")
Back to Journals » Cancer Management and Research » Volume 11
Knockdown of CHPF suppresses cell progression of non-small-cell lung cancer
Authors Hou XM, Baloch Z , Zheng ZH, Zhang WH, Feng Y, Li DD, Wu XA, Yang SH
Received 24 October 2018
Accepted for publication 7 March 2019
Published 24 April 2019 Volume 2019:11 Pages 3275—3283
DOI https://doi.org/10.2147/CMAR.S192036
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rituraj Purohit
Xiao-Ming Hou,1,2 Zulqarnain Baloch,3,4 Zhan-Hong Zheng,3,4 Wen-Hui Zhang,1,2 Ying Feng,5 Duan-Duan Li,3,4 Xin-An Wu,2,6 Shi-Hua Yang3,4
1Department of Oncology, The First Hospital of Lanzhou University, Lanzhou 730000, People’s Republic of China; 2Key Laboratory of Biotherapy and Regenerative Medicine of Gansu Province, The First Hospital of Lanzhou University, Lanzhou 730000, People’s Republic of China; 3College of Veterinary Medicine, South China Agricultural University, Guangzhou 510642, People’s Republic of China; 4Key Laboratory of Comprehensive Prevention and Control for Severe Clinical Animal Diseases of Guangdong Province, South China Agricultural University, Guangzhou 510642, People’s Republic of China; 5Department of Pathology, The First Hospital of Lanzhou University, Lanzhou 730000, People’s Republic of China; 6Department of Pharmacy, The First Hospital of Lanzhou University, Lanzhou 730000, People’s Republic of China
Purpose: The aim of the present study was to explore the role of CHPF in non-small-cell lung cancer (NSCLC) and to develop an shRNA vector-based therapy to repress the expression of CHPF gene in NSCLC cell lines.
Methods: In this study, we used immunohistochemical staining to verify the expression of CHPF in NSCLC tissue. Then, we determined the expression of CHPF gene in different NSCLC cell lines with RT-PCR and Western blotting. Specific CHPF shRNA was used to knockdown the expression of CHPF. Celigo image cytometry, cell cycle analysis, and flow cytometry assay were performed.
Results: The results showed that expression level of CHPF was higher in NSCLC tissues than normal lung tissues. Further, we established that CHPF expression knockdown in NSCLC cells could substantially restrain the cell proliferation, apoptosis, and cell cycle in vitro.
Conclusion: On the basis of these results, we concluded that CHPF expression has an important role in the progression of human NSCLC cells. Therefore, its interference could possibly be used as a potential therapeutic target against NSCLC.
Keywords: NSCLC, CHPF, progression, knockdown
Introduction
Lung cancer is a major cause of cancer-related death globally.1 According to WHO estimates, lung cancer constitutes 13% of total cancer incidence and was responsible for 19.4% of cancer mortality in 2012.2,3 Among them, most lung cancers are non-small-cell lung cancer (NSCLC). Despite the tremendous progresses in diagnostic and therapeutic methods in recent years, NSCLC still remains the foremost reason of cancer-related morbidity and mortality in the world. The malignant biological behavior of cancer, particularly NSCLC, is regulated by multiple genes, the occurrence and increased tumorigenesis of NSCLC is associated with the loss of activity of tumor-suppressor genes and high expression of tumor-related genes.
