")
Back to Journals » OncoTargets and Therapy » Volume 11
Knockdown of KLK11 reverses oxaliplatin resistance by inhibiting proliferation and activating apoptosis via suppressing the PI3K/AKT signal pathway in colorectal cancer cell
Authors Zhang Y, Xu Z, Sun Y, Chi P, Lu X
Received 16 September 2017
Accepted for publication 4 December 2017
Published 16 February 2018 Volume 2018:11 Pages 809—821
DOI https://doi.org/10.2147/OTT.S151867
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Yiyi Zhang,* Zongbin Xu,* Yanwu Sun,* Pan Chi, Xingrong Lu
Department of Colorectal Surgery, Fujian Medical University Union Hospital, Fuzhou, People’s Republic of China
*These authors contributed equally to this work
Introduction: Kallikrein 11 (KLK11) plays a crucial role in drug-resistance to oxaliplatin (L-OHP) in the treatment of metastatic colorectal cancer (mCRC). The study aimed to investigate the role of KLK11 in chemoresistance, and to clarify the mechanism underlying reverse of L-OHP resistance by knockdown of KLK11.
Materials and Methods: Resistance to oxaliplatin was induced in HCT-8 (HCT-8/L-OHP) colorectal adenocarcinoma cell lines by exposing cells to increasing concentrations of L-OHP. MTT, RT-qPCR, and Western blot were used to evaluate the resistance to L-OHP. We then knocked down KLK11 in HCT-8/L-OHP cells to explore the mechanism through which KLK11 reverses L-OHP resistance. The mRNA and protein expression of KLK11 in tissues from mCRC patients were detected by RT-qPCR and immunohistochemistry.
Results: The drug resistance index (RI) of HCT-8/L-OHP cell line to L-OHP, 5-Fluorouracil (5-FU), Irinotecan (CPT-11), Vincristine (VCR) and Cis-diamminedichloroplatinum (CDDP) were 10, 5.35, 3.23, 1.28, and 6.64, respectively. Increased expression of multi-drug resistant genes ABCC1, ABCB1, GSTP1 and ERCC1 were detected in HCT-8/L-OHP cell line. Moreover, the activated PI3K/AKT pathway was related to L-OHP-resistance. Knockdown of KLK11 in HCT-8/L-OHP cell reversed L-OHP-resistance by inhibiting cell growth and activating apoptosis via suppressing the PI3K/AKT signaling pathway. Moreover, high expression of KLK11 in chemoresistant-patients was associated with lymph node metastases and histopathology.
Conclusion: KLK11 was highly expressed in chemoresistant-patients and L-OHP-resistant cell lines. Moreover, L-OHP resistance was associated with activated PI3K/AKT signal pathway. Knockdown of KLK11 can reverse L-OHP resistance by blocking PI3K/AKT signaling pathway.
Keywords: colorectal cancer, kallikrein 11, oxaliplatin, drug resistance, apoptosis
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-related deaths in the world.1 Chemotherapy, together with surgery, radiotherapy and immunotherapy, constitutes the most common treatment for patients with CRC. Oxaliplatin (L-OHP), a third-generation platinum compound, is a promising therapeutic drug for the treatment of advanced CRC, typically in combination with 5-fluorouracil (5-FU) and leucovorin (FOLFOX/CapeOX).2 Unfortunately, approximately 50% of patients with CRC develop resistance to chemotherapeutic agents, such as oxaliplatin, and this thus results in disease recurrence.3 However, studies focused on these issues are limited, and the exact mechanism responsible for resistance to L-OHP remains largely unclear. Exploring the underlying mechanism reversal of L-OHP resistance has generated huge research interest. The phosphatidylinositil-3-kinase (PI3K)/AKT pathway plays a crucial role in tumorigenesis, and is associated with the proliferation, invasion, and metastasis of cancer cells.4 In addition, accumulating evidence has demonstrated that the PI3K/AKT pathway is involved in the drug resistance of cancer cells.5–7 However, whether the PI3K/AKT pathway is associated with L-OHP resistance in CRC has not yet been elucidated.
Human tissue kallikreins (KLKs), a family of 15 closely related serine proteases, have been demonstrated to possess protease activities, and are involved in various physiological processes, such as regulating apoptosis, proliferation, and cytokines.8 Human kallikrein 11 (KLK11) is a member of the human KLK gene family and was originally isolated from the human hippocampus.9 Previous studies have shown that KLK11 is expressed in human brain, skin, gastric, breast, prostate, ovarian, and intestinal tissues.10–15 Alexopoulou et al16 demonstrated that the expression of KLK11 mRNA was upregulated and it could be used as a novel prognostic biomarker of CRC, whereas Talieri et al did not provide similar results.17 In our previous study, we have identified that the differential mRNA expression of KLK11 was associated with chemosensitivity in patients with CRC. Furthermore, knockdown of KLK11 increased oxaliplatin sensitivity, and knockdown of KLK11 inhibited cell proliferation in human CRC cell lines.18
In this study, we successfully established the L-OHP resistance CRC cell line HCT-8/L-OHP, and demonstrated that the activated PI3K/AKT/apoptosis signal pathway was involved in L-OHP resistance. KLK11 is highly expressed in the HCT-8/L-OHP cell line and chemotherapy-resistant patients. Moreover, knockdown of KLK11 in the oxaliplatin-resistant CRC cell line HCT-8/L-OHP reverses oxaliplatin resistance by inhibiting cell proliferation and inducing cell apoptosis via suppressing the PI3K/AKT pathway.
Materials and methods
Patients, chemotherapy and follow-up
The Fujian Medical University Union Hospital Ethics Committee approved this study, and all patients provided written informed consent for the scientific use of the clinical tissue samples.
A total of 55 patients with metastatic colorectal cancer (mCRC) who received the neoadjuvant FOLFOX4/CapeOX chemotherapy regimen between January 2013 and December 2013 were identified from our prospective CRC database. The medical data of all patients was supplied by the Ethics Committee, and had already been de-identified. The Response Evaluation Criteria in Solid Tumors (RECIST) criteria were used to assess patient response following completion of the three cycles of chemotherapy.19 Among these patients, complete response (CR; two patients) and partial response (PR; 23 patients) were included in the chemotherapy-sensitive group (25 patients); stable disease (SD; eight patients) and progressive disease (PD; 22 patients) were included in the chemotherapy-resistant group (30 patients). Patient follow-up lasted until death or until the cutoff date of January 31, 2017.
Collection of surgical specimens
All eligible patients underwent R0 resection of primary colorectal tumors, which were documented pathologically, following the completion of neoadjuvant chemotherapy. Partial surgical specimens were harvested and cryopreserved in liquid nitrogen for further experiments.
Immunohistochemical analysis
The concentration of KLK11 protein in the paraffin–wax-embedded samples from 55 patients with mCRC was measured using the immunohistochemical streptavidin–biotin complex method.20 Phosphate-buffered saline (PBS) was used for the negative control, and the image of the positive control was from GE Healthcare Life Sciences. The criteria21 were used as follows: the percentage of positive cells for each of the sections and the color was determined on the basis of the intensity score. The intensity score as follows: 0), no staining; 1), light yellow staining; 2), brown staining; and 3), deep brown staining. Furthermore, the percentage of positive cells was scored as follows: 0, <5% stained cells; 1, 5%–25% stained cells; 2, 25%–50% stained cells; 3, 50%–75% stained cells; and 4, >75% stained cells. The mean value was calculated for each case with the aforementioned scoring methods and the final score was obtained by multiplying these two scores. The expression of KLK11 was qualitatively determined by the final score: 0, for negative (−); 1–3, for weakly positive (+); 4–7, for positive (++); and 8–12, for strongly positive (+++). All analyses were conducted in a double-blind manner.
