")
Back to Journals » OncoTargets and Therapy » Volume 11
Klotho ameliorates sepsis-induced acute kidney injury but is irrelevant to autophagy
Authors Chen X, Tong H, Chen Y, Chen C, Ye J, Mo Q, Zhao G, Hong G, Zheng C, Lu Z
Received 14 November 2017
Accepted for publication 21 December 2017
Published 19 February 2018 Volume 2018:11 Pages 867—881
DOI https://doi.org/10.2147/OTT.S156891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Xinxin Chen,1 Huan Tong,2 Yu Chen,3 Chaosheng Chen,1 Jingjing Ye,2 Qingfei Mo,2 Guangju Zhao,2 Guangliang Hong,2 Chenfei Zheng,1 Zhongqiu Lu2
1Department of Nephrology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China; 2Emergency Department, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China; 3Department of Nephrology, Wenzhou Hospital of Traditional Chinese Medicine Affiliated with Zhejiang Chinese Medical University, Wenzhou, Zhejiang, China
Background: The role of Klotho (KL) in sepsis-induced acute kidney injury (AKI) and the potential relationship between KL and autophagy in septic AKI were investigated.
Materials and methods: A murine model of sepsis-induced AKI was established by cecal ligation and puncture (CLP). Mice undergoing CLP and immortalized proximal tubular epithelial human HK-2 cells that were exposed to lipopolysaccharide (LPS) were treated with recombinant KL, autophagy stimulator rapamycin (Rap), and autophagy suppressor 3-methyladenine (3-MA).
Results: Autophagy activation and KL reduction reached maximum levels in mice 24 hours after CLP. Recombinant KL and/or Rap significantly attenuated CLP-induced renal dysfunction (P<0.05) and partially restored endogenous renal KL expression (P<0.05). Recombinant KL had no impact on CLP-induced autophagy and apoptosis, whereas Rap significantly stimulated autophagy and reduced apoptosis in mice. 3-MA significantly exacerbated renal dysfunction, increased apoptosis, and inhibited autophagy in mice with CLP-induced AKI (all P<0.05). In LPS-treated HK-2 cells, Rap significantly enhanced autophagy and reduced apoptosis (all P<0.05), whereas recombinant KL had no impact, and 3-MA inhibited autophagy and significantly increased apoptosis (P<0.05).
Conclusion: Recombinant KL alleviates renal dysfunction and restores renal KL expression in mice with sepsis-induced AKI, but the underlying mechanism may not be related to autophagy induction.
Keywords: acute kidney injury, autophagy, klotho, septic AKI, Rapamycin, 3-MA
Background
Sepsis, a severe infection-induced systemic inflammatory response syndrome, is a common complication associated with severe trauma, burns, hypoxia, reperfusion injury, and major surgery.1 Severe sepsis can lead to septic shock and multiple organ dysfunction syndrome, which ultimately causes mortality.1 In the event of sepsis, the kidney is the most commonly affected organ.1 The incidences of acute kidney injury (AKI) in patients with mild, moderate, and severe sepsis are 19%, 23%, and 51%, respectively, and the mortality associated with AKI is 70%.2–4 Therefore, prompt management of AKI is essential for reducing mortality and improving the prognosis in patients with sepsis. Understanding the mechanism underlying sepsis-induced AKI may facilitate the development of novel preventive therapies for AKI.
Klotho (KL), a transmembrane protein, is mainly expressed in renal tubular epithelial cells.5 KL has multiple functions, such as suppressing ageing, regulating phosphate metabolism, and acting as a co-receptor for FGF23.5–8 The extracellular domain of KL can be cleaved by a protease and released into blood, urine, and cerebrospinal fluid.9–13 Secreted KL is involved in the regulation of ion channel activity and the oxidative stress response.14–16 Urinary KL levels are dramatically reduced in patients with AKI, and KL expression is also decreased in mice with ischemia-reperfusion injury (IRI).17–19 In addition, injection of recombinant KL increases endogenous KL expression and attenuates kidney damage in rats with AKI.17 Therefore, KL may have a protective effect on kidney function. The cellular mechanism underlying KL-mediated renoprotection may relate to autophagy stimulation. In vitro, overexpression of KL can induce autophagy in hepatocellular carcinoma and gastric cancer cells.20,21 Moreover, autophagy can protect the kidney from toxin-mediated AKI and ischemia-reperfusion-induced AKI.22,23
To investigate the role of KL in sepsis-induced AKI, we established a mouse model of sepsis-induced AKI by cecal ligation and puncture (CLP) and investigated the effects of administration of recombinant KL on endogenous KL expression and kidney function in mice with CLP-induced AKI. An in vitro cell line model, human proximal tubular epithelial cell line HK-2, was also used to investigate the role of KL in autophagy.
Materials and methods
Reagents
Recombinant truncated mouse KL, which contains the entire extracellular domain (Ala35–Lys982) without transmembrane and intracellular domains, was purchased from R&D Systems, Inc. (Minneapolis, MN, USA [catalog number: 1819-KL]). Recombinant human KL protein was also purchased from R&D Systems, Inc. (catalog number: 5334-KL). KL biological activity has been previously characterized.24 Rapamycin (Rap; V900930) and 3-methyladenine (3-MA; M9281) were from Sigma-Aldrich Co. (St Louis, MO, USA). For in vivo animal experiments, Rap was dissolved in saline containing 0.2% carboxymethyl cellulose and 0.25% Tween-80 (CMC solution). CMC solution was injected into mice as a control. For in vitro cell line experiments, Rap was dissolved in DMSO; therefore, DMSO was used as a control.
Animals
All animal experiments were performed in strict accordance with the international guidelines for the care and use of laboratory animals. The protocol for animal maintenance and experiments was approved by the Institutional Animal Care and Use Committee (IACUC) of Wenzhou Medical University (approval no: wydw2016-0079). Male BALB/c mice aged 6–8 weeks and weighing 18–22 g were purchased from Shanghai Laboratory Animal Co. (SLAC), Ltd. (license: SCXK [HU] 2012-0002). The mice were housed in the Laboratory Animal Center of Wenzhou Medical University (license: SYXK (ZHE) 2010-0150) at 25°C and 40%–60% humidity under a 12-hour light/dark cycle and had free access to food and water. The mice habituated to their environment for 7 days before they were used for experiments. Every effort was made to minimize the number and suffering of the mice.
CLP in mice
Mice were randomized into the following nine groups (n=5 in each group): normal (N), sham (S), CLP model (CLP), CLP model treated with KL (CLP+KL), CLP model treated with Rap (CLP+Rap), CLP model treated with KL and Rap (CLP+KL+Rap), CLP model treated with CMC solution (CLP+CMC), CLP model treated with 3-MA (CLP+3-MA), and CLP model treated with KL and 3-MA (CLP+KL+3-MA). CLP-induced sepsis in mice was established as previously described.25,26 Briefly, the mice underwent 1 cm midline laparotomy under anesthesia (intraperitoneal injection of pentobarbital sodium, 50 mg/kg). The caecum was ligated at 1 cm from the end with a 4-0 silk suture. A 22-gauge needle was used to perforate the caecum with two holes near the ligation site. A small amount of cecal content was squeezed out of the caecum through the two holes to induce sepsis. After the ligated and punctured caecum was returned to the peritoneal cavity, the muscle layer and epidermal layer were sutured, and 1 mL of 0.9% sterile saline solution (37°C) was administered subcutaneously. Successful establishment of CLP-induced sepsis was confirmed by the following criteria: 1) survived 6 hours after the surgery; 2) developed the characteristic symptoms of sepsis, including diarrhea, piloerection, and hypoergia; and 3) exhibited a peak in mortality at 24 hours, with 40%–60% mortality at 72 hours after the surgery. Sham-operated mice underwent laparotomy and bowel manipulation without ligation or puncture. None of the mice received antibiotic treatment. The mice were allowed free access to food and water before and after the surgery. Recombinant mouse KL (0.01 mg/kg) was administered by intraperitoneal injection 1 hour after CLP surgery, as previously described.24,27 Rap (1 mg/kg) or 3-MA (30 mg/kg) was administered by intraperitoneal injection 2 hours before CLP surgery to activate or suppress autophagy, respectively.28
The mice were sacrificed by cervical dislocation at 3 hours, 6 hours, 12 hours, 1 day, 2 days, 3 days, and 5 days after CLP surgery. Blood samples were collected, and plasma was stored at −80°C. Both kidneys of each mouse were removed, and each kidney was cut into four pieces. Three pieces were instantly snap-frozen in liquid nitrogen and stored at −80°C. The remaining piece was fixed in 4% paraformaldehyde and 2.5% glutaraldehyde in PBS and then prepared as paraffin-embedded or epon-embedded tissue blocks for light microscopy or electron microscopy, respectively.
