCR products heated and cooled slowly on a Bio-Rad C1000 thermocycler as follows: 1: 95°C for 10 mins; 2: 95°C to 85°C ramping at ‐2.0°C/s; 3: 85°C for 1 min; 4: 85°C to 75°C ramping at ‐0.3°C/s; 5: 75°C for 1 min; 6: 75°C to 65°C ramping at ‐0.3°C/s; 7: 65°C for 1 min; 8: 65°C to 55°C ramping at ‐0.3°C/s; 9: 55°C for 1 min; 10: 55°C to 45°C ramping at ‐0.3°C/s; 11: 45°C for 1 min; 12: 45°C to 35°C ramping at ‐0.3 °C/s;13: 35°C for 1 min; 14: 35°C to 25°C ramping at ‐0.3°C/s; 15: 25°C for 1 min. After reannealing, the samples were immediately kept on ice and 1/10 of the total volume of each reaction mixture 1.5 M of MgCl2 was added. Next, 1 µL of Surveyor Enhancer S and 1 µL of Surveyor Nuclease were added to the products followed by 60 mins incubation at 42°C. Then, stop solution was added in 1/10 volume of each product and digestion products clearly analyzed by 2% agarose gel. The indel percentage was calculated according to the gray value detected using ImageJ software (NIH, Bethesda, MD). The percentage of mutation was measured using the following formulas wherein a is the integrated intensity of the undigested PCR product and b and c are the integrated intensities of each cleavage product:
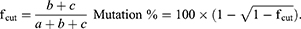
RNA Extraction And RT-qPCR Assay
Total RNA was extracted from cells using the YTA Total RNA Purification Mini Kit (Yekta Tajhiz, Tehran, Iran) 48 hrs after transfection, according to the supplier’s protocol. Complementary DNA (cDNA) was synthesized from 2 µg total RNA as a template by using random hexamer. Reverse transcription was performed using the RevertAid™ First-Strand cDNA Synthesis (Thermo Fisher Scientific, Germany) for 60 mins at 42°C followed by 70 mins at 5°C with RevertAid Reverse Transcriptase.
Quantitative Real-Time PCR (RT-qPCR) assay was carried out with Real Q Plus 2X Master Mix Green high ROX™ Kit (Ampliqon, Denmark). PCR amplification was performed to produce an amplicon of 249 bp in 10 μlit final reaction volume with the SYBR Green detection method using the Step one plus Real-Time PCR System (Applied Biosystems, USA). The sequences of the primer sets were used, including BIRC5-F, 5ʹ– CGCATCTCTACATTCAAG −3ʹ; BIRC5-R, 5ʹ– ATGTTCCTCTCTCGTGAT −3ʹ; GAPDH-F, 5ʹ– AAGCTCATTTCCTGGTAT −3ʹ; GAPDH-R, 5′-5ʹ– CTTCCTCTTGTGCTCTTG −3ʹ. The cycle conditions used for all genes consist of an initial step of 95°C for 15 mins followed by 40 cycles of 95°C for 30 s; 51.6°C for 30 s and 72°C for 30 s with one cycle melt curve stage of 95°C for 30 s; 60°C for 1 min; and finally, 95°C for 15 s. Relative quantification (RQ) of gene expression was calculated using the comparative Ct method and the equation 2−ΔΔCt after normalization the mRNA levels of target gene with an endogenous housekeeping gene, GAPDH.
Cell Viability Assay
Cell viability was analyzed by the MTT (3-(4, 5- methylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide) viability assay as previously reported.19 In brief, the cells were seeded in 96-well plates in triplicate at a density of 5 × 103 cells per well, 48 hrs after transfection of cells with CRISPR/Cas9 vectors. Next, 20 µL of the colorimetric MTT reagent was directly added to the wells with a final concentration of 1 mg/mL. Then, the cells were incubated for 4 h at 37°C, the medium was discarded completely and 100 µL Dimethyl sulfoxide was added and shacked for 10 min. The optical density (OD) was measured at 570 nm using a microplate reader (Stat Fax, Palm City, FL). The absorbance of control cells was considered as 100%. Percent of cell viability was calculated as = 100-(test OD/control OD) x100.
