")
Back to Journals » Cancer Management and Research » Volume 12
KLHL22 Regulates the EMT and Proliferation in Colorectal Cancer Cells in Part via the Wnt/β-Catenin Signaling Pathway
Authors Song Y, Yuan H, Wang J, Wu Y, Xiao Y, Mao S
Received 3 March 2020
Accepted for publication 30 April 2020
Published 27 May 2020 Volume 2020:12 Pages 3981—3993
DOI https://doi.org/10.2147/CMAR.S252232
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Yi Song,1,* Huiping Yuan,2,* Jia Wang,3 Yuhe Wu,4 Yuhong Xiao,5 Shengxun Mao1
1Department of General Surgery, The Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, People’s Republic of China; 2Department of Gastrointestinal Surgery, Cangzhou Hospital of Integrated Traditional Chinese and Western Medicine, Cangzhou, Hebei 061000, People’s Republic of China; 3Radiotherapy and Chemotherapy Department, Cangzhou Hospital of Integrated Traditional Chinese and Western Medicine, Cangzhou, Hebei 061000, People’s Republic of China; 4Basic Medical College, Gannan Medical University, Ganzhou, JiangXi 341000, People’s Republic of China; 5Department of Rehabilitation Medicine, The Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shengxun Mao Email [email protected]
Background: Colorectal cancer (CRC) is one of the most common aggressive malignancies. KLHL22 functions as a tumor suppressor, and previous findings have demonstrated that KLHL22 can regulate the development of breast cancer and CRC. However, few studies have investigated the role of KLHL22 in CRC cell epithelial-to-mesenchymal transition (EMT) and proliferation. The current study aimed to detect the role of KLHL22 in CRC cell proliferation and EMT and to elucidate the probable molecular mechanisms through which KLHL22 is involved with these processes.
Materials and Methods: Transwell invasion, MTT, immunohistochemistry and Western blotting assays were performed to evaluate the migration, invasion and proliferation abilities of CRC cells, and the levels of active molecules involved in the Wnt/β-catenin signaling pathway were examined through Western blotting analysis. In addition, the in vivo function of KLHL22 was assessed using a tumor xenograft model.
Results: KLHL22 expression was weaker in CRC tissues than in nonmalignant tissues and could inhibit cell invasion, migration, and proliferation in vitro. Furthermore, the regulatory effects of KLHL22 on EMT were partially attributed to the Wnt/β-catenin signaling pathway. The in vivo results also showed that KLHL22 modulated CRC tumorigenesis.
Conclusion: KLHL22 can regulate the activity of GSK-3β to influence the level of PI3K, and this regulation promotes EMT inhibition partially through the Wnt/β-catenin signaling pathway.
Keywords: colorectal cancer, KLHL22, Wnt, EMT
Introduction
Colorectal cancer (CRC), the third most common type of malignant tumor worldwide, has resulted in a steady increase in cancer-related mortality in recent decades .1 Even after undergoing surgical R0 resection, many patients suffer from metastasis and postoperative recurrence after several years, and recurrence develops in metastatic niches in the liver, lungs, brain, bones and other tissues through lymphatic and hematogenous vessels and becomes the leading cause of death due to CRC.2 The targets of distal metastasis can influence the mortality rate of the patient, so in depth investigations about the molecular mechanisms that underlie CRC development are urgently needed.
Kelch-like family member 22 (KLHL22) plays a significant role in regulating the activation of mTORC1 and the downstream events in mammals and nematodes.3 Although few studies have examined the function of KLHL22 in cancer, the relationship between KLHL22 expression and tumorigenesis has been revealed in breast cancer, and a study conducted by Chen demonstrated that KLHL22 could promote tumorigenesis and aging through amino acid-dependent mTORC1 signaling in breast cancer cells.3 It has been reported that KLHL22 could regulate the ubiquitylation of PLK1 with Cullin 3 (CUL3) to influence the regulator of mitosis and control chromosome alignment.4 In addition, it could bind to or regulate some other kinases, such as Nek2 and CDK1, that influence kinetochore-microtubule attachment and other mitotic processes at multiple levels, which means that KLHL22 may regulate cancer cell progression.5 However, little is known regarding the function of KLHL22 in controlling CRC progression. Recently, we found that the levels of the protein encoded by KLHL22 are lower in clinical pathological tissue specimens from patients than in normal tissues. Therefore, we aimed to characterize the role of KLHL22 in CRC and the mechanisms by which KLHL22 influences tumors.
Epithelial-to-mesenchymal transition (EMT) is a physiological process that is a crucial event in numerous developmental processes, but it could trigger cancer cell metastasis and invasion by causing cells to lose their epithelial characteristics and adhesive contacts with their neighboring cells.6–8 The most prominent characteristics of an EMT event are decreasing expression of epithelial markers, such as E-cadherin and tight junction proteins, and the acquisition of mesenchymal markers, such as N-cadherin and vimentin. EMT can be mediated by a network of EMT-inducing transcriptional factors, such as Snail1/2, Twist1 and ZEB1/2.9,10 Some studies have indicated that EMT and invasive cancer formation can be regulated through signaling pathways, such as Notch, Wnt/β-catenin, TGF-β and NF-kB.11 The canonical Wnt signaling pathway, a critical mediator of tissue homeostasis and repair, is a key signaling cascade in the regulation of both carcinogenesis and EMT.12 Cytoplasmic β-catenin can be transported from the cytoplasm to the nucleus and can regulate the transcription of target genes linked to the induction of EMT, which depends on the accumulation of the extracellular Wnt ligand and the function of the “APC/Axin-2/GSK-3β complex”.13–15 Moreover, nuclear β-catenin can induce the transcription of cyclin D1 and C-myc, which are involved in proliferation.16
In the present study, we examined the efficacy of KLHL22-induced inhibition of CRC cell EMT and proliferation in vitro. We further assessed the tumor migration and invasion mechanisms of KLHL22 by decreasing the activity of the Wnt/β-catenin signaling pathway in CRC. The results of these experiments should provide novel insights into the effects of the KLHL22 and Wnt/β-catenin signaling pathways on CRC therapy.
