")
Back to Journals » OncoTargets and Therapy » Volume 11
Knockdown of CCNO decreases the tumorigenicity of gastric cancer by inducing apoptosis
Authors Li L, Cao Y, Zhou H, Li Y, He B, Zhou X, Nie Z, Liang L, Liu Y, Ye L
Received 4 June 2018
Accepted for publication 11 September 2018
Published 26 October 2018 Volume 2018:11 Pages 7471—7481
DOI https://doi.org/10.2147/OTT.S176252
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yao Dai
Lan Li,1,2,* Yu Cao,1 Hourong Zhou,2 Yu Li,3 Bing He,1,* Xia Zhou,4 Zhao Nie,5 Li Liang,6 Ying Liu,7 Limin Ye3
1Department of Emergency, West China Hospital, Sichuan University, Chengdu 610041, China; 2Department of General Practice, Guizhou Provincial People’s Hospital, Guiyang 610041, China; 3Department of Gastroenterology, Guizhou Provincial People’s Hospital, Guiyang 610041, China; 4Department of Emergency, Guizhou Provincial People’s Hospital, Guiyang 610041, China; 5Department of Medical Records and Statistics, Guizhou Provincial People’s Hospital, Guiyang 610041, China; 6Medical Department, Guizhou Provincial People’s Hospital, Guiyang 610041, China; 7Department of Emergency, the Hospital affiliated to Southwest Medical University, Sichuan 646000, China
*These authors contributed equally to this work
Purpose: Recently, Cyclin O (CCNO) has been reported to be a novel protein of the cyclin family. However, the clinical significance and functional roles of CCNO in human cancer, including gastric cancer (GC), remain largely unexplored. In this study, we investigated the clinical and functional roles of CCNO in GC.
Methods: We analyzed CCNO expression patterns in GC patients. To investigate the role of CCNO in malignancy of GC, we used lentivirus-delivered short hairpin RNA to knockdown CCNO expression in GC cell lines. Then multiparametric high-content screening and MTT incorporation assay were used to assess the cell proliferation capability. Cell apoptosis was detected by flow cytometry and Caspase 3/7 assays. Furthermore, the effect of CCNO on tumorigenicity of GC was also determined in vivo. Finally, microarray analysis was performed to elucidate the molecular mechanisms by which shCCNO inhibited the malignancy of GC cells.
Results: The analysis from The Cancer Genome Atlas database revealed elevated CCNO mRNA expression in GC tissue than in the adjacent normal tissue. Immunohistochemical studies also showed that stronger cytoplasmic staining of CCNO was detected in GC tissues. Downregulation of CCNO in GC cells efficiently, through infection with the lentivirus-mediated specific short hairpin RNA, could significantly induce cell apoptosis and inhibit the proliferative properties both in vitro and in vivo. Microarray analysis further revealed 652 upregulated genes and 527 downregulated genes in the shCCNO group compared with control, and indicated that CCNO knockdown could inhibit the malignancy of GC cells through inducing genome-wide gene expression changes.
Conclusion: Our work is the first to reveal that elevated CCNO expression is closely associated with human GC development and that CCNO knockdown could efficiently inhibit the malignant properties of GC cells by inducing cell apoptosis. Therefore, CCNO could be used as a potential biomarker for prognosis or even as a therapeutic target in human GC.
Keywords: Cyclin O, gastric cancer, apoptosis, proliferation, tumorigenicity
Introduction
Gastric cancer (GC) is the most common cause of cancer-induced deaths in the world, representing an estimated 8.8% of the total deaths from cancer.1,2 The highest GC incidence and mortality rates exist in Eastern Asia.3 In spite of the recently increased systematic therapy and early diagnoses, the prognosis of GC is still poor. In particular, in the advanced stage when the disease is usually diagnosed, the 5-year survival rate is only 30%.4–6 Therefore, the molecular mechanisms underlying GC need to be further clarified, and more effective therapies aiming at specific molecular targets are urgently required.7
Cyclin O (CCNO), a cyclin-like DNA glycosylase that removes the cytosine deamination or misincorporated uracil on DNA, consists of three exons located on chromosome 5q11 encoding a1, 053-bpcDNA, and a 350-amino-acid protein.8,9 Nowadays, CCNO has been referred to as a cyclin-like protein containing two cyclin box regulatory elements predicted to function as protein-binding domains.9,10 It has been reported that CCNO is expressed in the S phase, colocalizing with proliferating cell nuclear antigen protein at the replication foci, while degraded at the S/G2 transition stage. In addition, in mouse lymphoid cells, CCNO is also associated with DNA damage-induced apoptosis.11 During oocyte meiotic maturation, CCNO was critical for the dephosphorylation of CDC2 (Tyr15), whereas the CCNO effects on CDC2 could be rescued by overexpression by CCNB1.12 Recently, it is well known that CCNO was a key regulator of oocyte meiotic maturation and multiple motile cilia formation linked to severe airway disease.13,14 However, studies concerning the role of CCNO in cancer are rarely reported. Previous studies have shown that CCNO is highly expressed in cervical cancer, and downregulation of CCNO could inhibit cell growth of human cervical cancer cells.15 However, the clinical significance of CCNO in human GC remains largely unknown, and the functional roles of CCNO in the pathogenesis of GC need to be clarified further.
The Cancer Genome Atlas Project (TCGA), which is supported by the National Cancer Institute of Health, provides whole exome sequence, whole genome sequence, RNA expression, methylation, proteomic, and clinical datasets.16 Recently, comprehensive and integrative genomic analysis of the TCGA data has further revealed the DNA methylation patterns, gene mutation and expression patterns, and clinical phenotypes involved in cancer.17–20 Through an analysis of the TCGA database, we found a set of known GC-related genes that were differentially expressed between GC and normal tissues. Moreover, we are the first ones to reveal that CCNO shows a higher fold-change in GC tissues compared with paired normal tissues, which indicates that elevated CCNO expression is closely related to human GC development, and that downregulation of CCNO may inhibit the malignant properties of GC.
