")
Back to Journals » Cancer Management and Research » Volume 12
Knockdown of IKKβ Inhibits Tumor Development in a Leptomeningeal Metastasis Mouse Model and Proliferation of Lung Cancer Cells
Authors Liu Y, Li Y, Li Z, Li C, He J, Bu H
Received 2 March 2020
Accepted for publication 24 June 2020
Published 20 July 2020 Volume 2020:12 Pages 6007—6017
DOI https://doi.org/10.2147/CMAR.S252184
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Yakun Liu,1,2,* Yuanyuan Li,1,2,* Zhongyao Li,1,2 Chunyan Li,1– 3 Junying He,1– 3 Hui Bu1– 3
1Department of Neurology, The Second Hospital of Hebei Medical University, Shijiazhuang, Hebei 050000, People’s Republic of China; 2Neurological Laboratory of Hebei Province, Shijiazhuang, Hebei 050000, People’s Republic of China; 3Institute of Cardiocerebrovascular Disease, Shijiazhuang, Hebei 050000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hui Bu
Department of Neurology, Second Hospital of Hebei Medical University, Shijiazhuang 050000, Hebei, People’s Republic of China
Tel/ Fax +86 13831106903
Email [email protected]
Objective: This study will explore the role of IKKβ in the leptomeningeal metastasis (LM) of lung cancer cells.
Methods: In vitro studies were conducted in mouse Lewis lung carcinoma (LLC) cells with IKKβ knockdown. Cell proliferation was explored using CCK-8 and tumor colony-forming assays. The expression of the extracellular signal-regulated kinase (ERK) signaling pathway was detected by Western blot. Tumor cell apoptosis was identified through Western blot detection of Bax and Bcl-2. The migration of tumor cells was identified by a wound healing assay. In vivo studies used an LM mouse model developed by injecting LLC cells with or without IKKβ knockdown into the cisterna magna. Mouse brain and spinal samples were sectioned for hematoxylin and eosin staining.
Results: In vitro: IKKβ knockdown inhibits tumor cell proliferation, initiates apoptosis, and attenuates cell migration. In vivo: IKKβ knockdown inhibits tumor growth in the LM mouse model. In addition, the in vitro results showed that IKKβ knockdown attenuated the expression of ERK phosphorylation in LLC cells.
Conclusion: Blocking the NF-κB signaling pathway by IKKβ knockdown in LLC cells inhibited tumor growth in the LM mouse model. IKKβ supports leptomeningeal tumor progression by promoting cancer cell proliferation and migration and inhibiting cancer cell apoptosis, and these actions may be correlated to ERK signaling.
Keywords: leptomeningeal metastasis, non-small-cell lung cancer, NF-κB signaling, IKKβ, shRNA
Introduction
Leptomeningeal metastasis (LM) is a devastating, late-stage complication of various solid tumors and is characterized by the diffuse dissemination of malignant tumor cells into the cerebrospinal fluid (CSF) and leptomeninges.1,2 Metastasis to this fluid-filled space can quickly spread to the entire central nervous system, leading to progressive neurological dysfunction and death.3 LM occurs in 3–5% of patients with advanced non-small cell lung cancer (NSCLC).4 Its incidence has increased, which may be due to improved outcomes brought about by new molecular therapies.5 However, the present treatment options, including intra-CSF chemotherapy, radiation therapy, and molecularly targeted therapies, are not effective. Thus, patients with LM from NSCLC have a poor prognosis, with a median overall survival range of 3 to 11 months.4,6,7 Novel and effective therapies are needed to manage LM in patients with NSCLC.
Epidermal growth factor receptor (EGFR)-mutant lung cancer is more frequently associated with central nervous system metastasis, including brain metastasis and LM, than NSCLC with wild-type EGFR. A recent study with a retrospective chart review also reported that EGFR recurrently mutated in the CSF of patients with NSCLC LM.8 Although EGFR tyrosine kinase inhibitors (EGFR-TKIs) have exhibited remarkable clinical efficacy on LM patients, most patients acquire resistance to EGFR-TKIs, leading to progression of the disease.9
Nuclear factor-κB (NF-κB) is a family of transcription factors that can form different heterodimers or homodimers and bind to consensus DNA sequences at promoter regions of the target genes.10 Constitutive NF-kB activation has been reported in a variety of tumors, including lung cancer tumors.11,12 Tumor progression in the lentiviral-vector-mediated mouse model is attenuated when shRNA knocks down IKKβ in tumor cells. This reduces ERK activation and cell proliferation.13 Several mechanisms have been identified, and it has been shown that an activated IKK/NF-kB signaling pathway in tumor cells induces target genes that modulate apoptosis, cell cycle, cell invasion, and metastatic growth.14
Along with its role in primary tumors, NF-kB signaling in the pre-metastatic niche may establish an environment for seeding primary tumor cells that contribute to lung metastasis. Compared to the parenchyma of other major organ metastasis sites, the CSF-filled leptomeningeal space lacks protein, glucose, and cytokine content and is a markedly different metastatic microenvironment for malignant cells.3 NF-kB signaling has been involved in EGFR-TKI resistance, and genetic or pharmacologic inhibition of NF-kB enhances erlotinib-induced apoptosis in EGFR-mutant lung cancer models. The activation of NF-kB through the overexpression of IKKβ shielded lung cancer cells from EGFR-TKI treatment.15
It is unclear whether IKK/NF-kB activity in lung cancer cells enhances their ability to thrive in leptomeninges. The present study describes a simple and replicable intracisternal injection method to induce LM in mice. Using this model, the impact of IKKβ knockdown on Lewis lung cancer (LLC) cells was shown to block the NF-κB signaling pathway and reduce tumor development in the pia mater space—suggesting a possible adjuvant therapy for pial meningeal metastasis.