Materials and Methods
RNA Sequencing Data and Bioinformatics Analysis
Gene expression data with clinical information from the colon adenocarcinoma (COAD) projects (490 cases, workflow type: HTSeq-Counts) were collected from TCGA. The exclusion criteria were normal COAD samples and an overall survival of <30 days. Then, level 3 HTSeq-Counts data were transformed into transcripts per million reads (TPM) for the following analyses. The TPM data for 430 patients with COAD were used for further analyses. Unavailable or unknown clinical features in 362 patients were regarded as missing values, and the data contained 321 colorectal cancer tissue samples and 41 normal tissue samples. Because the data were downloaded freely from the TCGA database, approval of the ethics committee of Nanchang University was not needed.
Gene Set Enrichment Analysis (GSEA)
GSEA (http://www.broadinstitute.org/gsea/index.jsp) was used to investigate the mechanisms related to KLHL22 expression in CRC patients. The 362 colorectal cancer samples from TCGA-COAD were divided into low and high expression groups by the median expression of KLHL22. One thousand permutations for gene sampling were utilized to verify statistical significance and to ensure the accuracy of the results. The significantly different inclusion was normalized with a P<0.05 and a false discovery rate (FDR) <25%. GSEA was conducted based on two groups, and significantly enriched pathways related to malignant cancer development were chosen according to the normalized enrichment score (NES). Correlative biological processes, cellular components and molecular functions were determined.
Human Tissue Samples and Cell Lines
In our study, we collected tissue samples from 80 colorectal cancer patients (40 males and 40 females over 65 years old) who came from the Second Affiliated Hospital of Nanchang University (Nanchang, China). Before starting this study, we have informed all patients about the use of human specimens in the study and obtained consent from each patient. All the patients comprehend it and signed paper informed consent.In addition, the ethics committee of the Second Affiliated Hospital of Nanchang University also provided consent for the study. Cell lines were obtained and authenticated from Shanghai Cell Biology (Shanghai, China). All cell lines were tested and were negative for mycoplasma contamination. HCT116, SW480 and LOVO cells were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS) (Solarbio, Beijing, China) and maintained at 37 °C in a humidified atmosphere with 5% CO2. In this experiment, we used TRIzol Reagent (Thermo Fisher, New York, USA) to extract the total RNA from tissue samples and cultured cells and utilized the PrimeScript RT kit (Vazyme, Nanjing, China) and SYBR Premix Ex Taq (China, Chengdu Atomic Energy) to perform quantitative RT-PCR. The following primer sequences were used: KLHL22 Forward) 5ʹ-AAGGGATCTTGTGGCTGT-3ʹand(reverse) 5ʹ-TGTTGTTCACGCAGTGTGG-3ʹ.
Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded patient-derived tumor specimens were collected, and specimens were deparaffinized and stained with an antibody against KLHL22. An anti-KLHL22 rabbit polyclonal antibody (1:150, Thermo Fisher, USA) was coated onto the prepared sections and placed at 4 °C overnight; then, the samples were washed three times with phosphate-buffered saline (PBS) and placed in the corresponding primary secondary antibody for 1 hour. Next, the peroxidase reaction was performed by incubating the samples with 3,3ʹ-diaminobenzidine (DAB) (DAKO, Shanghai, China), and the tissue sections were stained with hematoxylin. The IHC intensity was scored as 0 (no staining), 1 (weak staining), 2 (moderate staining), or 3 (strong staining).
Western Blot Analysis
The collected tissues and cultured cells were lysed with RIPA lysis buffer (CWBIO, Beijing, China), and the supernatant was collected by centrifugation and stored at −20 °C until use. Protein concentrations were determined by a BCA Protein Assay Kit. Proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and blotted with antibodies recognizing KLHL22 (1:1000, Thermo Fisher, US), Twist1 (1:1000), Snail1 (1:1000), LEF (1:1000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), GSK-3β (1:1000), β-catenin (1:1000), vimentin (1:1000), E-cadherin (1:1000) and N-cadherin (1:1000) (Proteintech, Hubei, China). An antibody against GAPDH (1:5000) was used as a loading control. Following primary antibody incubation, membranes were washed three times with TBST, and the appropriate horseradish peroxidase-conjugated secondary antibody was added for a 2-hour incubation at room temperature. Membranes were developed using an ECL Western blotting detection reagent (Proteintech, Hubei, China) and were exposed to radiographic film.