Apoptosis And Necrosis Assay
Forty-eight hours after transfection, cells were harvested and apoptosis was analyzed as previously reported.20 The cells were stained for Annexin V and PI according to the manufacturer’s instructions. Briefly, cells were harvested, washed and incubated with Annexin V and PI for 15 min in dark place. Next, after washing with 300 µL binding buffer, cells were gently resuspended in 300 µL of binding buffer, kept on ice and subjected to FACS analysis (FACS Calibur, Beckman Dickinson, San Jose, CA) within an hour. Flow cytometry data were analyzed by FCS Express software (De Novo Software, Los Angeles, CA).
Statistical Analysis
Arithmetic means and standard deviations were calculated and statistical significance was defined as P≤0.05 using Student’s t-test.
Results
The Design And Validation Of sgRNA Targeting BIRC5
In the present study, we first retrieved the sequence of BIRC5 gene (NCBI Reference Sequence (RefSeq): NG_029069.1) and then performed the designation of sgRNAs. We designed three constructs to express sgRNAs of the CRISPR/Cas9 nuclease toolkit. All of these sgRNAs are in principle composed of 20 nt in length, preceded by the canonical trinucleotide 5ˊ-NGG, the protospacer adjacent motif (PAM). The plasmid-based sgRNA expression constructs possess the U6 RNA polymerase III promoter, which, in turn, prefers a guanine (G) nucleotide as the first base of its transcript; thus, an extra G residue is added at the 5′ of the sgRNA. To minimize the off-target mutations, the DNA target site should entirely match the PAM motif as well as the 12 bp seed sequence closest to the PAM; the remaining bases are thought to be less important as their mismatches could be tolerated. In our study, sequences that contained the PAM motif inside exon 1 and/or 2 of BIRC5 were identified. Then, to determine whether or not candidate sequences were unique in the genome, they were analyzed by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The schematic features of DNA target sequences and their corresponding sgRNAs are depicted in Figure 1A and B.
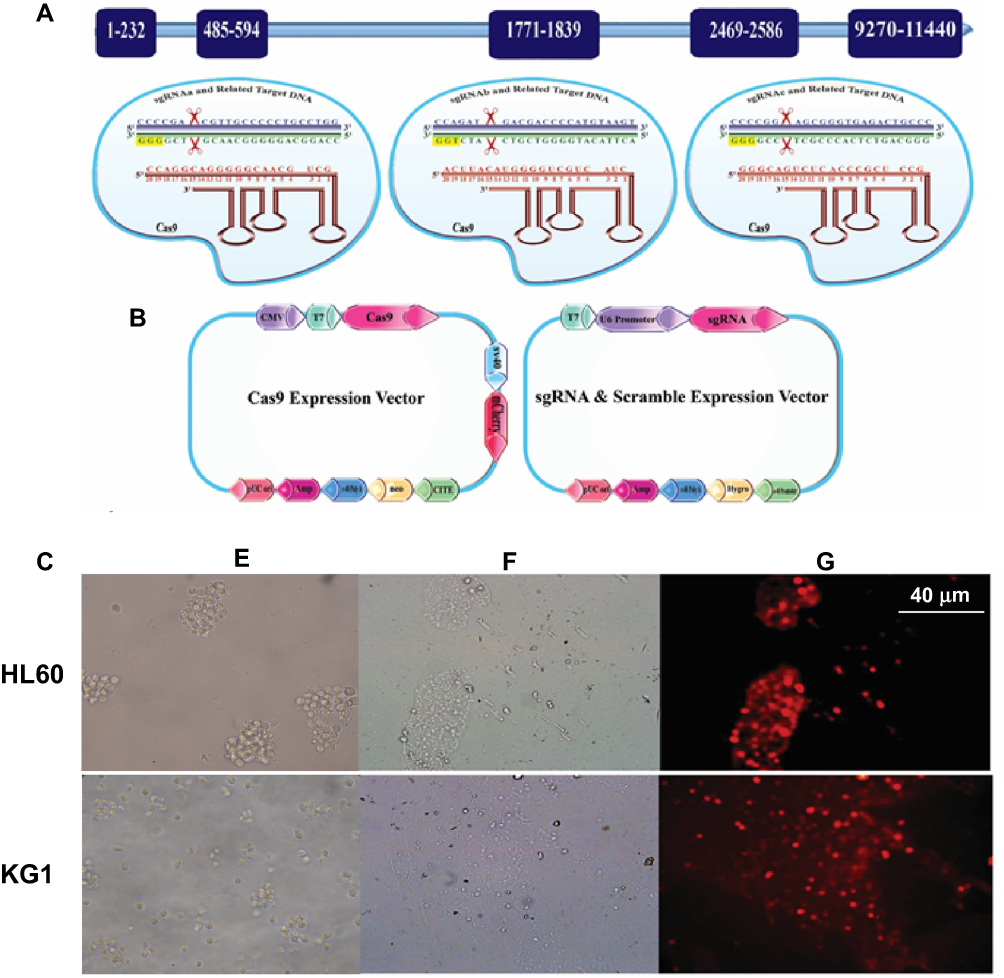 |
