")
Back to Journals » OncoTargets and Therapy » Volume 12
Knockdown Of TRIM31 Enhances Colorectal Cancer Radiosensitivity By Inducing DNA Damage And Activating Apoptosis
Authors Zhang H , Deng Y, Liang L, Shen L, Zhu J, Wang Y, Zhang J, Zhang Z
Received 15 May 2019
Accepted for publication 19 September 2019
Published 3 October 2019 Volume 2019:12 Pages 8179—8188
DOI https://doi.org/10.2147/OTT.S215769
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianmin Xu
Hui Zhang,1,2,* Yun Deng,1–3,* Liping Liang,1,2 Lijun Shen,1,2 Ji Zhu,1,2 Yaqi Wang,1,2 Jing Zhang,1,2 Zhen Zhang1,2
1Department of Radiation Oncology, Fudan University Shanghai Cancer Center, Shanghai, People’s Republic of China; 2Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, People’s Republic of China; 3Cancer Institute, Fudan University Shanghai Cancer Center, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhen Zhang
Department of Radiotherapy, Fudan University Shanghai Cancer Center, 270 Dong An Road, Shanghai 200032, People’s Republic of China
Tel +86-21-64175590-5210
Fax +86-21-64174774
Email [email protected]
Purpose: Biomarkers that predict radiosensitivity are essential for personalized radiotherapy. We performed microarray analysis for rectal cancer patients between those with good response and poor response to preoperative radiotherapy and found that patients with lower expression of tripartite motif-containing protein 31 (TRIM31) showed a better response. In this study, we confirmed the effects of TRIM31 on radiosensitivity by knockdown of TRIM31 in colorectal cancer cells.
Methods and materials: Human colorectal cancer cell lines HT-29 and SW480, which are TRIM31 stably knocked-down, were used for analysis. We studied the level of DNA damage and the change of relative proteins after irradiation in TRIM31-knockdown cells. Flow cytometry was used to test for apoptosis, cell cycle stage, and reactive oxygen species (ROS) levels after irradiation. Cell survival was measured by cloning assay. Proteins related to DNA damage were evaluated by Western blotting.
Results: The percentage of apoptotic cells and the levels of ROS were elevated, and the survival fraction was reduced in TRIM31-knockdown cells. The expression levels of the DNA damage proteins phosphorylated ataxia-telangiectasia mutation (P-ATM), DNA protein kinases (DNA-PKs), and γ-H2AX were higher in TRIM31-knockdown cells.
Conclusion: Knockdown of TRIM31 increases DNA damage and radiosensitivity in colorectal cancer cells.
Keywords: colorectal cancer, radiosensitivity, TRIM31
Introduction
Colorectal cancer (CRC) is the third most frequently diagnosed cancer and the third leading cause of cancer-related deaths in the United States.1 The incidence of CRC is rising in East Asia, especially in the more economically developed cities, as a result of Western-style diets, obesity, and smoking.2,3 Approximately 50% of CRC is in the rectum, and most rectal cancer is locally advanced or metastatic at the time of diagnosis. As the standard treatment for patients with locally advanced rectal cancer, preoperative chemoradiotherapy (CRT) can downgrade the tumor and even achieve a pathological complete response (pCR) and improve the local control.4,5 Our previous phsssase II studies6,7 showed that preoperative chemoradiotherapy was tolerable, effective and improved local control, and a high proportion of unresectable rectal cancer patients became resectable after preoperative chemoradiotherapy with many patients experiencing pCR. However, nearly 20–30% of the patients were not radiosensitive and could not benefit from preoperative chemoradiotherapy. In these patients, chemoradiotherapy even caused deterioration and greater toxicity. Therefore, it is urgent to screen for those patients who will benefit from chemoradiotherapy and those who will not. Biomarkers for radiosensitivity in rectal cancer will play an important role in this screening.
In our preliminary study, we performed a microarray analysis between completely responsive and non-responsive rectal cancer patients who underwent preoperative chemoradiation and screened for differentially expressed genes. Tripartite motif-containing protein 31 (TRIM31) was among them. We found that TRIM31 expression was much lower in the responsive patients than that in the non-responsive patients, suggesting that TRIM31 might play a critical role in the regulation of chemoradiosensitivity.
TRIM31 is a members of the TRIM family proteins, which are characterized by a common tripartite motif.8 The tripartite motif consists of a RING domain, one or two B-box domains, and a coiled-coil domain. Some TRIM family proteins function as E3 ubiquitin ligases in ubiquitin-mediated protein degradation pathways,8 including proteins involved in tumor development and progression.9 TRIM31 is highly expressed in the gastrointestinal tract and regulates tumor cell proliferation.10,11 In this study, we used rectal cancer cell lines to further study the effect of TRIM31 on the radiosensitivity of rectal cancer. We hypothesized that TRIM31 regulates radiosensitivity in colorectal cancer by interacting with proteins in cell damage pathways. TRIM31 may provide a novel biomarker for personalized chemoradiation treatment.