Chondroitin sulfate (CS) is found naturally in the body and consists of a chain of alternating sugars (N-acetylgalactosamine and glucuronic acid).4 Biosynthesis of CS is instigated by the adding of xylose to serine residues in a core protein, followed by the chronological addition of two residues, Gal and single residue of GlcA, to develop the tetrasaccharide linkage structure GlcAb1-3Galb1-3Galb1-4Xylb1-O-Ser.5 The synthesis of CS requires the promotion of molecules such as CHPF. Until now, four genes, CHSY1, CHPF, CHSY3, and CHPF2, have been identified to play an important role in CS biosynthesis. It has also been reported that each combination of these four enzymes is also able to control CS biosynthesis.6 In recent publications, the relationship between CHPF genes and human diseases has been explored.7,8 CHPF is a kind of protein-coding gene. Kitagawa et al isolated CHPF by catalog-probing with the CHSY1 sequence.5 CHPF gene has at least four exons over 5 kb in length. CHPF consists of 775-amino-acid protein with approximately 85 kD molecular mass and encloses three potential N-glycosylation locations. The sequence CHPF gene has important N-terminal hydrophobic sections and is envisaged a type II trans-membrane protein.5 CHPF gene has 23% and 57% similar sequence identity ashuman CHSY1 and CHPF2, respectively.6
RNAi is a significant technological advancement in the field of modern biology, and has been widely used in in vitro and in vivo experiments to effectively knockdown target genes' expression.9 siRNAs have already been effectively used in different types of mammalian cell lines, and have successfully caused 80% downregulation of candidate gene expression.10 siRNAi technology provides a short-term suppression of target gene expression. Therefore, alternative approaches have been introduced such as shRNAs, to obtain long-term suppression of target gene expression.11
Recently, it has been verified that CHPF expression plays an important role in initiation and development of colorectal cancer.7 However, CHPF expression's role in the progression of NSCLC remains unclear. Therefore, in this study, we examined the role of CHPF in NSCLC and then designed shRNA-CHPF-Lenti vector to knockdown the CHPF gene in NSCLC cell lines. At the end we explored the effect of the knockdown of CHPF in NSCLC cell lines on cell proliferation.
Methods
Tissue specimens
A total of 90 pairs of NSCLC tissues and adjacent tissues were obtained from The Hospital of Lanzhou University. All patients were requested to sign informed consent. No patient received any prior chemotherapy or radiotherapy. The protocols were approved by the Ethics Committee of The Hospital of Lanzhou University.
Immunohistochemical staining and evaluation
Formalin-fixed paraffin-embedded tissue block was incised into 4 μm thick sections. All incised sections were deparaffinized with xylene and rehydrated with graded alcohol. After incubating with 3% H2O2 for 20 minutes at room temperature to satiate the activity of endogenous peroxidase, the sections were further processed in sodium citrate buffer (pH =6.0) for heat-induced antigen retrieval. After washing several times, the pieces were incubated in 10% goat serum at room temperature for 30 minutes to block nonspecific binding. Then, the slides were incubated with primary anti-CHPF (Santa Cruz Biotechnology Inc., Dallas, TX, USA) antibody at 4°C overnight. After the primary antibody was washed off, anti-rabbit peroxidase-conjugated secondary antibody and DAB reagent were employed in the detection procedure.
Two qualified, independent pathologists who were blinded to the clinical and pathological data of the tumors. The staining of slides was observed and photographed using an Olympus microscope system (Olympus Corporation, Tokyo, Japan). The percentages of positive tumor cells were semi-quantitatively graded as 0 (<5%), 1 (5%–25%), 2 (26%–50%), and 3 (>50%). The staining intensity of tumor cells was scored according to criteria reported previously. The two scores were multiplied to acquire the final immunoreactive score. High expression of CHPF in tumor cells was defined as immunoreactive scores ≥4.
Cell culture
The human NSCLC cell lines H460, H1299, H1688, 95-D, and A549 were purchased from the Shanghai Branch of the Chinese Academy of Sciences. All cells were grown in DMEM supplemented with 10% FBS (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) in a 37°C incubator with 5% CO2. In this study, transfection was performed using Genechem Transfection Reagent (Genechem Co., Ltd., Shanghai, People’s Republic of China) according to the manufacturer’s instructions.
RNA extraction and RT-PCR
In this study, total RNA was extracted with Trizol (Invitrogen, Thermo Fisher Scientific) by following the manufacturer’s instructions. An ultraviolet spectrophotometer was used to measure the concentration. An amount of 5 μg RNA was reverse transcribed by PrimeScript™ RT reagent Kit (Takara, Japan). according to the manufacturer's instructions. We used Premier Primer 5.0 software to design the PCR primers. The primer sequences are shown in Table 1. We used SYBR_Green PCR Master Mix™ Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific) to perform RT-PCR. The PCR conditions were 30 minutes at 37°C, 5 minutes at 83°C. PCR was performed in 10 μL reactions with the following reaction conditions: 2 minutes pre-denaturation at 95°C, 30 cycles of 15 seconds denaturation at 95°C, 20 seconds at 60°C. The relative gene expression levels were measured and compared by 2−ΔΔCT analysis program.