Assessment of mouse renal function
Mouse renal function was assessed by blood urea nitrogen (BUN) and serum creatinine (Scr) levels. Blood samples were collected 1 day after CLP surgery. Blood samples were centrifuged at 2,500 rpm for 10 min to separate the serum. BUN and Scr concentrations were measured using a kit (Nanjing Jiancheng Biology Engineering Institute, Nanjing, China, C013-2 and C011-2) according to the manufacturer’s instructions.
Light microscopy of mouse kidney specimens
Paraffin-embedded kidney specimens were sectioned (3 μm thickness). Tissue sections were stained with H&E and periodic acid-Schiff (PAS) according to the standard protocols and observed under a light microscope (BX53; Olympus Corporation, Tokyo, Japan).
Electron microscopy
Approximately 1 mm3 epon-embedded kidney specimens were fixed in 1% osmium tetroxide, stained with uranyl acetate and lead citrate, and cut into ultrathin sections. The sections were observed under a Hitachi H7500 electron microscope (Hitachi Ltd., Tokyo, Japan).
Immunohistochemistry (IHC)
Paraffin-embedded kidney specimens were sectioned (3 μm thickness) and incubated with 3% H2O2 for 10 min. After tissue sections were washed with PBS, they were boiled in 10 mM citrate buffer (pH 6.0) for 4 min for antigen retrieval and then blocked with 10% goat serum in PBS (Zhongshan Golden Bridge Biotechnology, Beijing, China [PV-9001]) at room temperature for 1 hour. The sections were incubated with anti-KL (1:100, Abcam, Cambridge, UK [ab203576]) and anti-CASP3 (1:100, Cell Signaling Technology, Inc., Danvers, MA, USA, [9664]) at 4°C overnight. After washing, the tissue sections were incubated with HRP-conjugated secondary antibodies (Zhongshan Golden Bridge Biotechnology [PV-9001]). Color was developed using a DAB kit (Zhongshan Golden Bridge Biotechnology [ZLT-9018]). The tissue sections were counterstained with hematoxylin, dehydrated, and mounted on glass slides. Tissue sections that were incubated with secondary antibody alone were used as a negative control for IHC staining. Images at 200× magnification were acquired using a biological imaging microscope (BX53; Olympus Corporation). The intensity of positive staining, which appeared brown, was determined using Image-Pro plus 6.0 image analysis software. The mean optical density (MOD) was calculated to represent the intensity. MOD values increased as protein expression increased.
Cell culture
Human HK-2 cells, an immortalized proximal tubular epithelial cell line derived from normal human kidney, were obtained from Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai, China, SCSP-511). The cells were cultured in DMEM/F-12 (Thermo Fisher Scientific, Waltham, MA, USA [11330032]) supplemented with 10% FBS (Thermo Fisher Scientific [10099141]) and 1% penicillin-streptomycin (100 units/mL) in a humidified incubator with 5% CO2 at 37°C. Culture media were replaced with fresh media every 2 to 3 days. HK-2 cells were cultured in serum-free and penicillin-streptomycin-free media for 24 hours before analyses. Confluent HK-2 cells (70%–80% confluency) were detached by 0.05% trypsin/EDTA (Thermo Fisher Scientific [25300062]).
HK-2 cells were incubated with 0, 1, 5, 10, 50, or 100 μg/mL lipopolysaccharide (LPS; Sigma-Aldrich Co. [L4391]) for 3, 6, 12, or 24 hours. HK-2 cells were incubated with the following eight drug combinations: no drug control, LPS, LPS+KL, LPS+Rap, LPS+KL+Rap, LPS+DMSO, LPS+3-MA, or LPS+KL+3-MA. Recombinant human KL (0.4 μg/mL) was added 1 hour after LPS incubation.18,24,27 Rap (1 μM) and 3-MA (5 mM) were added 30 min before LPS treatment to activate or suppress autophagy, respectively.28 Rap was dissolved in DMSO (Sigma-Aldrich Co. [D2650]). DMSO was used as a control. Both the culture media and cells were collected.
Apoptosis analysis by flow cytometry
HK-2 cells were seeded in 6-well plates at a density of 2×106 cells per well. After reaching 80% confluency, the cells were treated with the eight drug combinations. The cells were then trypsinized and incubated in 500 μL of binding buffer containing 5 μL of Annexin V-FITC and 5 μL of propidium iodide (BD Biosciences, San Jose, CA, USA [556547]) in the dark for 15 min. The cells were analyzed with a flow cytometer (FACSCalibur, BD, Franklin Lakes, NJ, USA). Flow cytometry data were analyzed using FlowJo software (Tree Star, San Carlos, CA, USA).
Western blot
Mouse renal tissues were homogenized, and HK-2 cells were lysed in RIPA buffer containing protease and phosphatase inhibitors (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, sodium orthovanadate, sodium fluoride, EDTA, and leupeptin, pH 7.4; Beyotime, Shanghai, China [P0013B]). The lysate was centrifuged at 14,000 g at 4°C for 30 min, and supernatants were collected. The protein concentrations of the supernatants were measured using a BCA Protein Assay Kit (Thermo Fisher Scientific Pierce, Waltham, MA, USA [23235]) according the manufacturer’s protocol. Proteins in the supernatants were separated with 8% or 12% SDS-PAGE (tissue sample: 50 μg/well; cell lysate sample: 20 μg/well). The proteins were transferred to a PVDF membrane (Merck Millipore, Billerica, MA, USA). The membrane was blocked with 5% non-fat milk in Tris-buffered saline with Tween 20 (TBS-T) buffer at room temperature for 1 hour and then incubated with primary antibodies, including anti-KL (1:1,000, Abcam [ab203576]), anti-MAP1LC3B (1:1,000, Cell Signaling [3868]), and anti-P62 (1:1,000, Cell Signaling [8025]), at 4°C overnight. β-actin was used as the loading control. After the membrane was washed with TBS-T, it was incubated with HRP-conjugated secondary antibodies (1:2,000, Cell Signaling, [7074]) at room temperature for 1 hour. After three washes with TBS-T, protein signals were visualized using an enhanced chemiluminescence kit (Pierce [34095]) according to the manufacturer’s instructions. Protein signals were quantified by densitometry using Image-Lab 3.0 image analysis software (Bio-Rad Laboratories Inc., Hercules, CA, USA). The expression levels of all proteins were evaluated relative to the level of β-actin.
Immunofluorescence assay
HK-2 cells cultured on coverslips were fixed in 4% polyoxymethylene for 15 min and then blocked in 1% bovine serum at room temperature for 60 min. After incubation with anti-LC3B (1:400, Cell Signaling [3868]) overnight at 4°C, the coverslips were incubated with donkey anti-rabbit IgG-conjugated FITC secondary antibody (1:200; Santa Cruz Biotechnology Inc., Dallas, TX, USA) for 1 hour at room temperature. After the coverslips were extensively washed with PBS, they were mounted with ProLong Gold Antifade Reagent with DAPI (Thermo Fisher Scientific) and viewed under a fluorescence microscope (Olympus Corporation).
Statistical analysis
All in vitro experiments were repeated at least three times. The results are expressed as the mean ± SD. Student’s t-test or one-way ANOVA was used to compare values among the different groups. A 2-sided P-value was used, and P<0.05 was considered statistically significant. SPSS 23.0 statistical analysis software was used.
Ethics statement
The experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Wenzhou Medical University (approval no: wydw2016-0079), whose guidelines and protocols were followed for the welfare of the animals. These were also in line with international guidelines for the care and use of laboratory animals.