Methods And Materials
Cell Cultures And Construction
Two colorectal cancer cell lines (SW480 and HT-29), obtained from the American Type Culture Collection (ATCC, MD, USA), were cultured. HT-29 was grown in McCoy’s 5A medium, and SW480 was cultured in Dulbecco’s modified Eagle’s medium (Life Technologies, Grand Island, NY, USA). All media were supplemented with 10% fetal bovine serum, 100 U/mL penicillin G, and 100 mg/mL streptomycin. All cultures were maintained in a humidified atmosphere of 5% CO2 at 37°C. The pLKO.1 cloning vector (Addgene: 10878) was used to express shRNAs against TRIM31. In brief, 21-bp oligos targeting TRIM31 were ligated into the pLKO.1 cloning vector to generate the pLKO.1-shTRIM31 construct. pLKO.1-control containing scrambled non-target shRNA was used as a control. Both SW480 and HT-29 cell lines were stably infected with pLKO.1-shTRIM31 and pLKO.1-control lentiviral plasmids and were named HT-29 shTRIM31, SW480 shTRIM31, and control. We confirmed the knockdown efficiency of HT-29 shTRIM31 and SW480 shTRIM31 both at the protein and mRNA levels of TRIM31.
Irradiation
All X-ray radiation was delivered using the Xstrahl Small Animal Radiation Research Platform (SARRP) 200 (Xtrahl life Sciences, Suwannee, GA) with a dose rate of 3.845 Gy/min (200 KV and 13 mA).
Apoptosis Analysis
To test whether TRIM31 affected tumor radiosensitivity through apoptosis and related pathways, we first measured the proportion of apoptotic cells after X-ray exposure using an Annexin V/PI Apoptosis Detection Kit (BD Bioscience, CA, USA). Briefly, HT-29 shTRIM31, SW480 shTRIM31, and control cells were seeded in 6-well plates overnight. After 24 h of exposure to 0 Gy and 6 Gy radiation, the cells and the supernatant were harvested by trypsinization and centrifuged at 1500 r/min for 8 min before being washed twice with cold PBS and suspended in binding buffer. The cell suspension was stained with anti-human Annexin antibody and propidium iodide (PI) in the dark for 30 min and immediately analyzed by flow cytometry (Beckman Coulter, CA, USA). The experiments were repeated three times independently.
Cell Cycle Analysis
The effect of TRIM31 on the cell cycle redistribution of irradiated HT-29 and SW480 cells was assessed using propidium iodide (PI) staining by flow cytometry. Cells were seeded in 6-well plates at 50% confluency, and the next day a single dose of 4 Gy was delivered. Cells were then collected at designated time points using trypsin and washed twice with pre-chilled PBS. After ethanol precipitation at −20 °C overnight, the cells were suspended and stained with PI solution (containing 50 μg/mL propidium iodide, 10 μg/mL RNAse, and 0.02% Triton X-100) for 30 min in the dark at room temperature. Samples were acquired using flow cytometry (Beckman Coulter), and cell cycle distribution was analyzed using Modfit software. The experiments were repeated three times independently.
Reactive Oxygen Species
To test the level of reactive oxygen species (ROS) after radiation in HT-29 and SW480 cells, the fluorescence probe DCFH-DA (Beyotime Biotechnology, Jiangsu, China) was used. Cells were seeded in 6-well plates at 5×105/well overnight and then exposed to 0 Gy or 4 Gy. The cells were collected by trypsinization 24 h after irradiation and washed twice with cold PBS. The production of ROS was measured by performing flow cytometry using the oxidation-sensitive probe DCFH-DA according to the manufacturer’s instructions. Briefly, the probe was diluted with non-FBS culture medium at a working concentration of 10 μM. The cells were suspended with 500 μl of DCFH-DA working solution and incubated at 37°C in the dark for 20 min, then were slightly mixed every 5 min. The cells were then washed three times with cold PBS to remove unbound probe and immediately analyzed by flow cytometry. The experiments were repeated three times independently.
Clonogenic Survival Assay
Colony formation was used to evaluate the radiosensitivity in HT-29 and SW480 cells. A specific number of cells (approximately 100 clones per well after harvest) were seeded in 6-well plates in triplicate after being trypsinized into single-cell suspensions. After attaching to the bottom of the plates overnight, the cells were irradiated with a series of doses (0, 2, 4, 6, and 8 Gy for SW480 cells, and 0, 2, 4, 6, 8, and 10 Gy for HT-29 cells). Cells were continually incubated for two weeks after irradiation, then fixed with 4% paraformaldehyde and stained with 1% crystal violet. The colonies containing more than 50 cells were counted. The plating efficiency (PE) was the percentage of seeded cells that grew into colonies. The survival fraction (SF) was calculated as SF=PEirradiated/PEcontrol, and a survival curve was fitted using a linear-quadratic model (LQ model) with the equation S=exp−(αD+βD2), where S denotes survival probability, D (Gy) is the radiation dose, and α (Gy−1) and β (Gy−2) are constants. The sensitization enhancement ratio at the surviving fraction of 10% (SER10) was calculated as follows: SER10=Dcontrol/DshTRIM31. The experiments were repeated three times independently.
Immunofluorescence Staining For γ-H2AX
HT-29 cells were seeded and cultured overnight on glass cover slides in 24-well plates at a concentration of 5×104 per well. At different time points after 8 Gy radiation, cells were fixed with 4% paraformaldehyde for 30 min at room temperature and then permeabilized using 0.2% Triton X-100 for 10 min on ice. After being blocked with 5% BSA for 1 h, the cells were incubated with anti-γ-H2AX fluorescent antibody (Abcam, Cambridge, UK) for 2 h at room temperature and washed with PBS three times, and nuclei were stained with DAPI. Images were captured using confocal microscopy (×63 oil) (Leica, Wetzlar, Germany).