Materials and Methods
Cell Culture
Mouse LLC cells were purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, China) and kept in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco Life Technologies Corporation, US, cat. no: 12,100–038), supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). These cells were routinely cultured as monolayers in 25-cm flasks (Corning, NY, US) at 37°C in a 5% CO2 humidified atmosphere and passaged for further experiments.
Virus Production and Transduction in Cells
Lentiviral vectors (GV248) containing IKKβ shRNA or negative-control shRNA were obtained from GeneChem (Shanghai, China). The lentivirus particles were produced by co-transfecting the HEK293T cell line with the lentiviral plasmid and packing plasmid (GeneChem). The siRNA sequence used was 5′-CTGGTTACAGATGGAGGATGA-3′. A quantitative PCR (qPCR) assay was performed to determine the vector titer, which was 6 × 108 TU/mL. For cell transfection, LLC cells seeded in six-well plates (3 × 104 cells/well) were transduced with the constructed lentiviral shRNA particles (Lv-shIKKβ or Lv-shCon) in complete medium with 5 μg/mL polybrene. The medium was replaced with fresh medium at 16 hours after infection. At 72 hours after transfection, the tumor cells were stably infected with viral particles, and cells that expressed green fluorescent protein (GFP) were isolated via selection in the presence of 2 μg/mL puromycin.
Quantitative Real-Time PCR
Total RNA isolated from LLC cells using an RNA rapid extraction kit (Generay Biotech Co., Ltd., GK3016) was reverse-transcribed using a Moloney murine leukemia virus reverse transcriptase (Promega, M1705) with random primers. The qPCR was carried out in triplicate using the LightCycler480 II (Roche) through the SYBR Premix Ex Taq (TaKaRa, DRR041B), according to the manufacturer’s instructions. The real-time protocol involved denaturation maintained at 95°C for 30 seconds and 40 amplification cycles (denaturation at 95°C for five seconds; annealing and extension at 60°C for 30 seconds). The results were analyzed for the relative expression of mRNAs normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
The primers used in the present study were IKKβ, TAGTAGAGCGGATGATGGCA (forward), and CTTCTCCCTGAGTCTTCGGTA (reverse); GAPDH, TGGTGAAGGTCGGTGTGAAC (forward) and GCTCCTGGAAGATGGTGATGG (reverse).
Cell Viability Assay
The proliferation assays were performed with LLC cells (with or without IKKβ shRNA or control shRNA transfection) using a Cell Counting Kit-8 (CCK-8, Beyotime Institute of Biotechnology, Nanjing, China), according to the manufacturer’s instructions. Then, 3 × 103 cells were seeded in 96-well plates. After incubating for 36 hours, 10 μL CCK-8 solution with 100 μL medium was added to each well at 37°C. After two hours, the absorbance at 450 nm was measured. All experiments were performed in triplicate.
Colony-Forming Assays
LLC cells (500 cells/well) transfected with Lv-shIKKβ or Lv-shCon were seeded in six-well plates and cultured in DMEM. After incubation for nine days (the medium was replaced every two to three days), the cells were washed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 30 minutes at room temperature. The cells were stained with May-Grünwald-Giemsa (MGG) for ten minutes. The cell colonies in each group were photographed and counted under a microscope.
Wound Healing Assay
Next, 5 × 105 cells were plated into six-well plates and allowed to grow until confluence, when a scratch in the monolayer was made using a sterile 10-μL micropipette tip. These cells were grown in serum-free medium until the end of the experiment. The wells were washed with D-Hank’s solution three times to remove the cell debris before imaging the same area at the specified time points. The wounds were measured by width at three points and averaged.
Cell Migration and Invasion Assays
Transwell chambers containing 8.0-μm pores on a polycarbonate membrane in 24-well plates (Corning, NY, US) were used to assess cell migration and invasion. At post-transfection 96 hours, the cells were serum starved. For the invasion assay, 100 μL to 200 μg/mL Matrigel was diluted in serum-free DMEM, placed in the upper chamber, and left in an incubator for one hour to solidify. Then, 1×105 cells/well were plated in 200 μL of serum-free media in the upper chamber, and 500 μL of 10% FBS DMEM was added to the bottom chamber. After incubation for 24 hours at 37°C, each insert was washed three times in PBS, and cells on the lower surface were fixed with 4% paraformaldehyde and stained with MGG. To quantitate the cell movement, cells in five random fields from each well were counted and averaged.
Western Blot
For protein extraction, cells were lysed in ice-cold immunoprecipitation (IP) buffer containing phosphatase inhibitors, proteinase inhibitors, and phenylmethanesulfonyl fluoride (PMSF). Whole-cell lysates were centrifuged at 10,000 × g for five minutes at 4°C, and the protein concentrations were determined using a BCA protein assay kit. The protein extracts were separated into 10% SDS-PAGE gels and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were incubated overnight at 4°C with these primary antibodies: anti-β-actin (60008–1, Proteintech, Wuhan, China), anti-GFP (Protein Tech Group, Inc., Chicago, US), anti-IKKβ (ab178870, Abcam, Cambridge, UK), anti-p-Erk1/2 (#4370, Cell Signaling Technology), anti-Bcl-2 (YM3041, Immunoway), and anti-bax (YT0458, Immunoway). Membranes were incubated for one hour at room temperature with IRDyeTM700DX anti-mouse IgG, IRDyeTM800DX anti-rabbit IgG, or IRDyeTM800DX anti-goat IgG (Rockland, Maine, US) conjugated secondary antibody (1:3000). The bands of interest were detected using an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE, US).