In this study, we further confirmed the CCNO protein expression patterns in GC patient tissues through immunohistochemistry (IHC). Consistent with the results from our TCGA analysis, a stronger cytoplasmic staining of CCNO was detected in GC tissues compared with adjacent ones. We also used lentivirus-mediated specific short hairpin RNA (shRNA) to downregulate CCNO expression and investigated its role in cell proliferation. CCNO knockdown could significantly induce cell apoptosis and strikingly inhibit the proliferative property of GC cells, both in vitro and in vivo. Microarray analysis further revealed that CCNO knockdown inhibits the malignancy of GC cells by inducing genome-wide gene expression changes. Our findings demonstrated that elevated CCNO expression is closely related to human GC development, and CCNO may be a potential therapeutic target for the treatment of human GC.
Methods
Date sources and bioinformatics
TCGA RNA-Seq (V2) and corresponding clinical data were obtained from the TCGA website (https://cancergenome.nih.gov/), following the approval of this project by the consortium. In the TCGA database, 443 samples are available with data including 416 mRNA chips or RNA seq data samples. Among these, 26 GC patients have data from matched tumor and surrounding normal tissue. The expression spectrum analysis was based on RNA seq data from these 26 paired samples.
Clinical samples
In total, 80 patients were enrolled in this study. GC specimens and their corresponding normal gastric tissues (adjacent to them) were collected from the Department of Gastrointestinal Surgery, the Guizhou Provincial People’s Hospital (Guiyang, China), between March 2014 and June 2016. These patients did not receive chemotherapy or radiotherapy prior to the surgery. Written informed consent was obtained from the patients before surgery. The study protocol was approved by the Medical Ethics Review Committee of the Guizhou Provincial People’s Hospital, Guiyang, China.
Immunohistochemical analysis
Next, GC and normal tissue paraffin sections were deparaffinized with xylene and rehydrated with gradient concentrations of ethanol. Microwave heating was used to retrieve antigens. Endogenous peroxidase was blocked by incubation in 0.3% H2O2. The slides were incubated with primary anti-CCNO antibody (1:1,000, ab47682, Abcam, Cambridge, UK) overnight and secondary antibody for 30 minutes. Finally, all slides were stained with 3,3′-diaminobenzidine and then counterstained with hematoxylin. Semiquantitative evaluation of IHC staining of CCNO was carried out using an immunoscore based on the percentage of stained cells and on staining intensity, as previously described. The intensity score was defined as follows: 0, no appreciable staining; 1, weak intensity; 2, moderate intensity; 3, strong intensity; 4, very strong intensity. The fraction score was based on the proportion of positively stained cells (0%–100%). The mean of the immunoscores from ten microscopic high-power fields was recorded. The histologic classification of the specimens was independently determined by two pathologists.
Cell culture and lentivirus vector construction and transfection
The complementary DNA sequence of CCNO was designed from the full-length CCNO sequence (GenBank no NM_021147) by GeneChem Co. Ltd. (Shanghai, China). The most effective double-stranded CCNO-targeting shRNA interfering sequence (5′-GTACTTCCTTGACTCACAT-3′) was synthesized and inserted into the lentiviral vector pGCSIL-GFP (GeneChem). A negative control shRNA (5′-TTCTCCGAACGTGTCACGT-3′) was used, which showed no homology to human genes.
Human GC cell lines SGC-7901, AGS, and human renal epithelial 293 T cells were purchased from the Chinese Academy of Sciences (Shanghai, China), and cultured in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Ausbian, Vian-Saga Co., Ltd, Shanghai, China). Cells were plated in six-well plates (2×105 cells/mL), and infected with shCCNO lentivirus or negative control lentivirus until cell fusion reached 30%. The infected cells expressing green fluorescent protein were observed using fluorescence microscopy (IX71, Olympus, Tokyo, Japan) in order to identify the infection efficiency.
Total RNA isolation and real-time quantitative polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen, Shanghai, China) according to the manufacturer’s protocol, and used for reverse transcription later. Next, 2.0 μg of total RNA from each sample was reverse transcribed using M-MLV Reverse Transcription Kit (Promega, Madison, WI, USA), and used as a template for quantitative polymerase chain reaction (qPCR). qPCR was performed using an ABI Step One Real-time Detection System (Applied Biosystems, Foster City, CA, USA) using SYBR Green (Takara, Dalian, China). All samples were examined in triplicate. The primer sequences are listed in Table 1.
 | Table 1 Primer sequences for real-time PCR |
Western blot analysis
Cells were lysed using ice-cold lysis buffer (100 mM Tris, pH 6.8, 4% SDS, 2% MCE, 20% glycerol) kept on ice for 15 minutes. The lysates were centrifuged at 12,000g and 4°C for 15 minutes, and the supernatants were collected. Protein concentration was determined by the BCA protein assay kit (Beyotime, Shanghai, China). Equal amounts of total protein from each treatment were separated using 10% SDS-PAGE, and transferred onto polyvinylidene difluoride (PVDF) membranes (IPVH00010 Millpond). The membranes were subsequently incubated with primary antibodies overnight at 4°C. Western blots were developed using horseradish peroxidase-conjugated secondary antibodies, and detected by enhanced chemiluminescence (ECL) reagent (ECL-Plus/Kit; M3121/1859022 Thermo Fisher Scientific).
Cell proliferation assays
GC cells infected with shCCNO lentivirus or a control were seeded at a density of 2,000 cells/well into the 96-well plates and incubated at 37°C with 5% CO2 for 5 days. The cells in the plates were counted via multiparametric high-content screening (HCS), using the Cellomics Array Scan™ HCS automated reader (Cellomics, Inc., Pittsburgh, PA, USA) for each day’s analysis.
Furthermore, MTT incorporation assay using an MTT kit was also used to assess DNA synthesis in proliferating cells. The absorbance of each well at 490 nm was monitored by using a microplate reader (M2009PR, Tecan Infinite). All experiments were conducted in triplicate.
Detection of apoptosis by flow cytometry
Annexin V-APC Apoptosis Detection Kit (88-8007, eBioscience, San Diego, CA, USA) was used to monitor apoptosis according to the manufacturer’s instructions. GC cells infected with shCCNO lentivirus or control were seeded in 96-well plates at a density of 2,000 cells/well. The cells were harvested when the degree of fusion was 85%. Cells were washed with PBS buffer and stained with binding buffer containing Annexin V-APC at room temperature in the dark for 15 minutes. Cells were then analyzed using flow cytometry (Guava easyCyte HT, Millipore, Darmstadt, Germany). All experiments were performed in triplicate.