 | Table 1 Primers used in this study for PCR |
Lentiviral vector and cell transfection
The shRNA against CHPF (NM_024536; GenBank) gene was cloned into lentiviral gene transfer vector with GFP (Genechem Co., Ltd.). The sequence of CHPF shRNA was 5ʹ-CTGGCCATGCTACTCTTTG-3ʹ. The the non-targeted control mock lentivirus (LV-NC virus) and recombinant CHPF targeting lentivirus (LV-CHPF-shRNA virus) were made and transfected according to the manufacturer’s instructions for NSCLC cell lines. In short, after the lentivirus was constructed, 293T cells were seeded in 6-well plates and cells were infected to test its efficacy when cell growth reached 70%–80% confluency. After 24, 48, and 72 hours' transfection, viruses were collected. The infected cells were named shCtrl group and shCHPF group, respectively.
For lentivirus transfection, NSCLC cells were cultivated in 6-well plates and negative control (shCtrl) lentivirus or the CHPF-shRNA-lentivirus was dropped according to multiplicity of transfection. Cultured cells were inspected using a fluorescence microscope (MicroPublisher 3.3; Olympus) after 72 hours of transfection. The cells were collected to determine the knockdown efficiency by quantitative RT-PCR.
Western blotting
Total protein was extracted from cells by lysing in RIPA buffer. The total cell protein extract was determined and measured with BCA Protein Assay kit (Beyotime Biotechnology, Shanghai, People's Republic of China). Equal amounts of cell lysate were separated on 10% SDS-PAGE (Bio-Rad Laboratories Inc., Hercules, CA, USA) and transferred to a PVDF membrane (EMD Millipore, Billerica, MA, USA). The PVDF membrane was blocked with Tris-buffered saline/Tween with 5% non-fat milk for 2 hours at room temperature or overnight at 4°C and incubated with primary antibody and then with secondary antibody. ECL Western Blotting Substrate kit (Pierce, Thermo Fisher Scientific) was used to visualize protein bands. We purchased the primary anti-flag antibody from Sigma Biotechnology (Sigma-Aldrich Co., St Louis, MO, USA) and mouse anti-GAPDH from Santa Cruz Biotechnology Inc. Goat anti-mouse IgG and the secondary antibody was purchased from Santa Cruz Biotechnology Inc.
Cell growth assay
Cells in logarithmic growth phase were digested with trypsin (Sangon Biotech, Shanghai, People's Republic of China) and resuspended. Then, the cells were seeded in a 96-well plate at a density of 2,000 cells/100 μL/well. The cells were incubated overnight at 37°C and 5% CO2. From the second day after plating, we continuously measured the number of cells over 5 days using a Celigo image cytometer (Nexcelom Bioscience, Lawrence, MA, USA). Statistics were performed on the data to plot the proliferation curve of the cells. The cell proliferation ratio was calculated based on the cell count and the time-point, and a growth curve based on the cell proliferation factor was drawn.
Cell cycle assay
The NSCLC cells from shCHPF and shCtrl groups were inoculated in 6-well plates and cultured for 48 hours. Next, we used ice-cold PBS to wash the cells and fixed them in a 70% (v/v) ice-cold ethanol solution at 4°C overnight. Then, we used flow cytometry (EMD Millipore) to analyze the cells.
Cell apoptosis was analyzed using the Annexin V-allophycocyanin kit (Annexin V-APC; Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were collected and processed as previously mentioned. The cell concentration was adjusted to 1×106/mL by using staining buffer. Finally, 100 μL of suspension was stained with 5 μL Annexin V-APC.
Human apoptotic protein array
Human Apoptosis Antibody Array kit (Ray-Biotech, GA, USA) was used to perform apoptosis array test. NSCLC cells treated with shRNA were seeded in 6-well plates for 72 hours. Treated cells were collected after 72 hours and centrifuged at 2,500 rpm, for 5 minutes. Collected cells were washed twice using ice-cold PBS. We again performed centrifugation for 5 minutes at 2,500 rpm, then the supernatant was discarded. Cell protein extraction was completed and about 500 mg extracted proteins were incubated with the human apoptosis array overnight. Chemiluminescence detection was performed by scanning the membrane on Odyssey FC Imaging System (LI-COR, USA).
