")
Back to Journals » OncoTargets and Therapy » Volume 11
Knockdown of KIF26B inhibits breast cancer cell proliferation, migration, and invasion
Authors Gu S , Liang H, Qi D, Mao L, Mao G, Qian L, Zhang S
Received 22 January 2018
Accepted for publication 10 April 2018
Published 29 May 2018 Volume 2018:11 Pages 3195—3203
DOI https://doi.org/10.2147/OTT.S163346
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Shudong Gu,1,* Haibin Liang,2,* Donghui Qi,3 Liyan Mao,4 Guoxin Mao,1 Li Qian,1 Shu Zhang5
1Department of Oncology, Affiliated Hospital of Nantong University, Nantong 226001, China; 2Department of General Surgery, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200092, China; 3Medical College of Nantong University, Nantong 226001, China; 4Department of Endoscopic Diagnosis and Treatment of Digestive Diseases, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200092, China; 5Department of Pathology, Affiliated Hospital of Nantong University, Nantong 226001, China
*These authors contributed equally to this work
Background: Kinesin family member 26B (KIF26B) plays a key role in the development and progression of many human cancers. However, the role and underlying mechanisms of KIF26B in breast cancer cells remain unknown.
Materials and methods: In this study, we inhibited the expression of KIF26B in MDA-MB-231 and MCF-7 cells using lentivirus-delivered shRNA.
Results: Lentivirus-mediated KIF26B knockdown significantly suppressed cell proliferation, colony formation, migration, and invasion. Furthermore, cell cycle analyses revealed that the percentage of cells in the G0/G1 phase was significantly increased in KIF26B knockdown cells. Moreover, the knockdown of KIF26B significantly promoted cell apoptosis via the upregulation of cleaved caspase-3 and Bax.
Conclusion: Our data indicate that KIF26B plays a pivotal role in tumor growth and metastasis in breast cancer cells and may be a potential therapeutic target for treating breast cancer.
Keywords: KIF26B, breast cancer cell, proliferation, migration, invasion
Introduction
Breast cancer is one of the most frequent malignant tumors that occurs in women; worldwide, there are more than 1.7 million new cases each year.1–3 Although improvements in basic and clinical research have decreased the mortality rates of breast cancer in recent years, its prognosis remains poor.4 This may be attributed to its incomplete diagnosis, frequent recurrence, and high chemoresistance, as well as the adverse effects of surgeries.5–7 Therefore, it is important to identify novel therapeutic targets and factors that could be potential biomarkers for the early diagnosis and prognosis of patients with breast cancer.
The kinesin superfamily proteins (KIFs) are a conserved class of molecular motor proteins with microtubule binding and ATPase activities.8 The kinesin family member 26B (KIF26B) gene belongs to the N-type kinesin family and encodes a 21,088-amino acid protein.9 An increasing amount of evidence demonstrates the importance of KIF26B in the regulation of many physiological events, including brain function, developmental patterning, and tumorigenesis. Uchiyama et al10 reported that KIF26B is essential for kidney development because it regulates the adhesion and polarization of mesenchymal cells; moreover, the transcription of KIF26B is regulated by Sall1. In the neuronal system, KIF26B transports Abelson interactor protein 1 (Abi-1) to different cellular compartments, particularly to the postsynaptic density of excitatory synapses.11 Recently, a number of studies have reported that the abnormal expression of KIF26B plays a key role in the development or progression of a number of human cancers, including esophageal adenocarcinoma,12 colorectal cancer,13 and gastric cancer.14 Wang et al15 demonstrated that KIF26B was overexpressed in breast cancer tissues and that breast cancer patients with high KIF26B expression had a shorter survival time. However, the biological role of KIF26B and its mechanism of regulation in breast cancer have not been investigated. Thus, whether KIF26B regulates the proliferation or metastatic ability of breast cancer cells and its potential mechanism are unknown.
In the current study, we investigated the effects of KIF26B knockdown on breast cancer cell proliferation, migration, and invasion, as well as breast cancer cell apoptosis and cell cycle distribution. We also investigated the potential mechanism of KIF26B.
Materials and methods
Cell lines and cell culture
MDA-MB-231, SK-BR-3, BT-474, MCF-10A, and MCF-7 cells were purchased from the Chinese Academy of Science Cell Bank (Shanghai, China). The cells were cultured in high-glucose DMEM containing 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) and supplemented with antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin; Sigma-Aldrich Co., St Louis, MO, USA) at 37°C and 5% CO2 in a humidified incubator.
RNA isolation and quantitative reverse transcription polymerase chain reaction assays
Total cellular RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. First-strand cDNA was synthesized using the PrimeScript RT reagent kit (Takara, Shiga, Japan). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using SYBR Green Premix Ex Taq (Takara) according to the manufacturer’s protocol. The relative expression levels of genes were calculated using the ΔΔCt method, and GAPDH was used as the endogenous reference gene. The following primer sequences were used: KIF26B forward, 5′-GCTGCGTGTTCTGTTTCGG-3′ and reverse, 5′-TTCCTTGCGTTCGTTTATGAG-3′; Bax forward, 5′-CCCGAGAGGTCTTTTTCCGAG-3′ and reverse, 5′-CCAGCCCATGATGGTTCTGAT-3′; Bcl-2 forward, 5′-GGTGGGGTCATGTGTGTGG-3′ and reverse, 5′-CGGTTCAGGTACTCAGTCATCC-3′; cyclin D1 forward, 5′-TGGAGCCCGTGAAAAAGAGC-3′ and reverse, 5′-TCTCCTTCATCTTAGAGGCCAC-3′; and GAPDH forward, 5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse, 5′-AGGGGCCATCCACAGTCTTC-3′. Three independent experiments were performed.