Caspase activity assays
Caspase 3/7 assays (Promega) were performed according to the manufacturer’s instructions. The GC cells were plated overnight in 96-well plates at a density of 10,000 cells/well after infection with shCCNO lentivirus or control for 3 days. To measure caspase 3/7 activity, 100 μL of caspase-Glo 3/7 reagent was added to each well for 2 hours with constant shaking at room temperature. Luminescence was measured using a Tecan infinite M2009PR microplate reader.
In vivo studies in mice
Five-weeks-old BALB/c nude mice were obtained from the Laboratory Animal Center of Chongqing Medical University (Chongqing, China). These mice were maintained in a specific pathogen-free unit under isothermal conditions. All procedures were approved by the Committee on Animals of the Chongqing Medical University, Chongqing, China, and all procedures were performed in accordance with the guidelines of the Committee on Animals of the Chongqing Medical University (Chongqing, China). AGS cells (4×106) infected with shCCNO lentivirus or control were suspended in 100 μL PBS. Next, the cells were implanted subcutaneously into the right flank of nude mice to form xenograft tumors (10 mice per group). Tumor size was determined twice a week for 24 days using calipers, and tumor volume was calculated on the basis of width (x) and length (y) as follows: x2y/2 (x<y). After the mice were sacrificed, the tumor nodes were resected and measured.
Microarray analysis
Total RNA was extracted in triplicate from AGS cells infected with shCCNO lentivirus or control and subjected to hybridization on the GeneChip® PrimeView™ Human Gene Expression Array (Affymetrix 901838, Thermo Fisher Scientific) according to the manufacturer’s instructions. Images of hybridized microarrays were scanned using a GeneChip scanner 3000 (Affymetrix). Array data were normalized using log-scale robust multiarray analysis, and analyzed by R-Project software. Microarray hybridization, scanning, and analysis were performed by Genechem Corporation, LTD (Guizhou, China).
Statistical analysis
Data are presented as the SD. Statistical analyses were performed using SPSS 22.0 statistical software (SPSS, Chicago, IL, USA). For functional in vitro and in vivo assays, statistical significance between two groups was determined using a two-sided Student’s t-test. For all the analyses, values of P<0.05 were considered significant.
Results
Upregulation of CCNO is associated with the development of human GC