Reverse transcription-quantitative PCR
Total RNA from cells and patient tissues were isolated using TRIzol reagent (Thermo Fisher Scientific, Inc., Carlsbad, CA, USA) according to the manufacturer’s instructions, and 1 μg total RNA was used for reverse transcription reaction using M-MLV Reverse Transcriptase Product (Promega, Madison, WI, USA). Quantitative PCR (qPCR) was conducted using an ABI 7500 real-time PCR system (Thermo Fisher Scientific, Inc.), and KLK11, ABCB1, ABCC1, GSTP1, and ERCC1 mRNA levels were assessed by real-time quantitative polymerase chain reaction (RT-qPCR), with GAPDH used as an internal control. PCR amplification was done by denaturation at 94°C for 5 s, and annealing and extension at 62°C for 40 s for 40 cycles. The relative expression level of KLK11 was calculated using the ΔΔCq method. All PCR amplification was carried out in triplicate and repeated in three independent experiments. RT-qPCR analysis was conducted using primers as follows: GAPDH Forward 5′GGGAAACTGTGGCGTGAT3′, Reverse 5′GAGTGGGTGTCGC TGTTGA3′; ABCC1, Forward 5′ATCTTGGTCACGCACAGCAT3′, Reverse 5′GCATAGGTACGCAGGAACTCA3′; ABCB1, Forward 5′AACACCACTGG AGCATTGACTA C3′, Reverse 5′ATTACAGCAAGCCT GGAACCTAT3′; GSTP1, Forward 5′ACCGTGGTCTATTTCCCAGTTC3′, Reverse 5′AGGTGACGCAGGATG GT ATTG3′; ERCC1, Forward 5′TTTGGCGACGTAATTCCCG3′, Reverse 5′T CCGCTGGTTTCTGCTCATAG3′; KLK11 Forward 5′CCTAA GCCAAGACCCTC TACG A3′, Reverse 5′AACAAACCAGGTGTTGTCATTCC3′.
Cell culture and reagents
The human colon cancer HCT-8 cell lines were purchased from Shanghai Genechem Co., Ltd (Shanghai, People’s Republic of China). Resistance to L-OHP was induced in a colonic cancer cell line (HCT-8/L-OHP) created from the parental HCT-8 cell line as described further. Parental and HCT-8/L-OHP were cultured in RPMI-1640 (Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS; Hangzhou Sijiqing Bio-Engineering Material Co. Ltd., Hangzhou, People’s Republic of China), 100 U/mL penicillin and 100 mg/mL streptomycin (Thermo Fisher Scientific, Inc.) in 5% CO2 at 37°C.
Establishment of the HCT-8/L-OHP cell line
Cells from the human colon cancer cell line HCT-8 were seeded in a 25 mL culture flask, and 2 μmol/L (1/3 half maximal inhibitory concentration [IC50]) L-OHP solution was added into the medium; the medium was changed after 48-h incubation. When cells recovered to normal growth and subculture, treatment with the same drug concentration was repeated. The cell culture with L-OHP was repeated 81 times (L-OHP concentration increased by 1 μmol/L every three subcultures). The HCT-8 cell line from the last subculture, which was resistant to 30 μmol/L L-OHP, was cultured in a complete culture medium containing 4 μmol/L L-OHP, and was defined as HCT-8/L-OHP. The period for the establishment of the drug-resistant cell line was 10 months.
Gene silencing with the lentivirus encoding specific RNAi
In order to silence KLK11, the RNA interference (RNAi) was generated by ligating synthetic oligonucleotides (Thermo Fisher Scientific, Inc.) against the target genes into the AgeI and EcoRI sites of a pLKO.1-TRC cloning vector (designed by Shanghai GeneChem Co., Ltd. [Shanghai, People’s Republic of China]). The sequences of the KLK11 RNAi were as follows: KLK11-RNAi (41637), 5′-AGCACCAGAAGTGTGAGAA-3′ (KD1); KLK11-RNAi (41638) 5′-AGGAGACGATGAAGAACAA-3′ (KD2); KLK11-RNAi (41640), 5′-TCTGG CAACAGGGCTTGTA-3′ (KD3); and KLK11-Control, 5′-TTCTCCGAACGT GTCACGT-3′ (control groups [CON]). Lentiviral virions were produced by co-transfection of HEK293T cells with 5 μg pLKO.1-puro vector and 5 μg packaging and envelope vectors by using Lipofectamine 2000 (Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocol. Lentivirus was harvested 48 h after transfection. HCT-8/L-OHP cells were infected with lentivirus containing KLK11-RNAi or CON for 24 h. Then 2 days later, the virus-infected cells were selected by 4 μg/mL puromycin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 48 h and subjected to the requisite assays.
Cell viability assay
Cell viability was quantified using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, 3×103 HCT-8 cells (CON or KLK11 RNAi) were seeded in 96-well plates and 20 μL MTT solution (5 mg/mL; Sigma-Aldrich) was added to each well 24, 48, 72, 96, 120, 144, and 168 h later. The optical density was measured using a microplate reader (Thermo Fisher Scientific, Inc.) at 595 nm.
For drug sensitivity, cells were plated in 96-well plates at 3×103 cells per well; when the cells reached 60% confluence, the medium was removed and replaced with fresh medium containing varying concentrations of L-OHP, 5-fluorouracil (5-FU), irinotecan (CPT-11), vincristine (VCR), and cis-diamminedichloroplatinum (CDDP) and incubated for 48 h. The optical density was then measured, and the cell viability was calculated.
Caspase-3 activity analysis
The activity of caspase-3 was measured using a Caspase-3 Assay kit (Beyotime Biotechnology, Co., Ltd, Shanghai, People’s Republic of China) according to the manufacturer’s instructions. In brief, 2×107 cells were harvested, resuspended in 100 μL chilled cell lysis buffer, and incubated on ice for 15 min. Then, 40 μL reaction buffer was added to each sample, along with 10 μL N-Acetyl-Asp-Glu-Val-Asp-p-Nitroanilide (DEVD-pNA) (2 mM) substrate and the mixture was incubated for 2 h at 37°C. The optical density was measured at 405 nm using a microplate reader (Thermo Fisher Scientific, Inc.).
Apoptosis assay
Apoptosis detection was undertaken using an Annexin V Apoptosis Detection kit I (BD Biosciences, Franklin Lakes, NJ, USA). Cells were seeded in six-well plates (4×105 well) and incubated for 12 h and then 10 μmol/L L-OHP was added; after 48 h, the cells were collected and washed with PBS. Then, 5 μL annexin V and propidium iodide was added to the cell suspension and incubated at room temperature in the dark for 30 min. The volume was then made up to 500 μL and the cells were analyzed using a FACS Calibur flow cytometer (BD Biosciences).
Western blot assay
Total protein was extracted from cells using RIPA buffer (Thermo Fisher Scientific, Inc.) and quantified using the BCA protein assay kit according to the manufacturer’s instructions (Pierce, USA). Totally, 10 μg protein was loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis for electrophoresis. After transfer, membranes were blocked with tris-buffered saline with Polysorbate 20 (TBST) containing 5% nonfat milk for 0.5 h at room temperature, and incubated with primary polyclonal antibodies raised in rabbits against KLK11, PI3K, AKT, pAKT, Bax, Bcl-2, and GAPDH (Abcam, Cambridge, UK), excision repair cross-completion1 (ERCC1), multidrug-resistant protein (MRP, ABCC1), glutathione S-transferase-P1 (GSTP1), and P-glycoprotein (P-gp, ABCB1). Signals were detected after incubation in horseradish peroxidase (HRP)-conjugated secondary antibodies with the addition of the ECL substrate kit (Thermo Fisher Scientific, Inc.). GAPDH acted as the reference protein for loading control.
Statistical analysis
Statistical analysis was conducted using SPSS version 20.0 (SPSS, Inc., Chicago, IL, USA). Categorical variables were expressed as numbers with percentages and compared using a chi-square test or Fisher’s exact test when appropriate. Normally distributed data were described by means ± standard deviations and analyzed using Student’s t-tests. Statistical significance was considered to be indicated by P<0.05.