Colony formation assays
MDA-MB-231 and MCF-7 cells were infected for 72 h and seeded in 6-well plates at a density of 500 cells/well. After culture for 14 days, the cells were fixed with 4% paraformaldehyde for 20 min and stained with 0.1% crystal violet (Beyotime, Shanghai, China) for 25 min. After the cells were washed with PBS and air dried, the colonies were manually counted and photographed under a microscope.
In vitro Transwell cell migration/invasion assay
Migration and invasion assays were performed as described by Xie and Wang.16 Briefly, 4×104 and 1×105 lentivirus-transfected MDA-MB-231 and MCF-7 cells, respectively, were seeded in the upper chambers of 8-μm pore size inserts with or without Matrigel (Corning Incorporated, Corning, NY, USA) in a 24-well plate. Then, the cells were fixed in 4% paraformaldehyde and stained with 0.1% crystal violet. Cells that had penetrated the membranes were quantified in five random microscopic fields at 100× magnification under a microscope. Three independent experiments were performed.
Cell apoptosis assay
Apoptosis was performed using an annexin V/propidium iodide (PI) apoptosis kit according to the manufacturer’s protocol (Thermo Fisher Scientific). Briefly, MDA-MB-231 and MCF-7 cells were infected with KIF26B shRNA or negative control RNA. Then, the cells were placed in 6-well culture plates (5×104 cells/mL). After 48 h, the cells were harvested by trypsinization without EDTA, washed with cold PBS, and resuspended in 100 μL of binding buffer at a density of 1×106 cells/mL. Then, 5 μL of annexin V-fluorescein isothiocyanate (FITC) and 5 μL of a PI working solution (100 μg/mL) were added to the cells, gently mixed, and then incubated in the dark for 15 min at room temperature. Afterward, 400 μL of binding buffer was added, and the cells were immediately analyzed by flow cytometry. This experiment was repeated three times.
Cell cycle analysis
Breast cancer cells were seeded in 60-mm culture plates. After transfection with KIF26B shRNA or control RNA for 48 h, the cells were harvested by trypsinization, washed twice with cold PBS, and fixed in 70% ethanol at 4°C overnight. After fixation, the cells were rinsed with PBS and incubated with 10 mg/mL RNase and 1 mg/mL PI (Sigma-Aldrich Co.) at 37°C for 30 min in the dark. The cell cycle distribution was measured and analyzed using Cell Quest acquisition software (BD Biosciences, San Jose, CA, USA). Each experiment was performed in triplicate.
Lentivirus-mediated RNA interference
The siRNA sequence targeted to KIF26B was 5′-CGGACAGCCTCTCCTATTA-3′, as reported previously,15 and the negative control sequence was 5′-TTCTCCGAACGTGTCACGT-3′. Recombinant lentiviruses expressing KIF26B shRNA or negative control shRNA were provided by GeneChem (Shanghai, China). Cells were infected with lentivirus for 72 h and then selected with puromycin for 7 days. The expression level of KIF26B was analyzed by qRT-PCR and western blotting.
Western blot analysis
Total protein was isolated from the cultured cell lines using radioimmunoprecipitation assay lysis buffer (Beyotime Biotechnology, Shanghai, China). The protein concentration in the cell extracts was measured using a bicinchoninic acid protein assay kit (Beyotime Biotechnology) with bovine serum albumin as a standard. Equal amounts of protein (40 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred onto polyvinylidene difluoride (PVDF) membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked in 5% fat-free milk and then incubated with the following primary antibodies at 4°C overnight: anti-KIF26B (Abcam, Cambridge, UK), anti-GAPDH (Abways, Shanghai, China), anti-Bcl-2 (Cell Signaling Technology, Danvers, MA, USA), anti-Bax (Cell Signaling Technology), anti-cyclin D1 (Cell Signaling Technology), anti-p-Rb (ser780) (Cell Signaling Technology), anti-cleaved caspase-3 (Cell Signaling Technology), anti-E-cadherin (Cell Signaling Technology), anti-N-cadherin (Cell Signaling Technology), anti-Vimentin (Cell Signaling Technology), and anti-Snail (Cell Signaling Technology); then, the membranes were incubated with the secondary detection antibody (Cell Signaling Technology) for 1 h at room temperature. Protein bands were visualized using an enhanced chemilumescent (ECL) reagent (Pierce Biotechnology, Waltham, MA, USA).
Statistical analysis
All statistical analyses were carried out using SPSS 19.0 software (IBM Corporation, Armonk, NY, USA). The statistical significance of the qRT-PCR results for KIF26B gene expression and the results for cell proliferation, colony formation, cell cycle, migration, invasion, and apoptosis were calculated using Student’s t-tests (two-tailed). All values are expressed as the mean±SD unless otherwise stated. A p-value <0.05 was considered to be statistically significant.