Results
Autophagy was maximally activated 1 day after CLP in mice
One day after CLP, H&E and PAS staining revealed vacuolar degeneration, loss of brush borders, exposure of the basement membrane, and infiltration of the interstitium by inflammatory cells in proximal tubular epithelial cells, indicating obvious renal tissue damage (Figure 1A). Electron microscopy showed autophagic vacuoles engulfing abnormal mitochondria and other cytoplasmic components 1 day after CLP (Figure 1A). CLP affected the expression of autophagy-associated proteins, including LC3 and P62, in a time-dependent manner. The ratio of LC3-II/LC3-I protein levels peaked 1 day after CLP and then gradually decreased (Figure 1B), whereas P62 protein levels changed in the opposite manner. P62 protein levels were the lowest 1 day after CLP and increased at later times (Figure 1B). In addition, compared with the sham and normal control groups, the CLP group exhibited a gradual decrease in KL protein levels after CLP that reached the lowest levels 1 day after CLP and recovered at later times (Figure 1C). These data clearly indicate that CLP induces maximal autophagy activation and maximal KL reduction 1 day after CLP in mice.
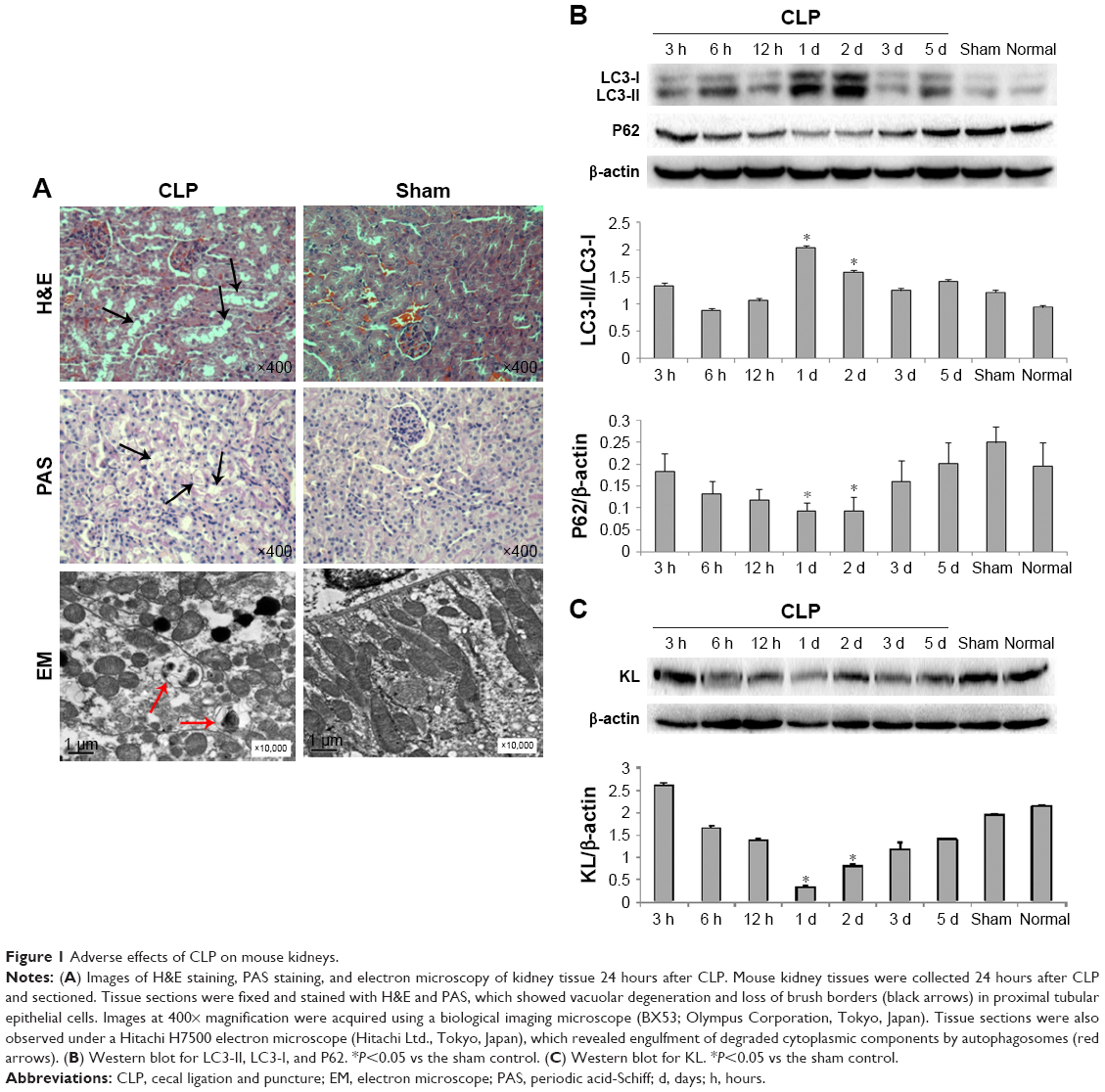 | Figure 1 Adverse effects of CLP on mouse kidneys. |
Recombinant KL alleviated CLP-induced renal dysfunction and damage without impacting autophagy and apoptosis
Compared with the no treatment control, treatment with KL alone (0.01 mg/kg) or Rap (1 mg/kg) alone significantly reduced Scr and BUN levels in mice with CLP, and KL+Rap further synergistically reduced Scr and BUN levels (all P<0.05, Figure 2A), suggesting that both Rap-mediated autophagy activation and KL may alleviate CLP-induced renal dysfunction. By contrast, Scr and BUN levels were significantly increased in the 3-MA (30 mg/kg), an autophagy suppressor, group compared with those in the no treatment control group, and KL reduced Scr and BUN levels only slightly in the presence of 3-MA (P<0.05, Figure 2A), suggesting that autophagy suppression by 3-MA may decrease CLP-induced renal dysfunction and block beneficial KL-mediated effects. H&E staining (Figure 2B) also showed that CLP-induced renal tissue damage was attenuated by KL alone, Rap alone, and KL+Rap, whereas 3-MA blocked the beneficial effects of KL.
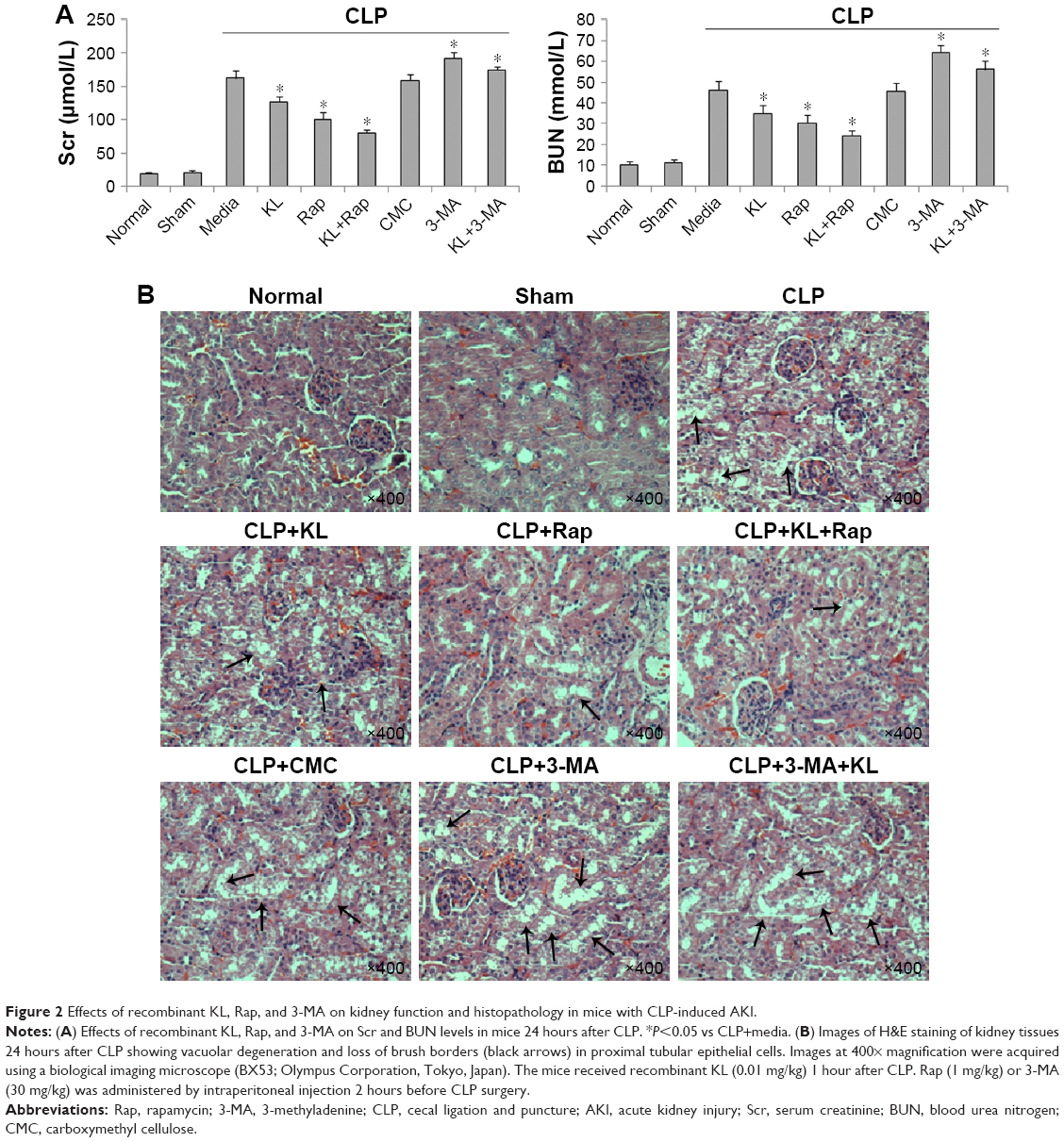 | Figure 2 Effects of recombinant KL, Rap, and 3-MA on kidney function and histopathology in mice with CLP-induced AKI. |
Compared with the sham control group, the CLP+media, CLP+KL, CLP+Rap, CLP+KL+Rap, and CMC groups exhibited a significant increase in the ratio of LC3-II/LC3-I levels and a significant reduction in P62 levels (all P<0.05, Figure 3A), suggesting that CLP stimulated autophagy. Notably, compared with CLP+media or CLP+KL, Rap significantly increased LC3-II/LC3-I (all P<0.05, Figure 3A), indicating further autophagy stimulation in addition to CLP-induced autophagy. The ratios of LC3-II/LC3-I and P62 levels in the CLP+3-MA and CLP+KL+3-MA groups were similar to those in the sham control group (Figure 3A). These data demonstrate that Rap activates autophagy and that 3-MA suppresses autophagy in mice with CLP, whereas KL does not further increase CLP-induced autophagy. Electron microscopy revealed obvious autophagy in the CLP, CLP+KL, CLP+Rap, and CLP+KL+Rap groups, with milder autophagy in the CLP+3-MA and CLP+KL+3-MA groups (Figure 3B).