Figure 1 (A) Illustration of the sgRNA sequences-directed CRISPR/Cas9 system and their corresponding regions in BIRC5 gene as well as the vectors used in this study. PAM sequences are labeled in yellow; sgRNAs in red, as well as sense and anti-sense strands of the BIRC5 gene are shown in navy blue and dark green, respectively. (B) SV40 Poly A, simian vacuolating virus 40 polyadenylation signal sequence; CITE, EMC virus Cap-Independent Translation Enhancer sequence; CMV, cytomegalovirus promoter; U6 promoter, U6 small nuclear RNA promoter, which drives transcription by RNA polymerase III; neo, Neomycin; Hygro, Hygromycin; pUC ori, origin of replication that “UC” stands for the “University of California”. (C) Assessment of transfection efficiency using mCherry visualization. HL60 and KG1 cell lines were cotransfected with Lipofectamine 3000 and visualized under phase contrast or fluorescence microscopy 2 days post-transfection. Untransfected (E), Transfected cells visualized under phase-contrast microscopy (F), and transfected cells visualized under fluorescence microscopy (G) 2 days post-transfection. |
Optimizing Cas9/SgRNAs Delivery Of Leukemic Cells
We next aimed at optimizing conditions for the co-transfection of Cas9/sgRNA expression plasmids leukemic cells via lipofectamine. For this purpose, we used reporter plasmids mCherry as a reporter gene in Cas9 and scramble vectors. As shown in Figure 1C, fluorescence micrographs of mCherry-transfected cells indicated that Lipofectamine 3000 is an appropriate reagent for delivering Cas9 or scramble in leukemic cells.
Verification Of The Effect Of sgRNAs Targeting Human BIRC5 Gene
To characterize the functionality of Cas9 nuclease, as well as their respective sgRNAs on BIRC5 gene, we used the SURVEYOR nuclease assay. This method is a mismatch-specific DNA endonuclease for indel mutations identification through the generation of double-strand breaks (DSBs) and subsequent NHEJ DNA repair mechanism. Genomic DNA was isolated from transfected cells and screened for the presence of site-specific gene modification by PCR. As shown in Figure 2, both of the duplex DNA strands were catalyzed at 3 bp upstream of the PAM. Moreover, our data show that PCR amplicon of 868 bp in BIRC5 gene was broken into several smaller fragments, where sgRNAa, sgRNAb, and sgRNAc were cleaved at their 176 bp, 625 bp, and 269 bp positions, respectively (Figure 2). According to these results, we found that all of sgRNAs successfully triggered site-specific cleavage in the BIRC5 gene locus.
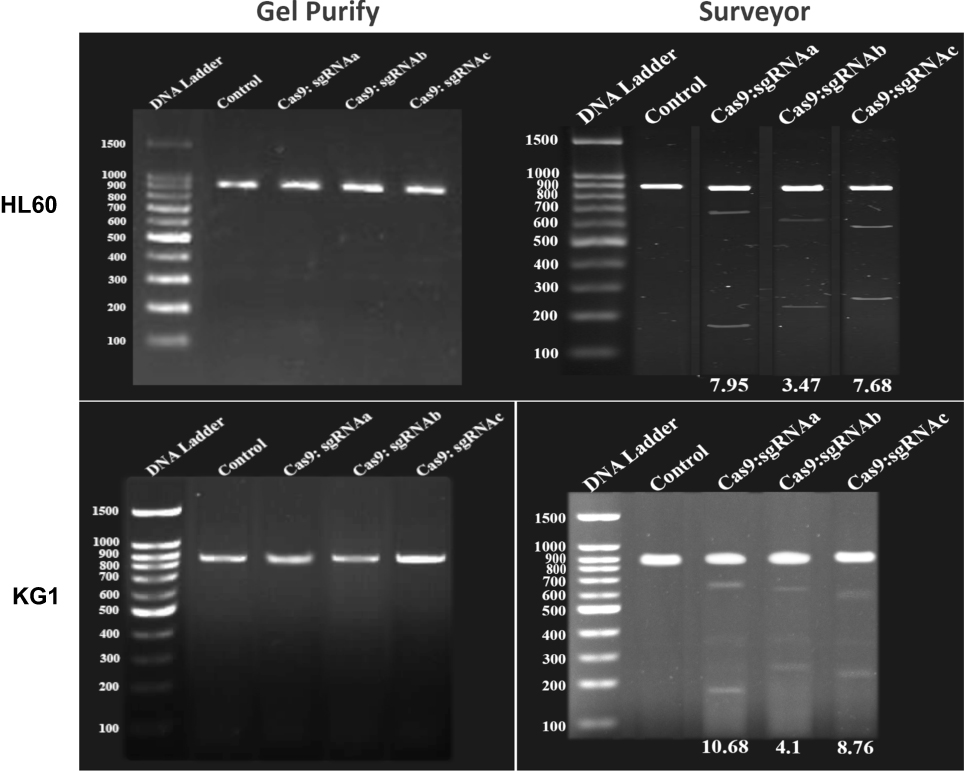 |