Immunofluorescence Staining
LLC cells treated with IKKβ shRNA or control shRNA were plated on coverslips coated by laminin in a 24-well plate. After incubation for 36 hours, the cells were fixed with 4% paraformaldehyde and washed three times in PBS. Cell permeabilization and blocking were performed using 0.3% Triton X-100 PBS containing 10% horse serum for 30 minutes at room temperature. After incubation overnight at 4°C with antibodies against IKKβ (ab178870, Abcam, Cambridge, UK), the cells were stained with goat anti-rabbit IgG secondary antibodies (Jackson ImmunoResearch, West Grove, PA, US) at room temperature for one hour and Hoechst (Invitrogen, Carlsbad, CA, US) for nuclear staining for 15 minutes. The labeled cells were immediately imaged by confocal microscopy.
LM Mouse Model
Six- to eight-week-old C57BL/6 mice were used (purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd.). The animals were anesthetized by intraperitoneal injection of 2% pentobarbital sodium (3.3 µL/g) and sacrificed. All animal experiments were approved by the ethics committee of the Second Hospital of Hebei Medical University, and all work was carried out in compliance with the laboratory animal management regulations provided by the Ministry of Science and Technology of the People’s Republic of China (under the NIH Guide for the Care and Use of Laboratory Animals). An injection into the cisterna magna (CM) of the anesthetized recipient mice was performed, as described in a previous study (Reijneveld et al, 1999). Each anesthetized mouse was placed prone with its neck draped over a 10-mL needle cylinder.
The occiput was palpated, and a midline incision was made in the skin of the nape of the neck, exposing the midline. A 25-μL microsyringe fitted with a 31G beveled (cutting) needle was introduced at an angle between the external occipital protuberance and the first cervical vertebra. The needle was advanced 4 mm before withdrawing 10 μL of LLC cell suspension (1 × 107 cells/mL). Mice were randomly assigned to one of two treatment groups in which LLC cells were infected with IKKβ shRNA or control shRNA. The dynamic change in the bodyweight of the mice after CM injection was recorded daily, and the neurological manifestations were observed and evaluated. Mouse brain and spinal samples collected at the symptomatic time were fixed with 4% paraformaldehyde, paraffin-embedded, and sectioned for hematoxylin and eosin (H&E) staining.
Statistical Analysis
All data are presented as mean ± standard deviation (SD) from at least three independent experiments. The statistical analysis was performed using the SPSS 21.0 software. Data from the different groups were analyzed using a Student’s t-test, one-way ANOVA, or Mann–Whitney U-test. The statistics were considered significant when P < 0.05.
Results
IKKβ Knockdown in Lung Cancer Cells by Lentiviral-Vector-Mediated shRNA