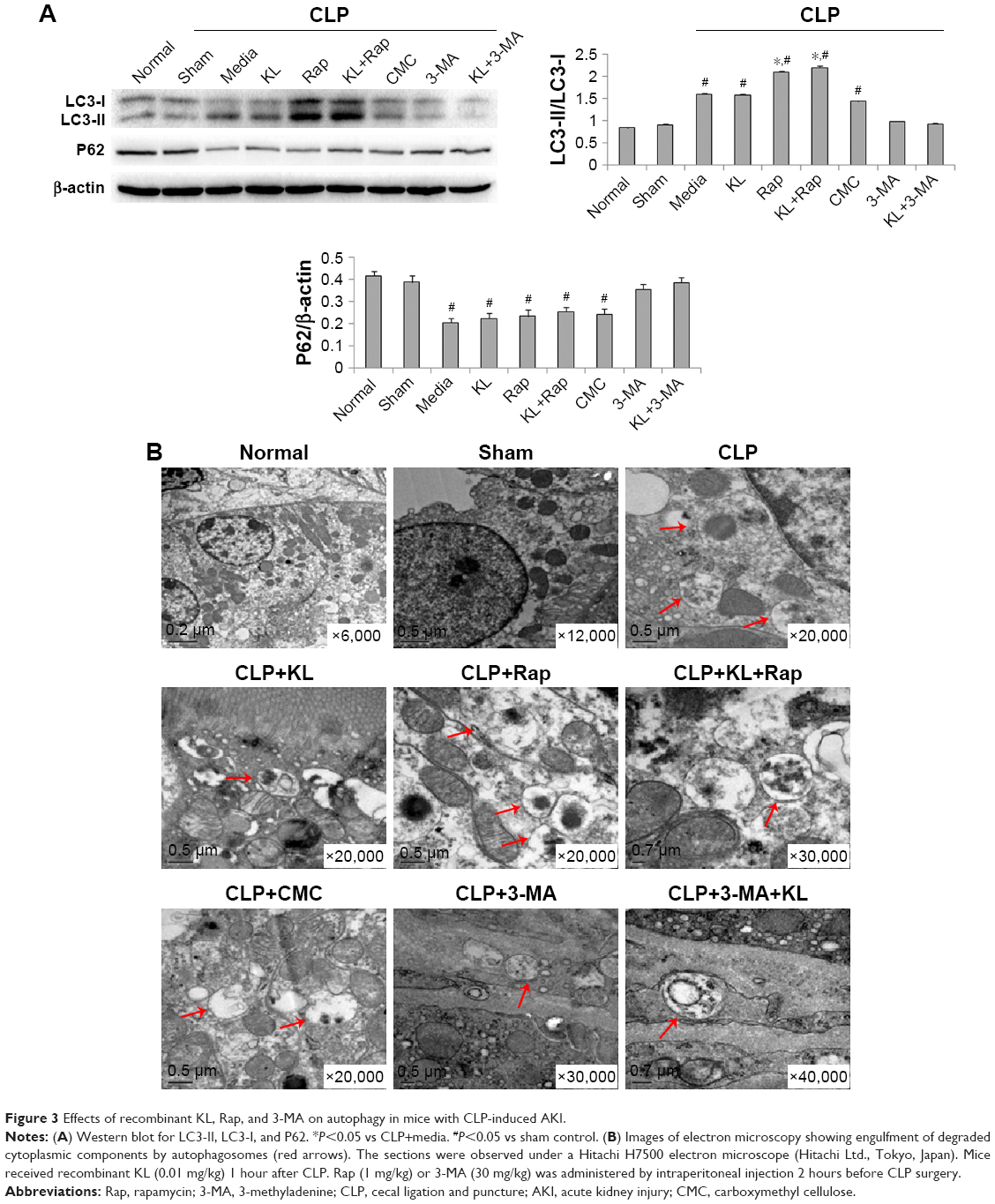 | Figure 3 Effects of recombinant KL, Rap, and 3-MA on autophagy in mice with CLP-induced AKI. |
In mice with CLP, compared with the no treatment control, treatment with KL alone, Rap alone, or KL+Rap significantly increased endogenous KL levels, whereas treatment with 3-MA alone or KL+3-MA did not affect endogenous KL levels (all P<0.05, Figure 4). In addition, CLP significantly increased CASP3 expression in the kidney (Figure 5). In mice with CLP, recombinant KL had no impact on CASP3 expression, Rap significantly reduced CASP3 levels, and 3-MA significantly increased CASP3 expression (all P<0.05, Figure 5). These data indicate that recombinant KL may not affect apoptosis in the kidneys of mice with CLP, whereas Rap may reduce apoptosis, and 3-MA may induce apoptosis.