Gene Knockdown and Overexpression Assays
We purchased all lentiviruses containing short hairpin RNA (shRNA) specific for KLHL22 or GSK-3β and lentiviruses overexpressing KLHL22 and GSK-3β RNA from GeneChem, Inc. (Shanghai, China). All cells were maintained in RPMI-1640 (Sigma), supplemented with 10% (v/v) fetal calve serum, 100 U/mL penicillin and 100 mg/mL streptomycin in a humidified atmosphere of 5% CO2 at 37°C。Preliminary transfected cells were repeatedly screened with G418 to obtain stable transfected homologous cell lines. HCT116 and SW480 cells were infected with lentivirus to produce stable KLHL22-silenced cell lines, while LOVO cells were infected with lentivirus to construct stable KLHL22-enhanced cell lines. Transfection reagent was used for all virus production, and infection was carried out with polybrene. Target KLHL22 shRNA sequences: forward primer, 5ʹ-TGTAAGGGATCTTGTGGCTGT-3ʹ and reverse primer, 5ʹ-GTGTTGTTCACGCAGTGTGG-3ʹ. Target GSK-3β shRNA sequences: forward primer, 5ʹ-ACCCATTTCTGCCCTCCAAG-3ʹ and reverse primer: 5ʹ-AGCGAGCGATGTATTTCTCCTT3ʹ.
Invasion and Migration Assay
Invasion and migration assays were performed using transwell plates (8 µm; Corning, Inc.). In brief, the cells were suspended in serum-free medium and incubated in the upper transwell chambers coated with or without Matrigel basement membrane matrix (BD, Biosciences, USA), and medium containing 10% FBS was placed as a chemical attractant in the bottom of the chamber. After incubation for 24 hours, the remaining cells were fixed with 4% methyl alcohol for 20 minutes, stained with 0.1% crystal violet and observed via optical microscopy.
Cell Scratch Assays
Cells were cultured in 6-cm Petri dishes at a concentration of 1 x 106 cells per well and incubated for 12 hours to ensure 100% confluence. The next day, a scratch was made in each well using a needle, and then, the cells were washed twice with PBS and incubated in serum-free medium. Typical images were obtained via light microscopy immediately after the scratches were made (0 hour), and the scratches were subsequently imaged every 12 hours thereafter.
Immunofluorescence
We detected the expression of the E-cadherin and Twist1 proteins in scramble and lentivirus-infected cells by immunofluorescence. First, the cells were grown on cell creeping slides to 50–60% saturation for 24 hours. Afterwards, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 (Beyotime, Zhenjiang, Jiangsu, China) and blocked with 10% goat serum at room temperature for 30 minutes. One of the following antibodies was added and incubated at 4 °C overnight: anti-E-cadherin (1:200) and anti-Twist1 (1:200) (China Hubei Protein Technology Co, Ltd.). After the primary antibody incubation, cell creeping slides were incubated with a secondary antibody (DyLight 488-conjugated mouse anti-rabbit IgG; 1:200) at room temperature for 2 hours. Finally, cell nuclei were counterstained with DAPI, and we obtained images using a fluorescence microscope (Olympus Corporation, Tokyo, Japan).
In vivo Tumorigenesis
All animal experiments were performed according to protocols supported by the Second Affiliated Hospital of Nanchang University and obeyed the National Institutes of Health guidelines for the use of experimental animals.(Approval No.): 2019060715.In our study, eight-week-old male athymic BALB/c nude mice were raised. HCT116 and SW480 cells transduced with KLHL22 shRNA or normal HCT116 and SW480 cells (1 x 10^6 cells per mouse) were subcutaneously inoculated into nude mice. Another group of nude mice was infected with treated LOVO cells with KLHL22 overexpression or normal LOVO cells in the same way. After 8 weeks, the mice were humanely euthanized and the subcutaneous tumor tissues were extracted. Tumor volume was measured every week using the formula: volume =2 ×b/2, where a is the major diameter and b is the minor diameter vertical to a.