Results
Inhibition of KIF26B in the MCF-7 and MDA-MB-231 cell lines
To investigate the expression of KIF26B in a large cohort of patient samples, we retrieved and analyzed the expression of KIF26B in The Cancer Genome Atlas database from breast cancer using the online web portal UALCAN (http://ualcan.path.uab.edu). As shown in Figure 1A, KIF26B was significantly upregulated in luminal, HER2-positive, and triple-negative breast cancer tissues compared with normal tissues. In addition, KIF26B showed highest expression in luminal breast tumors. In order to examine the functional role of KIF26B in breast cancer cells, we performed qRT-PCR analysis of KIF26B in 4 breast cancer cell lines and the normal breast cancer cell line MCF-10A. As shown in Figure 1B, the mRNA level of KIF26B in these cell lines was differentially expressed, with its high expression levels in MDA-MB-231 and MCF-7 cells and lowest level in MCF-10A cells. To examine the underlying role of KIF26B in tumorigenesis in breast cancer, lentiviral RNA interference (shKIF26B) was used to suppress KIF26B in MCF-7 and MDA-MB-231 cells; an empty lentiviral vector was used as a negative control (shNC). The transfection efficiency was approximately 92% in MCF-7 cells and 95% in MDA-MB-231 cells (Figure 1C). Furthermore, both the mRNA and protein levels of KIF26B were significantly downregulated in shKIF26B compared to the shNC group or the control group in the MCF-7 and MDA-MB-231 cell lines (Figure 1D and E, p<0.01).
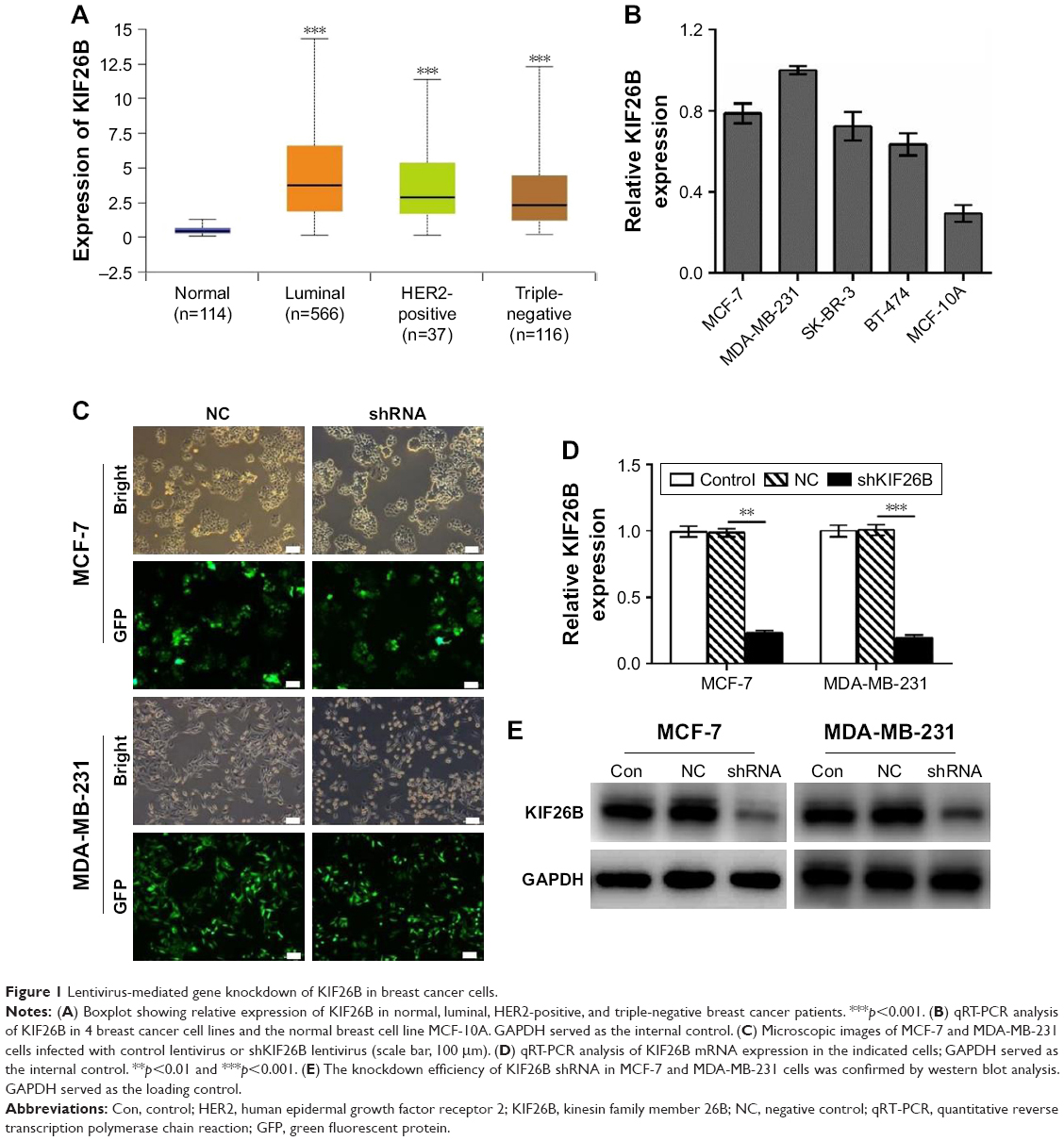 | Figure 1 Lentivirus-mediated gene knockdown of KIF26B in breast cancer cells. |
Knockdown of KIF26B decreases the proliferation and colony formation capacities of breast cancer cells
To identify the potential effect of KIF26B on cell proliferation, we performed CCK8 assays in MCF-7 and MDA-MB-231 cells. The results showed that KIF26B knockdown MCF-7 and MDA-MB-231 cells had a significantly lower proliferation rate than that of control cells (p<0.01, p<0.05, Figure 2A). Furthermore, colony formation assays revealed that smaller and fewer colonies were observed in the shKIF26B group compared with the shNC group (p<0.01, Figure 2B and C). These data indicate that KIF26B might play a crucial role in breast cancer cell proliferation.