Because the NF-κB pathway plays an important role in tumor cell development, and IKKβ is the critical kinase responsible for NF-κB activation, the effect of IKKβ inhibition on lung cancer cells was explored. LLC cells were transduced with a lentiviral-vector-mediated shRNA, targeting IKKβ. After cell transfection for 72 hours (Supplementary Figure 1), the expression level of IKKβ was tested in sorted cells to confirm the transfection efficiency by qRT-PCR analysis and Western blot. The results showed that the mRNA and protein levels of IKKβ were markedly decreased in cells treated with IKKβ shRNA compared to cells from control shRNA (Figure 1A–C). The immunofluorescence analysis revealed that the LLC cells expressing GFP reduced the levels of IKKβ when treated with IKKβ shRNA, based on the decreased fluorescence intensity of IKKβ (Figure 1D and E).
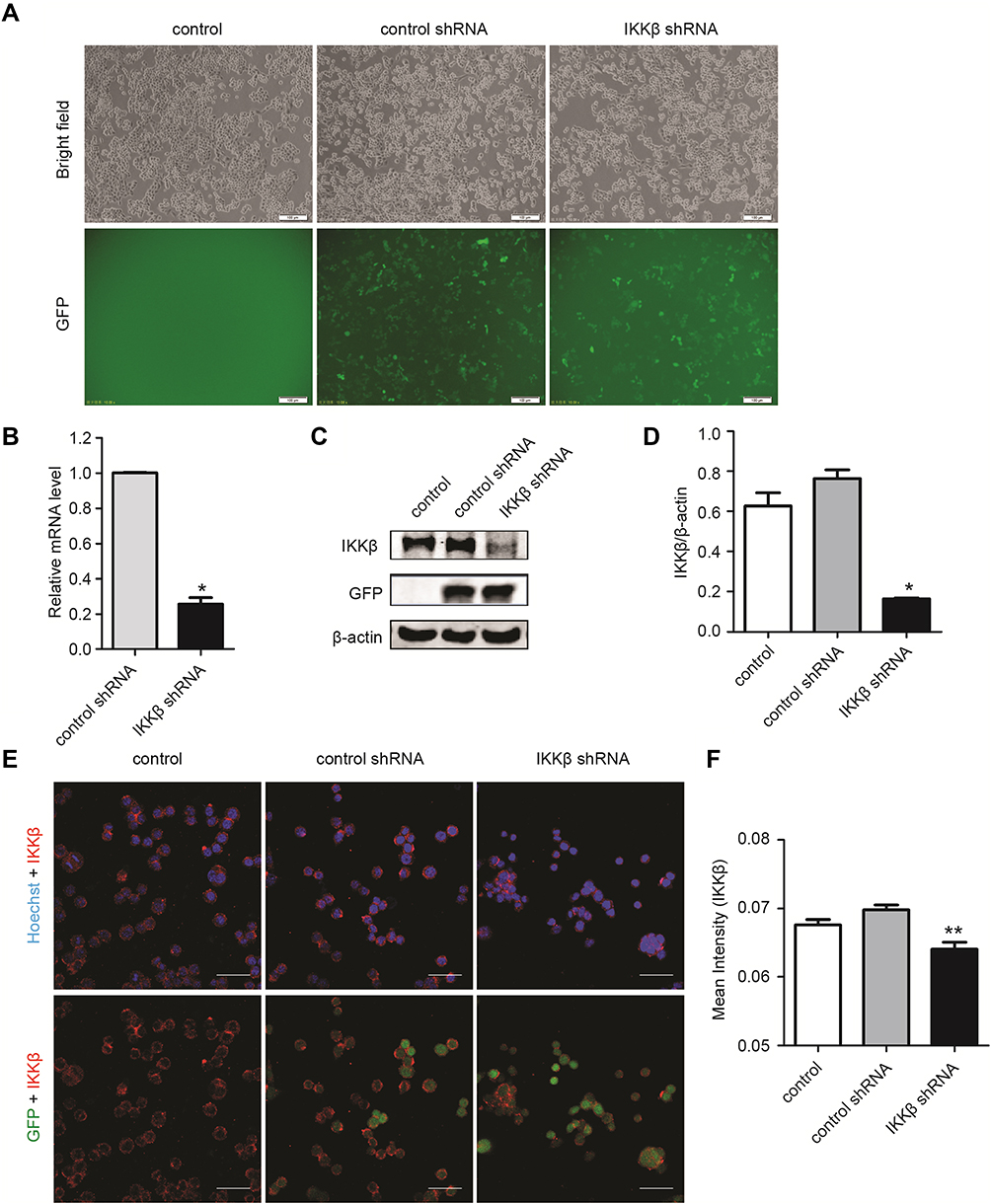 |
Figure 1 The knockdown of IKKβ with lentivirus-delivered shRNA (small hairpin RNA) reduced the endogenous expression in Lewis lung carcinoma (LLC) cells. (A) qRT-PCR analysis of IKKβ knockdown efficiency in LLC cells treated with control shRNA or IKKβ shRNA. (B, C) The levels of IKKβ were evaluated by Western blot in uninfected and lentivirus-infected LLC cells (B). The intensity of the bands was evaluated by ImageJ software (C). (D) mRNA expression of KKβ in each group. (E) The representative images of LLC cells transfected with control shRNA or IKKβ shRNA and stained by immunofluorescence for IKKβ (red) and Hoechst (blue) (scale bars = 50 μm). (F) Quantification of (E). The mean intensity of IKKβ was determined by ImageJ software. The results were the combined means ± standard deviation (SD) from three independent experiments. *P < 0.05, **P < 0.01, compared with control. |
These results confirmed that lentivirus-delivered shRNA effectively downregulated the endogenous IKKβ expression in LLC cells.
IKKβ Knockdown Inhibits Tumor Growth in the LM Mouse Model
To determine whether the knockdown of IKKβ could affect tumor LM, a mouse model was first modified to induce LM through intracisternal injection of LLC cells. The H&E staining for histological analysis showed the diffuse infiltration of malignant tumor cells to the leptomeninges and subarachnoid space in the mice that received the LLC injection, and the tumor cells were primarily observed at the base of the brain and cervical cord (Figure 2C).
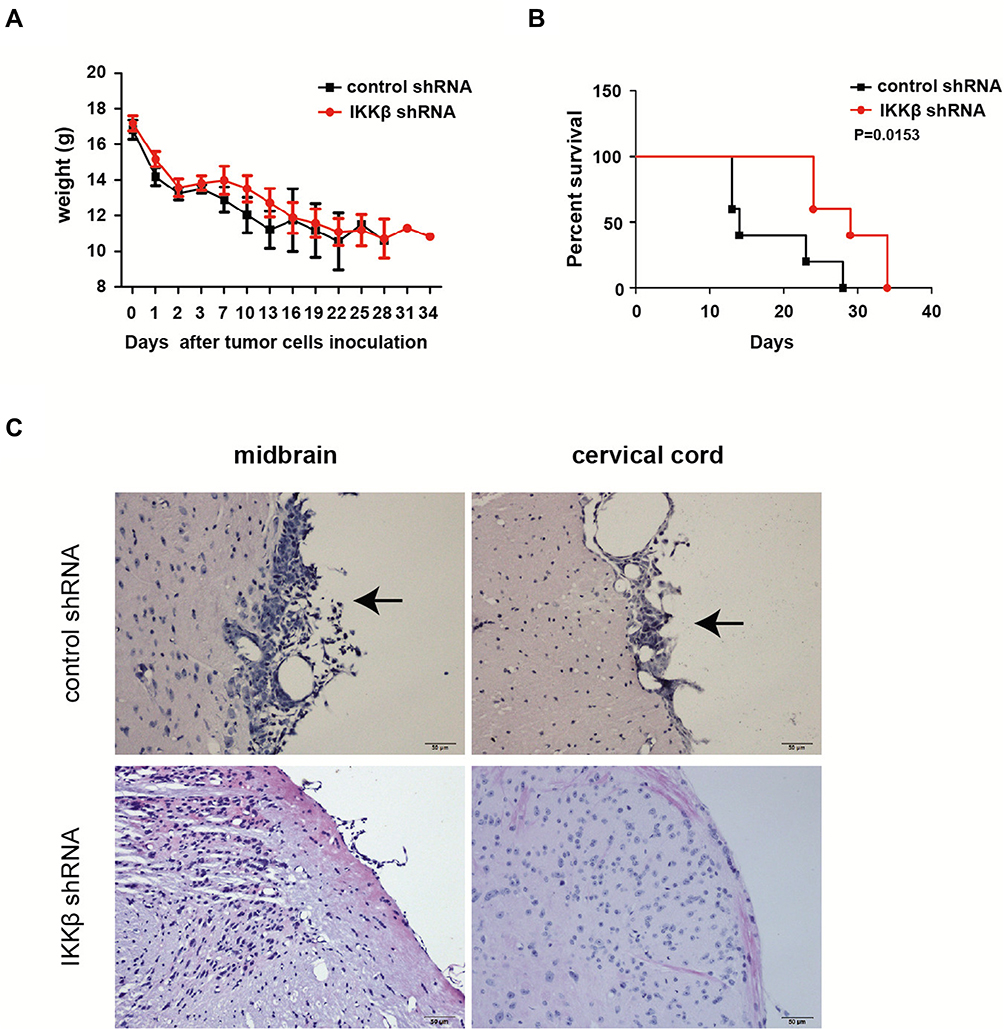 |
Figure 2 IKKβ knockdown inhibited tumor growth in a mouse model of LM of lung cancer cells. (A) body weight changes in mice after intrathecal inoculation of Lewis lung carcinoma (LLC) cells into the cisterna magna. (B) The survival times recorded for mice in the two groups. The median survival of mice administered with IKKβ knockdown LLC cells was significantly longer, when compared to control mice (29 vs 14 days, log-rank analysis: P = 0.0153). (C) representative images of brain or spinal cord sections from mice with LM resulting from the intracisternal administration of LLC cells treated with control shRNA (small hairpin RNA) or IKKβ shRNA. LLC cells with IKKβ knockdown inhibited tumor infiltration in leptomeninges, when compared to cells transfected with control shRNA. The arrows indicate the presence of a tumor in the histopathological sections obtained from the control shRNA group at the indicated levels. The arrows show that tumor cells metastasis to the meninges occurred at these sites. The histopathological sections were observed and analyzed by a pathologist. (paraffin section, H&E × 20, scale bars = 50 μm). |