Statistical Analysis
The data are expressed as the mean ± standard deviation of at least three independent experiments that were performed in triplicate. The data were analyzed using a Mann–Whitney U-test with Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). We also used SPSS 17.0 software (SPSS, Inc., Cary, NC, USA) for data analyses. Significant differences were considered at p < 0.05 (*).
Results
Unlike Matched Adjacent Normal Tissues, Colorectal Cancer Tissues Negatively Regulate KLHL22 (Figure 1A–E)
The level of the KLHL22 protein in a multitude of CRC tissues was assessed by IHC, as shown by the figure: KLHL22 showed weak staining in tumor tissues and exhibited strong staining in matched adjacent noncancerous tissues. As a result, the expression of KLHL22 in CRC tumor tissues was significantly decreased compared with that in nontumor tissues (1A). In addition to measuring the protein levels in tumor tissues and their corresponding normal tissues, we used Western blotting, and the results were similar to those of IHC (Figure 1B). To further investigate the role of KLHL22 in colorectal tumors and subsequent experiments, we selected three CRC cell lines (LOVO, SW480, and HCT116) to examine the expression of KLHL22 in each cell line and found that the LOVO cell line exhibited weak KLHL22 expression, whereas the SW480 and HCT116 cell lines exhibited high KLHL22 expression (Figure 1C). To investigate the role of KLHL22 in CRC progression, we first evaluated the mRNA expression level of KLHL22 in TCGA, and the results showed that KLHL22 expression was significantly lower in CRC than in normal tissues (p< 0.05) (*) (Figure 1D). Besides, the expression of KLHL22 in 160 CRC tissues and matched adjacent normal tissues was detected by qRT-PCR, as is shown that KLHL22 expression level in CRC tissues was lower than that of adjacent normal tissues, of which showed statistically significant difference. (Figure 1E)
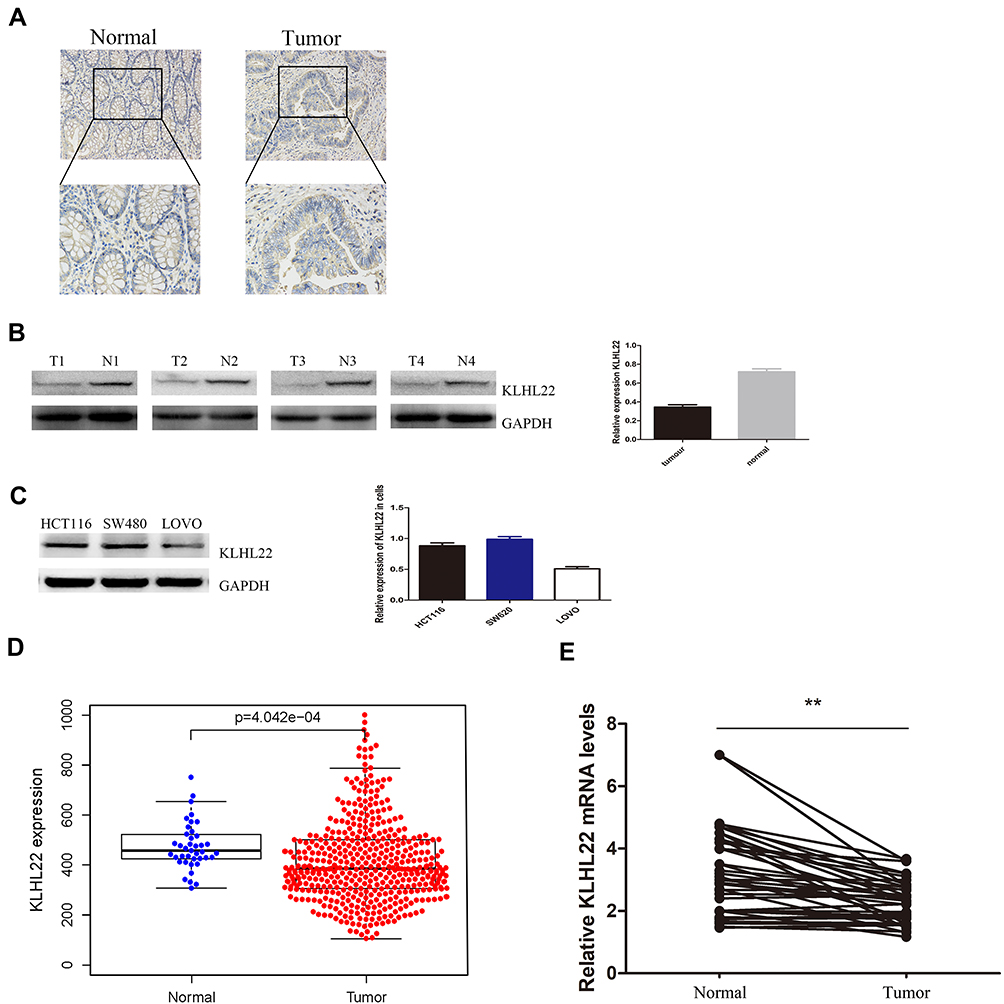 |
Figure 1 KLHL22 expression in CRC tissues and three CRC cell lines (LOVO, HCT116, and SW480). (A) Immunohistochemical staining of KLHL22 in CRC tissues. Original magnification, ×200, ×400; bar = 100 um. KLHL22 protein is lower expression in tumor tissue than in adjacent normal tissue. (B) KLHL22 protein expression in representative samples of CRC tissues and paired adjacent non-cancerous tissues. KLHL22 is significantly decreased in cancerous tissues than to adjacent non-cancer tissues. (C) KLHL22 protein expression in three CRC cell lines. In the LOVO cell lines, KLHL22 protein expression was lower than in the HCT116 and SW480 cell lines. (D) Gene set enrichment analysis for high KLHL22 expression group and low KLHL22 expression group. (E) KLHL22 mRNA expression levels in 160 CRC tissues and paired non-cancerous tissues, calculated using the 2—ΔΔct method. **p < 0.01, Mann–Whitney U-test. In the CRC tissues, KLHL22 mRNA expression levels were lower than in the corresponding normal tissues. |
Therefore, we silenced KLHL22 expression in the HCT116 and SW480 cell lines by transfecting the cells with shRNA constructs. In addition, lentiviral vectors were constructed to enhance the expression of KLHL22 in the LOVO cell line. Therefore, we established the stable KLHL22 knockdown cell lines (HCT116-shRNA and SW480-shRNA) and stable KLHL22 overexpression (LOVO-KLHL22) cell line for use in the following experiments.
GSEA in Two Independent Groups with Different KLHL22 Levels. (Figure 2)
To further explore the function of KLHL22 in affecting the prognosis of colorectal cancer patients, GSEA was performed in TCGA-COAD samples. These samples in the high KLHL22 expression group were enriched in the cell cycle.