Statistical analysis
Statistical analysis was performed with SPSS 19.0 program (IBM Corporation, Armonk, NY, USA). One-way ANOVA was applied for comparisons between multiple groups and the Student–Newman–Keuls method was used for comparisons between two groups. All data were presented as mean ± SD, and P<0.05 was considered statistically significant.
Results
CHPF is highly expressed in NSCLC
We determined the expression of CHPF in NSCLC tissues using immunohistochemistry. We found that CHPF protein was mainly localized in the cytoplasm of cells (Figure 1). Of the 90 NSCLC samples, the expression rate of CHPF in NSCLC tissues was 55.6% (50/90), which was significantly higher than that in adjacent tissues (28/90; P=0.001). According to immunohistochemistry results, 90 patients with NSCLC were divided into CHPF low expression and high expression groups. At the same time, we analyzed the relationship between clinicopathological features and CHPF expression in patients with NSCLC. The results suggest that high expression of CHPF is associated with tumor size and TNM stage in NSCLC patients (Table 2).
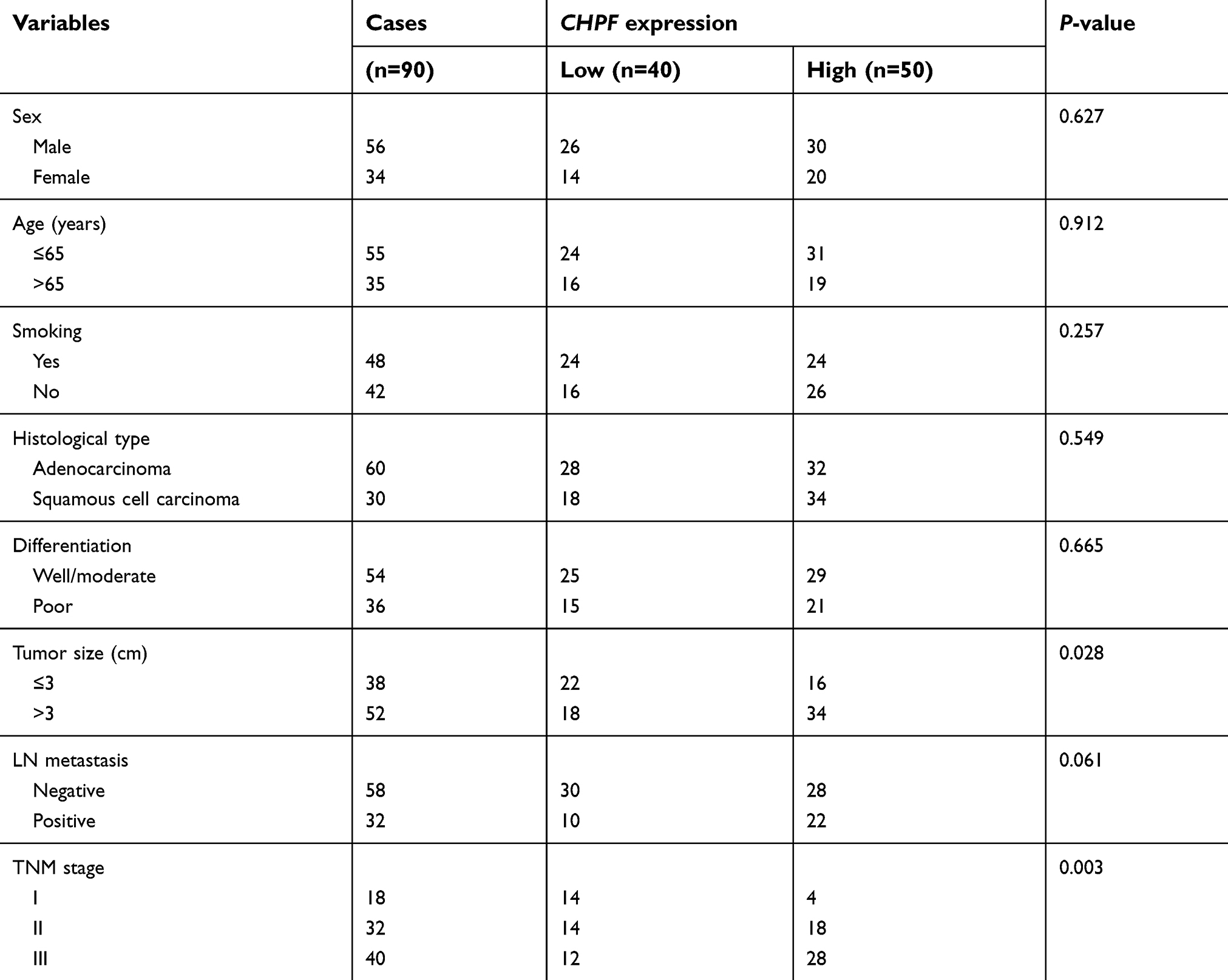 | Table 2 The correlations between CHPF expression and clinicopathological features in NSCLC patients |
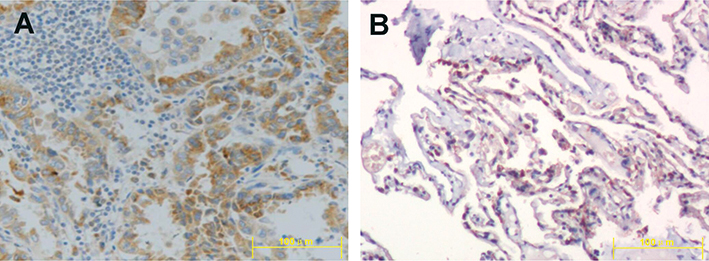 | Figure 1 Expression of CHPF in non-small-cell lung cancer tissue (A) and adjacent tissue (B) ×200. |