To further discern the role of IKKβ blocking on tumor LM, mice were randomly assigned into groups, in which LLC cells transfected with IKKβ shRNA or control shRNA were intracisternally injected. The animals in these two groups were sacrificed simultaneously after cancer cell inoculation. The tumor progression was evaluated by pathological manifestations via H&E staining. The results revealed that LLC cells with IKKβ knockdown inhibited the tumor infiltration in the leptomeninges, when compared to cells transfected with control shRNA (Figure 2). Furthermore, it could be seen that the median survival time of mice administered with IKKβ knockdown LLC cells was significantly longer compared to control mice (29 vs 14 days, log-rank analysis: P = 0.0153, Figure 2).
IKKβ Knockdown Inhibits Tumor Cell Proliferation in vitro
To determine the effect of IKKβ knockdown on LLC cell viability, a CCK-8 assay was performed on LLC cells transfected with IKKβ shRNA or control shRNA. The results revealed that the number of viable cells in the Lv-shIKKβ treated group significantly decreased in contrast to those in the control and Lv-shCon groups (P < 0.05, Figure 3A and B), indicating that the knockdown of IKKβ could decrease the viability of lung cancer cells.
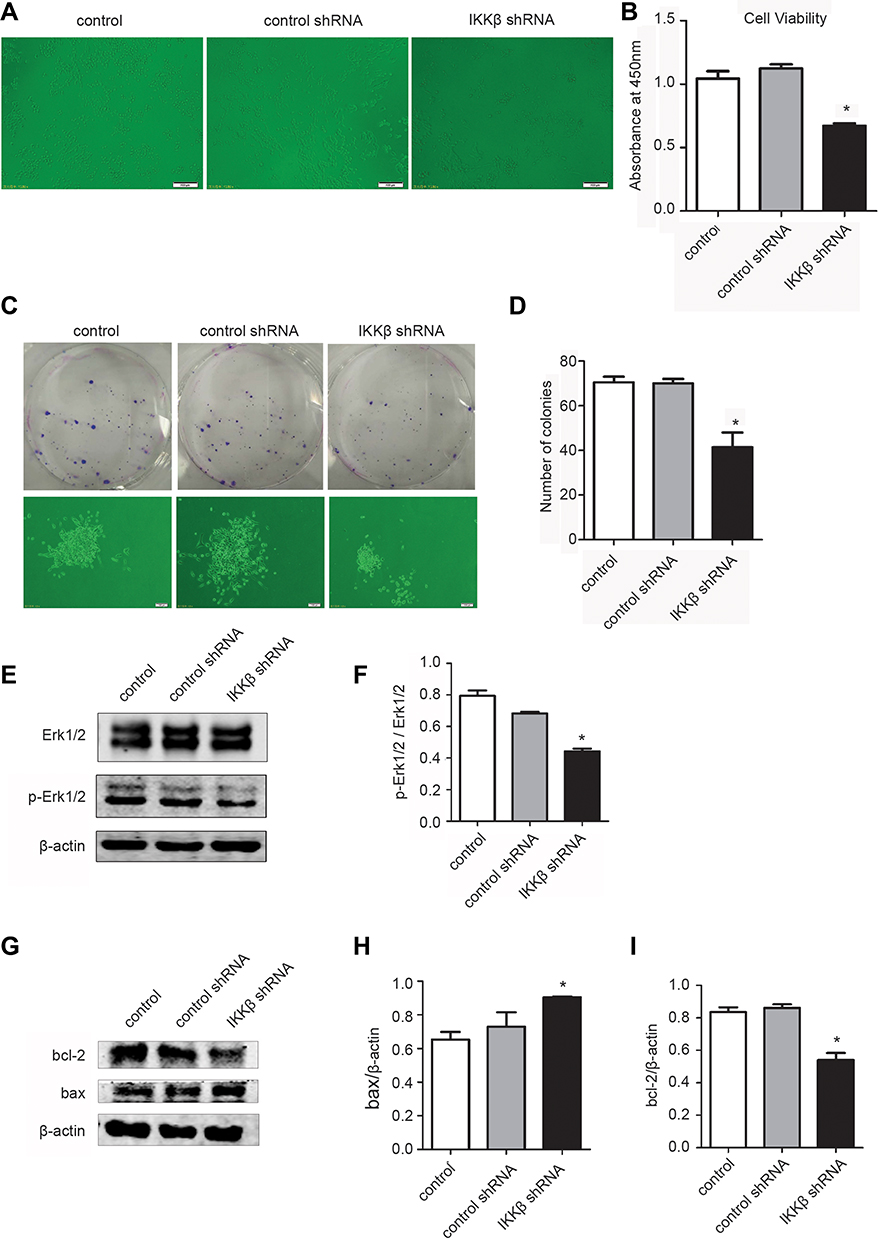 |
Figure 3 The knockdown of IKKβ inhibited tumor cell proliferation and initiated apoptosis in vitro. (A, B) The cell viability of uninfected and lentivirus-infected Lewis lung carcinoma (LLC) cell lines was measured by CCK-8 assay. The cell morphology was observed using an inverted microscope (A), and absorbance was detected at 450 nm (B). (C, D) representative images present the typical colony formation by LLC cells untreated or treated with control shRNA (small hairpin RNA) or IKKβ shRNA at nine days after cell plating. The colony-forming number of each group was assessed. (E, F) The levels of phosphorylated extracellular signal-regulated kinase (p-ERK1/2) were determined by Western blot in LLC cells with different treatments, and the statistical analysis of the expression of p-ERK1/2 and β-actin is presented. (G–I) Western blot bands and statistical analysis of Bax, Bcl-2, and β-actin from uninfected and lentivirus-infected LLC cell lines. The results were the combined means ± standard deviation (SD) of three independent experiments. *P < 0.05, compared with control shRNA cells. |
A tumor colony-forming assay was performed using LLC cells. Cancer cells transfected with Lv-shIKKβ formed approximately 41% fewer colonies when compared to control cells transfected with control shRNA (Figure 3C and D). This suggests that the knockdown of IKKβ with shRNA in lung tumor cells significantly reduced cell growth.