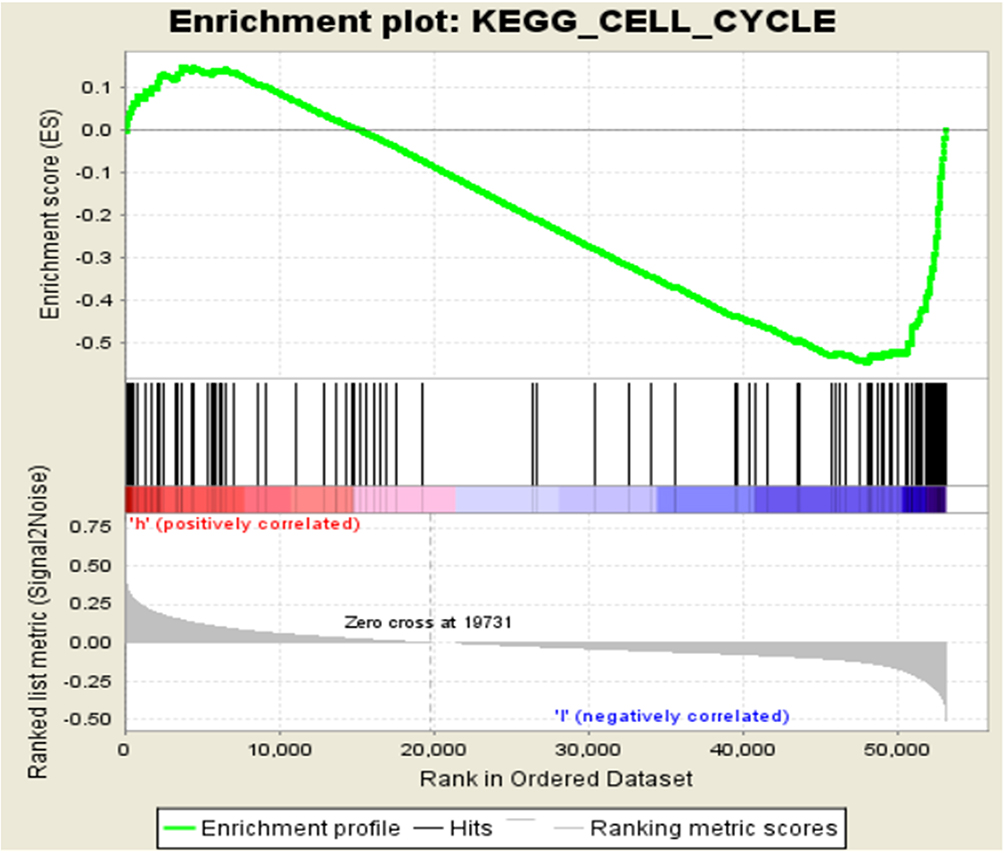 |
Figure 2 The relative mRNA expression of KLHL22 in TCGA cohort (normal=41, CRC=321). |
KLHL22 Knockdown Encourages CRC Invasion in vitro, Whereas KLHL22 Overexpression Inhibits CRC Invasion (Figure 3)
To determine whether KLHL22 expression restrains cell proliferation capacity, we examined the proliferation of transduced and nontransduced cells using cell migration and invasion. The results revealed that the migratory and invasion capacities were enhanced in cells when KLHL22 expression was suppressed (Figure 3A and B). And that KLHL22 overexpression decreased cell migratory and invasion (Figure 3C) (p < 0.05).We also performed wound scratch assays, and the results showed that the KLHL22 knockdown cell lines exhibited enhanced migration (Figure 3D and E) and KLHL22-overexpressing cells exhibited decreased migration (Figure 3F). Representative fields of the wound-healing process were monitored at 0 and 12 hours (p < 0.01).In general, alterations in the cell migration and invasion capacity indicate that EMT is induced in response to the downregulated expression of KLHL22 in CRC cells.
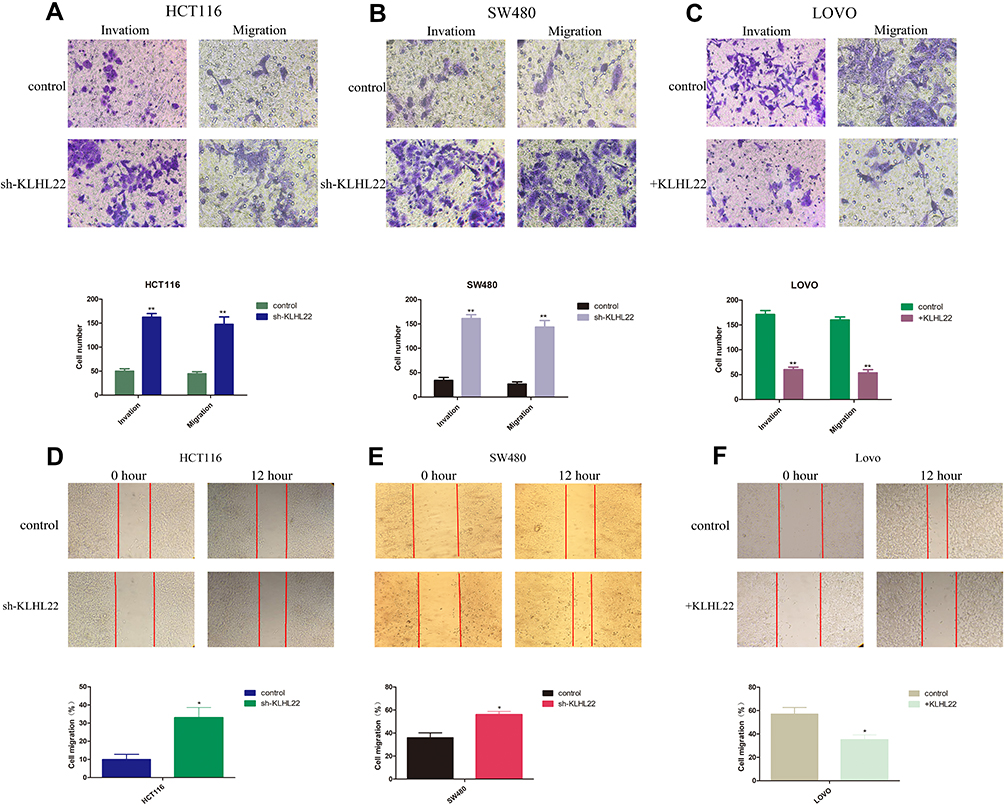 |
Figure 3 KLHL22 acts as a tumor inhibitor affecting CRC migration and invasion. Migration and invasion were demonstrated in three CRC cell lines that were transduced as the following pictures. (A, B) The results revealed that the migratory and invasion capacities were enhanced in cells when KLHL22 expression was suppressed. (C) And that KLHL22 overexpression decreased cell migratory and invasion. (p < 0.05). (D, E) Migration was measured using a cell scratch assay. HCT116 and SW480 cells transduced with KLHLL22-shRNA migrated more faster than scramble control cells. (F) LOVO cells with +RHBDF1 migrated more slowly than normal cells or scramble control cells. (*p<0.05,**p < 0.01 by Student’s t-test.). |
Increased KLHL22 Expression Contributes to the Inhibition of EMT in CRC Cells (Figure 4+Figure 5)
The transwell assay suggested that KLHL22 deregulates cell migration and invasion in CRC cells, indicating a link between KLHL22 expression and EMT. Herein, we further detected the levels of EMT markers, including the epithelial marker E-cadherin and the mesenchymal markers N-cadherin, vimentin, Twist1 and Snail1. We measured the levels in all transduced and nontransduced cells by Western blotting. As shown in the Figure 4, KLHL22 knockdown resulted in a dramatic decrease in the E-cadherin level while