CHPF mRNA expression was determined in the following cell lines: H460, H1299, H1688, 95-D, and A549 by RT-PCR (Figure 2A). The CHPF protein expression level was detected in these cell lines by using Western blotting (Figure 2B). Our observation indicated that CHPF mRNA expression was high in all A549 cell lines. Therefore, we used A549 cell lines for subsequent functional experiments.
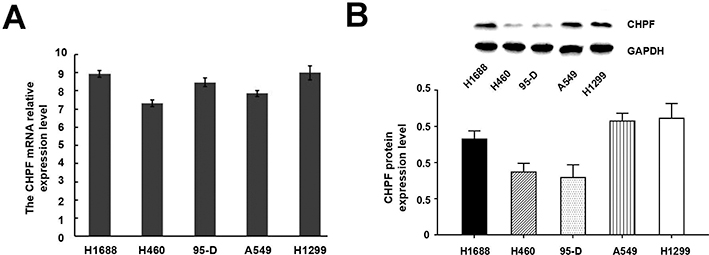 | Figure 2 Expression of CHPF in five non-small-cell lung cancer cell lines. (A) CHPF mRNA was detected by RT-PCR; (B) CHPF protein was detected by Western blotting. |
Knockdown efficiency determined by Western blot analysis
To explore the role of CHPF, we constructed the shCHPF with recombinant lentivirus and transfected the shCHPF into NSCLC A549 cell lines. Then, the transfection efficiency was observed by fluorescence microscope. The percentage of infected cells was >80% for shCtrl and shCHPF lentivirus 3 days post-transfection (Figure 3). A549 cell lines were infected with CHPF-shRNA or shCtrl lentivirus. In order to verify the efficiency of knockdown, the expression of CHPF protein was determined by Western blotting. We found that the expression of CHPF protein was decreased as compared to negative control (Figure 4).