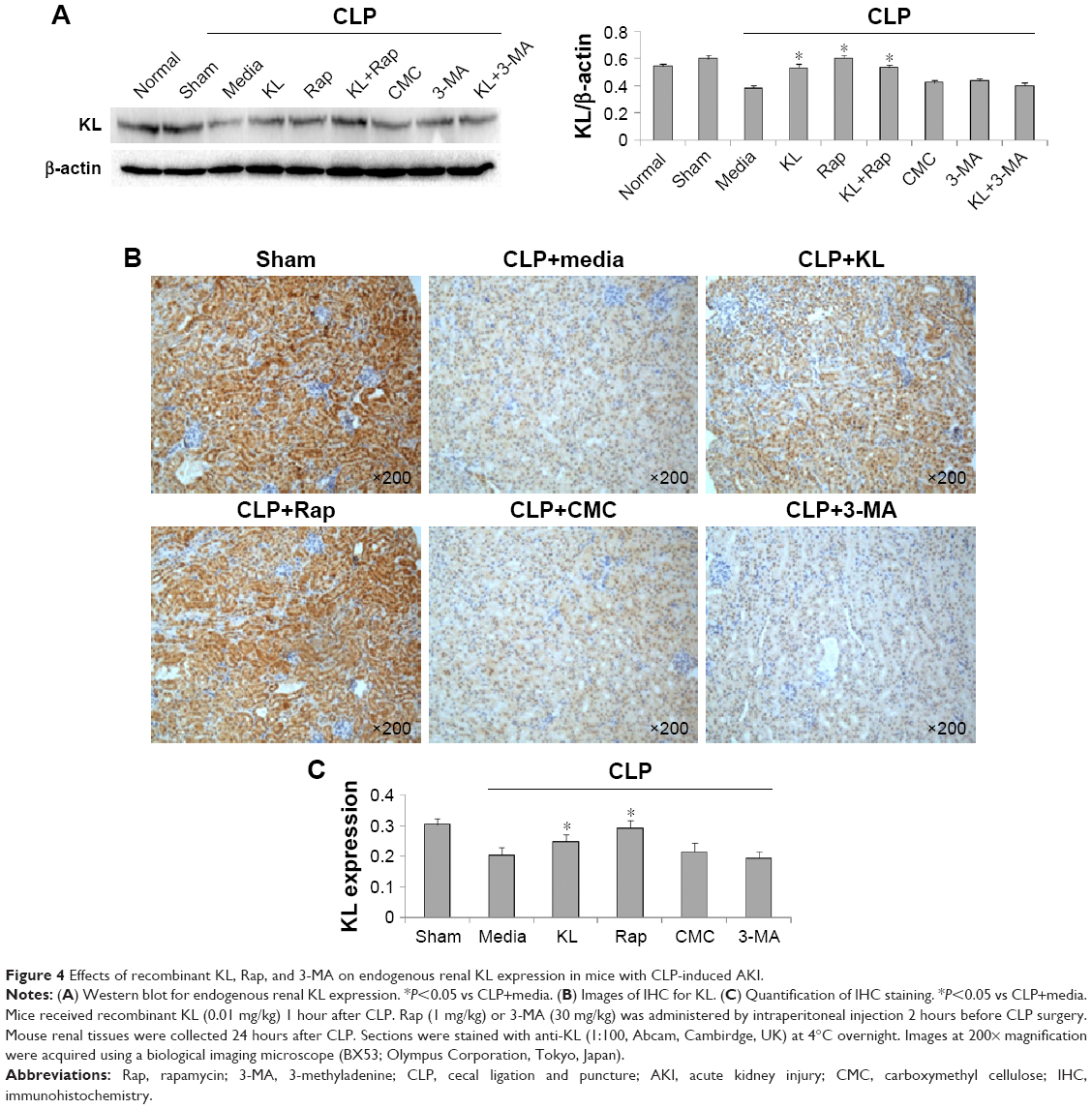 | Figure 4 Effects of recombinant KL, Rap, and 3-MA on endogenous renal KL expression in mice with CLP-induced AKI. |
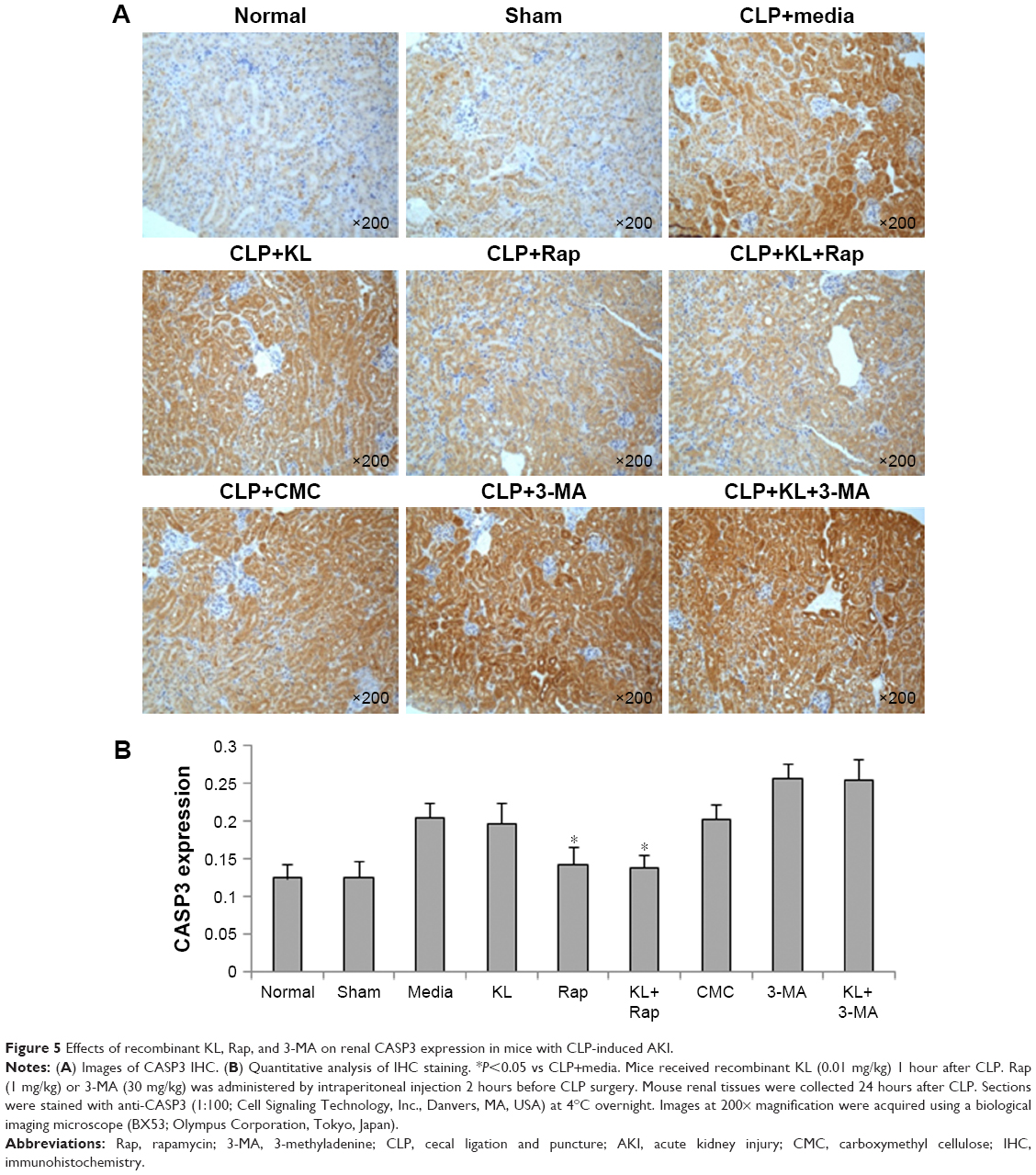 | Figure 5 Effects of recombinant KL, Rap, and 3-MA on renal CASP3 expression in mice with CLP-induced AKI. |
Recombinant KL did not appear to affect autophagy in LPS-treated HK-2 cells
To further investigate the association between autophagy and KL-mediated renoprotection, we treated HK-2 cells with LPS to induce autophagy. LPS reduced KL expression in HK-2 cells in a dose- and time-dependent manner. The dose response of HK-2 cells to LPS showed that 10 μg/mL LPS maximally reduced KL expression, increased the LC3-II/LC3-I ratio, and decreased P62 levels (Figure 6A). A time-course assay revealed that 24-hour treatment with 10 μg/mL LPS resulted in maximal effects (Figure 6B). Therefore, 24-hour treatment with 10 μg/mL LPS appears to maximally reduce KL expression while inducing autophagy in HK-2 cells.