Results
KLK11 is associated with resistance to L-OHP and poor prognosis in mCRC patients
A total of 55 patients were included in the present study, including 25 patients in the chemotherapy-sensitive group, and 30 patients in the chemotherapy-resistant group. No statistical differences were observed between the two groups in terms of age, gender, American Society of Anesthesiologists (ASA) score, tumor location, gross type, tumor differentiation, pathological T stage, pathological N stage, the number of primary distant metastasis sites, and pretreatment carcinoembryonic antigen (CEA) level (Table 1). Kaplan–Meier analysis was used to evaluate the contribution of chemoresistance to the low survival rate in patients with mCRC. With a median follow-up period of 23 months (range 8–46 months), the 3-year overall survival (OS) rate was 3.3% in the chemotherapy-resistant group, lower than 20% in the chemotherapy-sensitive group; however, this difference was not significant (P=0.204, Figure 1D).
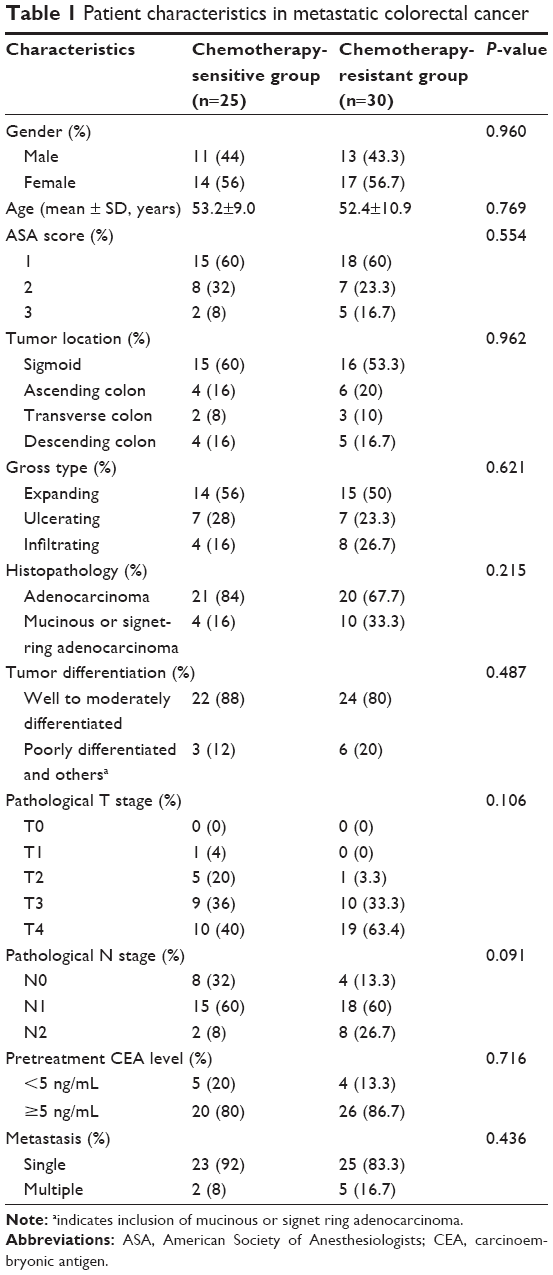 | Table 1 Patient characteristics in metastatic colorectal cancer |
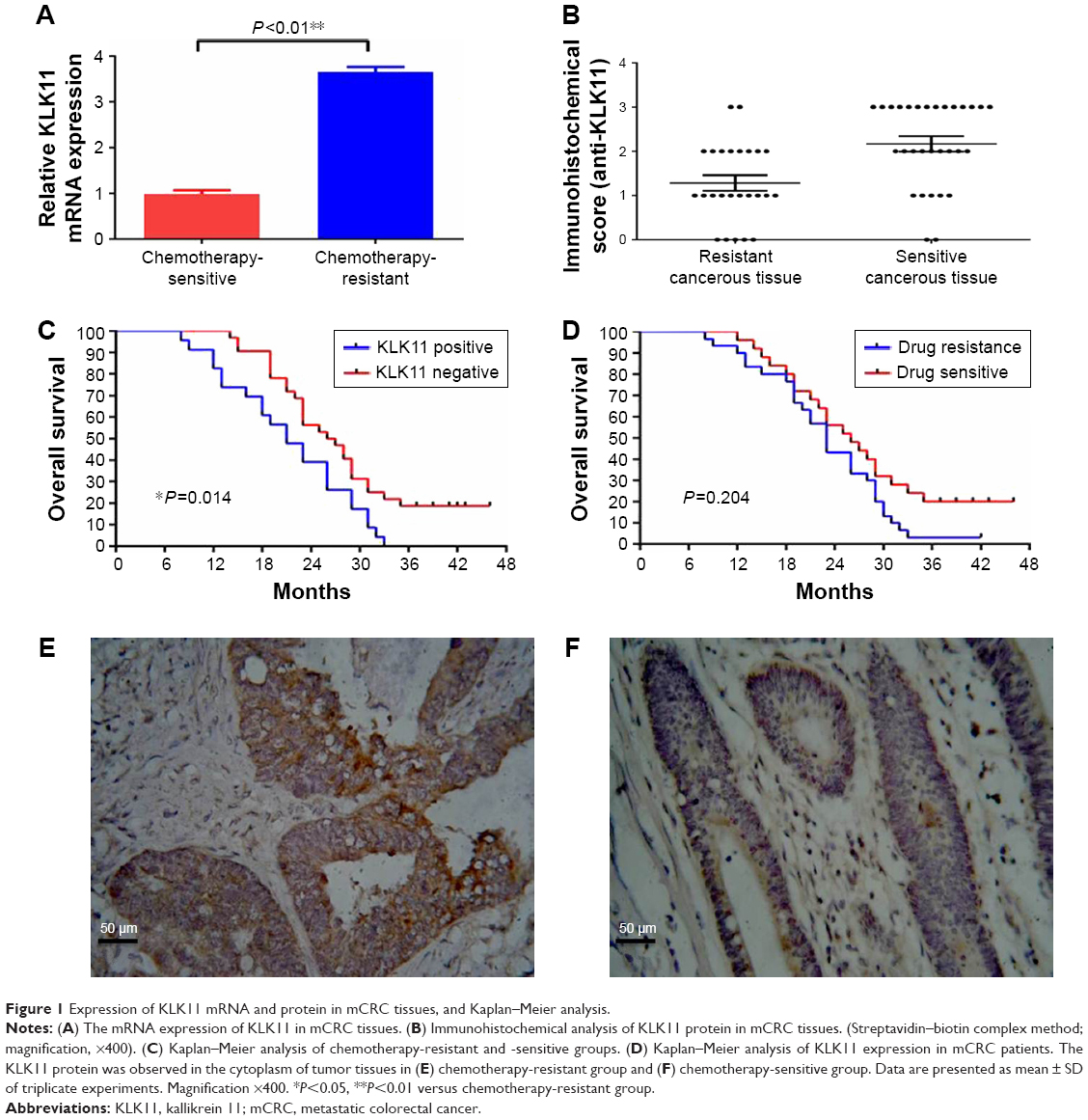 | Figure 1 Expression of KLK11 mRNA and protein in mCRC tissues, and Kaplan–Meier analysis. |
The relative KLK11 mRNA expression levels in the chemotherapy-sensitive and -resistant groups were 0.988±0.082 and 3.658±0.103, respectively (P<0.01, Figure 1A). Immunohistochemical analysis demonstrated that the KLK11 protein was observed in the cytoplasm (Figure 1E and F) of tumor tissues in the chemotherapy-resistant group, with lower or no expression in the chemotherapy-sensitive group (P<0.01, Figure 1B). The numbers of grades −, +, ++, and +++ in the chemotherapy-resistant group were 1, 10, 11, and 8, respectively, and that of grades −, +, ++, and +++ in the chemotherapy-sensitive group were 7, 14, 4, and 0, respectively. These results indicated that higher expression of KLK11 was associated with the chemosensitivity to L-OHP-containing chemotherapy.