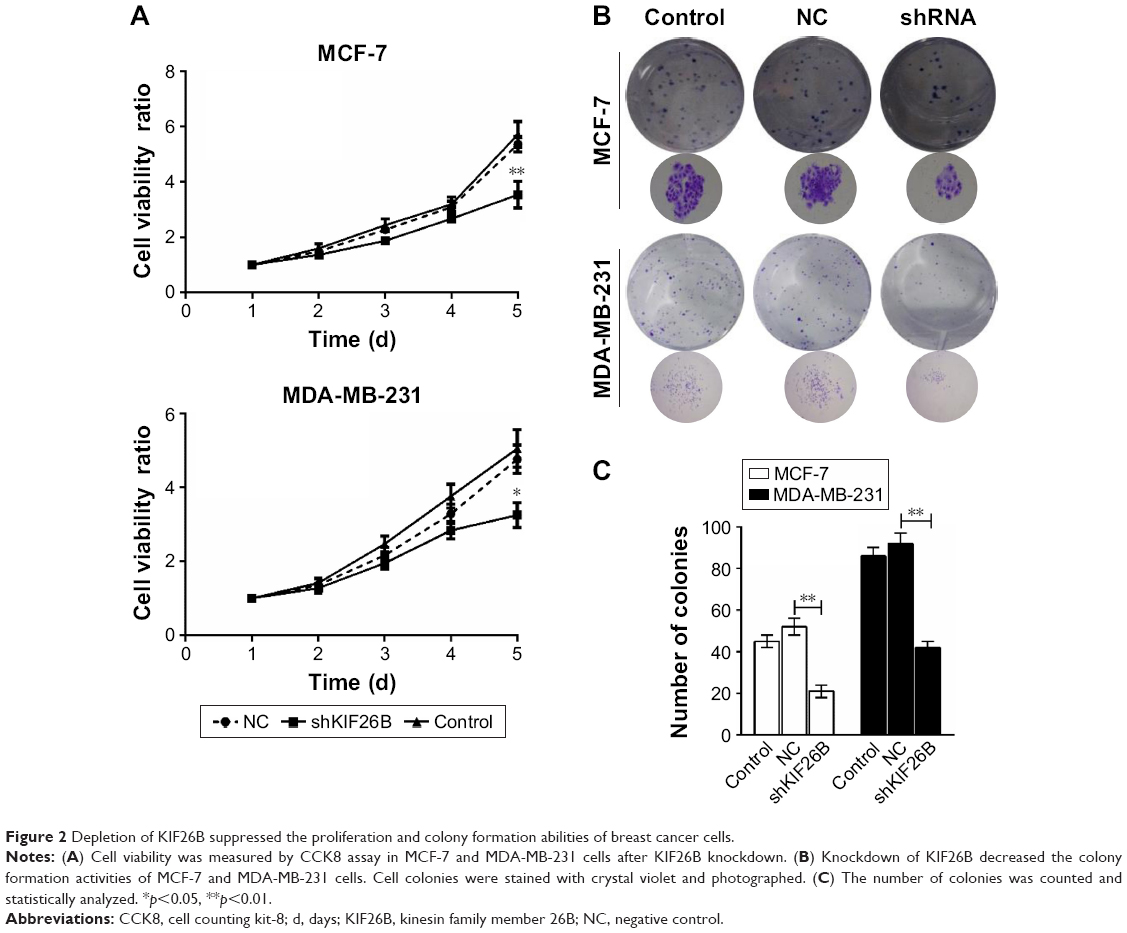 | Figure 2 Depletion of KIF26B suppressed the proliferation and colony formation abilities of breast cancer cells. |
Knockdown of KIF26B induces breast cancer cell apoptosis
Next, we examined the effects of KIF26B knockdown on cell apoptosis by flow cytometry (Figure 3A). As shown in Figure 3B, KIF26B knockdown significantly reduced the number of surviving cells and increased the number of apoptotic cells compared with those in the shNC group. To further investigate the underlying molecular mechanism of shKIF26B-induced apoptosis in MCF-7 and MDA-MB-231 cells, we measured the expression of apoptosis-related proteins after KIF26B knockdown. As shown in Figure 3C, lentivirus-mediated knockdown of KIF26B led to the upregulation of Bax and cleaved caspase-3 expression and the downregulation of Bcl-2 expression, which agreed with our previous flow cytometry results. Consistent with the western blot results, knockdown of KIF26B results in the increased mRNA levels of Bax and reduced mRNA levels of Bcl-2 (Figure 3D). The results suggest that the knockdown of KIF26B promotes cell apoptosis by regulating apoptosis-related proteins.