To further analyze the mechanisms by which suppression of cell growth was caused by the knockdown of IKKβ in LLC cells, tumor cells treated with Lv-shIKKβ or Lv-shCon were lysed and subjected to Western blot analysis to examine the expression of related regulators. Because the ERK signaling pathway is critical for cell proliferation, the investigators explored whether IKKβ knockdown might affect the activation of ERK signaling. As shown in (Figure 3E and F), LLC cells in the Lv-shIKKβ group exhibited an attenuated expression of ERK phosphorylation when compared to the control group, indicating that the IKKβ downregulation influences the proliferative capacity of tumor cells partly by suppressing ERK activation.
IKKβ Knockdown Initiates Tumor Cell Apoptosis in vitro
To determine whether the inhibition of IKKβ in LLC cells may promote the apoptosis of tumor cells, the expression levels of apoptotic signal proteins were assessed. Compared to untreated LLC cells or cells infected with control shRNA, IKKβ knockdown cells had a higher pro-apoptotic protein expression of Bax and a reduced anti-apoptotic protein expression of Bcl-2 (Figure 3G–I). These data may suggest that downregulation of IKKβ in lung tumor cells could initiate cell apoptosis.
IKKβ Knockdown Attenuates the Migration of Lung Cancer Cells in vitro
A wound healing assay was performed on LLC cells transfected with control or IKKβ shRNA. The results showed that 24 hours after the wound was created, cells in neither of the two groups could close the wound. However, LLC cells containing IKKβ shRNA were slower to close the wound, with an observed reduction of approximately 48% (Figure 4A and B).
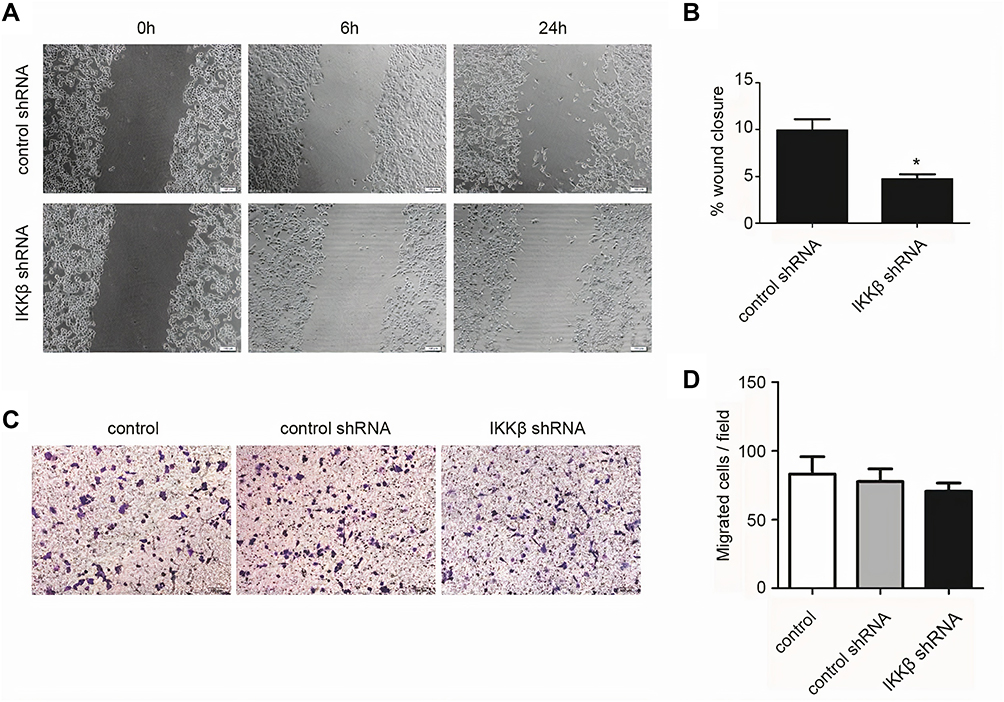 |
Figure 4 Knockdown IKKβ inhibited wound healing and Matrigel invasion in Lewis lung carcinoma (LLC) cells. (A, B) Wound healing assays were performed on LLC cells infected with either control shRNA (small hairpin RNA) or IKKβ shRNA. The representative images are presented at the time of the scratches and at 6 and 24 hours after. The arrows show that a visible wound was still present in the IKKβ shRNA plate at 24 hours post-scratch. A graphical representation of the data from the 24-hour time point is shown on the right from at least three independent experiments (mean ± standard deviation [SD], *P < 0.05). (C, D) The Transwell cell Matrigel invasion assay examining the directional invasion of LLC cells towards IKKβ knockdown is shown. A total of 1 × 105 LLC cells infected with control shRNA or IKKβ shRNA were seeded into the top chamber of the Transwell units, while 500 μL of 10% FBS DMEM was added to the bottom chamber. The number of migrated cells on the lower surface of the top chamber was counted at 24 hours. The representative images are present from five randomly chosen fields per filter (scale bar =100 μm). The quantification analysis of the LLC cell invasion assay. The data are presented as mean ± standard deviation (SD, n = 3). *P < 0.05. |
Since Matrigel is a protein matrix that comprises collagen and laminin and that acts as a model basement membrane, the ability of LLC cells to degrade the basement membrane was measured and used as an indicator of the invasive potential of cancer cells. A thin layer of Matrigel was applied and allowed to solidify on top of the insert, and LLC cells with control or IKKβ shRNA were plated on top of the protein layer, while the bottom chambers contained media, with 10% serum as the sole chemoattractant. Although LLC cells with IKKβ shRNA appeared to invade the Matrigel less compared to controls (Figure 4C and D), this effect did not reach statistical significance.