We further evaluated whether KLK11 expression was relevant with regard to the clinicopathological features of patients with mCRC (Table 2). Noticeably, the overexpression of KLK11 was significantly associated with lymph node metastasis and histopathology (mucinous or signet-ring adenocarcinoma), but was not associated with age, gender, ASA score, tumor location, gross type, pathological T stage, the number of primary distant metastasis sites, and pretreatment CEA level. Kaplan–Meier analysis was used to determine the prognostic value of KLK11 in patients with mCRC. The 3-year OS rate was 0% in patients with high KLK11 expression, compared to 18.8% in patients with low KLK11 expression. (P=0.014, Figure 1C).
 | Table 2 Clinical association between KLK11 and the clinicopathological factors of patients with mCRC |
Characterization of HCT-8/L-OHP cell lines
After continuous exposure to L-OHP for 286 days, the resistant phenotypes of the resultant HCT-8/L-OHP cells were verified by exposure of cells to the L-OHP concentrations at which the resistant cells were continuously propagated in the final stages. Thus, the parental HCT-8 and HCT-8/L-OHP cells were exposed to a range of L-OHP concentrations (10, 20, 40, 80, and 160 μmol/L) and the HCT-8/L-OHP cells were significantly less sensitive to L-OHP (P<0.01, Figure 2A). As shown in Figure 2F, the IC50 of parental HCT-8 was 5.78±0.05 μmol/mL, whereas the IC50 of L-OHP–resistant HCT-8/L-OHP cells increased to 62.33±1.87 μmol/mL, with drug resistance index (RI) of 10.78, indicating that HCT-8/L-OHP cells were significantly resistant to L-OHP (P<0.01).
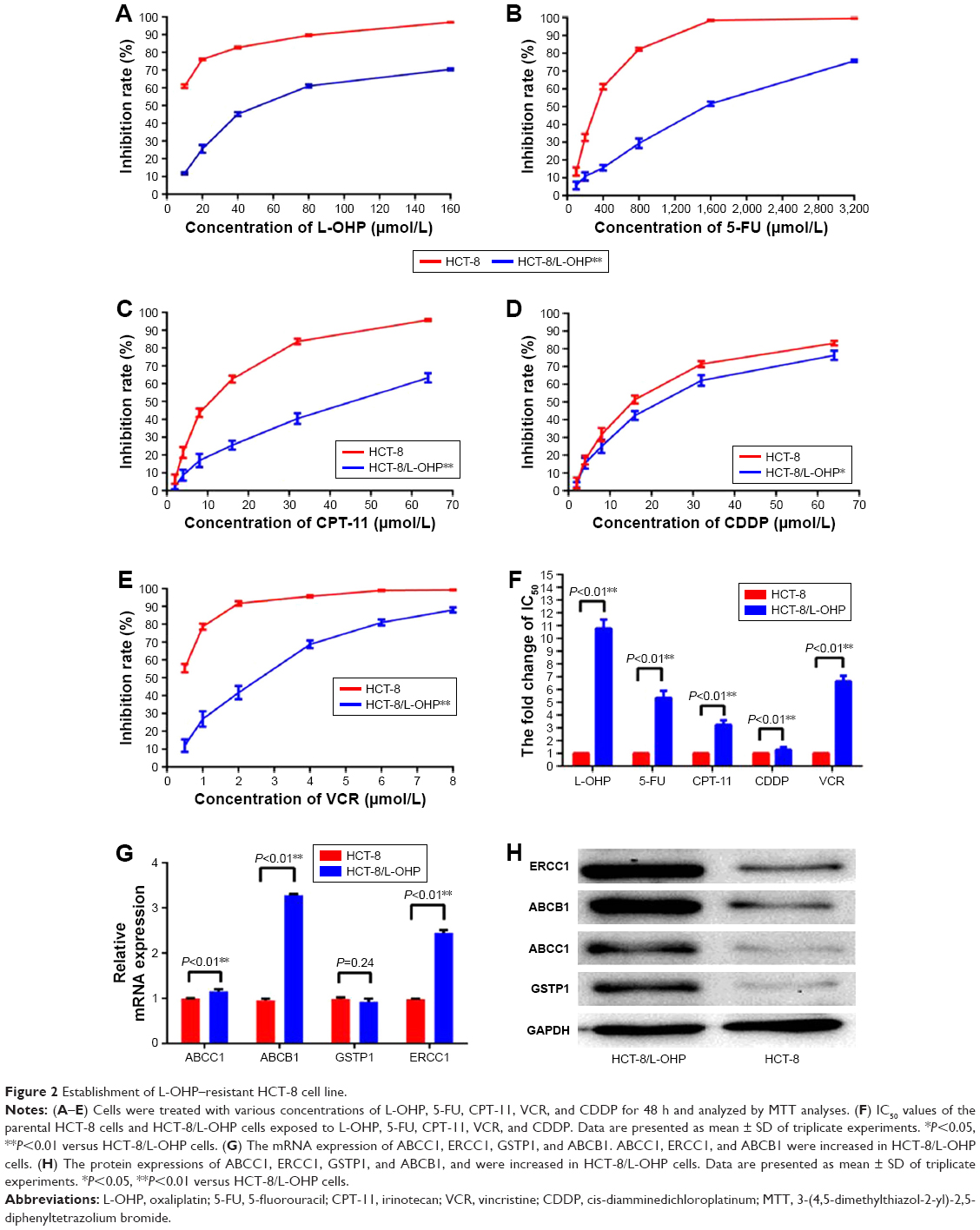 | Figure 2 Establishment of L-OHP–resistant HCT-8 cell line. |
Further, the cells were tested for sensitivity to a range of 5-FU, CPT-11, VCR, and CDDP concentrations (Figure 2B–E), and the HCT-8/L-OHP cells were significantly less sensitive to multiple anti-tumor drugs (P<0.01). As shown in Figure 2F, the IC50 of the parental HCT-8 cell line to 5-FU, CPT-11, VCR, and CDDP was 296.45±10.63, 12.45±0.96, 15.33±0.68, and 0.44±0.03 μmol/mL, respectively. Meanwhile, the IC50 of L-OHP–resistant HCT-8/L-OHP cells to 5-FU, CPT-11, VCR, and CDDP elevated to 1,587.22±16.96, 40.21±1.28, 19.56±0.95, and 2.92±0.18 μmol/mL, respectively. This demonstrated that the L-OHP–resistant cell line HCT-8/L-OHP was a multidrug resistance (MDR) cell line, not only resistant to L-OHP. To further validate whether the HCT-8/L-OHP was an MDR cell line, four multidrug-resistance-related genes – ABCB1, ABCC1, GSTP1, and ERCC1 – were determined by RT-qPCR and Western blot analysis. As shown in Figure 2G and H, the mRNA expressions of ABCC1, ERCC1, and ABCB1 were increased in resistant cells of HCT-8/L-OHP (P<0.01), and the protein expressions of ABCB1 (P=0.24), ABCC1, GSTP1, and ERCC1 were all increased in the HCT-8/L-OHP cell line. These results confirmed that the L-OHP–resistant cell line HCT-8/L-OHP was also an MDR cell line.
The high expression of KLK11 in the HCT-8/L-OHP cell line and stable knockdown of KLK11 specifically inhibits KLK11 mRNA and protein expression.