Figure 2 CRISPR/Cas9 -mediated cleavage at BIRC5 locus in AML cells. (Left) PCR detection and (Right) Surveyor assay of CRISPR/Cas9 activity in HL60 and KG1 cell lines. The numbers on the left represent the sizes of the DNA Ladder. The numbers at the bottom of the gel indicate mutation percentages measured by band intensities. |
Evaluation Of Survivin Expression At mRNA Levels
To confirm that BIRC5 gene was thoroughly disrupted in these leukemic cells, we next examined the expression of survivin at mRNA level by RT-qPCR. We observed that survivin is highly expressed in these cells confirming the previous reports14 and disruption of BIRC5 by CRISPR/Cas9 leads to a significant reduction (30 fold) of survivin in these cells. However, the scramble did not show any significant difference in the expression of survivin in both cell lines (Figure 3).
 |
Figure 3 BIRC5 expression was quantitatively evaluated via RT-qPCR 48 h after cotransfection using sgRNAa, sgRNAb, sgRNAc and Cas9 vectors. Relative expression values were normalized assigning the value of the cells in control groups to 1.0. Error bars represent mean ± s.d. of biological replicates from one experiment (P<0.0001). |
The Effect Of CRISPR/Cas9-Mediated BIRC5 Knockout On Cells Survival And Apoptosis
Next, we examined cell proliferation in CRISPR/Cas9-mediated BIRC5 knockout HL60 and KG1 by MTT assay and found that the disruption of BIRC5 resulted in a significant decline of cell viability compared to control and scramble DNA (Figure 4).
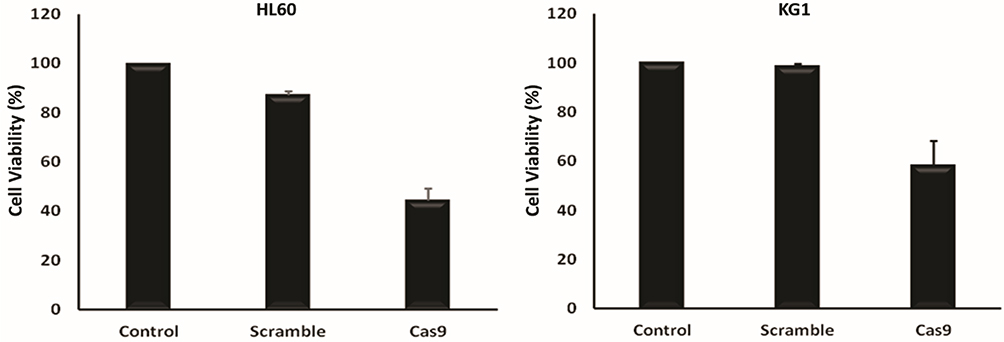 |
Figure 4 Proliferation of the various groups–cotransfected HL60 and KG1 cells via sgRNAa, sgRNAb, sgRNAc and Cas9 vectors was quantified using an MTT assay 48 hrs after transfection. The viability of the untreated cells was considered as 100%, and the viability of other groups is presented as the percentage of the untreated cells. Data were mean ± s.d. of three independent experiments (P<0.0001). |
Having shown that CRISPR/Cas9-mediated BIRC5 knockout reduces cell viability, we sought to examine if disruption of BIRC5 by CRISPR/Cas9 could lead to the induction of apoptosis. To do this, the cells were stained with Annexin V and PI, and the apoptosis was detected by flow cytometry. As demonstrated in Figure 5, CRISPR/Cas9-mediated BIRC5 knockout shows a remarkable induction of apoptosis in both cell lines. However, scramble plasmid did not induce apoptosis in these leukemic cells.