Immunoblot Analysis
The total proteins of HT-29 shTRIM31 and control cells were collected with lysis buffer (Thermo Scientific, Waltham, MA, USA), including 1× protease inhibitor cocktail mix (Roche, Basel, Switzerland) and 1× PhosSTOP (Roche) at denoted times following irradiation. The protein concentration was measured with BCA Protein Assay Reagent (Thermo Scientific). Equal amounts of proteins were loaded onto 7.5–15% Tris-Glycine gels and then electrophoretically transferred to a PVDF membrane (Roche). The PVDF membrane was blocked with 5% BSA in TBS-Tween20 (0.1%, v/v) for 1 h and then incubated with the primary antibody at room temperature for 2 h or overnight at 4°C, followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibody (Millipore, Darmstadt, Germany) at a dilution of 1:5000 for 1 h at room temperature. Protein bands were visualized through chemiluminescence (Thermo Scientific) and protein bands images were captured using a Chemiscope3300 mini (Clinx, Shanghai, China). The primary antibodies used at a dilution of 1:1000 were as follows: rabbit anti-ATM, anti-phospho-ATM, rabbit anti-phospho-γ-H2AX, rabbit anti-DNA-PK (Cell Signaling Technology, Danvers, MA, USA), and mouse anti-GAPDH antibody (Santa Cruz Biotechnology, Dallas, Texas, USA). The experiments were repeated three times independently. Densitometrical analysis was performed on the software of Image J software.
Statistical Analysis
Statistical analysis was carried out using Graphpad InStat 7 software (GraphPad Software, San Diego, CA, USA). Data showing comparisons between two groups were assessed using Student’s t-test. Comparisons among more than two groups were analyzed using ANOVA with the appropriate post hoc testing. P<0.05 was considered statistically significant.
Results
TRIM31 Knockdown Radiosensitized SW480 And HT-29 Cells
We knocked down TRIM31 using a lentivirus particle shTRIM31 and confirmed the knockdown efficiency both at the protein and mRNA levels. As shown in Figure 1A and B, the knockdown efficiencies of HT-29 and SW480 were 88% and 85%, respectively, and the protein level of TRIM31 further confirmed successful knockdown (Figure 1C and D). We first performed clonogenic survival assays to check if TRIM31 could modulate radiosensitivity in HT-29 and SW480 cells. As shown in Figure 1E and F, knockdown of TRIM31 significantly radiosensitized HT-29 and SW480 cells with enhancement ratio (SER10) of 1.28 and 1.64, respectively. These data indicate that TRIM31 is a potential candidate of radiosensitizing colorectal cancer cells.
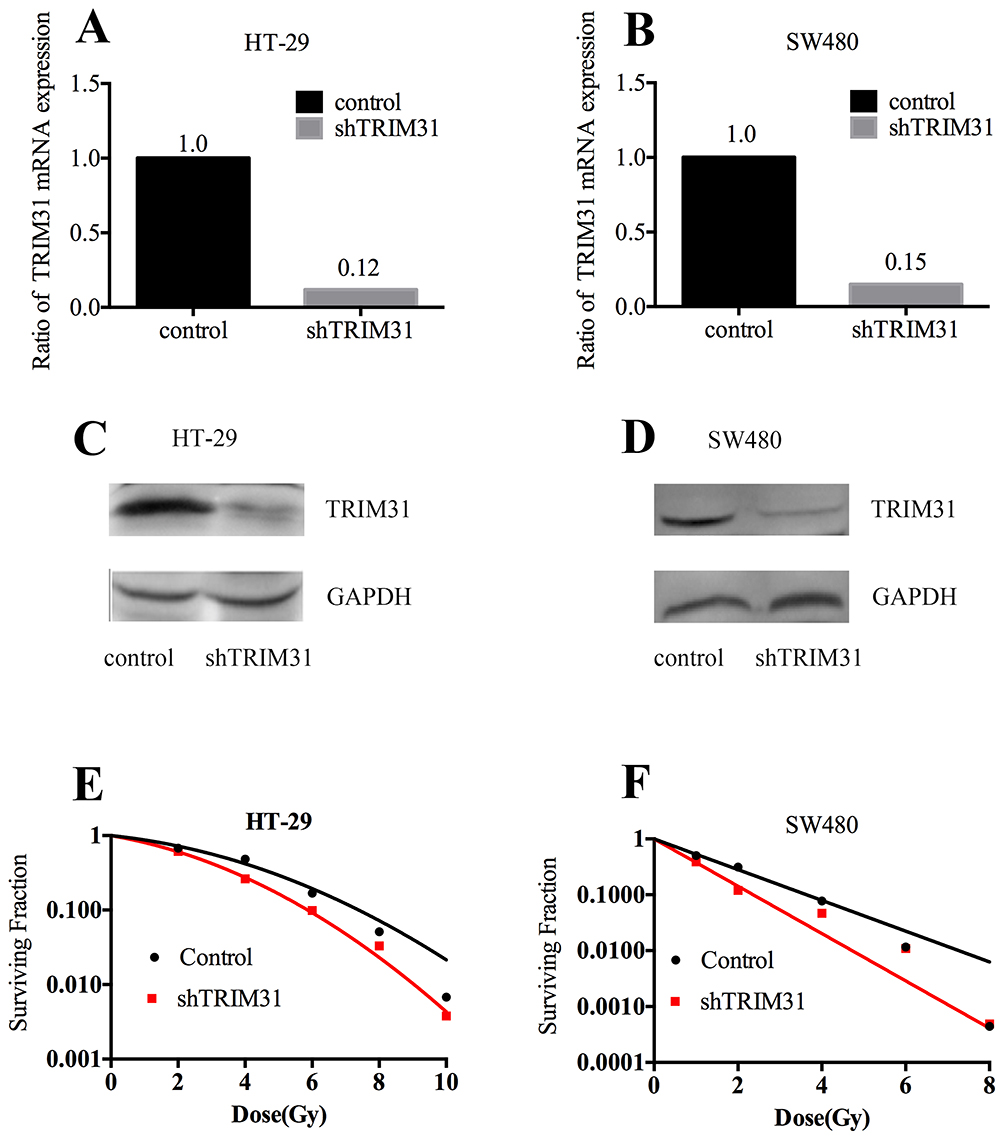 |