Our previous study showed that high expression of KLK11 was associated with resistance to FOLFOX4 chemotherapy in patients with CRC. To explore whether the expression of KLK11 was associated with L-OHP resistance, we detected the expression of KLK11 in the HCT-8/L-OHP cell line and parental cell line HCT-8 by RT-qPCR and Western blot. The expression of KLK11 was significantly increased in the HCT-8/L-OHP cell line, when compared to the parental cell line (P<0.01, Figure 3A, C and D). To determine whether KLK11 was involved in the progression of L-OHP resistance, three different lentivirus-based RNAi (KLK11 KD1, KLK11 KD2, and KLK11 KD3) were employed to downregulate KLK11 expression. The mRNA and protein levels of KLK11 in stably transfected HCT-8/L-OHP cells were confirmed using RT-qPCR and Western blot analysis (P<0.01, Figure 3B–D). The mRNA expression and protein expression levels of KLK11 were significantly decreased in KLK11 KD2 groups in HCT-8/L-OHP cells. The subsequent assays were conducted with KLK11 KD2, which is, furthermore, referred to as KLK11-RNAi. These results suggested that the lentivirus-mediated KLK11-RNAi targeting KLK11 could effectively knockdown KLK11 expression in colorectal L-OHP–resistant cells. Together, these results demonstrated that the expression of KLK11 was increased in the HCT-8/L-OHP cell line. Moreover, we established a stable KLK11 knockdown HCT-8/L-OHP KD2 cell line.
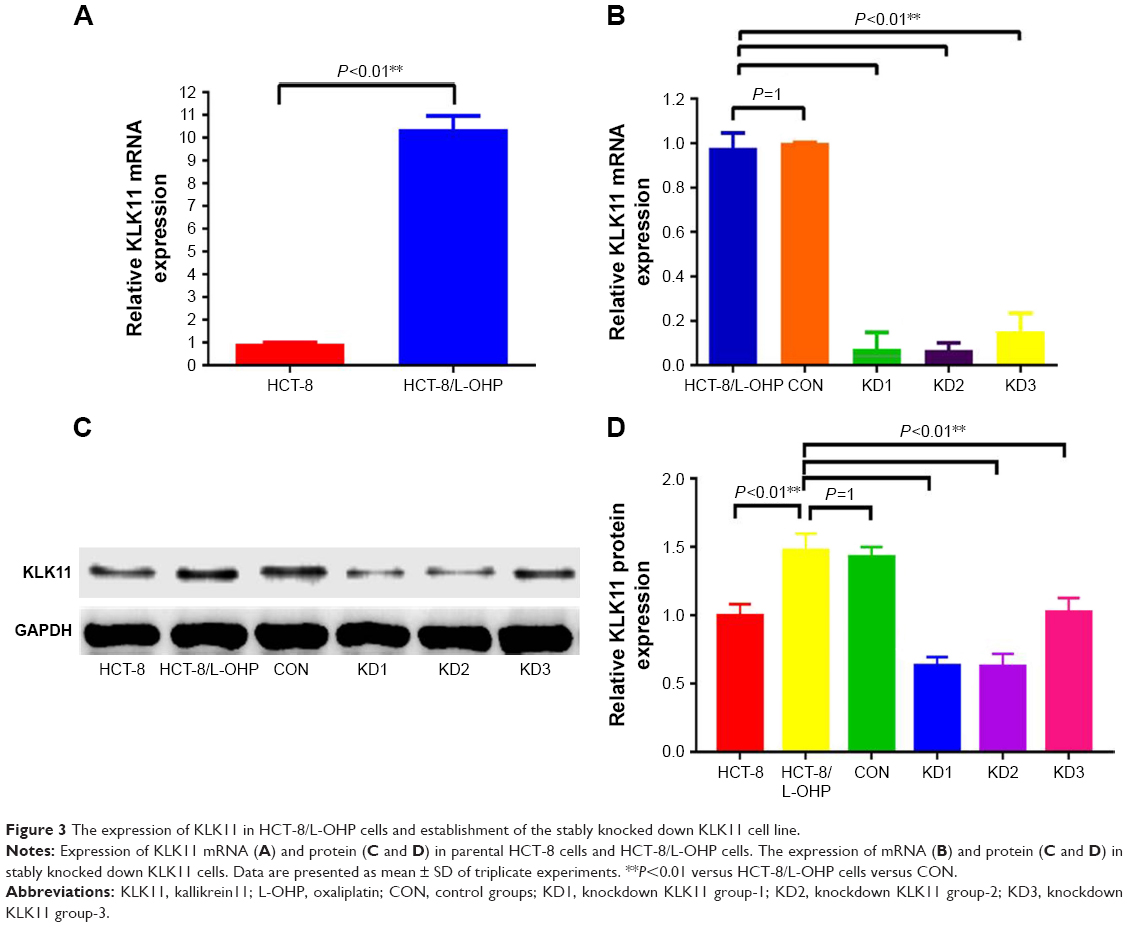 | Figure 3 The expression of KLK11 in HCT-8/L-OHP cells and establishment of the stably knocked down KLK11 cell line. |
Downregulation of KLK11 enhanced sensitivity of HCT-8/L-OHP cells to L-OHP and L-OHP–induced apoptosis in vitro
Our initial result showed that knockdown of KLK11 gene reversed L-OHP resistance in the CRC cell line 18. However, whether KLK11 played an important role in L-OHP–resistant CRC cells was unclear. Herein, parental HCT-8, HCT-8/L-OHP, control, and HCT-8/L-OHP cells with stable KLK11 silencing were treated with 10, 20, 40, 80, and 160 μmol/L L-OHP for 48 h. MTT assays showed that HCT-8/L-OHP cell lines developed a significant increase in resistivity to L-OHP, compared with the parental cell lines (P<0.01). Meanwhile, knockdown of KLK11 in HCT-8/L-OHP cell lines led to a significant reduction in resistance to L-OHP in a dose-dependent manner, compared with HCT-8/L-OHP cell lines and CON (P<0.01, Figure 4A).