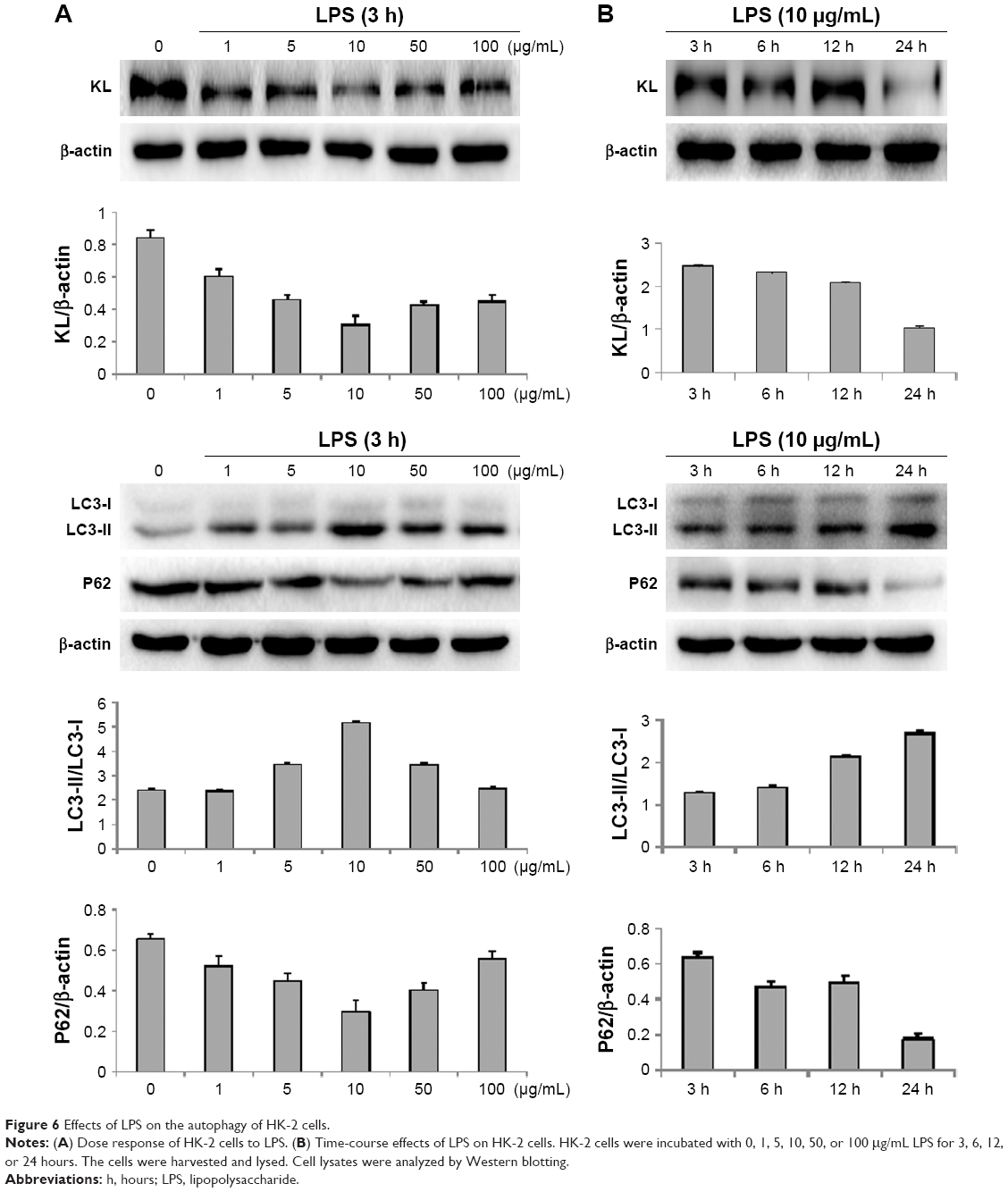 | Figure 6 Effects of LPS on the autophagy of HK-2 cells. |
Recombinant human KL (0.4 μg/mL) was added after HK-2 cells were incubated with LPS at 10 μg/mL for 1 hour, and the cells were then incubated for another 23 hours. Similar to the results from the animal model, recombinant KL did not further increase LPS-induced autophagy, whereas Rap significantly increased the LC3-II/LC3-I ratio and decreased P62 levels (Figure 7A). In addition, recombinant KL and Rap increased endogenous KL levels only slightly in LPS-treated HK-2 cells (Figure 7B). These findings indicate that exogenous recombinant KL may not further enhance LPS-induced autophagy and may not substantially affect endogenous KL expression in LPS-treated HK-2 cells. Furthermore, an apoptosis assay revealed that Rap significantly reduced apoptosis, whereas 3-MA increased apoptosis of LPS-treated HK-2 cells (all P<0.05, Figure 8). By contrast, recombinant KL did not affect apoptosis in LPS-treated HK-2 cells (Figure 8).
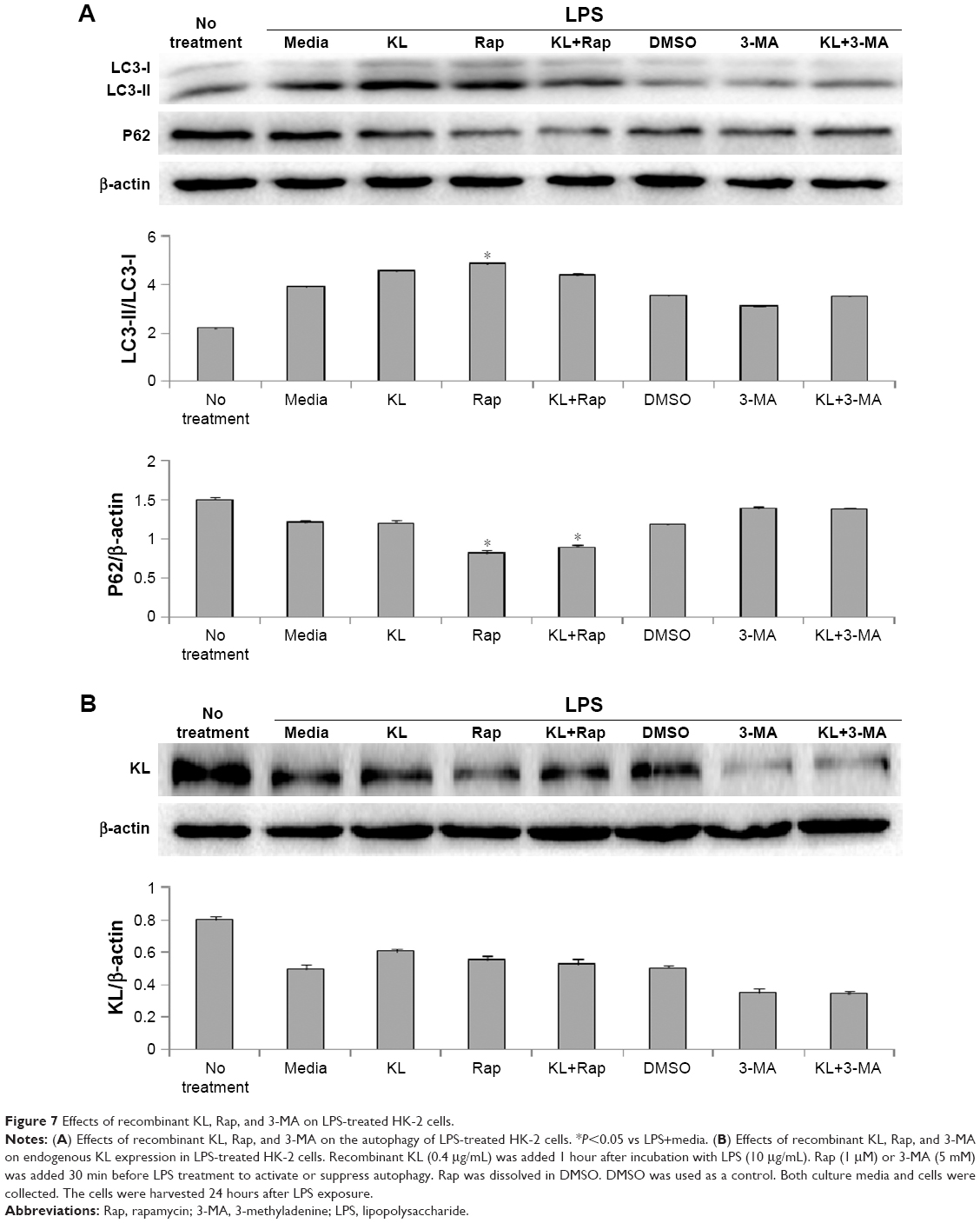 | Figure 7 Effects of recombinant KL, Rap, and 3-MA on LPS-treated HK-2 cells. |
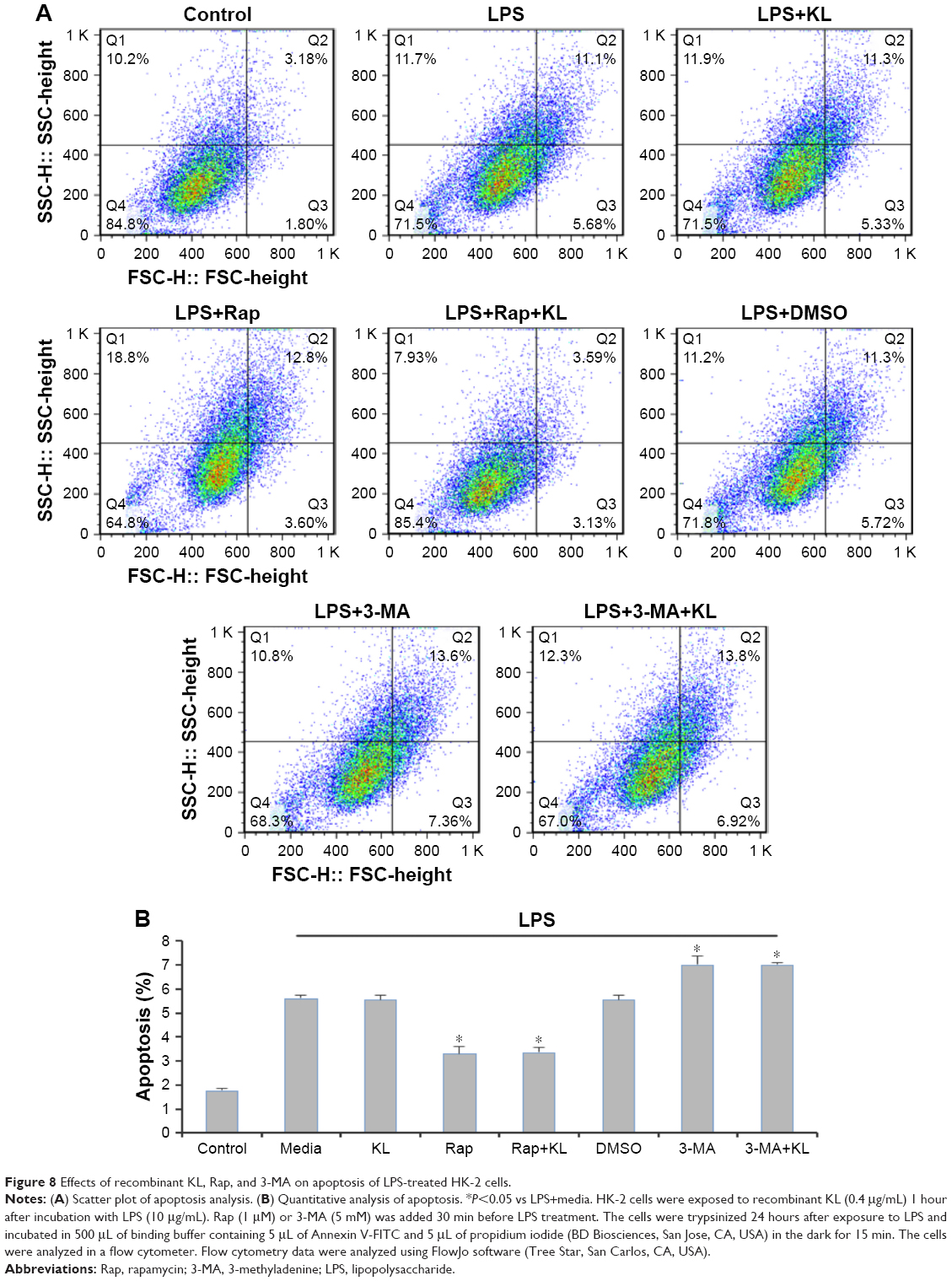 | Figure 8 Effects of recombinant KL, Rap, and 3-MA on apoptosis of LPS-treated HK-2 cells. |
Autophagy-related signal in HK-2 cells following different treatments