contributing to a statistically significant increase in the vimentin, N-cadherin, Twist1 and Snail1 level (Figure 4A and B). In contrast, in cells in which KLHL22 expression was upregulated, the expression levels of vimentin, N-cadherin, Twist1 and Snail1 were decreased, and the expression level of E-cadherin was increased (Figure 4C). (p < 0.05). From these results, we can conclude that the upregulation of KLHL22 could inhibit EMT in CRC, while in contrast, the downregulation of KLHL22 promotes EMT. Through these experimental results, we initially linked the expression of KLHL22 to the EMT process.
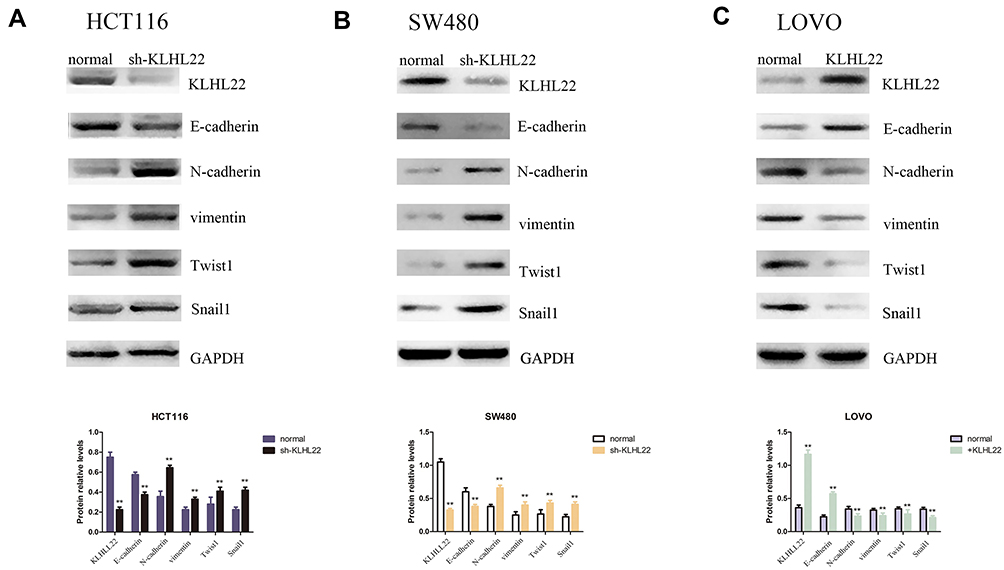 |
Figure 4 Cells after transduction with KLHL22 shRNA or +KLHL22 lentivirus to regulate the expression of EMT markers. We performed a Western blotting assessment of E-cadherin, N-cadherin, vimentin, Twist1 and Snail1 protein expression. (A, B) The results showed that the expression levels of N-cadherin, vimentin, Twist1 and Snail1 were increased and that E-cadherin expression was reduce when HCT116 and SW480 cells KLHL22 gene silenced by Lentivirus. (C) However, when KLHL22 protein was overexpressed in LOVO cells, the expression levels of N-cadherin, vimentin Twist1 and Snail1 decreased, and the expression levels of E-cadherin increased. GAPDH was used as the loading controls. (**p < 0.01 by Student’s t-test). |
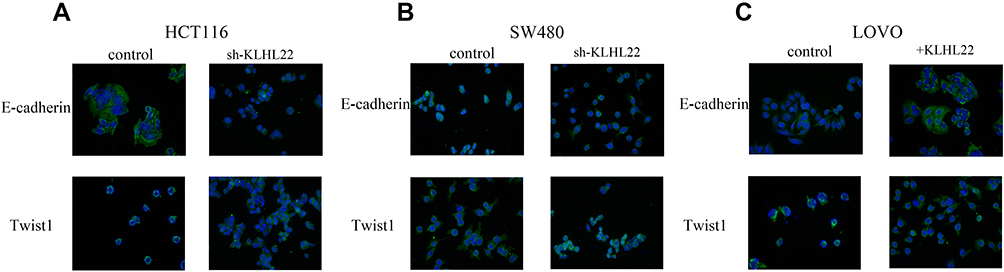 |
Figure 5 Immunofluorescence staining was performed to detect E-cadherin and Twist1 expression in cells after transduction with KLHL22 shRNA or +KLHL22 lentivirus, and images were compared with those of normal cells. (A, B) In the HCT116 and SW480 cells, there by enhance EMT, KLHL22 knockdown suppressed E-cadherin expression and increased Twist1 expression. (C) In the LOVO cells, KLHL22 overexpression increased E-cadherin expression and suppressed Twist1, through the same method. |
To reinforce our hypothesis, we examined the expression of E-cadherin and Twist1 in treated or untreated cell lines via immunofluorescence. The results are shown in Figure 5. Downregulation of KLHL22 expression in the HCT116 and SW480 cell lines resulted in a decrease in E-cadherin and an increase in Twist1 expression (Figure 5A and B). Inversely, KLHL22 overexpression in the LOVO cell line inhibited Twist1 expression and elevated E-cadherin expression (Figure 5C). Therefore, we further confirmed that KLHL22 is required for the physiological process of EMT and that KLHL22 plays a significant role in CRC EMT.