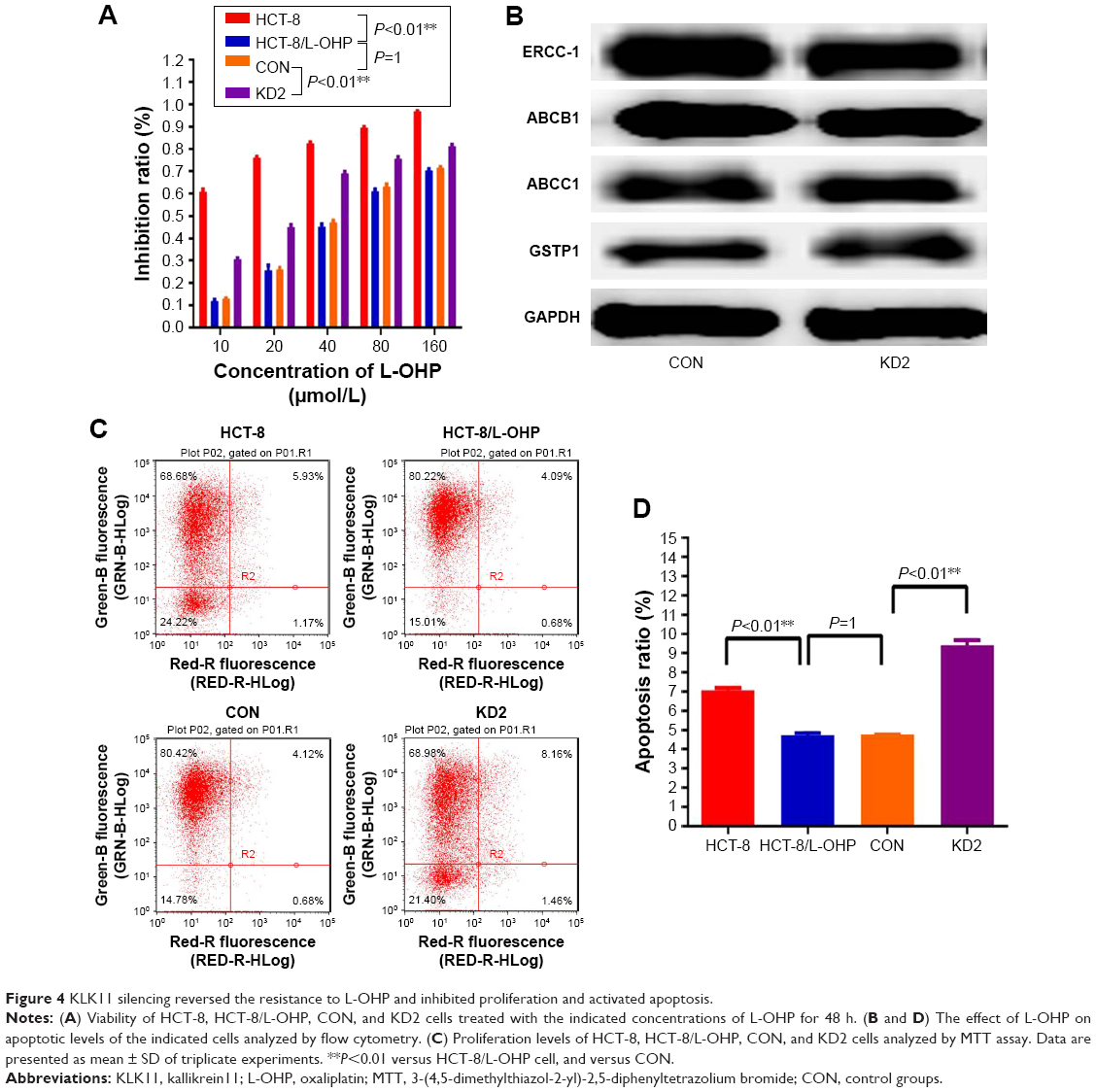 | Figure 4 KLK11 silencing reversed the resistance to L-OHP and inhibited proliferation and activated apoptosis. |
To clarify the mechanism underlying enhanced sensitivity to L-OHP, we detected the drug-resistance markers after knocking down KLK11 in L-OHP–resistant cells by Western blotting (Figure 4B); the results demonstrated that knockdown of KLK11 did not regulate the expression of ABCB1, ABCC1, ERCC1, and GSTP1.
Furthermore, flow cytometric analysis showed that the apoptosis rate of HCT-8/L-OHP cell lines was significantly lower than that in the parental cell lines, whereas HCT-8/L-OHP KD2 cell lines had a significantly higher apoptosis rate than HCT-8/L-OHP and CON groups (P<0.01, Figure 4C and D). Taken together, these results suggested that the L-OHP–resistant HCT-8/L-OHP cell line decreased the chemosensitivity, and knockdown of KLK11 could increase the chemosensitivity to L-OHP by enhancing apoptosis in vitro.
Downregulation of KLK11 expression inhibits cell growth and activates the PI3K/AKT apoptosis signaling pathway
To explore the biological function of KLK11 in L-OHP–resistant HCT-8/L-OHP cell progression, MTT assays were used to examine the proliferative ability of parental HCT-8, HCT-8/L-OHP, control, and HCT-8/L-OHP KD2 cells on days 2, 3, 4, 5, 6, and 7. As shown in Figure 5A, the proliferation rates of HCT-8/L-OHP cells started to increase and enhance as compared with parental cell lines HCT-8 (P<0.01), and the proliferation of HCT-8/L-OHP KD2 cells decreased and reduced, when compared with those of the HCT-8/L-OHP and CON groups (P<0.01).
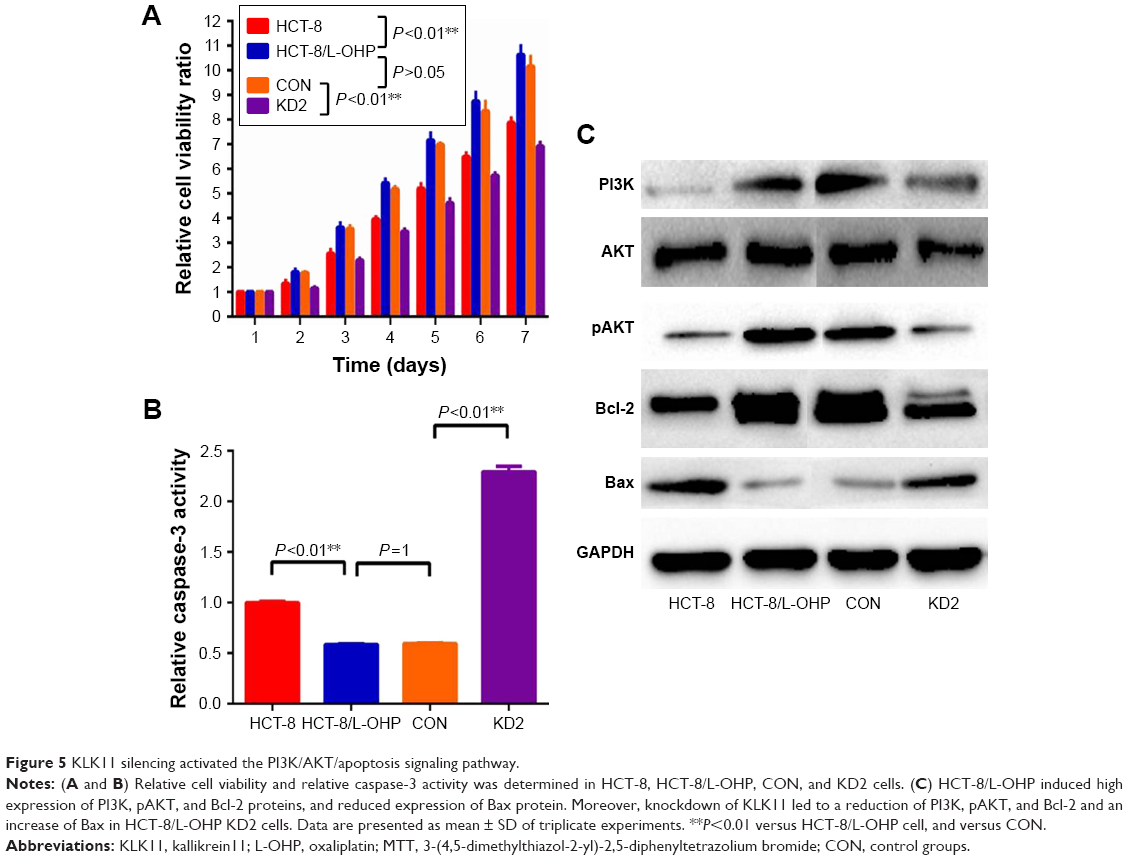 | Figure 5 KLK11 silencing activated the PI3K/AKT/apoptosis signaling pathway. |
To clarify the mechanism underlying the regulation of cell growth, we detected the PI3K/AKT and apoptosis signaling pathway proteins – Bcl-2 and Bax.22 The results showed that HCT-8/L-OHP cells induced a high expression of PI3K, pAKT, and Bcl2 proteins, and reduced expression of Bax protein. Moreover, knockdown of KLK11 led to a reduction of PI3K, pAKT, and Bcl-2 and an increase of Bax in HCT-8/L-OHP KD2 cells (Figure 5C). These results demonstrated that the caspase-3 activity was significantly reduced in HCT-8/L-OHP cell lines as compared with the parental cell lines. Meanwhile, the HCT-8/L-OHP KD2 cell line had a significantly higher caspase-3 activity rate than that in HCT-8/L-OHP cell lines and CON (P<0.01, Figure 5B). Together, these data demonstrated that the activated PI3K/AKT signaling pathway played an important role in L-OHP resistance, and downregulation of KLK11 reversed L-OHP resistance via suppression of the PI3K/AKT signaling pathway.