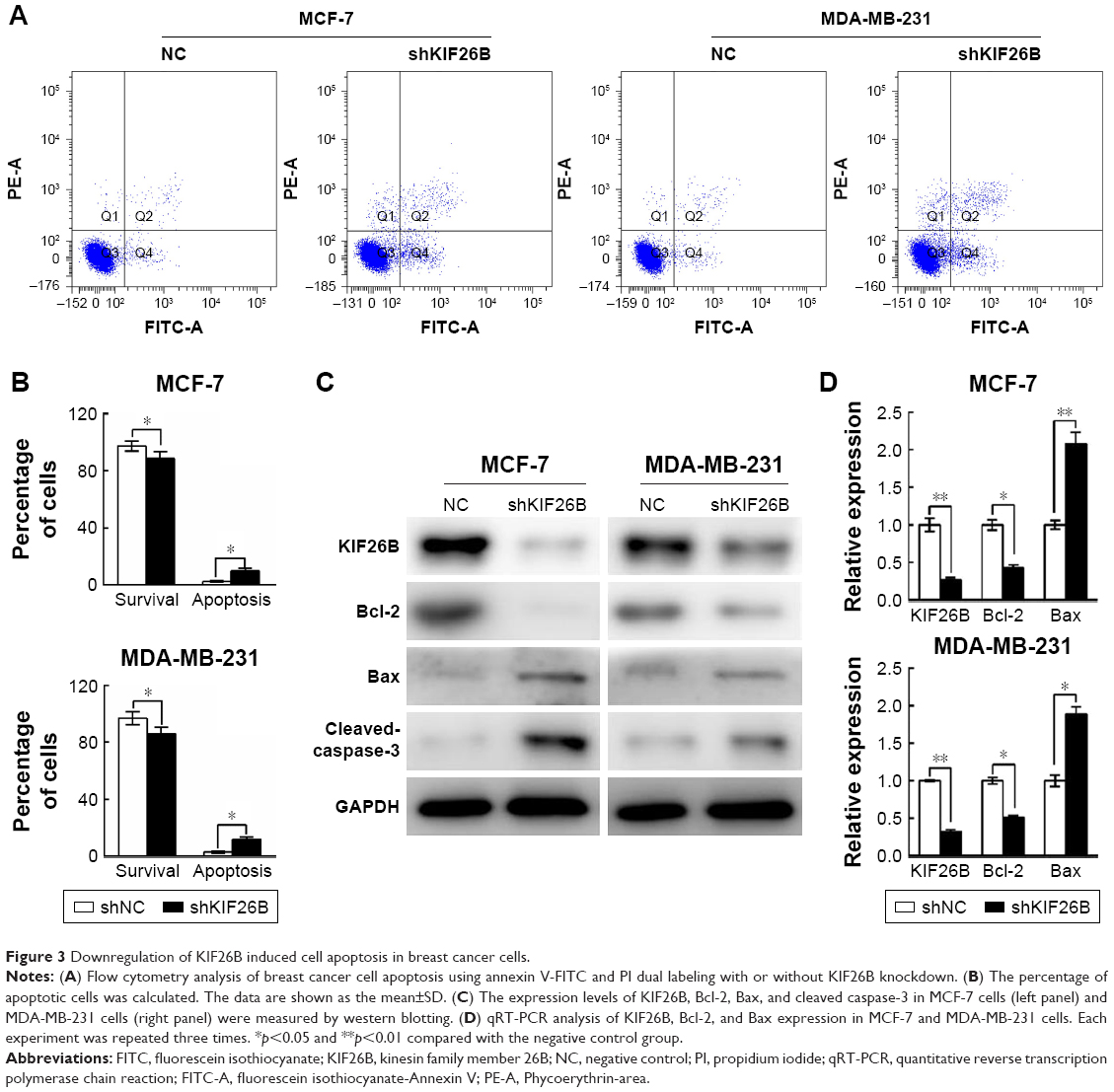 | Figure 3 Downregulation of KIF26B induced cell apoptosis in breast cancer cells. |
Knockdown of KIF26B arrests the cell cycle in the G0/G1 phase
To examine the reason for the shKIF26B-mediated inhibition of cell proliferation, we analyzed the cell cycle by flow cytometry. Compared to the shNC group, the percentage of cells in the G0/G1 phase was significantly increased in shKIF26B-transfected MCF-7 and MDA-MB-231 cells (Figure 4A and B), suggesting that KIF26B knockdown induced G0/G1 phase arrest. In addition, we observed a dramatic decrease in the protein expression of cyclin D1 and p-Rb in MCF-7 and MDA-MB-231 cells after the downregulation of KIF26B (Figure 4C), which is consistent with the G0/G1 phase arrest induced by shKIF26B. Consistently, the mRNA level of cyclin D1 showed a significant decrease after knockdown of KIF26B (Figure 4D). Taken together, these data suggest that G0/G1 phase arrest might contribute to the suppression of cell proliferation induced by the knockdown of KIF26B.