Following treatment with recombinant KL, Rap and 3-MA, the LC3-II fluorescence signal varied among the different groups. There were no marked changes in the HK-2 group. However, after the cells were exposed to LPS, the fluorescent signal exhibited the largest increase in the HK-2+LPS+Rap group and the smallest increase in the HK-2+LPS+3-MA group; the signal was comparable among the HK-2+LPS, HK-2+LPS+DMS and HK-2+LPS+KL groups (Figure 9).
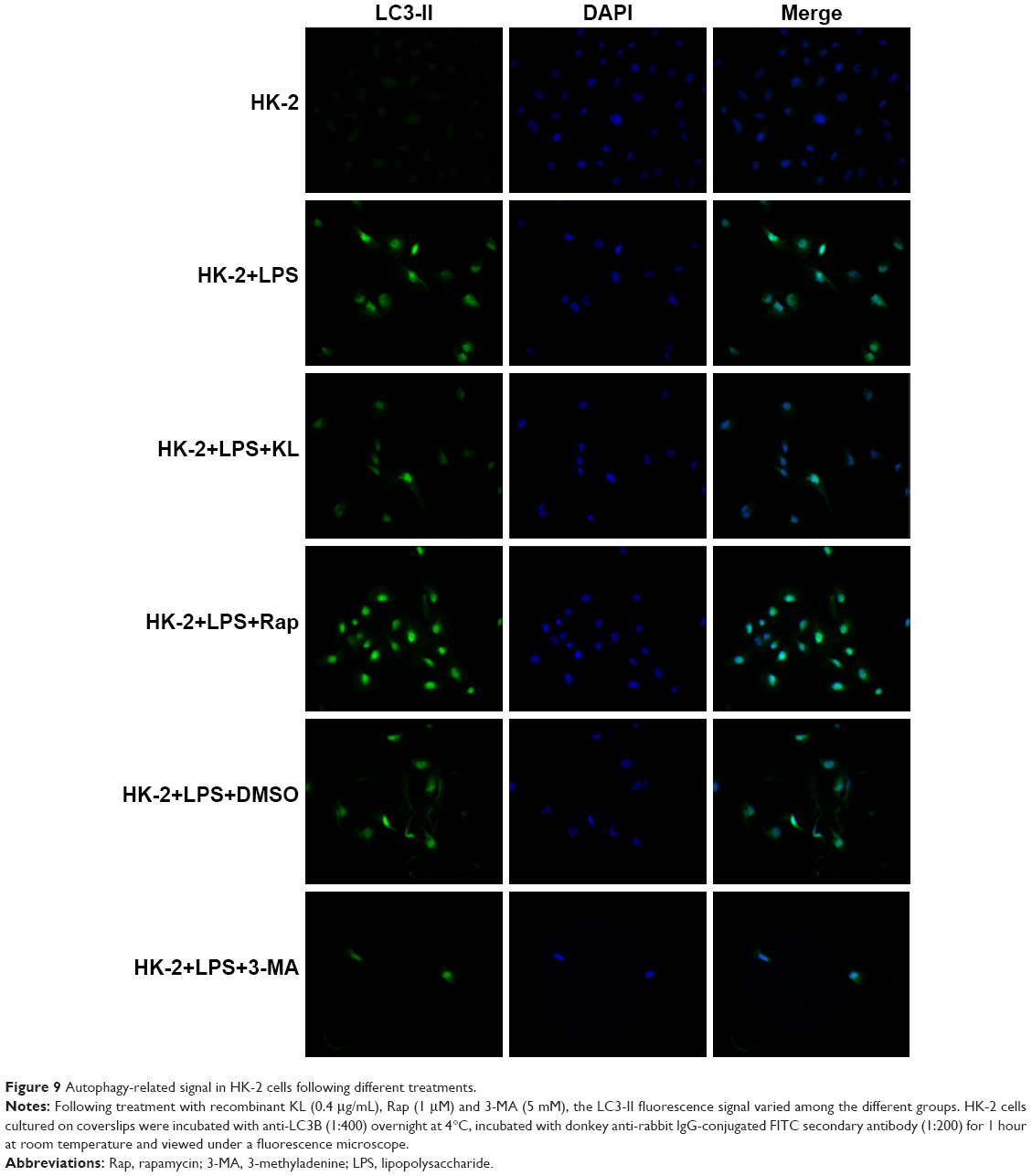 | Figure 9 Autophagy-related signal in HK-2 cells following different treatments. |
Discussion
The current study established a mouse model of CLP-induced AKI. In the mouse model, autophagy activation and KL protein reduction in the kidney reached maximal levels 24 hours after CLP. These CLP-associated adverse effects appeared to be acute and spontaneously partially resolved 5 days after CLP. A similar acute response in the kidney was observed in a rat model of IRI.17 Hu et al used temporary bilateral renal arterial occlusion to establish IRI-induced AKI and found that Scr levels were the highest, and KL protein expression was the lowest 24 hours after IRI induction, suggesting the development of AKI.17 Therefore, 24 hours after CLP may be an optimal time to investigate the molecular mechanisms underlying sepsis-induced AKI and test the effects of agents on sepsis-induced AKI. Here, we tested recombinant KL, Rap, and 3-MA.