Discussion
This study found that blocking the NF-κB signaling pathway by knocking down IKKβ in LLC cells inhibited tumor growth in an LM mouse model. IKKβ supports leptomeningeal tumor progression by promoting cancer cell proliferation and migration and inhibiting cancer cell apoptosis.
NF-κB is a key anti-apoptotic transcription factor,16 and it has been reported that tumor cells may rely on the NF-κB pathway to avoid apoptosis.17 In a mouse model of melanoma, the conditional ablation of IKKβ increased tumor cell apoptosis.18 Thus, the protein expression levels of apoptotic signaling were assessed to investigate the role of IKKβ in regulating cancer cell apoptosis. It was found that IKKβ knockdown LLC cells had a higher pro-apoptotic protein expression of Bax and a reduced anti-apoptotic protein expression of Bcl-2. Therefore, the present data imply that IKKβ might be a regulator involved in the proliferation and apoptosis of LLC cells.
The CSF-filled leptomeningeal space is a favorable tumor microenvironment enhanced by the blood-brain barrier’s impermeability to immune cells.19 Tumor cell invasion of leptomeninges can lead to local inflammation and impaired CSF resorption.20 In the clinic, it was found that inflammatory cells, especially activated monocytes, increased in the CSF of patients with LM, which may indicate there was a potential link between inflammation and tumors in the leptomeningeal space.
Evidence has suggested that chronic inflammation in the tumor microenvironment benefits cancer cells.21 NF-κB activity has been linked to tumor promotion and progression in inflammatory cancers.13 The constitutive activity of NF-κB drives chronic inflammation, which boosts the development and progression of multiple diseases, including cancer.22 Therefore, reduced tumorigenesis in leptomeninges might arise from a suppressed microenvironment caused by blocking the NF-κB pathway in cancer cells, primarily through the knockdown of IKKβ, a critical kinase in the NF-κB activation cascade.
A recent study showed that cancer cells within the CSF could upregulate the production of complement component 3 (C3), which helps to disrupt the choroidal blood-CSF barrier, allows the entry of plasma growth factors into the CSF, and promotes cancer cell growth.3
Cancer cells can grow and proliferate in the CSF environment and infiltrate the leptomeningeal space; a change in cell motility after treatment with IKKβ shRNA or control shRNA might affect LLC cell growth in the leptomeninges. As expected, the wound healing assay revealed that the knockdown of IKKβ attenuated the migration of LLC cells. The Transwell cell invasion assay appeared to reveal that Matrigel had less invasion of LLC cells transfected with IKKβ shRNA compared to controls, but this effect did not reach statistical significance. These results might partly explain the differences in the pathological changes in the leptomeninges between the two treatment groups.
The intrathecal inoculation of NSCLC cells into the CM of mice is the most commonly used strategy to create LM models.9,23 Mouse LLC cells have been shown to grow within the CSF.3 In the present study, the LM model was established by implanting 10 μL of cell suspension containing 1.5 × 105 LLC cells into the CM,24 with slight modifications. Evidence suggests that pathological LM involvement primarily occurs in the basal cisterns of the brain, posterior fossa, and cauda equina.25
In the present model, leptomeningeal tumor invasion was primarily observed at the base of the brain and cervical cord. Using this model, it was found that LLC cells with IKKβ knockdown by shRNA inhibited tumor infiltration in leptomeninges and significantly prolonged mouse survival when compared to cells transfected with control shRNA; this suggests that blocking the NF-κB signaling pathway by knocking down IKKβ in cancer cells can inhibit tumor growth in the LM model.
To investigate the underlying mechanism of the anti-tumor activities of IKKβ knockdown in the LM mice models, experiments were performed in vitro. A previous study indicated that knocking down IKKβ in tumor cells reduces ERK activation and cell proliferation, attenuating tumor progression in a mouse lung cancer model.13
Recent studies have confirmed the constitutive activity of the transcription factor NF-κB in mouse and human glioblastoma multiforme (GBM) models. The tumor cell proliferative capacity decreased, and mice survival was extended through inhibition of NF-κB in tumor cells caused by knocking down IKKβ or by using a small peptide targeting NEMO (the NEMO-binding domain).26 The present result showed that lentivirus-mediated RNAi efficiently suppressed IKKβ expression in LLC cells, resulting in a significant reduction in cell viability and proliferation, and this partly affected the ERK activation.