TCGA Research Network has been established to generate a comprehensive catalog of genomic abnormalities driving tumorigenesis. In order to identify GC-related candidate genes, we firstly analyzed the 26 GC patients from the TCGA database, which could provide RNA expression and clinical datasets. Through a careful review of the literature, we found a set of known GC-related genes that were differentially expressed between GC and normal tissues. Furthermore, we also first revealed that CCNO showed higher expression levels in GC compared with the adjacent normal tissue (Figure 1A and Table 2). Furthermore, the transcription levels of CCNO mRNA were increased to more than 1.5-fold, in 14 out of 26 GC tissue specimens, compared with those in their adjacent tissues (Figure 1B).
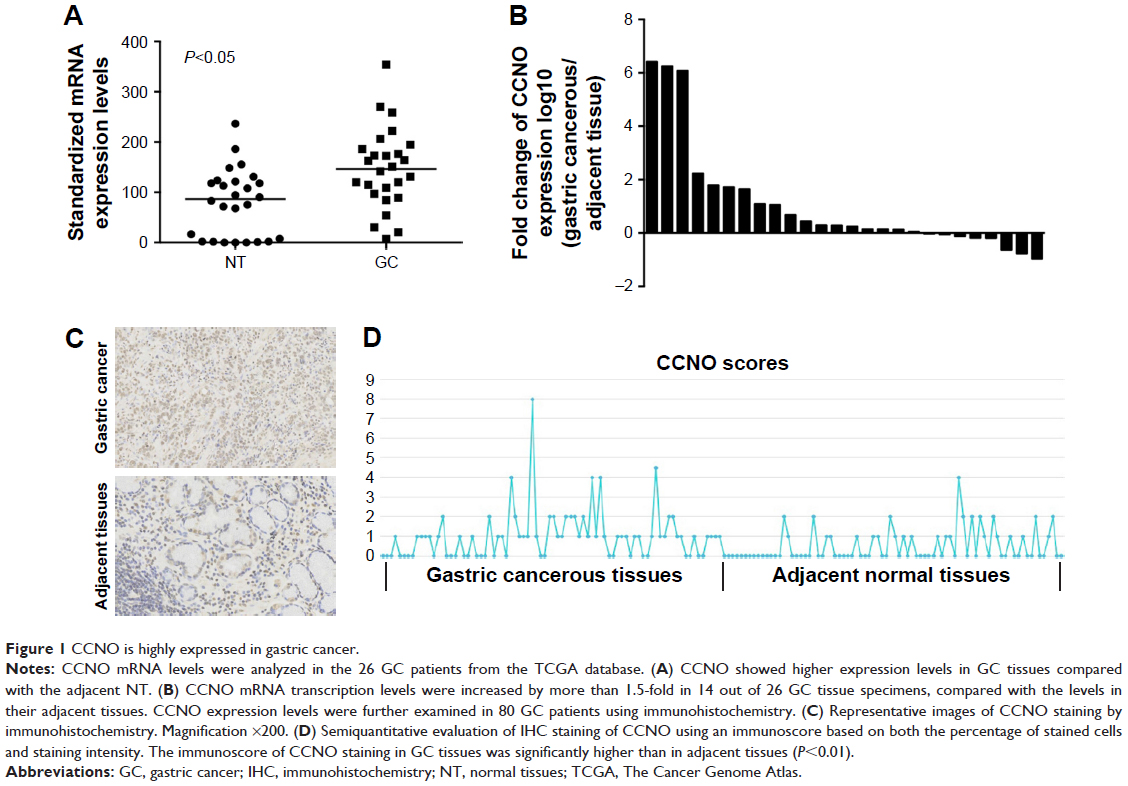 | Figure 1 CCNO is highly expressed in gastric cancer. |
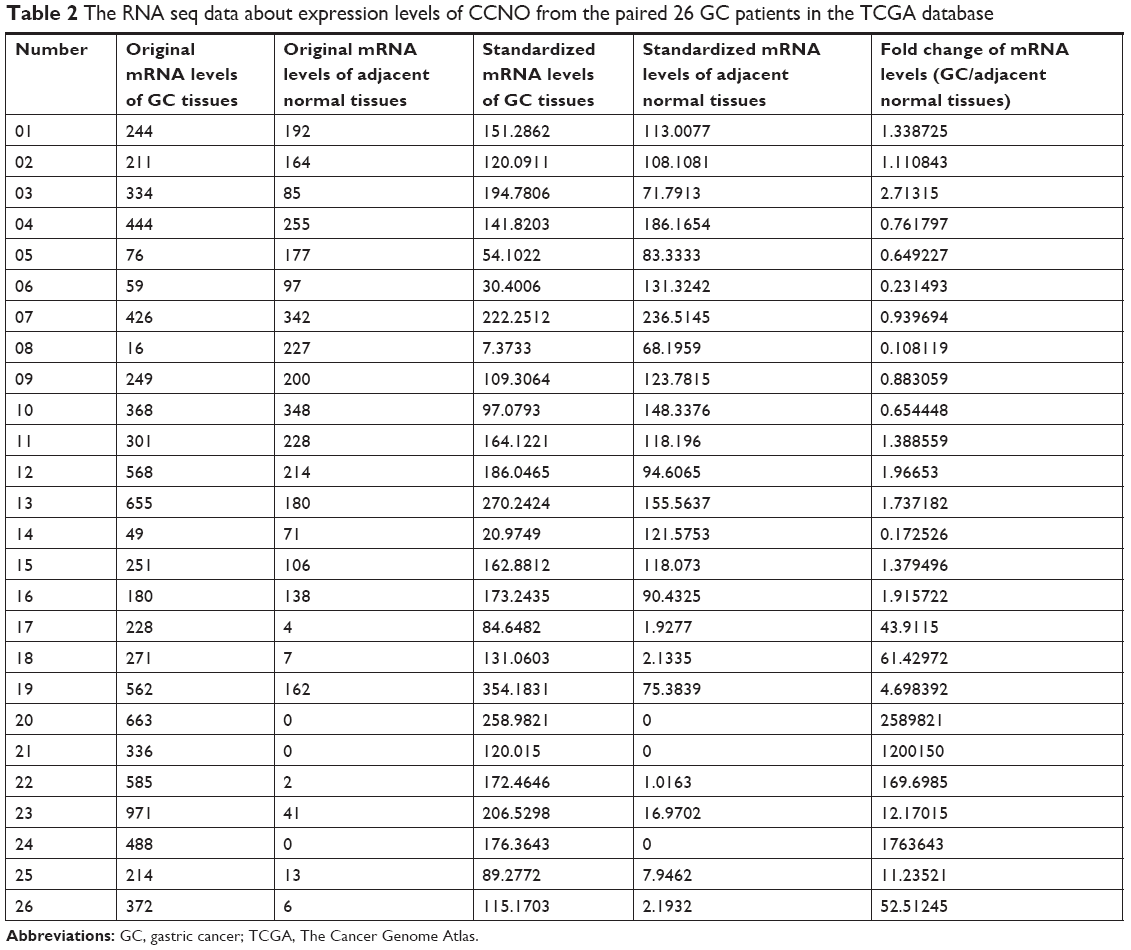 | Table 2 The RNA seq data about expression levels of CCNO from the paired 26 GC patients in the TCGA database |
In addition, CCNO protein expression levels were further examined in 80 GC tissues and their matched adjacent normal gastric mucosal tissues using IHC. Consistent with the results from TCGA analysis, IHC studies also showed that strong cytoplasmic staining of CCNO was detected in 64% of gastric tumor tissues, whereas only faint CCNO staining was observed in 34% of their adjacent tissues (P<0.01, Figure 1C). Further confirmed through semiquantitative evaluation based on both the percentage of stained cells and staining intensity, the immunoscore of CCNO staining in GC tissues was significantly higher than in adjacent tissues (P<0.01, Figure 1D). These data indicated that elevated CCNO expression is related to human GC development. Therefore, we chose CCNO as a candidate oncogene for further investigation.
Knockdown of CCNO suppresses the proliferative capability of gastric cancer cells
To investigate the role of CCNO in malignancy of GC, we used lentivirus-delivered shRNA to knockdown CCNO expression in AGS and SGC-7901 cell lines, both of which expressed high levels of CCNO. After infection with the lentivirus, CCNO mRNA levels were assessed by real-time PCR. The results showed that both AGS and SGC-7901 GC cells infected with the lentivirus expressing shCCNO exhibited significantly reduced CCNO transcripts compared with the cells infected with control lentivirus-expressing shNC (Figure 2A). Moreover, decreased CCNO protein levels were also detected in shCCNO-treated cells (Figure 2B). In order to monitor the growth of GC cells, HCS assays were performed for >5 days. As shown in Figure 2C, CCNO knockdown significantly decreased the total cell numbers 3 days after infection and slowed the growth rate of both AGS and SGC-7901 cells. Furthermore, DNA synthesis as analyzed by MTT assays also demonstrated a significant inhibition of cell proliferation in both cell lines treated with shCCNO compared with that in the control cells (P<0.05, Figure 2D).