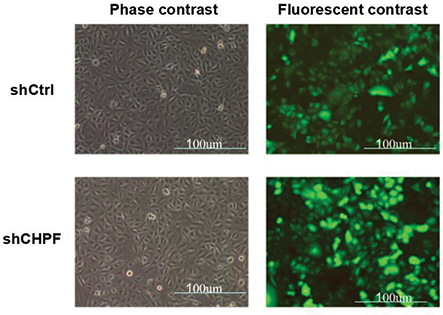 | Figure 3 Observation of the transfection efficiencies using fluorescence microscopy. |
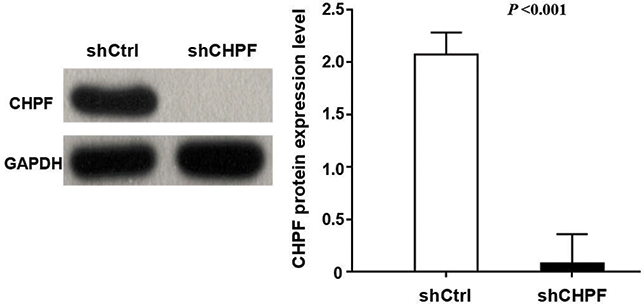 | Figure 4 Western blotting-verified transfection efficiency of A549 cell lines. |
Knockdown of CHPF inhibits cell proliferation
In this study, we analyzed the effect of CHPF expression on NSCLC cell growth by Celigo image cytometer for 5 days. The results showed that control cells' growth increased during 5 days, while growth of CHPF-shRNA-trasfected cells decreased during the experiment period (Figure 5). It is suggested that the CHPF gene is significantly associated with the proliferative capacity of A549 cells.
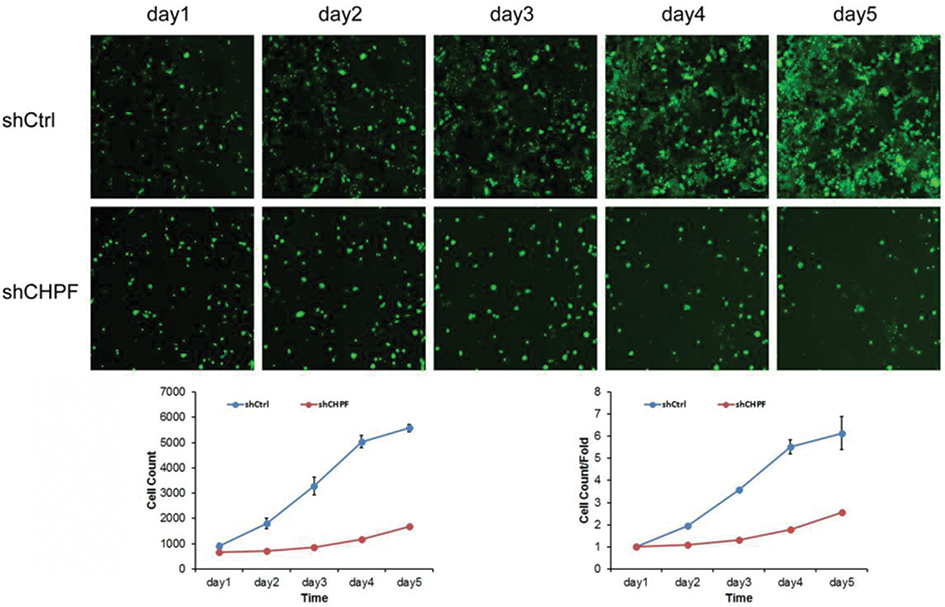 | Figure 5 Cell proliferation ability determined by Celigo assay. The proliferation of A549 cells was significantly inhibited after knockdown of CHPF. |
Effect of CHPF on cell cycle and cell apoptosis
Three days after shRNA lentivirus infection, compared with the shCtrl group, the number of cells in the G1 phase of shCHPF group A549 cells increased, the number of cells in the S phase decreased, and the number of cells in the G2/M phase decreased (Figure 6). It is suggested that the CHPF gene is significantly associated with the periodic distribution of A549 cells.
 | Figure 6 Cell cycle of A549 cell lines was examined by flow cytometry. The A549 cells were arrested in G1 phase after knockdown of CHPF. |
After 4 days of shRNA lentivirus infection, compared with the shCtrl group, the apoptosis of A549 cells in the shCHPF group increased significantly (Figure 7). The results suggested that CHPF gene is significantly associated with apoptosis of A549 cells.