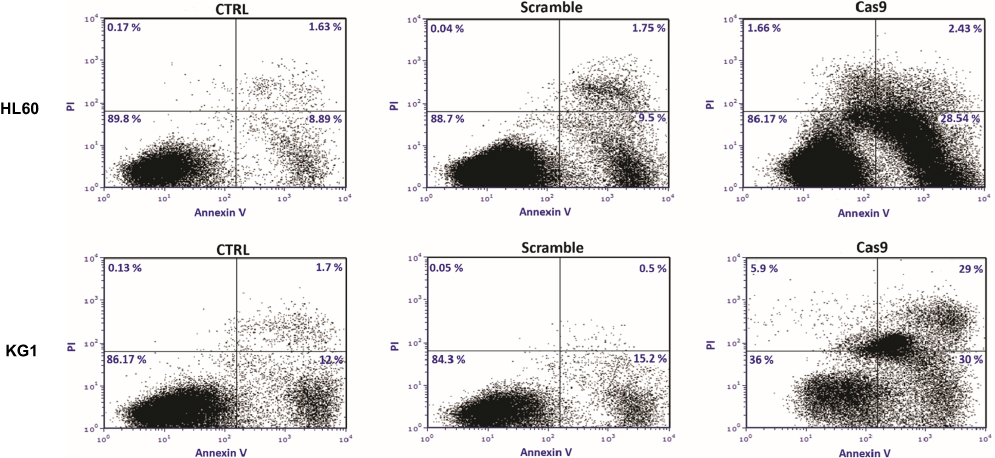 |
Figure 5 Targeting of survivin resulted in the induction of apoptosis in leukemic cell lines. 48 hrs after cotransfection of sgRNAa, sgRNAb, sgRNAc cells were stained with Annexin V and PI. The cells were subjected to flow cytometry analysis right away. Representative analysis of at least three flow cytometry analysis was shown. |
Discussion
The CRISPR/Cas9 technology has been employed on the genome manipulation, modification, and engineering in microorganisms, plants, animals, and humans for experimentally and therapeutically purposes. In the current study, we successfully disrupted the BIRC5 gene in two human leukemic cells and observed that this genome-editing toolkit is not only feasible and easy to handle but also capable of inducing apoptosis and cell death in these leukemic cells.
Previous studies clarified that BIRC5 gene is overexpressed in numerous malignancies such as leukemia and correlated with tumor progression and drug resistance.9 The most distinctive function of survivin is cell death regulation. This protein cannot directly interact with caspases; it associates with the X-linked inhibitor of apoptosis (XIAP) and represses the caspase-dependent apoptotic pathway. The emerging body of evidence indicates that survivin acts as anti-apoptotic protein, as well as the promotion of cancer cell invasion and migration, implying that survivin can be considered as a noticeable prognostic and metastatic factor in cancerous cells.21 Several lines of evidence made it apparent that high BIRC5 expression has been associated with substantially inferior, poor, and adverse clinical outcome in AML.10,11,22,23
Previous studies have demonstrated that the isoforms (−2B, -Ex3 and −3B, except survivin-2α) of survivin are overexpressed in a human AML-M3 cell line, NB4, as well as bone marrow samples from patients. However, their expression was declined after arsenic trioxide (ATO) treatment as a front-line therapy.10 In 2017, Pazhang et al11 have demonstrated survivin was downregulated in another AML-M3 cell line, HL60 following treatment with embelin and celastrol, two anti‑tumor agents, and leads to a considerable response improvement to chemotherapeutic agents. Accordingly, another study has revealed that pretreatment with survivin siRNA could synergistically sensitize leukemic cells U937 cells to anticancer drugs and induced apoptosis in these cells.9 Thus, survivin is a key player in leukemic cells survival and their resistance to chemo regiments; therefore, we hypothesized that BIRC5 knockout with CRISPR/Cas toolkit would result in the induction of apoptosis and cell death in HL60 and KG1. Herein, for the first time, we employed the CRISPR/Cas9 gene knockout system to disrupt the BIRC5 gene in two human leukemic cells. It is obvious that the transfection of non-adherent cells such as leukemic cells is rather difficult than adherent cell line. However, we co-transfected three DNA plasmids into HL60 and KG1 cell lines by lipofectamine 3000. Thus, this novel non-viral-mediated gene-editing method has many advantages of clinical applications such as low cost and safety. In line with our observations, a recent study employed Cas9:sgRNA gene knockout system to disrupt the PDL-1 gene in human lymphocytes.24 In accordance with our data and previously published reports, we envision that designing of CRISPR/Cas9 toolkit to disrupt overexpressed genes such as survivin in leukemic cells is a feasible and effective approach. Consistently, we have recently reported that CRISPR/Cas9 nickase (Cas9n) is an alternative