Discussion
Oxaliplatin resistance is a critical medical problem in CRC, and the underlying mechanism has not yet been fully elucidated. We previously revealed that KLK11 was associated with chemosensitivity to the FOLFOX4 regimen.23 Additionally, knockdown of KLK11 can inhibit cell proliferation and increase oxaliplatin sensitivity in human CRC.18 Herein, we demonstrated increased constitutive activity of the PI3K/AKT signaling pathway in CRC cancer cells with acquired resistance to L-OHP. In addition, we revealed that KLK11 expression was associated with chemoresistance and poor prognosis in patients with mCRC. More importantly, knockdown of KLK11 can reverse oxaliplatin resistance by inhibiting proliferation and activating apoptosis via suppressing the PI3K/AKT signal pathway in an L-OHP–resistant cell line.
KLK11 is expressed in numerous human malignancies and correlates with malignant behavior of tumors, including in CRC. It was reported that the expression level of KLK11 might be a novel prognostic marker of gastric cancer.24 Previous studies showed elevated expression of KLK11 in CRC specimens as compared with paired normal colon mucosa, and it was associated with worse overall survival.16,17 In our study, we evaluated the expression of KLK11 mRNA and protein in tissues from patients with mCRC, and demonstrated that the mRNA and protein expression of KLK11 was increased in tumor tissue from L-OHP–resistant patients. In addition, the expression level of KLK11 was associated with lymph node metastasis and histopathology (mucinous or signet-ring adenocarcinoma). It is well documented that lymph node metastasis indicates poor response to chemotherapy.25,26 Mucinous histological type has been shown to be an independent factor associated with poor prognosis, such as overall survival.27,28 The correlation between KLK11 expression and lymph node metastases and histopathology type may be associated with the response to chemotherapy in patients with mCRC. Moreover, patients with high KLK11 expression had a significantly poorer 3-year OS rate when compared to patients with low KLK11 expression. Together, these results demonstrated that KLK11 may be used as a biomarker to predict the response to L-OHP–containing chemotherapy and prognosis in patients with mCRC.
HCT-8 cell lines, a widely useful model system of CRC, may provide an easily manipulable discovery platform that can facilitate preclinical studies and thus uncover clinical obstacles. In our previous study, we found that knockdown of KLK11 in the HCT-8 cell line can enhance L-OHP sensitivity. In this study, we successfully established the L-OHP resistance cell line HCT-8/L-OHP, which not only showed stable resistance to L-OHP, but also showed resistance to 5-FU, CPT-11, VCR, and CDDP via increased expression of multidrug-resistant genes ABCC1, ABCB1, GSTP1, and ERCC1 indicating that they were associated with the mechanisms of MDR. In this study, we attempted to verify whether KLK11 was associated with L-OHP efficiency, and the HCT-8 cell line can be an appropriate experimental model to study the relationship between KLK11 and the L-OHP–resistance mechanism. Moreover, we believed the use of the same cell line would reduce errors caused by using different cell lines in investigating the same gene KLK11. PI3K is a lipid kinase and plays a critical role in cell growth and survival.29 Overexpression of AKT has been reported in CRC cell lines, and was closely related to chemoresistance and a poor prognosis for patients.30–32 In addition, our study revealed that the activated PI3K/AKT signal pathway was related with L-OHP resistance in CRC. More importantly, searching novel compounds that target PI3K/AKT signaling as a new potential therapeutic option for drug-resistance treatment is of much scientific interest.32–34 Therefore, we further clarified that the KLK11 regulated the PI3K/AKT signal pathway in the L-OHP–resistance cell line HCT-8/L-OHP and affected the L-OHP resistance.
Evidence from our previous study indicated that KLK11 was associated with L-OHP resistance. Therefore, we postulated that KLK11 might be an L-OHP–resistance-related gene. However, the underlying mechanism through which KLK11 regulates the L-OHP resistance remains elusive. Based on the successfully established L-OHP–resistant cell line HCT-8/L-OHP, we demonstrated that KLK11 was highly expressed in an L-OHP–resistant cell line, and knockdown of KLK11 in the L-OHP–resistance cell line inhibited cell growth and induced apoptosis. Meanwhile, we showed that the KLK11 knockdown cell also had increased caspase-3 activity through activation of the PI3K/AKT/apoptosis signaling pathway, which induced a reduction of the Bcl-2/Bax ratio.
KLK11, a member of the KLK family, is expressed in numerous normal tissues,35 and dysregulated expression of KLK11 is reported to be associated with various tumors.36 Given that KLK11 is associated with L-OHP resistance, we hypothesized that KLK11 is an L-OHP–resistance-related gene. As expected, lentivirus-mediated KLK11 silencing in HCT-8/L-OHP cells effectively enhanced the sensitivity to L-OHP. Moreover, knockdown of KLK11 suppressed the proliferation of HCT-8/L-OHP KD2 cells in vitro. Meanwhile, apoptosis in HCT-8/L-OHP KD2 cells was significantly upregulated compared with the CON. Taken together, these results suggested that KLK11 played an important role in L-OHP resistance in vitro.
Previous study has shown that KLK11 expression was associated with an inactivated PI3K/AKT pathway.37 To further elucidate how KLK11 mediates L-OHP resistance and cell growth, we investigated PI3K/AKT signaling pathway and Bcl-2, Bax, two crucial regulatory proteins related to apoptosis, which have both anti- and pro-apoptotic effects in the HCT-8/L-OHP KD2 cells.38–40 The result showed that the L-OHP–resistant cell line acquired resistance to apoptosis by activation of PI3K/AKT signaling pathway, upregulation of Bcl-2 and downregulation of Bax.41 The inactivated PI3K/AKT signaling pathway promoting apoptosis induced a reduction of the Bcl-2/Bax ratio, eventually triggering caspase-3 activation and inducing apoptosis.42 Similarly, we found that knockdown of KLK11 inactivated the PI3K/AKT signaling pathway, increased the Bax expression, and decreased the Bcl-2 expression, which led to a reduction of the Bcl-2/Bax ratio. Additionally, downregulation of KLK11 activated caspase-3 activity, and subsequently induced apoptosis of tumor cells. These results suggested that KLK11 silencing inactivated the PI3K/AKT signaling pathway, and induced apoptosis. There is considerable evidence supporting the hypothesis that inactivated apoptosis is associated with resistance to chemotherapy.43 Consistent with data mentioned above, downregulation of KLK11 in an L-OHP–resistant cell line resulted in the inhibition of cell growth and apoptosis, and thus reversed the L-OHP resistance in vitro.
In this study, we successfully established the L-OHP–resistant CRC cell line HCT-8/L-OHP, and demonstrated that the activated PI3K/AKT signaling pathway was related to L-OHP resistance. More importantly, we revealed differentially expressed KLK11 between the chemotherapy-sensitive and -resistant groups. KLK11 overexpression was associated with lymph node metastases, histopathology, and prognosis in patients with mCRC, indicating that KLK11 is involved in response to chemotherapy. Furthermore, downregulation of KLK11 could inhibit cell proliferation, induce apoptosis, and reverse L-OHP resistance via suppression of the PI3K/AKT signaling pathway in vitro. KLK11 may be used as a valid biomarker to predict resistance to L-OHP and can also offer a novel therapeutic approach for L-OHP–resistant CRC treatment.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No 81472777), Training Plan of Middle-aged and Young Talents of Fujian Province Health and Family Planning Commission (No 2013-ZQN-ZD-11), Science Foundation of the Fujian Province, (No 2017J01296), and National Clinical Key Specialty Construction Project (General Surgery) of China (No 2012-649).
Disclosure
The authors report no conflicts of interest in this work.