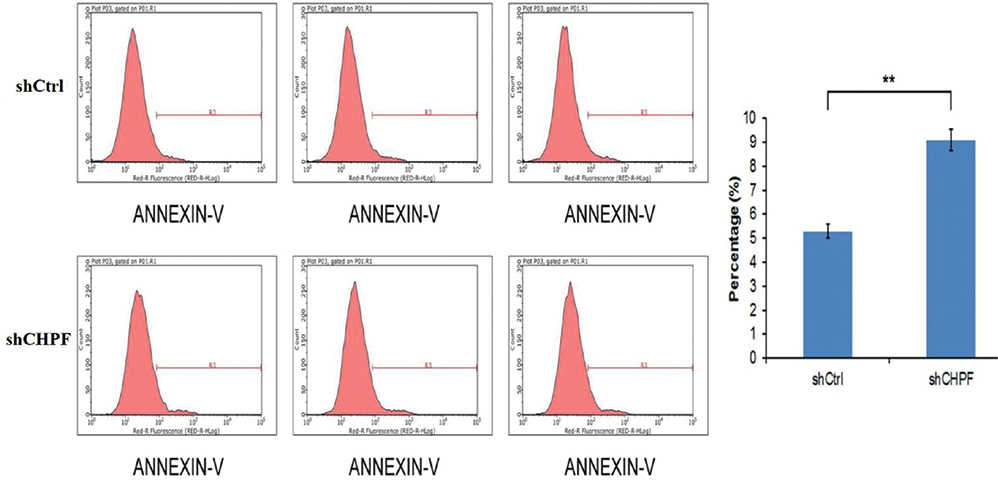 | Figure 7 Analysis of apoptosis of A549 cell lines using Annexin V-allophycocyanin kit. The apoptosis of A549 cells was significantly increased after knockdown of CHPF. |
Human apoptotic protein array
We used human apoptotic protein array to determine the semi-quantitative of apoptotic proteins in A549 cells. The concentration of signal for antigen-specific antibody spot was comparative to the antigen-relative concentration. Further, we observed that the positive control signal on all array images had comparable intensities. Therefore, evaluation of signal intensities for every antigen-specific antibody spot between the array membranes might be applied to count the relative protein expression differences. In A549 cells, the expression level of Caspase-3 protein was upregulated after silencing CHPF by RNAi, and other proteins showed no significant changes (Figure 8).
 | Figure 8 The human apoptotic protein array was used to determine apoptotic proteins. The Caspase-3 protein was upregulated after knockdown of CHPF. |
Discussion
During past decades, researchers have mainly concentrated on discovering potential biomarkers which can be used for treatment and diagnosis of cancers. However, the clinical applications of those molecular markers are very restricted. Therefore, scientists are continually concentrating on the identification of new potential genes. Exploring new potential candidate sites and understanding the related signaling pathways is very important. It has been previously confirmed that many genes such as EGFR, ALK or ROS1 have very important roles in the initiation of tumorigenesis and development of cancers, particularly NSCLC.12–14 RNAi technology has been widely used for gene silencing.11 The application of RNAi technology in cancer treatment has already been proven in in vitro and in vivo model experiments.15 In this study, shRNA technology effectively silenced the expression of CHPF in the NSCLC cell lines, which laid an important foundation for subsequent research.
Here, we have effectively knocked down the expression of CHPF gene in A549 cell lines by using RNAi method, and further, we examined the effect of CHPF expression knockdown on the regulation of cell proliferation, cell cycle, and apoptosis. According to the best of our knowledge, nobody has investigated the role of CHPF expression in NSCLC. At first we investigated the expression of CHPF in human NSCLC cell lines and tissues. We observed that CHPF expression was substantially increased in the NSCLC tissues from patients, compared to adjacent tissues. The elevated expression of CHPF supported that CHPF might contribute to proliferation of NSCLC. Our observation is consistent with results of Kalathas et al, in which they reported that CHPF expression was increased in colorectal cancer tissues compared to normal tissue.7 Therefore, a detailed study of sulfotransferases, glycosyltransferases, and dermatan sulfate epimerase in combination with chondroitin structure in NSCLC patients is needed.