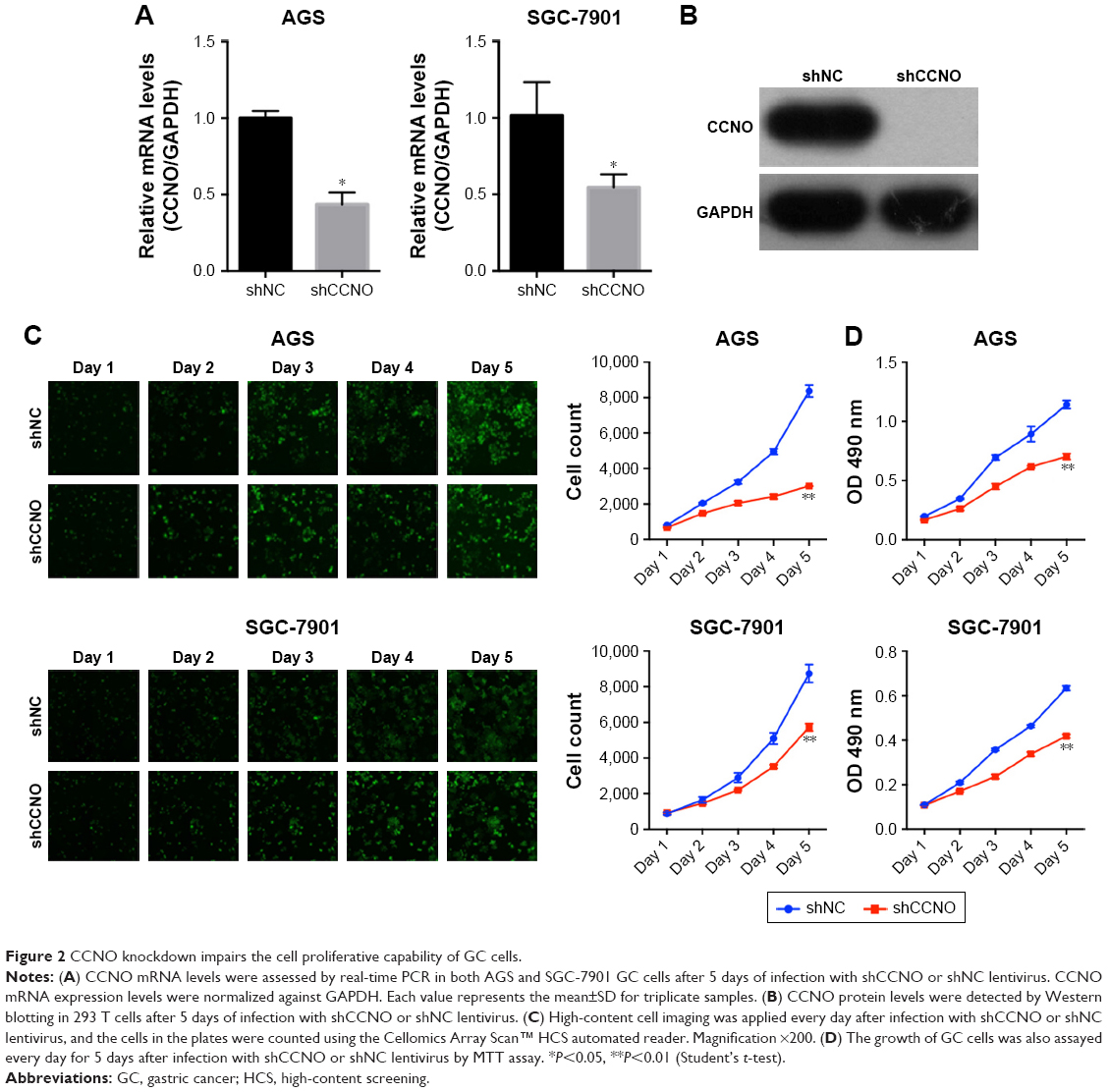 | Figure 2 CCNO knockdown impairs the cell proliferative capability of GC cells. |
Knockdown of CCNO induces apoptosis of GC cells
To assess how CCNO knockdown inhibited the proliferation of GC cells, flow cytometry with Annexin V staining was performed to quantitate the population of the apoptotic cells in AGS cells, 5 days after infection with shCCNO or shNC lentivirus. As shown in Figure 3A, cellular apoptosis was significantly increased in the shCCNO group compared with that in the shNC group (P<0.01). Furthermore, we analyzed caspase 3/7 activity using a Caspase-Glo 3/7 assay according to the manufacturer’s instructions. As shown in Figure 3B, in AGS cells, caspase 3/7 activation was strongly elevated after CCNO knockdown, when compared with that in the control group. Similar results were also observed in SGC-7901 cells. These results affirmed that CCNO knockdown could induce cell apoptosis and suppress cell proliferation in GC cells.
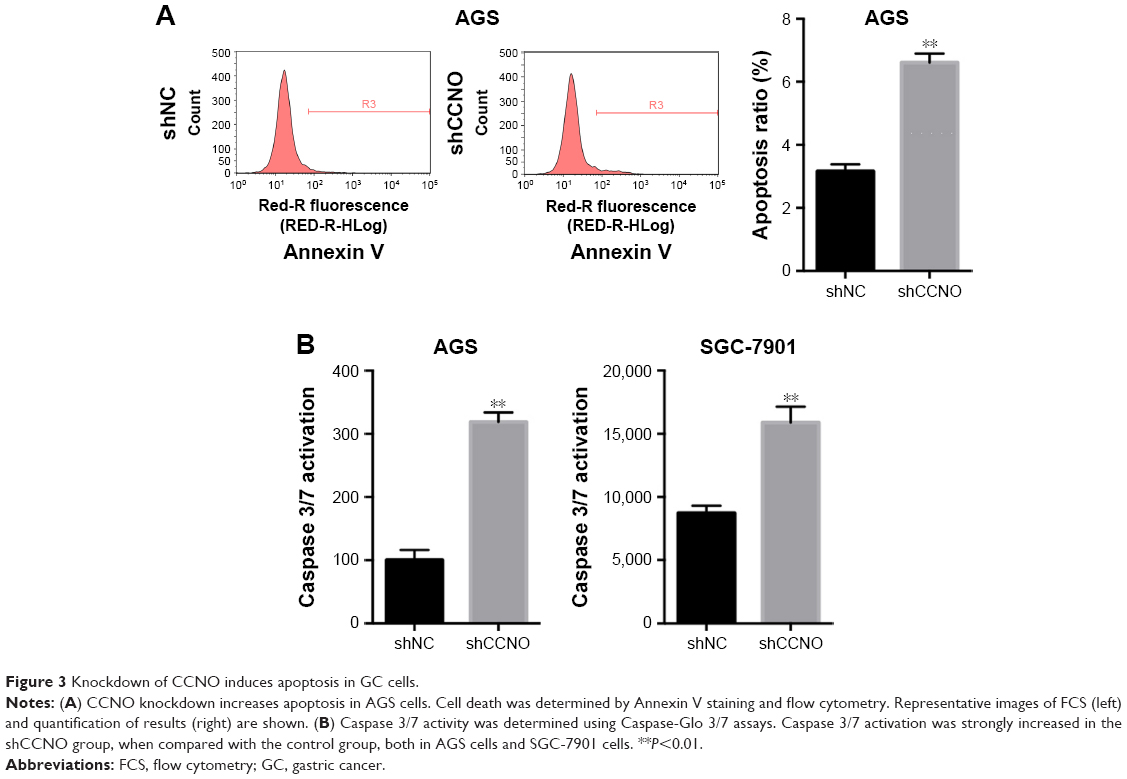 | Figure 3 Knockdown of CCNO induces apoptosis in GC cells. |
Knockdown of CCNO decreases the tumorigenicity of GC cells in vivo
A xenograft model in which BALB/c nude mice were subcutaneously injected with AGS cells was used to further determine whether CCNO knockdown could reduce the tumorigenicity of GC cells in vivo. In the control group, tumor nodules were detected in all of the mice (10/10) on Day 10, whereas tumor nodules in the CCNO knockdown group were detected in 8 of 10 mice. Moreover, xenografts were also significantly smaller in the shCCNO group than in the control group at every point in time (Figure 4A). At the end of the experiment, when the mice were sacrificed, the tumor weights were markedly reduced in the shCCNO group consistently (Figure 4B). These results demonstrate that infection of GC cells by shCCNO lentivirus inhibits their tumor-forming ability.