The Regulatory Effects of KLHL22, Which Promotes EMT, are Attributable to the Wnt/β-Catenin Signaling Pathway (Figure 6)
It is known from previous literature reports that the Wnt/β-catenin signaling pathway is a classical signaling pathway that induces EMT and cancer metastasis. Combined with the experimental results obtained from the previous experiments, we considered that KLHL22 could restrain tumor cell growth and migration through the Wnt/β-catenin pathway and could play a crucial role in the EMT process of CRC cells. To verify this possibility, we examined the expression of β-catenin in three cell lines using Western blotting. The results showed that downregulation of KLHL22 increased β-catenin expression in constructed cells (Figure 6A and B). In contrast, β-catenin protein levels were downregulated in cell lines treated with upregulated KLHL22 (Figure 6C). It is widely acknowledged that GSK-3β and LEF play an important role in Wnt/β-catenin signaling. In general, β-catenin was maintained at a relatively low level in cells with sturdy KLHL22 expression. Subsequently, we examined the activities of GSK-3β and LEF by Western blotting. As we hypothesized, the β-catenin and LEF levels were significantly increased in cells with downregulated KLHL22 expression. Moreover, in cells with upregulated KLHL22 expression, β-catenin and LEF expression was inhibited; interestingly, GSK-3β expression was increased (P<0.05). Therefore, we concluded that the regulatory effects of KLHL22, which inhibit EMT, are attributable to the Wnt/β-catenin signaling pathway.
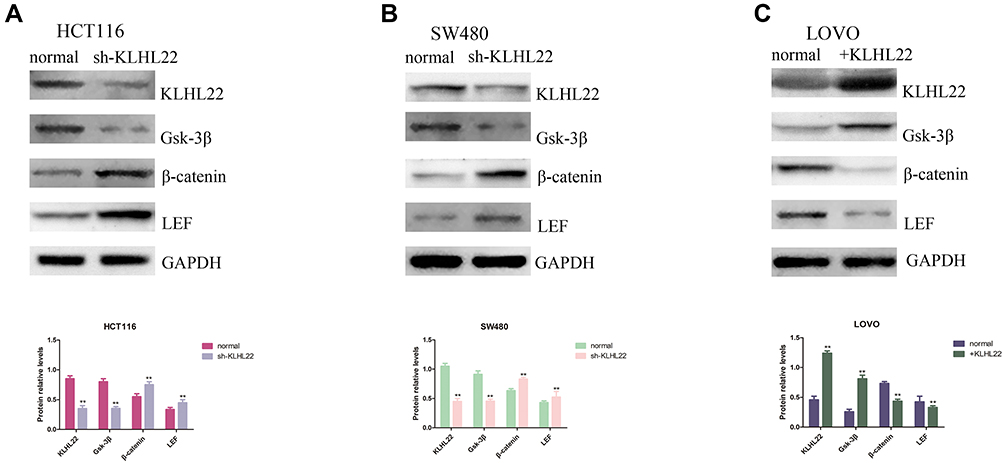 |
Figure 6 KLHL22 regulates the expression of GSK-3β markers. Through Western blotting, we assessed GSK-3β, β-catenin and LEF expression. (A, B) We show that the GSK-3β expression levels were reduced and that the β-catenin and LEF protein expression levels increased when the gene of KLHL22 knockdown in the HCT116 and SW480 cells. (C) The GSK-3β expression levels increased, but the β-catenin and LEF expression levels were reduced when the gene of KLHL22 overexpression in the LOVO cells. GAPDH was used as the loading controls. (**p < 0.01 by Student’s t-test.). |
In vitro Effects of GSK-3β Overexpression or Silencing in Cell Lines with KLHL22 Upregulation or Knockdown (Figure 7)
Through the above experiments, we have identified the tumor suppression function of KLHL22, further investigation is needed to address the mechanism by which KLHL22 influences tumor angiogenesis through the Wnt/β-catenin signaling pathway. It is known from previous reports that β-catenin is degraded by a complex consisting of APC, GSK-3β and Axin-2 and that GSK-3β is an important component of the β-catenin degradation complex. Therefore, we selected GSK-3β to test the underlying mechanism of KLHL22. As shown in the figure, KLHL22-shRNA HCT116 and SW480 cells led to enhanced GSK-3β expression, which promoted the degradation of β-catenin and thus inhibited the Wnt/β-catenin signaling pathway. Thus, GSK-3β was blocked or overexpressed in the stable KLHL22-expressing and KLHL22-knockdown cell lines, respectively, and affected the expression of proteins containing β-catenin, Twist1, LEF, Snail1, and E-cadherin. When KLHL22 was knocked down and GSK-3β was upregulated, the expression levels of β-catenin, Twist1, LEF and Snail1 marginally decreased, and the expression levels of E-cadherin increased; however, as GSK-3β was knocked down, the relative expression of β-catenin, Twist1, LEF, and Snail1 increased and that of E-cadherin significantly decreased (Figure 7). Moreover, the results revealed that the expression levels of downstream genes, namely, β-catenin, Twist1, LEF, and Snail1, were increased and that of E-cadherin was decreased following GSK-3β knockdown in the KLHL22-overexpressing LOVO cells (Figure 7C). In contrast, after upregulation of GSK-3β expression, downstream gene expression was reduced in KLHL22-silenced HCT116 and SW480 cells (Figure 7A and B). Based on the results of these experiments, we believe that KLHL22 silence, which promotes GSK-3β degradation, led to influence the level of β-catenin, Twist1, LEF and Snail1. In conclusion, we demonstrated that KLHL22 may regulate the level of GSK-3β in vitro.