Fan et al explored the role of CHPF in glioma, a lentiviral vector expressing CHPF shRNA was constructed and transfected into glioma U251 cells, which stably downregulated the expression levels of the CHPF gene in U251 cells in vitro.8 The results confirmed that CHPF promotes growth and inhibits apoptosis in glioma U251 cells. In different studies, researchers have reported that the shRNA expression vector transfected with different genes controlled the propagation, invasiveness, cell cycle, and apoptosis of tumor cells.15 These observations suggest that gene knockdown strategy has the potential to be used for the treatment of cancers. Yet, no report has used this strategy to repress the expression of CHPF gene in NSCLC. Therefore, we selected A549 cell lines to investigate the effect of CHPF knockdown in vitro, to explain its role in NSCLC progression. We found that lentivirus-mediated CHPF knockdown extensively repressed the proliferation of cancer cells and induced cell cycle arrest of NSCLC cells. We also observed that the cell growth rate and numbers in the shCHPF group were drastically decreased compared to the control group. Our results showed that CHPF gene can be used as a target for the treatment of NSCLC.
Conclusion
In the current study, we highlighted the role of CHPF in NSCLC progression. This gene could be used as a therapeutic target for curing NSCLC. But, our study also had some limitations. The main limitation is the lack of follow-up data for NSCLC patients and a poor assessment of whether CHPF could be a marker for prognosis in patients with NSCLC.
Acknowledgments
This work was financially supported by funding from Science and Technology Major Project of Guangdong Province (2014B020225007). The funders played no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Longo-Sorbello GS, Chen B, Budak-Alpdogan T, Bertino JR. Role of pemetrexed in non-small cell lung cancer. Cancer Invest. 2007;25(1):59–66. doi:10.1080/07357900601130748
3. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
4. Ogawa H, Shionyu M, Sugiura N, et al. Chondroitin sulfate synthase-2/chondroitin polymerizing factor has two variants with distinct function. J Biol Chem. 2010;285(44):34155–34167. doi:10.1074/jbc.M110.109553
5. Kitagawa H, Izumikawa T, Uyama T, Sugahara K. Molecular cloning of a chondroitin polymerizing factor that cooperates with chondroitin synthase for chondroitin polymerization. J Biol Chem. 2003;278(26):23666–23671. doi:10.1074/jbc.M302493200
6. Filipek-Górniok B, Holmborn K, Haitina T, et al. Expression of chondroitin/dermatan sulfate glycosyltransferases during early zebrafish development. Dev Dyn. 2013;242(8):964–975. doi:10.1002/dvdy.23981
7. Kalathas D, Theocharis DA, Bounias D, et al. Chondroitin synthases I, II, III and chondroitin sulfate glucuronyltransferase expression in colorectal cancer. Mol Med Rep. 2011;4(2):363–368. doi:10.3892/mmr.2011.431
8. Fan YH, Xiao B, Lv SG, Ye MH, Zhu XG, Wu MJ. Lentivirus-mediated knockdown of chondroitin polymerizing factor inhibits glioma cell growth in vitro. Oncol Rep. 2017;38(2):1149–1155. doi:10.3892/or.2017.5731
9. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. doi:10.1038/35888
10. Krueger U, Bergauer T, Kaufmann B, et al. Insights into effective RNAi gained from large-scale siRNA validation screening. Oligonucleotides. 2007;17(2):237–250. doi:10.1089/oli.2006.0065
11. Bernards R, Brummelkamp TR, Beijersbergen RL. shRNA libraries and their use in cancer genetics. Nat Methods. 2006;3(9):701–706. doi:10.1038/nmeth921
12. Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. doi:10.1016/j.cell.2007.11.025
13. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. doi:10.1038/nature05945
14. Liu S, Yang H, Jiang Y, Zhang T, Yan R, Zhang J. Evolution strategy of ROS1 kinase inhibitors for use in cancer therapy. Future Med Chem. 2018;10(14):1705–1720. doi:10.4155/fmc-2018-0033
15. Dykxhoorn DM, Lieberman J. Knocking down disease with siRNAs. Cell. 2006;126(2):231–235. doi:10.1016/j.cell.2006.07.007
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.