Figure 1 TRIM31 modulates radiosensitivity in HT-29 and SW480 colorectal cancer cells. TRIM31 knockdown efficiency at the mRNA level in HT-29 (A) and SW480 (B) cell lines. Ratio of TRIM31 mRNA expression = TRIM31 mRNA expression/TRIM31 mRNA expression in the control. Validation of TRIM31 knockdown at the protein level in HT-29 (C) and SW480 (D) cell lines. (E) HT-29 shTRIM31 and control cells received irradiation at 0 Gy, 2 Gy, 4 Gy, 6 Gy, 8 Gy, and 10 Gy. SER10= 1.28. (F) SW480 shTRIM31 and control cells were irradiated at 0 Gy, 2 Gy, 4 Gy, 6 Gy, and 8 Gy. SER10= 1.64. SER10=survival enhancement ratio at a surviving fraction of 0.10. |
TRIM31 Knockdown Increased Cell Death And Changed The Cell Cycle Distribution After Radiation In SW480 And HT-29 Cell Lines
The most severe and direct result for radiation is cell death, while more commonly, it causes cell damage and cell cycle arrest. Here, we first tested whether the radiosensitivity induced by TRIM31 knockdown was due to activating cell apoptosis. Apoptosis analysis was performed in HT-29 and SW480 cell lines with Annexin V-FITC/PI staining using flow cytometry. As shown in Figure 2A and B, knockdown of TRIM31 significantly increased the radiation-induced apoptosis in both HT-29 and SW480 cell lines compared to the control cells and was statistically different (Figure 2C and D, P<0.01). We investigated whether there were any differences in radiation-induced cell cycle arrest following TRIM31 knockdown. HT-29 cells received 4 Gy irradiation, and the DNA content was measured at 12 h, 24 h, and 48 h after irradiation. Before irradiation, the distribution of the cell cycles in both HT-29 shTRIM31 and control cells was basically the same, which means TRIM31 itself has no direct effect on the cell cycle. After 4 Gy irradiation, knockdown of TRIM31 in HT-29 cells resulted in increased radiation-induced G2/M arrest at 24 h and 48 h compared with the control (Figure 2E, P<0.01 at 24 h, and P<0.05 at 48 h). In addition, the proportion of cells in the S phase decreased at 24 h after irradiation (P<0.01). This might be one of the mechanisms by which TRIM31 modulates radiosensitivity in colorectal cancer cells. And the effect may be most obvious at 24 h after irradiation by both G2/M phase arrest and S phase inhibition. The potential mechanism may be that the expression of cell cycle proteins, which are targeted by TRIM31, changes obvious at 24 h after irradiation.
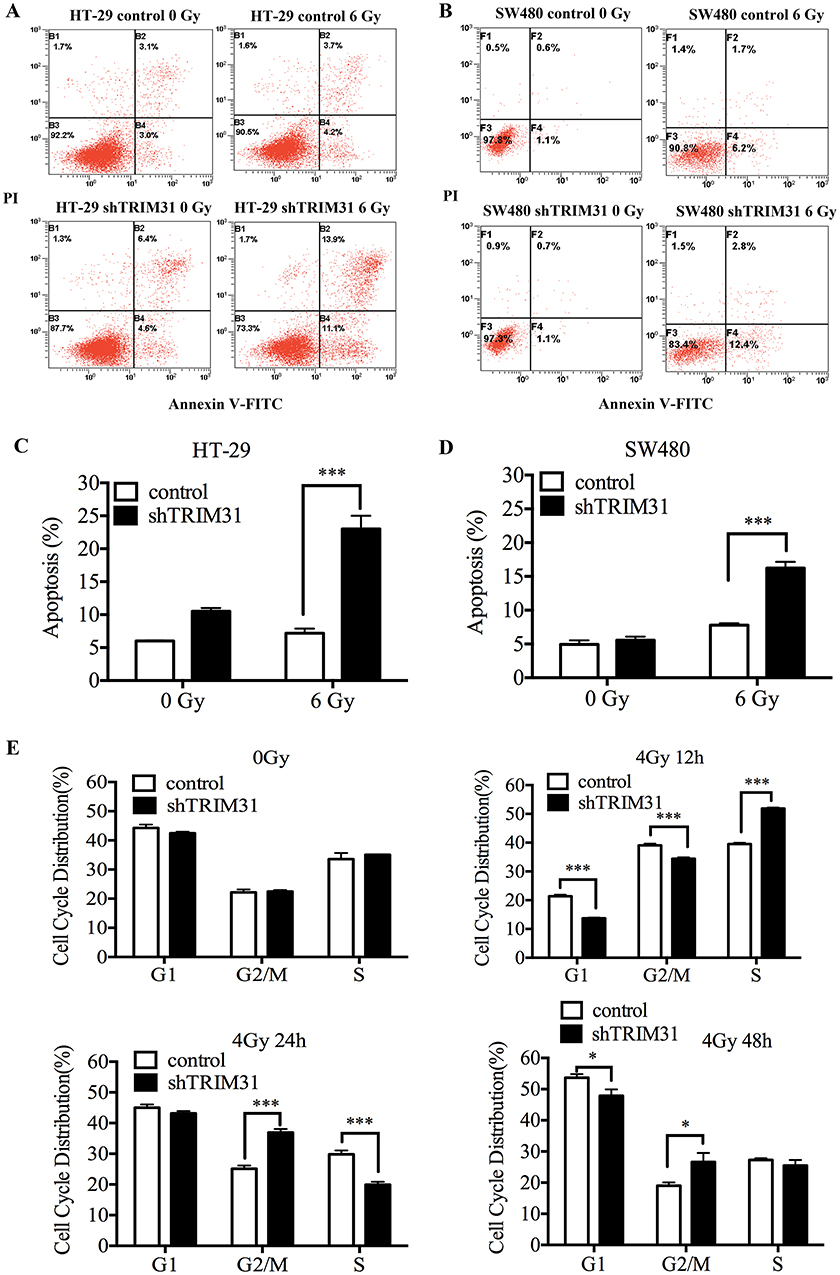 |
Figure 2 TRIM31 Knockdown increases cell death and changes the cell cycle distribution after radiation. The levels of apoptosis in HT-29 (A) and SW480 (B) cells before irradiation and 24 h following 6 Gy irradiation. Apoptosis (%) = early apoptosis (%) + late apoptosis (%). The difference between the two groups by statistical analysis in HT-29 (C) and SW480 (D), respectively. (E) Knockdown of TRIM31 increases the proportion of cells in G2/M phase and decreases those in S phase at 12h and 24h after irradiation. (*P<0.05; ***P<0.01). The experiments were repeated three times independently. |