References
Edwards BK, Noone AM, Mariotto AB, et al. Annual Report to the Nation on the status of cancer, 1975–2010, featuring prevalence of comorbidity and impact on survival among persons with lung, colorectal, breast, or prostate cancer. Cancer. 2014;120(9):1290–1314. | ||
André T, Boni C, Mounedji-Boudiaf L, et al; Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer (MOSAIC) Investigators. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350(23):2343–2351. | ||
Weitz J, Koch M, Debus J, Höhler T, Galle PR, Büchler MW. Colorectal cancer. Lancet. 2005;365(9454):153–165. | ||
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. | ||
Li B, Li J, Xu WW, et al. Suppression of esophageal tumor growth and chemoresistance by directly targeting the PI3K/AKT pathway. Oncotarget. 2014;5(22):11576–11587. | ||
Xiao ZM, Wang XY, Wang AM. Periostin induces chemoresistance in colon cancer cells through activation of the PI3K/Akt/survivin pathway. Biotechnol Appl Biochem. 2015;62(3):401–406. | ||
Lv L, Liu HG, Dong SY, et al. Upregulation of CD44v6 contributes to acquired chemoresistance via the modulation of autophagy in colon cancer SW480 cells. Tumour Biol. 2016;37(7):8811–8824. | ||
Borgoño CA, Diamandis EP. The emerging roles of human tissue kallikreins in cancer. Nat Rev Cancer. 2004;4(11):876–990. | ||
Yoshida S, Taniguchi M, Suemoto T, Oka T, He X, Shiosaka S. cDNA cloning and expression of a novel serine protease, TLSP. Biochim Biophys Acta. 1998;1399(2–3):225–228. | ||
Lundwall A. Old genes and new genes: the evolution of the kallikrein locus. Thromb Haemost. 2013;110(3):469–475. | ||
Mitsui S, Nakamura T, Okui A, Kominami K, Uemura H, Yamaguchi N. Multiple promoters regulate tissue-specific alternative splicing of the human kallikrein gene, KLK11/hippostasin. FEBS J. 2006;273(16):3678–3686. | ||
Kontos CK, Mavridis K, Talieri M, Scorilas A. Kallikrein-related peptidases (KLKs) in gastrointestinal cancer: mechanistic and clinical aspects. Thromb Haemost. 2013;110(3):450–457. | ||
Yousef GM, Obiezu CV, Luo LY, Black MH, Diamandis EP. Prostase/KLK-L1 is a new member of the human kallikrein gene family, is expressed in prostate and breast tissues, and is hormonally regulated. Cancer Res. 1999;59(17):4252–4256. | ||
Debela M, Beaufort N, Magdolen V, et al. Structures and specificity of the human kallikrein-related peptidases KLK 4, 5, 6, and 7. Biol Chem. 2008;389(6):623–632. | ||
Liu X, Quan B, Tian Z, et al. Elevated expression of KLK8 predicts poor prognosis in colorectal cancer. Biomed Pharmacother. 2017;88:595–602. | ||
Alexopoulou DK, Kontos CK, Christodoulou S, Papadopoulos IN, Scorilas A. KLK11 mRNA expression predicts poor disease-free and overall survival in colorectal adenocarcinoma patients. Biomark Med. 2014;8(5):671–685. | ||
Talieri M, Li L, Zheng Y, et al. The use of kallikrein-related peptidases as adjuvant prognostic markers in colorectal cancer. Br J Cancer. 2009;100(10):1659–1665. | ||
Xu Z, Chi P, Pan J, et al. Knockdown of KLK11 inhibits cell proliferation and increases oxaliplatin sensitivity in human colorectal cancer. Exp Ther Med. 2016;12(5):2855–2860. | ||
Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. | ||
Qiu JY, Liu P, Shi C, Han B. Low-grade myofibroblastic sarcomas of the maxilla. Oncol Lett. 2015;9(2):619–625. | ||
Lei KF, Liu BY, Zhang XQ, et al. Development of a survival prediction model for gastric cancer using serine proteases and their inhibitors. Exp Ther Med. 2012;3(1):109–116. | ||
Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011;278(3):403–413. | ||
Lu X, Pan J, Li S, et al. Establishment of a predictive genetic model for estimating chemotherapy sensitivity of colorectal cancer with synchronous liver metastasis. Cancer Biother Radiopharm. 2013;28(7):552–558. | ||
Wen YG, Wang Q, Zhou CZ, et al. Identification and validation of Kallikrein-ralated peptidase 11 as a novel prognostic marker of gastric cancer based on immunohistochemistry. J Surg Oncol. 2011;104(5):516–524. | ||
Sawayama H, Hayashi N, Honda S, et al. Treatment results of FOLFOX chemotherapy before surgery for lymph node metastasis of advanced colorectal cancer with synchronous liver metastasis: the status of LN metastasis and vessel invasions at the primary site in patients who responded to FOLFOX. Int J Clin Oncol. 2010;15(1):70–76. | ||
Ando K, Oki E, Saeki H, et al. Number of lymph node metastases may indicate the regimen for adjuvant chemotherapy in patients with stage III colorectal cancer. Anticancer Res. 2015;35(11): 6207–6211. | ||
Benedix F, Kuester D, Meyer F, Lippert H. [Influence of mucinous and signet-ring cell differentiation on epidemiological, histological, molecular biological features, and outcome in patients with colorectal carcinoma]. Zentralbl Chir. 2013;138(4):427–433. German. | ||
Hawk NN, Long TE, Imam MH, et al. Clinicopathologic features and outcome of young adults with stage IV colorectal cancer. Am J Clin Oncol. 2015;38(6):543–549. | ||
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. | ||
Lee MH, Hong SH, Park C, et al. Hwang-Heuk-San induces apoptosis in HCT116 human colorectal cancer cells through the ROS-mediated activation of caspases and the inactivation of the PI3K/Akt signaling pathway. Oncol Rep. 2016;36(1):205–214. | ||
Pandurangan AK. Potential targets for prevention of colorectal cancer: a focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pac J Cancer Prev. 2013;14(4):2201–2205. | ||
Danielsen S, Eide PW, Nesbakken A, Guren T, Leithe E, Lothe RA. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim Biophys Acta. 2015;1855(1):104–121. | ||
Moral M, Paramio JM. Akt pathway as a target for therapeutic intervention in HNSCC. Histol Histopathol. 2008;23(10):1269–1278. | ||
Wu Z, Doondeea JB, Gholami AM, et al. Quantitative chemical proteomics reveals new potential drug targets in head and neck cancer. Mol Cell Proteomics. 2011;10(12):M111.011635. | ||
Paliouras M, Borgono C, Diamandis EP. Human tissue kallikreins: the cancer biomarker family. Cancer Lett. 2007;249(1):61–79. | ||
Mavridis K, Scorilas A. Prognostic value and biological role of the kallikrein-related peptidases in human malignancies. Future Oncol. 2010;6(2):269–285. | ||
Paliouras M, Diamandis EP. Intracellular signaling pathways regulate hormone-dependent kallikrein gene expression. Tumour Biol. 2008;29(2):63–75. | ||
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. | ||
Pettersson F, Dalgleish AG, Bissonnette RP, Colston KW. Retinoids cause apoptosis in pancreatic cancer cells via activation of RAR-gamma and altered expression of Bcl-2/Bax. Br J Cancer. 2002;87(5):555–561. | ||
Yuan W, Xiaoyun H, Haifeng Q, et al. MicroRNA-218 enhances the radiosensitivity of human cervical cancer via promoting radiation induced apoptosis. Int J Med Sci. 2014;11(7):691–696. | ||
Xiang Z, Kang QJ, Xiang X. Gene and protein expression in the oxaliplatin-resistant HT29/L-OHP human colon cancer cell line. Genet Mol Res. 2015;14(3):11013–11022. | ||
Liu Z, Wang F, Zhou ZW, et al. Alisertib induces G2/M arrest, apoptosis, and autophagy via PI3K/Akt/mTOR- and p38 MAPK-mediated pathways in human glioblastoma cells. Am J Transl Res. 2017;9(3):845–873. eCollection 2017. | ||
Gillet JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47–76. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.