Conclusion
IKKβ can play a tumorigenesis role in cancers, and it was found that blocking the NF-κB signaling pathway by knocking down IKKβ in LLC cells inhibited tumor growth in an LM mouse model. Further investigation of the details and exact mechanisms, including tumor invasion and local inflammation response within the leptomeningeal space, should be performed with the hope of better understanding the pathogenesis of LM and developing potential supportive therapeutic strategies for this disease.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
All animal experiments were approved by Ethics Committee of the Second Hospital of Hebei Medical University and all work were carried out in compliance with the laboratory animal management regulations promulgated by the Ministry of Science and Technology of the People’s Republic of China, which are in accordance with the NIH Guide for the Care and Use of Laboratory.
Disclosure
The authors declare that they have no competing interests.
References
1. Chamberlain M, Soffietti R, Raizer J, et al. Leptomeningeal metastasis: a response assessment in neuro-oncology critical review of endpoints and response criteria of published randomized clinical trials. Neuro-Oncology. 2014;16(9):1176–1185. doi:10.1093/neuonc/nou089
2. Mack F, Baumert BG, Schafer N, et al. Therapy of leptomeningeal metastasis in solid tumors. Cancer Treat Rev. 2016;43:83–91. doi:10.1016/j.ctrv.2015.12.004
3. Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massague J. Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell. 2017;168(6):1101–1113.e1113. doi:10.1016/j.cell.2017.02.025
4. Remon J, Le Rhun E, Besse B. Leptomeningeal carcinomatosis in non-small cell lung cancer patients: a continuing challenge in the personalized treatment era. Cancer Treat Rev. 2017;53:128–137. doi:10.1016/j.ctrv.2016.12.006
5. Cheng H, Perez-Soler R. Leptomeningeal metastases in non-small-cell lung cancer. Lancet Oncol. 2018;19(1):e43–e55. doi:10.1016/S1470-2045(17)30689-7
6. Le Rhun E, Galanis E. Leptomeningeal metastases of solid cancer. Curr Opin Neurol. 2016;29(6):797–805. doi:10.1097/WCO.0000000000000393
7. Li YS, Jiang BY, Yang JJ, et al. Leptomeningeal metastases in patients with NSCLC with EGFR mutations. J Thorac Oncol. 2016;11(11):1962–1969. doi:10.1016/j.jtho.2016.06.029
8. Li Y, Liu B, Connolly ID, et al. Recurrently mutated genes differ between leptomeningeal and solid lung cancer brain metastases. J Thorac Oncol. 2018;13(7):1022–1102. doi:10.1016/j.jtho.2018.03.018
9. Nanjo S, Arai S, Wang W, et al. MET copy number gain is associated with gefitinib resistance in leptomeningeal carcinomatosis of EGFR-mutant lung cancer. Mol Cancer Ther. 2017;16(3):506–515. doi:10.1158/1535-7163.MCT-16-0522
10. Xia Y, Shen S, Verma IM. NF-kappaB, an active player in human cancers. Cancer Immunol Res. 2014;2(9):823–830. doi:10.1158/2326-6066.CIR-14-0112
11. Tang X, Liu D, Shishodia S, et al. Nuclear factor-kappaB (NF-kappaB) is frequently expressed in lung cancer and preneoplastic lesions. Cancer. 2006;107(11):2637–2646. doi:10.1002/cncr.22315
12. Meylan E, Dooley AL, Feldser DM, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462(7269):104–107. doi:10.1038/nature08462
13. Xia Y, Yeddula N, Leblanc M, et al. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat Cell Biol. 2012;14(3):257–265. doi:10.1038/ncb2428
14. Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. doi:10.1016/j.cell.2004.07.013
15. Bivona TG, Hieronymus H, Parker J, et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471(7339):523–526. doi:10.1038/nature09870
16. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2011;441(7092):431–443. doi:10.1038/nature04870
17. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2006;144(5):646–674. doi:10.1016/j.cell.2011.02.013
18. Yang J, Splittgerber R, Yull FE, et al. Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J Clin Invest. 2010;120(7):2563–2574. doi:10.1172/JCI42358
19. Lyle LT, Lockman PR, Adkins CE, et al. Alterations in pericyte subpopulations are associated with elevated blood-tumor barrier permeability in experimental brain metastasis of breast cancer. Clin Cancer Res. 2016;22(21):5287–5299. doi:10.1158/1078-0432.CCR-15-1836
20. Wang N, Bertalan MS, Brastianos PK. Leptomeningeal metastasis from systemic cancer: review and update on management. Cancer. 2018;124(1):21–35. doi:10.1002/cncr.30911
21. Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. 2016;27(8):1482–1492. doi:10.1093/annonc/mdw168
22. Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. doi:10.1038/nature02924
23. Guo Z, Wang Y, Wang Y, et al. AZD3759, a BBB-penetrating EGFR inhibitor for the treatment of EGFR mutant NSCLC with CNS metastases. Sci Transl Med. 2016;8:368ra172. doi:10.1126/scitranslmed.aag0976
24. Boyle R, Thomas M, Adams JH. Diffuse involvement of the leptomeninges by tumour–a clinical and pathological study of 63 cases. Postgrad Med J. 1980;56(653):149–158. doi:10.1136/pgmj.56.653.149
25. Kusne Y, Carrera-Silva EA, Perry AS, et al. Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFalpha in glioblastoma. Sci Signal. 2014;7(338):ra75. doi:10.1126/scisignal.2005196
26. Saito N, Hatori T, Murata N, et al. Comparison of metastatic brain tumour models using three different methods: the morphological role of the pia mater. Int J Exp Pathol. 2008;89(1):38–44. doi:10.1111/j.1365-2613.2007.00563.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.