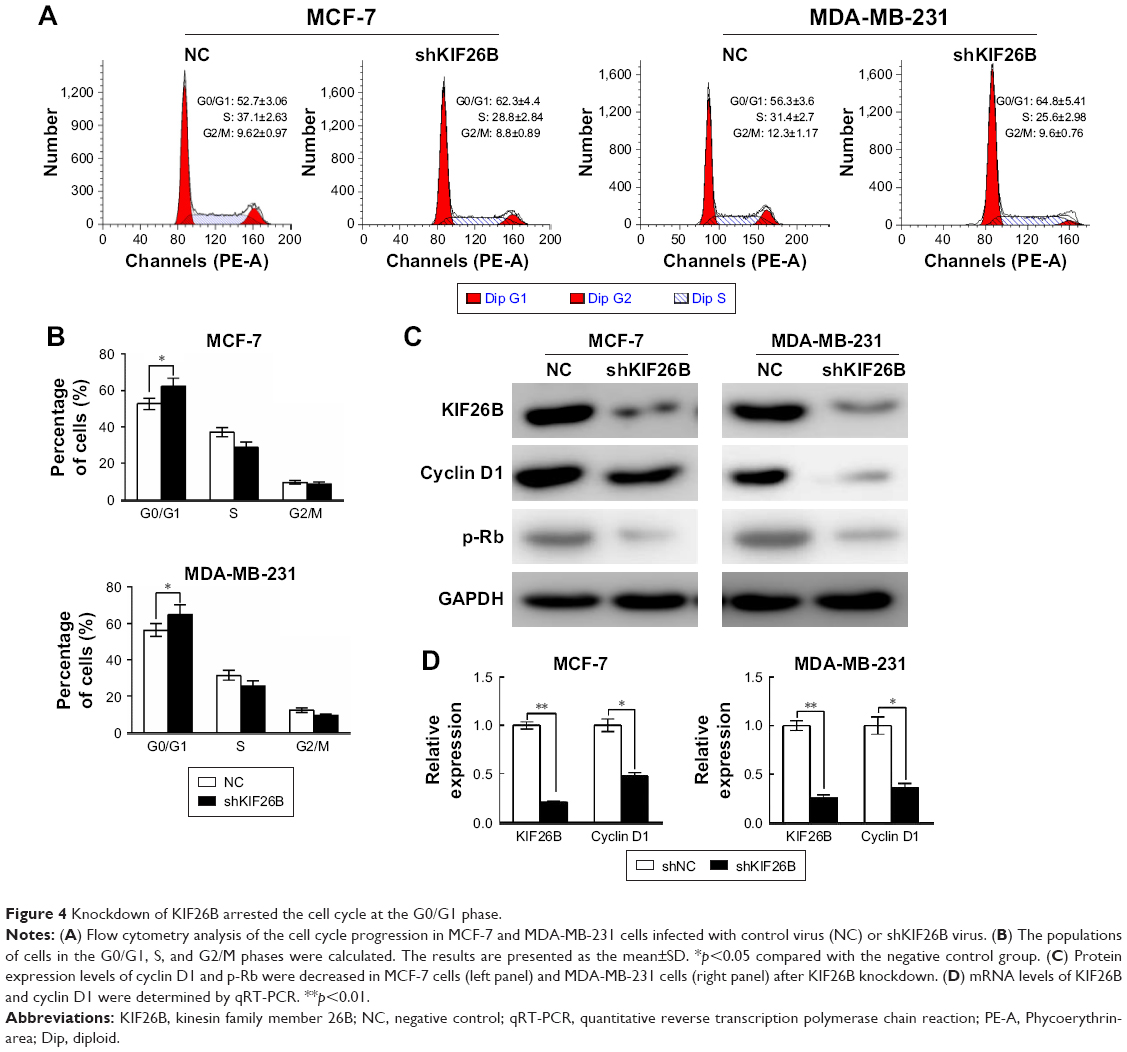 | Figure 4 Knockdown of KIF26B arrested the cell cycle at the G0/G1 phase. |
Knockdown of KIF26B inhibits tumor metastasis and invasion by modulating EMT-related genes
To investigate the effect of KIF26B on the migration and invasion of breast cancer cells, we conducted Transwell migration and Matrigel invasion assays. As shown in Figure 5A and B, cell migration and the invasive capabilities of KIF26B knockdown cells were significantly decreased compared with those of control cells. To explore the potential mechanisms underlying the shKIF26B-induced inhibition of migration and invasion, the expression of EMT-related proteins was detected by western blot. We found that KIF26B knockdown increased the expression of E-cadherin but decreased the expression of N-cadherin, vimentin, and Snail1 (Figure 5C). Thus, the above results suggest that KIF26B plays an important role in breast cancer cell migration and invasion.