In the current study, administration of recombinant KL reduced renal damage in the mouse model of CLP-induced AKI. The renoprotective effects of KL have been indicated in studies using other types of kidney injury animal models. In a rat model of doxorubicin hydrochloride-induced nephropathy, Sugiura et al demonstrated that erythropoietin-mediated attenuation of kidney damage was associated with increased KL expression in the kidney.19 KL-deficient transgenic rats show more severe kidney dysfunction and renal histological damage than wild-type mice after IRI, whereas transgenic rats overexpressing KL develop milder kidney damage after IRI.17 In addition, administration of recombinant KL to rats with IRI alleviates IRI-induced AKI.17
The molecular and cellular mechanisms underlying KL-mediated renoprotection remain unclear. Our results showed that exogenous recombinant KL and Rap significantly restored endogenous KL expression in mouse renal tissues. KL is a tumor suppressor in gastric cancer,29 and restoration of KL expression induces autophagy in gastric cancer and hepatocellular carcinoma cells.20,21 Therefore, in the current study, we hypothesized that KL might alleviate CLP-induced AKI by enhancing autophagy.
We observed maximal autophagy activation 24 hours after CLP. In a rat model of CLP-induced AKI, Hsiao et al also demonstrated that autophagy was activated 3 hours after CLP.30 Autophagy induction is also detected in kidney tissues during ischemia-reperfusion in mice.28,31 The role of autophagy in renal protection and recovery from AKI has been shown in previous studies. Kimura et al established proximal tubular epithelial cell-specific autophagy-deficient transgenic mice; compared with wild-type mice, transgenic mice exhibited obvious cellular hypertrophy and degeneration, developed more severe kidney dysfunction, and showed increased proximal tubule cell apoptosis during ischemia-reperfusion.22 Inhibition of autophagy by 3-MA enhances renal tubular cell apoptosis and deteriorates renal tissue damage, whereas stimulation of autophagy by Rap reduces apoptosis and tissue damage in mice during ischemia-reperfusion.28,31 Similarly, the current study also revealed that activation of autophagy by Rap improved kidney function, reduced apoptosis, and attenuated renal tissue injury, whereas suppression of autophagy by 3-MA exacerbated kidney dysfunction, increased apoptosis, and deteriorated renal tissue damage in mice with CLP-induced AKI. Our in vitro results also showed that activation of autophagy by Rap significantly reduced apoptosis and inhibition of autophagy by 3-MA-enhanced apoptosis in LPS-treated HK-2 cells. These findings clearly demonstrate that autophagy plays a protective role in AKI induced by CLP or ischemia-reperfusion.
Notably, our results did not show that KL attenuates CLP-induced AKI by enhancing autophagy. Administration of recombinant KL did not affect the expression levels of LC3-II, LC3-I, or P62 in the renal tissue of mice with CLP-induced AKI and LPS-treated HK-2 cells, indicating that recombinant KL may not enhance autophagy in our in vivo and in vitro AKI models. Therefore, the beneficial effects of KL on renal function in mice with CLP-induced AKI may not be associated with autophagy. Moreover, in both a mouse model in vivo and HK-2 cells in vitro, recombinant KL did not affect apoptosis.
Conclusion
In summary, the current study established a mouse model of CLP-induced AKI. The acute kidney responses, including renal dysfunction, renal KL expression reduction, and autophagy activation, were maximally affected 24 hours after CLP. Rap-mediated autophagy stimulation and 3-MA-mediated autophagy suppression reduced and exacerbated AKI in mice, respectively. Although recombinant KL attenuated renal dysfunction and increased endogenous KL expression in mice with CLP-induced AKI, it did not affect autophagy or apoptosis in the kidney. Therefore, KL-mediated renoprotection may be associated with mechanisms other than autophagy in sepsis-induced AKI.
Acknowledgments
This study was supported by the Zhejiang Province Natural Science Foundation (Grant No: LY16H150007, Xinxin Chen), the Zhejiang Province Medicine and Health Science and Technology Program (Grant No: 2016KYB188, Xinxin Chen), the Wenzhou Science and Technology Program (Grant No: Y20160311, Xinxin Chen) and the Key Construction Academic Subject (Medical Innovation) of Zhejiang Province (11-CX26).
Disclosure
The authors report no conflicts of interest in this work.
References
Soong J, Soni N. Sepsis: recognition and treatment. Clin Med (Lond). 2012;12(3):276–280. | ||
Chen LW, Chen W, Hu ZQ, et al. Protective effects of growth arrest-specific protein 6 (Gas6) on sepsis-induced acute kidney injury. Inflammation. 2016;39(2):575–582. | ||
Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351(2):159–169. | ||
Vincent JL, Atalan HK. Epidemiology of severe sepsis in the intensive care unit. Br J Hosp Med (Lond). 2008;69(8):442–443. | ||
Kuro-O M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. | ||
Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281(10):6120–6123. | ||
Kurosu H, Kuro-O M. The Klotho gene family as a regulator of endocrine fibroblast growth factors. Mol Cell Endocrinol. 2009;299(1):72–78. | ||
Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–774. | ||
Imura A, Tsuji Y, Murata M, Maeda R, et al. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007;316(5831):1615–1618. | ||
Li SA, Watanabe M, Yamada H, Nagai A, Kinuta M, Takei K. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct Funct. 2004;29(4):91–99. | ||
Imura A, Iwano A, Tohyama O, et al. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;565(1–3):143–147. | ||
Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310(5747):490–493. | ||
Hu MC, Shi M, Zhang J, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24(9):3438–3450. | ||
Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-O M, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A. 2008;105(28):9805–9810. | ||
Cha SK, Hu MC, Kurosu H, Kuro-O M, Moe O, Huang CL. Regulation of renal outer medullary potassium channel and renal K(+) excretion by Klotho. Mol Pharmacol. 2009;76(1):38–46. | ||
Kuro-O M. Klotho as a regulator of oxidative stress and senescence. Biol Chem. 2008;389(3):233–241. | ||
Hu MC, Shi M, Zhang J, Quinones H, Kuro-O M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010;78(12):1240–1251. | ||
Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309(5742):1829–1833. | ||
Sugiura H, Yoshida T, Mitobe M, Shiohira S, Nitta K, Tsuchiya K. Recombinant human erythropoietin mitigates reductions in renal klotho expression. Am J Nephrol. 2010;32(2):137–144. | ||
Shu G, Xie B, Ren F, et al. Restoration of klotho expression induces apoptosis and autophagy in hepatocellular carcinoma cells. Cell Oncol (Dordr). 2013;36(2):121–129. | ||
Xie B, Zhou J, Shu G, Liu DC, Zhou J, Chen J, Yuan L. Restoration of klotho gene expression induces apoptosis and autophagy in gastric cancer cells: tumor suppressive role of klotho in gastric cancer. Cancer Cell Int. 2013;13(1):18. | ||
Kimura T, Takabatake Y, Takahashi A, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22(5):902–913. | ||
Livingston MJ, Dong Z. Autophagy in acute kidney injury. Semin Nephrol. 2014;34(1):17–26. | ||
Guan X, Nie L, He T, et al. Klotho suppresses renal tubulo-interstitial fibrosis by controlling basic fibroblast growth factor-2 signalling. J Pathol. 2014;234(4):560–572. | ||
Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW 3rd, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24 (Suppl 1):52–57. | ||
Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4(1):31–36. | ||
Hu MC, Shi M, Cho HJ, et al. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int. 2013;84(3):468–481. | ||
Guan X, Qian Y, Shen Y, et al. Autophagy protects renal tubular cells against ischemia/reperfusion injury in a time-dependent manner. Cell Physiol Biochem. 2015;36(1):285–298. | ||
Wang L, Wang X, Wang X, et al. Klotho is silenced through promoter hypermethylation in gastric cancer. Am J Cancer Res. 2011;1(1):111–119. | ||
Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM, Hsu C. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37(3):289–296. | ||
Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176(3):1181–1192. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.