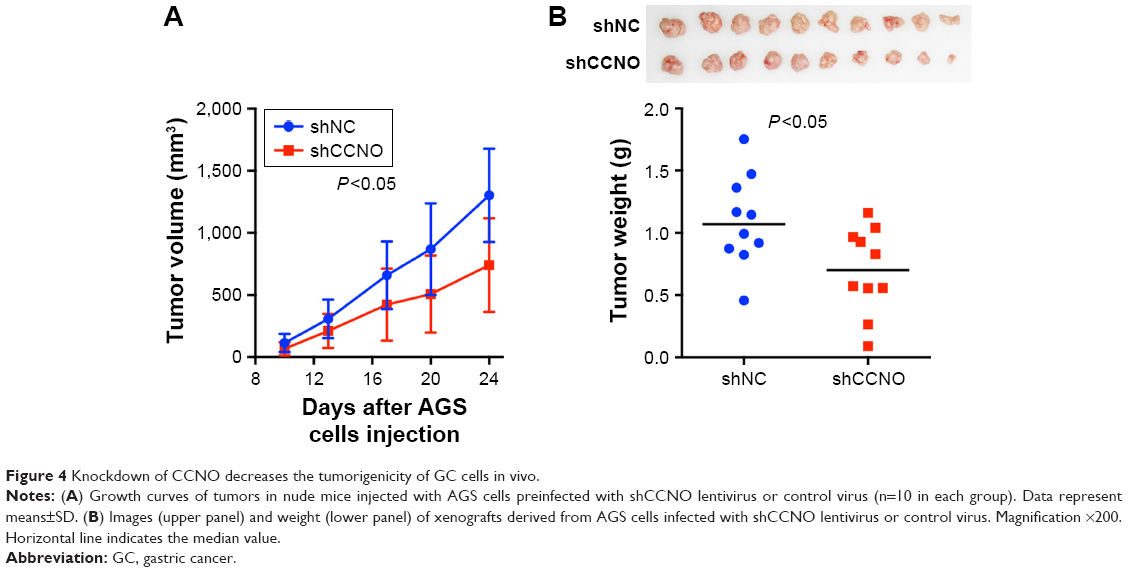 | Figure 4 Knockdown of CCNO decreases the tumorigenicity of GC cells in vivo. |
CCNO knockdown induces genome-wide gene expression changes in GC cells
To elucidate the molecular mechanisms by which shCCNO inhibited the malignancy of GC cells, microarray analysis was performed to compare gene expression in AGS cells infected with shCCNO and shNC lentivirus. The data revealed 652 upregulated genes and 527 downregulated genes in the shCCNO group compared with those in the control by at least a 1.5-fold change (P<0.05; Figure 5A). An ingenuity pathway analysis was performed and the differentially expressed genes were significantly enriched in several pathways based on a P<0.001 threshold. These pathways, including the cell cycle control of chromosomal replication, interferon signaling, and Jak/Stat and MAPK signaling pathways were altered following CCNO silencing (Table 3). Further disease and function analysis revealed that specific gene sets were significantly enriched, including those involved in cellular growth and proliferation, cell death and survival, cellular development, cancer, organism injury, and abnormalities (Figure 5B). Moreover, we verified the expression levels of certain important genes, which are closely associated with regulation of cell proliferation and the process of tumorigenesis. These genes, including TCF4, PCNA, GADD45B, E2F1, E2F2, BCL2L1, ETS1, WEE1, and MCM2, were significantly reduced at the mRNA level after CCNO knockdown, which was consistent with the microarray analysis (Figure 5C). Furthermore, suppressed protein levels of TCF4, ETS1, PCNA, and MCM2 were also observed in the shCCNO-treated GC cells (Figure 5D). These results indicate that CCNO knockdown inhibits the malignancy of GC cells by inducing the genome-wide gene expression change.