TRIM31 Knockdown Increased IR-Induced DNA Damage And ROS Production
Ionization radiation (IR) directly ionizes DNA molecules and induces DNA double-strand breaks (DSBs), which is the most critical trigger of genomic instability. To evaluate whether knockdown of TRIM31 could aggregate DSBs after irradiation, γ-H2AX foci, which are a marker of DSBs, were counted and analyzed in HT-29 cells at different times after 8 Gy irradiation using immunofluorescent staining, and the foci number from 500 nuclei were determined for individual samples. γ-H2AX foci were most obvious at 3 h after 8 Gy. Figure 3A shows foci staining in HT-29 cells at 3 h after 8 Gy, and we found that γ-H2AX foci were significantly increased in the HT-29 shTRIM31 group compared to the control group (P<0.001) (Figure 3B), which meant that in HT-29 shTRIM31 cells, ionizing radiation caused much more DNA damage.
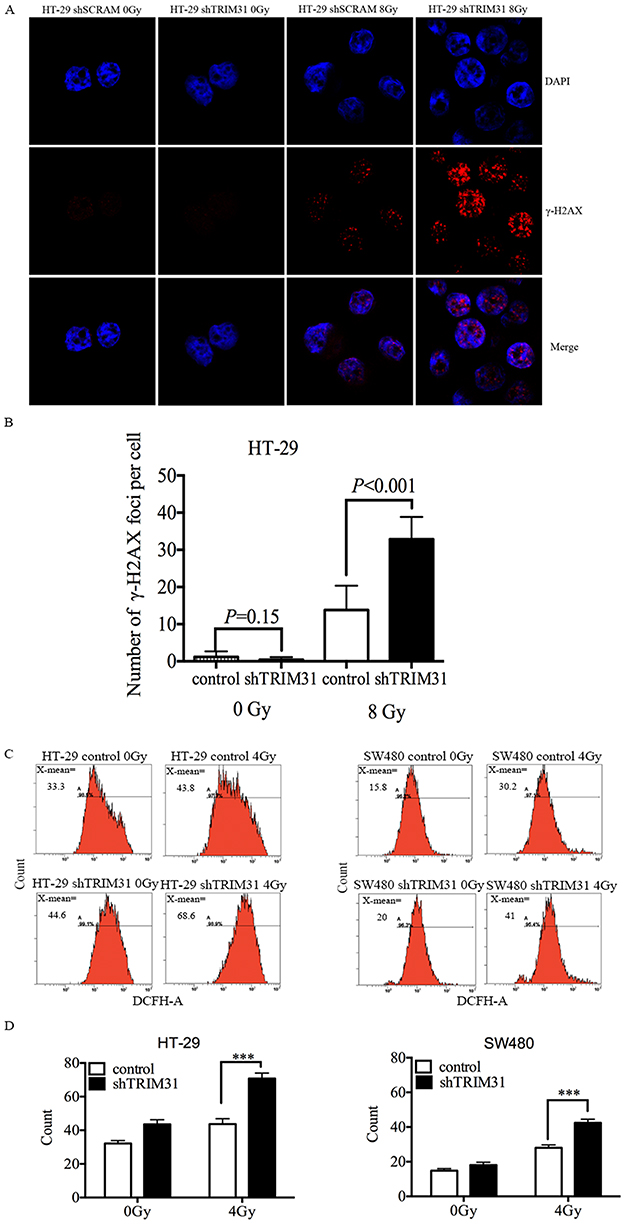 |
Figure 3 TRIM31 knockdown increases IR-induced DNA damage and ROS production. (A and B) Knockdown of TRIM31 increases γ-H2AX foci at 3 h after 8 Gy irradiation in HT-29 cells. HT-29 cells received 8 Gy irradiation and were stained with γ-H2AX and DAPI (indicates cell nuclei) at different times after irradiation, then the images of γ-H2AX and DAPI were merged, γ-H2AX foci were much more frequent in HT-29 shTRIM31 cells than in HT-29 shSCRAM cells at 3 h (A). The numbers of γ-H2AX foci were calculated for individual samples and compared between HT-29 shTRIM31 and control (B). (C and D) The levels of intracellular ROS for HT-29 and SW480 cells before irradiation and 24 h following 4 Gy irradiation. Cells were exposed to 0 and 4 Gy X-rays, and the fluorescent probe DCFH-DA was used to test ROS 24h after irradiation. Each cell line was seeded in triplicate (N=3). The differences between the two groups were determined by statistical analysis. ROS: reactive oxygen species. ***P<0.01. The experiments were repeated three times independently. |
IR can cause DNA damage directly and can also interact with atoms or molecules (mainly water) in cells to produce free radicals, such as ROS, which further exacerbates DNA damage. Therefore, we checked the ROS level in HT-29 and SW480 cells. As shown in Figure 3C and D, before irradiation, the basic levels of ROS were low in both cell lines, and knockdown of TRIM31 did not cause any difference, which indicated that TRIM31 itself did not influence ROS metabolism. When cells were irradiated, the levels of ROS increased in all cell lines after exposure to 4 Gy X-rays 24 h. The ROS level was much higher in HT-29 shTRIM31 cells than in control cells (70.86±2.01 vs 43.68±1.43, P<0.01) 24 h after exposure to 4 Gy. The results were similar in SW480 cells (Figure 3C and D), with higher ROS levels in SW480 shTRIM31 cells than in control (42.4±1.18 vs 28.04±0.89, P<0.01).