approach for knocking out the leukemic cells.25 Cas9n is a mutated form of Cas9 which stands in need of two heminuclease domain to create a double-stranded break at the target site.25 We envisioned that both CRISPR/Cas9 and CRISPRCas9n are feasible approaches to generate genetically modified human cells. However, on-target mutations of CRISPR/Cas9 are nearly twice that of CRISPR/Cas9n in HL60 cell lines, but there are equal in the KG1 cells. On the other hand, designing of sgRNAs for CRISPR/Cas9 is more feasible than CRISPR/Cas9n. Altogether, both systems are appropriate methods for editing or knocking out a gene/oncogene. However, each system might be more suitable for a certain type of cells or cancer.
Furthermore, we delineated an inverse correlation between survivin protein levels and apoptosis in leukemic cells. Having knocked out cells with CRISPR constructs, we detected an increasing number of programmed cell death, confirming the survivin invaluable function in cancer development, cell survival, and proliferation. Our data are in close agreement with previous ones which unraveled survivin/BIRC5 mechanistic events was the pivotal and ubiquitous nature of tumor progression and clinical manifestation, consequently emerged as an attractive therapeutic target in cancer treatment.26–29
Collectively, this study strengthens the idea that cationic lipid delivered efficiently Cas9: sgRNA into leukemic cells and that CRISPR/cas9 editing gene could be an appropriate method for disrupting the overexpressed gene such as BIRC5 which play an important role in the pathogenesis of cancers including leukemia. Further investigations of employing CRISPR/cas9, particularly in an animal model and preclinical studies, may pave the path for personalized medicine and cell therapy.
Author Contributions
All authors contributed toward data analysis, drafting and revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
This work was supported by grants from Kurdistan University of Medical Sciences to AJ to purchasing reagents for the thesis of MN as a PhD candidate in Molecular Medicine. The authors report no other conflicts of interest in this work.
References
1. Kim TK, Gore SD, Zeidan AM. Epigenetic therapy in acute myeloid leukemia: current and future directions. Semin Hematol. 2015;52(3):172–183. doi:10.1053/j.seminhematol.2015.04.003
2. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221.
3. O’Brien EC, Prideaux S, Chevassut T. The epigenetic landscape of acute myeloid leukemia. Adv Hematol. 2014.
4. Kantarjian H. Acute myeloid leukemia – major progress over four decades and glimpses into the future. Am J Hematol. 2016;91(1):131–145. doi:10.1002/ajh.24246
5. Liersch R, Carsten M€uller-Tidow WE, Berdel KU. Prognostic factors for acute myeloid leukaemia in adults – biological significance and clinical use. Br J Haematol. 2014;165:17–38. doi:10.1111/bjh.12750
6. Döhner H, Weisdorf DJ, CD B. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. doi:10.1056/NEJMra1406184
7. Bernasconi P, Farina M, Boni M, Irene Dambruoso CC. Therapeutically targeting SELF-reinforcing leukemic niches in acute myeloid leukemia: a worthy endeavor? Am J Hematol. 2016;91(5):507–517. doi:10.1002/ajh.24312
8. Béné MC, Grimwade D, Haferlach C, Haferlach T, Zini G, LeukemiaNet E. Leukemia diagnosis: today and tomorrow. Eur J Haematol. 2015;95(4):365–373. doi:10.1111/ejh.12603
9. Jafarlou M, Baradaran B, Shanehbandi D, et al. siRNA-mediated inhibition of survivin gene enhances the anti-cancer effect of etoposide in U-937 acute myeloid leukemia cells. Cell Mol Biol. 2016;62(6):44–49.