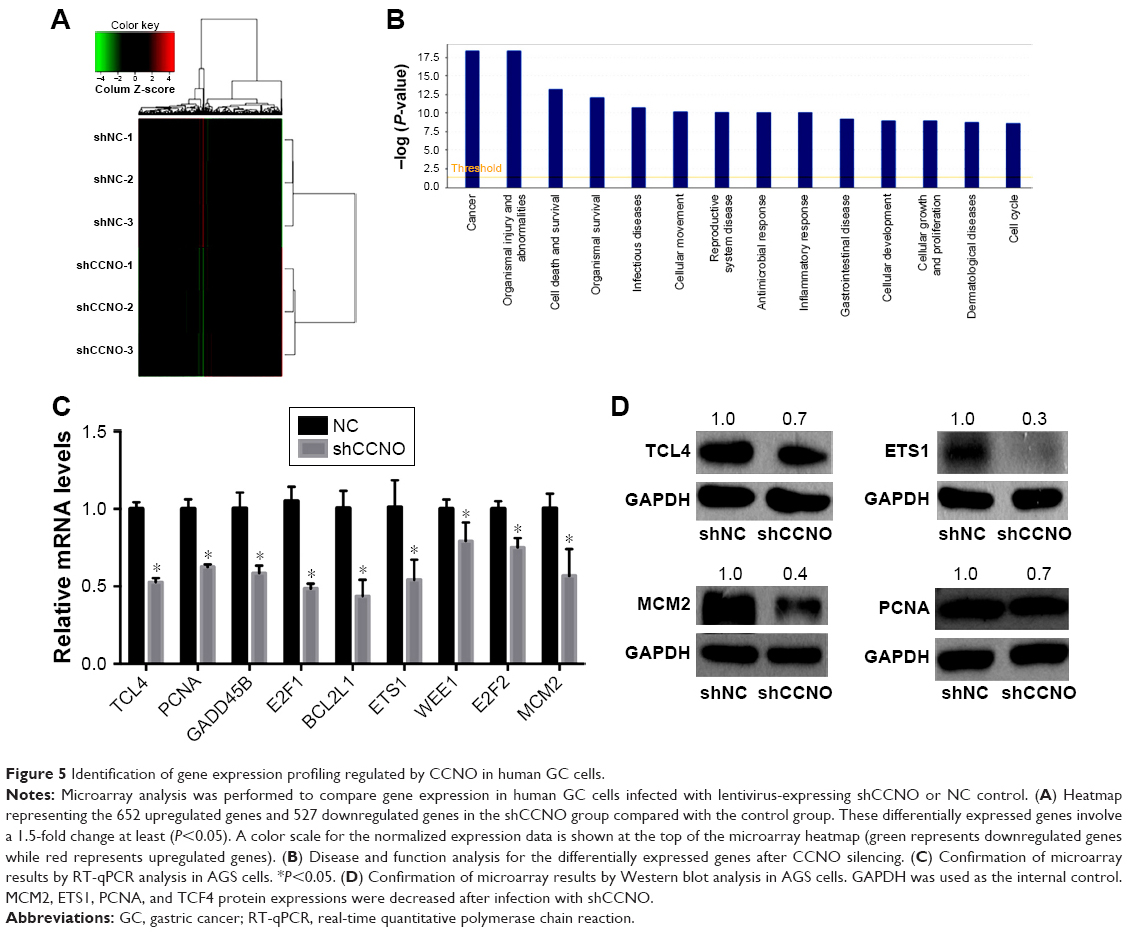 | Figure 5 Identification of gene expression profiling regulated by CCNO in human GC cells. |
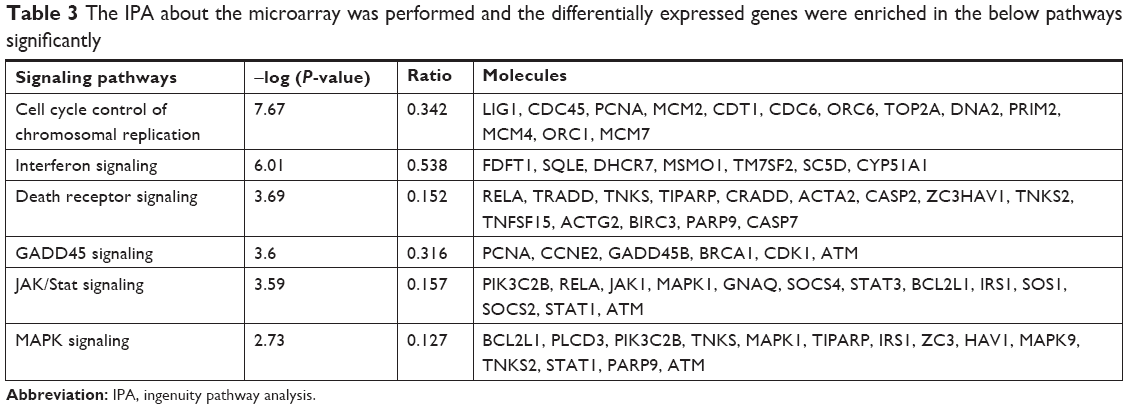 | Table 3 The IPA about the microarray was performed and the differentially expressed genes were enriched in the below pathways significantly |
Discussion
It has been reported that cyclins, which are key regulatory proteins complexing with and activating cyclin-dependent kinase (CDK) subunits, play important roles in the regulation of cell proliferation.21 Among cyclin molecules, previous studies have implicated the abnormal expression of cyclins A, B, D, and E and their pivotal roles in tumorigenesis and progression of human malignancies, including GC. CCNO, a novel cyclin protein family member containing a conserved cyclin-like domain, is located at the 5q11 region and consists of 350 amino acids. Previous studies mainly demonstrated various mutations and important functions in respiratory disease.13,22,23 However, studies concerning the role of CCNO in cancer are rarely reported. Therefore, the clinical significance and functional role of CCNO in human cancer, including GC, remains largely unexplored.
We are the first ones, in our study, to confirm both RNA and protein expression levels in GC patients. The analysis from the TCGA database revealed elevated CCNO mRNA expression in GC tissue than in the adjacent normal tissue. IHC studies also showed that stronger cytoplasmic staining of CCNO was detected in GC tissues. To our knowledge, these data are the first to demonstrate that elevated CCNO expression is closely associated with human GC development. We propose that high CCNO levels may be correlated with more aggressive tumor behavior and with poorer prognosis of human GC, which we need to verify further.
Moreover, in order to investigate the roles of CCNO in the malignant properties of GC cells, we used lentivirus-mediated specific shRNA to downregulate CCNO expression. Both qPCR and Western blotting confirmed that shCCNO lentivirus could efficiently reduce endogenous CCNO levels, and the HCS and MTT assays indicated that CCNO knockdown strikingly inhibited the proliferative properties of GC cells. Furthermore, CCNO knockdown could also reduce the tumorigenicity ability of GC cells in mice. Previous studies rarely report the role of CCNO in cancer. Our study is the first to reveal the proliferation-promoting effects of CCNO on GC cells, and that targeting CCNO may be a potential therapeutic strategy for treatment of human GC.