Knockdown TRIM31 Influenced DNA Damage And Repair Pathways
Based on the aforementioned data, we speculated that knockdown TRIM31 radiosensitized colorectal cancer cells by influencing DNA damage and repair pathways, so we measured the levels of proteins related to DNA damage and repair in HT-29 cells, including DNA damage sensors: ATM, DNA-PKs, and DNA damage sensor: γ-H2AX. ATM and P-ATM were much higher in HT-29 shTRIM31 than in the control group at 24 h after 8 Gy radiation (Figure 4A), and this was statistically significant. After 8 Gy radiation, the levels of DNA-PKs and γ-H2AX were elevated continuously in HT-29 shTRIM31 cells compared to control cells (Figure 4A), and the gray value difference was statistically significant between shTRIM31 and control at 24 h and 48 h for DNA-PK, and at 12 h, 24 h, and 48 h for γ-H2AX (Figure 4B), which meant that DNA repair was deficient and DSBs existed continually.
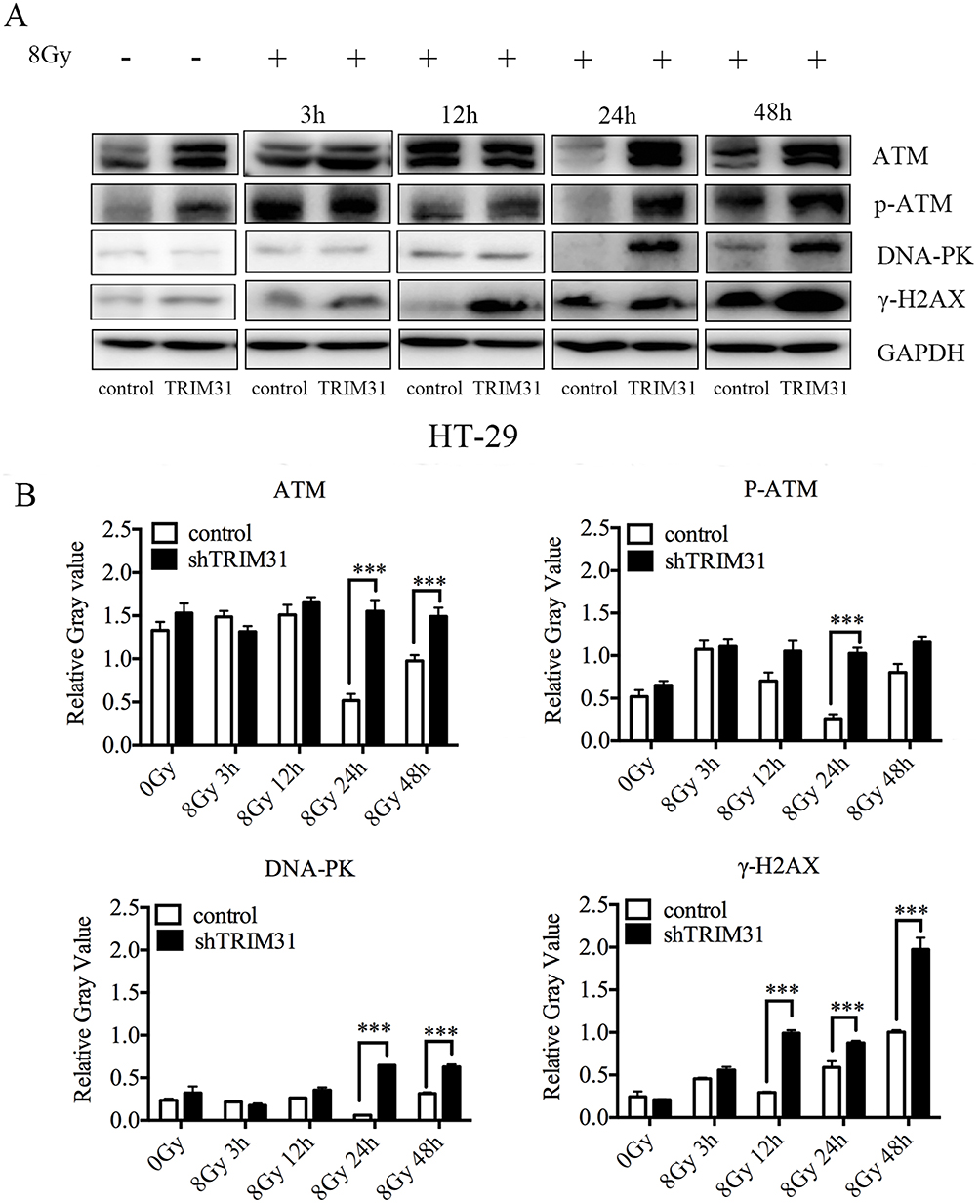 |
Figure 4 Knockdown of TRIM31 influences DNA damage and repair pathways. (A) Western blotting was used to investigate the expressions of proteins involved in DNA damage and repair, and densitometric analysis was performed using Image J software. (B) Relative gray value = Gtarget protein/GGAPDH (G: gray value). ***P<0.01. The experiments were repeated three times independently. |
Discussion
Exploiting molecular markers to predict radiosensitivity in rectal cancer is an essential strategy for the development of individual clinical therapies. TRIM31 is an E3 ubiquitin-protein ligase that has a tripartite motif containing a RING domain that confers ubiquitin ligase activity. It plays an important role in innate immune response, in particular by negatively regulating viral entry into host cells; it is also a negative regulator of Src-induced anchorage-independent cell growth in gastrointestinal tract, shows altered expression in certain tumors, and may be a negative regulator of cell growth.10,12–14 However, recently a study shows that TRIM31 promote glioma proliferation and invasion.15 In addition, it is reported that TRIM31 overexpression confers gemcitabine resistance on pancreatic cancer.16 TRIM31 also medicates inflammatory effect through the NF-κB pathway.17 However, there are no studies on the relationship between TRIM31 and radiosensitivity. We found that the expression of TRIM31 was lower in radiation-responsive rectal cancer patients, and knockdown TRIM31 radiosensitized colorectal cancer cells by aggregating DNA damage and inducing ROS production after radiation. During this process, TRIM31 may be involved in DNA damage and repair pathways through interacting with ATM.