10. Zaki Dizaji M, Seyed H G, Hosseini E, et al. Survivin isoform expression in arsenic trioxide-treated acute promyelocytic leukemia cell line and patients: the odd expression pattern of survivin-2α. Asia Pac J Clin Oncol. 2016.
11. Pazhang Y, Jaliani HZ, Mehdi Imani DH. Synergism between NF-kappa B inhibitor, celastrol, and XIAP inhibitor, embelin, in an acute myeloid leukemia cell line, HL-60. J Cancer Res Ther. 2017;12(1):155–160.
12. Sah NK, Khan Z, Khan GJ, Bisen PS. Structural, functional and therapeutic biology of survivin. Cancer Lett. 2006;244(2):164–171. doi:10.1016/j.canlet.2006.03.007
13. Huynh T, Walchli S, Sioud M. Transcriptional targeting of small interfering RNAs into cancer cells. Biochem Biophys Res Commun. 2006;350(4):854–859. doi:10.1016/j.bbrc.2006.09.127
14. Friedrichs B, Siegel S, Andersen MH, Schmitz N, Zeis M. Survivin-derived peptide epitopes and their role for induction of antitumor immunity in hematological malignancies. Leuk Lymphoma. 2006;47(6):978–985. doi:10.1080/10428190500464062
15. Horii T, Arai Y, Yamazaki M, et al. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci Rep. 2014;4:4513. doi:10.1038/srep04513
16. Nakade S, Tsubota T, Sakane Y, et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun. 2014;5:5560. doi:10.1038/ncomms6560
17. Morita S, Noguchi H, Horii T, et al. Targeted DNA demethylation in vivo using dCas9–peptide repeat and scFv–TET1 catalytic domain fusions. Nat Biotechnol. 2016;34(10):1060–1065. doi:10.1038/nbt.3658
18. Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10(10):957–963. doi:10.1038/nmeth.2649
19. Haghshenas V, Fakhari S, Mirzaie S, et al. Glycyrrhetinic Acid inhibits cell growth and induces apoptosis in ovarian cancer a2780 cells. Adv Pharm Bull. 2014;4(Suppl 1):437–441. doi:10.5681/apb.2014.064
20. Pirzadeh S, Fakhari S, Jalili A, Mirzai S, Ghaderi B, Haghshenas V. Glycyrrhetinic acid induces apoptosis in leukemic HL60 cells through upregulating of CD95/CD178. Int J Mol Cell Med. 2014;3(4):272–278.
21. Wang S, Yingqi X, Chan HF, et al. Nanoparticle-mediated inhibition of survivin to overcome drug resistance in cancer therapy. J Controlled Release. 2016;240:454–464. doi:10.1016/j.jconrel.2016.04.018
22. Feng W, Yoshida A, Ueda T. YM155 induces caspase-8 dependent apoptosis through downregulation of survivin and Mcl-1 in human leukemia cells. Biochem Biophys Res Commun. 2013;435:52–57. doi:10.1016/j.bbrc.2013.04.036
23. Serrano-López J, Serrano J, Figueroa V, et al. Cytoplasmic localization of wild-type survivin is associated with constitutive activation of the PI3K/Akt signaling pathway and represents a favorable prognostic factor in patients with acute myeloid leukemia. Haematologica. 2013;98:1877–1885. doi:10.3324/haematol.2013.083642
24. Su S, Hu B, Shao J, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep. 2016;6:20070. doi:10.1038/srep20070
25. Narimani M, Sharifi M, Hakhamaneshi MS, et al. BIRC5 gene disruption via CRISPR/Cas9n platform suppress acute myelocytic leukemia progression. Iran Biomed J. 2019;23:369–378. doi:10.29252/ibj.23.6.369
26. Shoeneman JK, Ehrhart EJ
27. Morrison DJ, Hogan LE, Condos G, et al. Endogenous knockdown of survivin improves chemotherapeutic response in ALL models. Leukemia. 2012;26:271–279.
28. Xue J, Xie X-J, Lin M-F. Expression and clinical significance of antiapoptotic gene (Survivin) in NB4 and acute promyelocytic leukemia cells. Sci World J. 2012;2012.
29. Mak CSL, Yung MMH, Hui LMN, et al. MicroRNA-141 enhances anoikis resistance in metastatic progression of ovarian cancer through targeting KLF12/Sp1/survivin axis. Mol Cancer. 2017;16(1). doi:10.1186/s12943-017-0582-2
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.