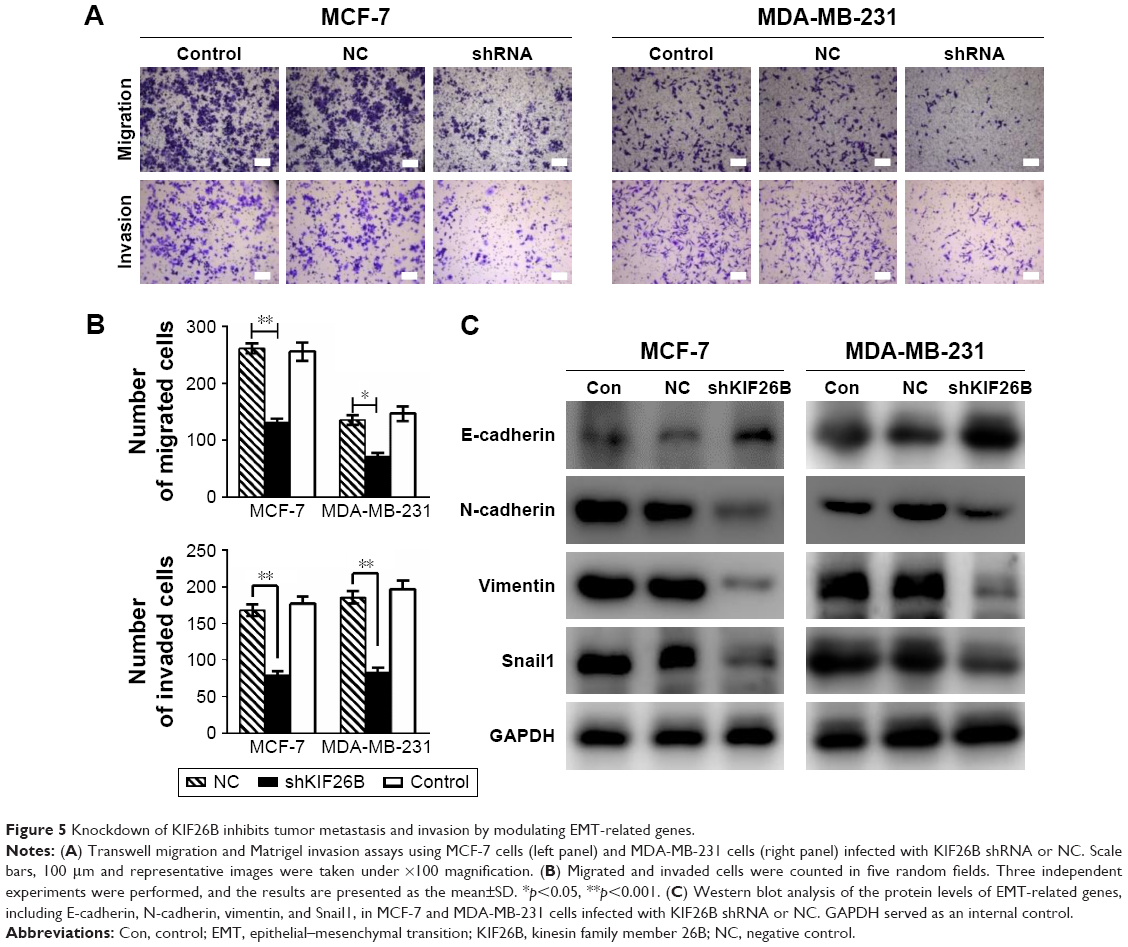 | Figure 5 Knockdown of KIF26B inhibits tumor metastasis and invasion by modulating EMT-related genes. |
Discussion
The KIFs play important roles in proliferation and migration of breast cancer cells. Previous reports demonstrated that elevated expression of KIF2A and KIF14 was positively associated with lymph nodes of patients with breast cancer.17,18 Knockdown of KIF3C in breast cancer cell lines inhibited epithelial–mesenchymal transition (EMT) and metastasis.19 In addition, silencing of KIF3C reduced proliferation of breast cancer cells via inducing G2/M phase arrest.19 It has been reported that KIF26B plays an important role in many human cancers.12–14 KIF26B is highly expressed in breast cancer tissues, and high KIF26B expression is correlated with poor prognosis.15 This evidence suggests that KIF26B might play an instrumental role in tumorigenesis in breast cancer cells. In this study, for the first time, we examined the oncogenic function of KIF26B and its underlying mechanism in breast cancer cells.
To better understand the underlying mechanism of shKIF26B-induced cell growth inhibition, we performed cell cycle distribution experiments after KIF26B knockdown and found that the knockdown of KIF26B induced G0/G1 phase arrest. It is well known that the activation of cell cycle phases is dependent on the regulation of cyclins and cyclin-dependent kinases (CDKs).20,21 The complex of cyclin D1 and CDK4 promotes the transition of cells from the G0/G1 phase to the S phase by phosphorylating the retinoblastoma tumor suppressor gene.22 Our results demonstrated that the knockdown of KIF26B resulted in a decrease in the expression of cyclin D1, which might be the reason for G0/G1 arrest.