The biologic effects of radiation, like cell death and redistribution of the cell cycle, involve many pathways, especially DNA damage repair pathways, including the ATM and P53 pathway.18,19 We first found that knockdown of TRIM31 significantly increased radiation-induced apoptosis, which explains why TRIM31 knockdown radiosensitized colorectal cancer cells in this study. Radiation-induced G2/M arrest was also very profound after TRIM31 downregulation. A previous study showed that TRIM31 overexpression decreases the protein expression of cyclin D1 and cyclin E,20 which are cell cycle regulators, which is consistent with our results showing that TRIM31 regulates the cell cycle. Cell cycle arrest allows injured cells to have enough time to repair DNA damage and avoid cell death. In this case, knockdown of TRIM31 should protect the cells from death. So, we hypothesized that this prolonged G2/M arrest was caused by persistent DNA damage without repair. Indeed, we found that γ-H2AX foci existed along with the activation of ATM and DNA-PK. In addition, we detected excessive ROS production in TRIM31-knockdown cells after irradiation, which could prompt and aggregate DNA damage. Radiation induces G2/M arrest by activating the ATM/Chk2 pathway, which is followed by the activation of P53, causing cell apoptosis; furthermore, the persistent DNA damage triggers the components of the DNA damage and repair pathway.21 In TRIM31-downregulated cells, the levels of ATM, P-ATM, and DNA-PKs were much higher compared to those in control cells. Both ATM and DNA-PK are DNA damage sensors and initiate DNA repair, and ATM can phosphorylate histone H2AX at DSBs, thereby regulating the DNA damage response.22 We noticed that unlike DNA-PK, even without radiation, TRIM31-knockdown cells still had a higher level of ATM protein, although without statistical significance, which meant that TRIM31 might regulate ATM by a different pathway. To discriminate the potential mechanisms by whichTRIM31 regulates ATM and the DNA-PK pathway, we applied mass spectrometry to identify its possible interaction factors, and we found that ATM was a candidate interaction proteins. TRIM31 is an E3 ubiquitin-protein ligase that can exert its role by promoting the degradation of its target proteins, we inferred that TRIM31might inhibit ATM expression via protein degradation, but this needs further study.