It has been demonstrated that cyclins play an important role in regulating the cell cycle and apoptosis.24,25 CCNO could regulate CDK2 kinase activity through the phosphorylation of the 81st serine residue,26 and induce the intrinsic apoptotic signaling pathway in lymphoid cells in concert with CDK2.11 In our study, flow cytometry revealed that cell apoptosis was significantly induced in the shCCNO group compared with that in the control. In addition, the Caspase-Glo 3/7 assay also affirmed increased caspase 3/7 activation by CCNO knockdown. Therefore, our results indicate that CCNO knockdown suppresses proliferation through inducing cellular apoptosis in GC cells, which is consistent with previous reports.
Microarray analysis performed using AGS cells infected with shCCNO and shNC lentivirus demonstrated genome-wide gene expression changes in GC cells after CCNO knockdown, including 652 upregulated genes and 527 downregulated genes. Gene expression profiling further revealed that CCNO silencing enriched expression of genes involved in cellular growth and proliferation, cell death and survival, cellular development, cancer, organism injury, and abnormalities. Several genes that have been reported to be involved in the process of tumorigenesis were also significantly reduced by CCNO, includingTCF4, PCNA, GADD45B, E2F1, E2F2, BCL2L1, ETS1, WEE1, and MCM2.27–31 Among these important genes, ETS1 and TCF4 are major downstream effectors of the MAPK and Wnt signaling pathways, respectively, which indicates that the inactivation of MAPK and Wnt signaling pathways may contribute to the suppressive effect of CCNO silencing in GC cells. However, the key molecules or pathways responsible for the malignant functions of CCNO require further investigation.
Conclusion
Our work is the first to reveal that elevated CCNO expression is closely associated with human GC development and that CCNO knockdown could efficiently inhibit the malignant properties of GC cells by inducing cell apoptosis. Therefore, CCNO may find application as a potential biomarker for prognosis or even as a therapeutic target in human GC.
Acknowledgment
This work was supported by the Young Grant of Guizhou Provincial People’s Hospital (project number: GZSYQN [2016] 19) and the Combined Science and Technology Grant of Guizhou Provincial People’s Hospital (project numbers: LH [2016] 7175 and LH [2016] 7176).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends – an update. Cancer Epidemiol Biomarkers Prev. 2016;25(1):16–27. | ||
den Hoed CM, Kuipers EJ. Gastric cancer: how can we reduce the incidence of this disease? Curr Gastroenterol Rep. 2016;18(7):34. | ||
Foo M, Leong T. Adjuvant therapy for gastric cancer: current and future directions. World J Gastroenterol. 2014;20(38):13718–13727. | ||
Ilson DH. Current progress in the adjuvant treatment of gastric cancer. Surg Oncol Clin N Am. 2017;26(2):225–239. | ||
de Vita F, di Martino N, Fabozzi A, et al. Clinical management of advanced gastric cancer: the role of new molecular drugs. World J Gastroenterol. 2014;20(40):14537–14558. | ||
Muller SJ, Caradonna S. Isolation and characterization of a human cDNA encoding uracil-DNA glycosylase. Biochim Biophys Acta. 1991;1088(2):197–207. | ||
Muller SJ, Caradonna S. Cell cycle regulation of a human cyclin-like gene encoding uracil-DNA glycosylase. J Biol Chem. 1993;268(2):1310–1319. | ||
Amirav I, Wallmeier J, Loges NT, et al. Systematic analysis of CCNO variants in a defined population: implications for clinical phenotype and differential diagnosis. Hum Mutat. 2016;37(4):396–405. | ||
Roig MB, Roset R, Ortet L, et al. Identification of a novel cyclin required for the intrinsic apoptosis pathway in lymphoid cells. Cell Death Differ. 2009;16(2):230–243. | ||
Ma JY, Ou-Yang YC, Luo YB. Cyclin O regulates germinal vesicle breakdown in mouse oocytes. Biol Reprod. 2013;88(5):1–9. | ||
Wallmeier J, Al-Mutairi DA, Chen CT, et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet. 2014;46(6):646–651. | ||
Stubbs JL, Vladar EK, Axelrod JD, Kintner C. Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat Cell Biol. 2012;14(2):140–147. | ||
Wang X. The impact of CCNO gene expression on the biological behavior of cervical cancer Hela cells. [doctoral dissertation]. Beijing: Peking Union Medical College; 2015. | ||
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7261):1061–1068. | ||
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | ||
Cancer Genome Atlas Research Network, Brat DJ, Verhaak RG, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. | ||
Cancer Genome Atlas Research Network, Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(4774):67–73. | ||
Weisenberger DJ. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J Clin Invest. 2014;124(1):17–23. | ||
Duman-Scheel M, Weng L, Xin S, du W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature. 2002;417(6886):299–304. | ||
Funk MC, Bera AN, Menchen T, et al. Cyclin O (CCNO) functions during deuterostome-mediated centriole amplification of multiciliated cells. Embo J. 2015;34(8):1078–1089. | ||
Villa M, Crotta S, Dingwell KS, et al. The aryl hydrocarbon receptor controls cyclin O to promote epithelial multiciliogenesis. Nat Commun. 2016;7:12652. | ||
Mori T, Anazawa Y, Matsui K, Fukuda S, Nakamura Y, Arakawa H. Cyclin K as a direct transcriptional target of the p53 tumor suppressor. Neoplasia. 2002;4(3):268–274. | ||
Liu J, Cui ZS, Luo Y, Jiang L, Man XH, Zhang X. Effect of cyclin G2 on proliferative ability of SGC-7901 cell. World J Gastroenterol. 2004;10(9):1357–1360. | ||
Kim DH, Park JH, Lee B, Jang KO, Chung IS, Han YS. Phosphorylation of cyclin O, a novel cyclin family protein containing a cyclin-like domain, is involved in the activation of cyclin-dependent kinase 2. Oncol Lett. 2014;8(6):2769–2775. | ||
Grossi V, Peserico A, Tezil T, Simone C. p38α MAPK pathway: a key factor in colorectal cancer therapy and chemoresistance. World J Gastroenterol. 2014;20(29):9744–9758. | ||
Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773(8):1213–1226. | ||
Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322–327. | ||
Anastas JN. Functional crosstalk between WNT signaling and tyrosine kinase signaling in cancer. Semin Oncol. 2015;42(6):820–831. | ||
Wickström M, Dyberg C, Milosevic J, et al. Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat Commun. 2015;6:8904. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.