Apoptosis is considered to be one of the main contributors to cancer development. In apoptosis progression, caspase-3 is a crucial executioner caspase that is activated by the cleavage of caspase-8 or caspase-9.23 Activated caspase-3 cleaves several downstream effectors, including the PARP protein, which eventually induces apoptosis. In the present study, cleaved caspase-3 was upregulated in the KIF26B knockdown group. Moreover, the proapoptotic Bax was increased, whereas the antiapoptotic Bcl-2 was decreased in KIF26B knockdown cells. These data suggest that the mitochondrial pathway was involved in shKIF26B-induced apoptosis.
EMT is a multistep plasticity process that plays a crucial role in cancer metastasis. EMT involves the loss of epithelial properties, such as decreased E-cadherin expression, and the gain of mesenchymal properties, such as increased N-cadherin and vimentin expression.24,25 In this study, we found that the knockdown of KIF26B inhibited cell metastasis by increasing the expression of the epithelial marker E-cadherin and by decreasing the expression of the mesenchymal markers vimentin, Snail1, and N-cadherin. Our findings indicate that KIF26B may drive EMT in breast cancer cells, resulting in metastasis.
Overexpression of KIF26B in breast cancer tissues has been reported.15 However, the mechanisms regulating KIF26B expression remain unknown. As Sall1 is a transcriptional activator of KIF26B,10 it would be interesting to investigate the relationship between KIF26B and Sall1 in breast cancer. Recently, Pu et al26 demonstrated that KIF26B is a direct target of miR-20a-5p and plays a key role in multidrug resistance in osteosarcoma. We believe that KIF26B may also contribute to multidrug resistance in breast cancer. In summary, KIF26B may be a promising target for breast cancer treatment. However, further studies are needed to fully understand the oncogenic role of KIF26B in breast cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. | ||
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. | ||
Fan L, Strasser-Weippl K, Li JJ, et al. Breast cancer in China. Lancet Oncol. 2014;15:e279–e289. | ||
Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–1167. | ||
Cuzick J, DeCensi A, Arun B, et al. Preventive therapy for breast cancer: a consensus statement. Lancet Oncol. 2011;12:496–503. | ||
Shimelis H, Mesman RLS, Von Nicolai C, et al. BRCA2 Hypomorphic missense variants confer moderate risks of breast cancer. Cancer Res. 2017;77:2789–2799. | ||
Weigelt B, Peterse JL, van ‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5:591–602. | ||
Miki H, Setou M, Kaneshiro K, Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci U S A. 2001;98:7004–7011. | ||
Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. | ||
Uchiyama Y, Sakaguchi M, Terabayashi T, et al. Kif26b, a kinesin family gene, regulates adhesion of the embryonic kidney mesenchyme. Proc Natl Acad Sci U S A. 2010;107:9240–9245. | ||
Heinrich J, Proepper C, Schmidt T, Linta L, Liebau S, Boeckers TM. The postsynaptic density protein Abelson interactor protein 1 interacts with the motor protein Kinesin family member 26B in hippocampal neurons. Neuroscience. 2012;221:86–95. | ||
Gu J, Ajani JA, Hawk ET, et al. Genome-wide catalogue of chromosomal aberrations in barrett’s esophagus and esophageal adenocarcinoma: a high-density single nucleotide polymorphism array analysis. Cancer Prev Res. 2010;3:1176–1186. | ||
Wang J, Cui F, Wang X, et al. Elevated kinesin family member 26B is a prognostic biomarker and a potential therapeutic target for colorectal cancer. J Exp Clin Cancer Res. 2015;34:13. | ||
Zhang H, Ma RR, Wang XJ, et al. KIF26B, a novel oncogene, promotes proliferation and metastasis by activating the VEGF pathway in gastric cancer. Oncogene. 2017;36:5609–5619. | ||
Wang Q, Zhao ZB, Wang G, Hui Z, Wang MH, Pan JF, Zheng H. High expression of KIF26B in breast cancer associates with poor prognosis. PLoS One. 2013;8:e61640. | ||
Xie Y, Wang B. Downregulation of TNFAIP2 suppresses proliferation and metastasis in esophageal squamous cell carcinoma through activation of the Wnt/β-catenin signaling pathway. Oncol Rep. 2017;37:2920–2928. | ||
Wang J, Ma S, Ma R, et al. KIF2A silencing inhibits the proliferation and migration of breast cancer cells and correlates with unfavorable prognosis in breast cancer. BMC Cancer. 2014;14:461. | ||
Corson TW, Gallie BL. KIF14 mRNA expression is a predictor of grade and outcome in breast cancer. Int J Cancer. 2006;119:1088–1094. | ||
Wang C, Wang C, Wei Z, et al. Suppression of motor protein KIF3C expression inhibits tumor growth and metastasis in breast cancer by inhibiting TGF-β signaling. Cancer Lett. 2015;368:105–114. | ||
Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8:149–160. | ||
Nigg EA. Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays. 1995;17:471–480. | ||
Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21:4773–4784. | ||
Zheng L, Zheng J, Wu LJ, Zhao YY. Julibroside J8-induced HeLa cell apoptosis through caspase pathway. J Asian Nat Prod Res. 2006;8:457–465. | ||
Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–641. | ||
Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. | ||
Pu Y, Yi Q, Zhao F, Wang H, Cai W, Cai S. MiR-20a-5p represses multi-drug resistance in osteosarcoma by targeting the KIF26B gene. Cancer Cell Int. 2016;16:64. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.