In summary, knockdown of TRIM31 after irradiation increased ROS production, exacerbated radiation-induced DNA damage, and prolonged G2/M arrest, consequently causing apoptosis and radiosensitization in colorectal cancer cells. During this process, TRIM31 may interact with ATM to regulate this series of events induced by radiation. Through their E3 ligase protein degradation function, TRIM family proteins are implicated in the negative regulation of the innate immune response, which provides a good opportunity to design drugs that target these family proteins, as there are many potential inhibitors, and the generally low expression of TRIM31 in human tissues could minimize the side effects of such drugs. In addition, colorectal cancer cells with inflammation infiltration have better survival, and thus TRIM31 may be a useful therapeutic target for colorectal cancer.
Conclusion
The expression of TRIM31 affects the radiosensitivity of colorectal cancer cells via complicated mechanisms that need further exploration to be clinically useful.
Acknowledgments
This study was supported by the grants from the National Natural Science Foundation of China (grant numbers 81372432, 81572955 and 81773357) and National Natural Science Foundation of Shanghai (grant number 13ZR1408100). We would like to thank Ran Hu, Guichao Li, Wang Yang and Ye Yao of the Fudan University Shanghai Cancer Center for their contributions to experiments in part of the study, which are not displayed in this paper. We thank LetPub for its linguistic assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi:10.3322/caac.21254
2. Li L, Ma BB. Colorectal cancer in Chinese patients: current and emerging treatment options. Onco Targets Ther. 2014;7:1817–1828. doi:10.2147/OTT.S48409
3. Center MM, Jemal A, Ward E. International trends in colorectal cancer incidence rates. Cancer Epidemiol Biomarkers Prev. 2009;18(6):1688–1694. doi:10.1158/1055-9965.EPI-09-0090
4. Sauer R, Liersch T, Merkel S, et al. Preoperative versus postoperative chemoradiotherapy for locally advanced rectal cancer: results of the German CAO/ARO/AIO-94 randomized phase III trial after a median follow-up of 11 years. J Clin Oncol. 2012;30(16):1926–1933. doi:10.1200/JCO.2011.40.1836
5. Zorcolo L, Rosman AS, Restivo A, et al. Complete pathologic response after combined modality treatment for rectal cancer and long-term survival: a meta-analysis. Ann Surg Oncol. 2012;19(9):2822–2832. doi:10.1245/s10434-011-2209-y
6. Zhu J, Lian P, Liu F, et al. Phase II trial of first-line chemoradiotherapy with intensity-modulated radiation therapy followed by chemotherapy for synchronous unresectable distant metastases rectal adenocarcinoma. Radiat Oncol. 2013;8:10. doi:10.1186/1748-717X-8-10
7. Zhu J, Liu F, Gu W, et al. Concomitant boost IMRT-based neoadjuvant chemoradiotherapy for clinical stage II/III rectal adenocarcinoma: results of a phase II study. Radiat Oncol. 2014;9:70. doi:10.1186/1748-717X-9-70
8. Ikeda K, Inoue S. TRIM proteins as RING finger E3 ubiquitin ligases. Adv Exp Med Biol. 2012;770:27–37. doi:10.1007/978-1-4614-5398-7_3
9. Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11(11):792–804. doi:10.1038/nrc3139
10. Watanabe M, Tsukiyama T, Hatakeyama S. TRIM31 interacts with p52(Shc) and inhibits Src-induced anchorage-independent growth. Biochem Biophys Res Commun. 2009;388(2):422–427. doi:10.1016/j.bbrc.2009.08.028
11. Sugiura T, Miyamoto K. Characterization of TRIM31, upregulated in gastric adenocarcinoma, as a novel RBCC protein. J Cell Biochem. 2008;105(4):1081–1091. doi:10.1002/jcb.21908
12. Li Y, Wu H, Wu W, et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014;24(6):762–765. doi:10.1038/cr.2014.46
13. Micale L, Chaignat E, Fusco C, Reymond A, Merla G. The tripartite motif: structure and function. Adv Exp Med Biol. 2012;770:11–25.
14. Ozato K, Shin DM, Chang TH, Morse HC
15. Zhou L, Deng ZZ, Li HY, et al. TRIM31 promotes glioma proliferation and invasion through activating NF-κB pathway. Onco Targets Ther. 2019;12:2289–2297. doi:10.2147/OTT.S183625
16. Yu C, Chen S, Guo Y, Sun C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics. 2018;8(12):3224–3236. doi:10.7150/thno.23259
17. Wang H, Yao L, Gong Y, Zhang B. TRIM31 regulates chronic inflammation via NF-κB signal pathway to promote invasion and metastasis in colorectal cancer. Am J Transl Res. 2018;10(4):1247–1259.
18. Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3(2):117–129. doi:10.1038/nrc992
19. Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31(7):402–410. doi:10.1016/j.tibs.2006.05.004
20. Li H, Zhang Y, Zhang Y, Bai X, Peng Y, He P. TRIM31 is downregulated in non-small cell lung cancer and serves as a potential tumor suppressor. Tumour Biol. 2014;35(6):5747–5752. doi:10.1007/s13277-014-1763-x
21. Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi:10.1146/annurev.biochem.73.011303.073723
22. Novotna E, Tichy A, Pejchal J, Lukasova E, Salovska B, Vavrova J. DNA-dependent protein kinase and its inhibition in support of radiotherapy. Int J Radiat Biol. 2013;89(6):416–423. doi:10.3109/09553002.2013.767993
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.