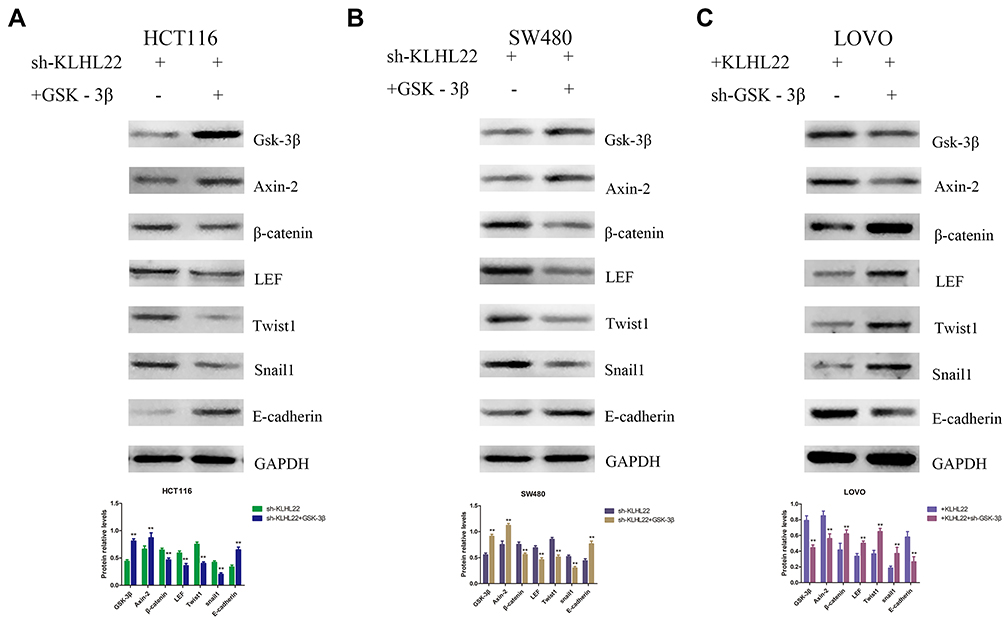 |
Figure 7 Effects of GSK-3β re-expression or silencing in stable KLHL22-expressing or KLHL22-silenced cell lines in vitro and vivo. We evaluated the expression of β-catenin, LEF, Twist1, Snail1 and E-cadherin via Western blotting. (A, B) The expression levels of β-catenin, LEF, Twist1, and Snail1 were reduced and that of E-cadherin was increased when GSK-3β and Axin-2 was overexpressed in KLHL22-silenced HCT116 and SW480 cells. (C) But the levels of these proteins showed the opposite trend when GSK-3β and Axin-2 was knocked down in KLHL22-overexpressing LOVO cells. GAPDH (cell marker) was used as a loading control. (**p < 0.01 by Student’s t-test.). |
KLHL22 Inhibits Tumorigenesis in vivo (Figure 8)
To further investigate whether KLHL22 inhibits tumor growth in vivo, we established a CRC xenograft mouse model by subcutaneously injecting HCT116-shRNA and SW480-shRNA cells or LOVO cells with stable KLHL22 expression into the right subcutaneous flank of nude mice. We measured the size of the growing tumors every week with a caliper. After 8 weeks of growth in nude mice, tumors of uneven size formed, and the size and weight of the tumors are shown in Figure 8. As shown in the tumor growth curve, all colorectal cancer cell lines successfully formed tumor xenografts, and the KLHL22-overexpressing cell line group exhibited a smaller tumor volume than the corresponding control group. In contrast, KLHL22-knockdown HCT116 and SW480 cells had a larger tumor volume than the corresponding control group (Figure 8A). Moreover, the average tumor volume of the group with increased KLHL22 expression was generally less than that of the group with decreased KLHL22 expression throughout the growth process (Figure 8B), and the above results were consistent with the in vitro results. Thus, KLHL22 inhibits the progression of colorectal tumors both in vitro and in vivo.
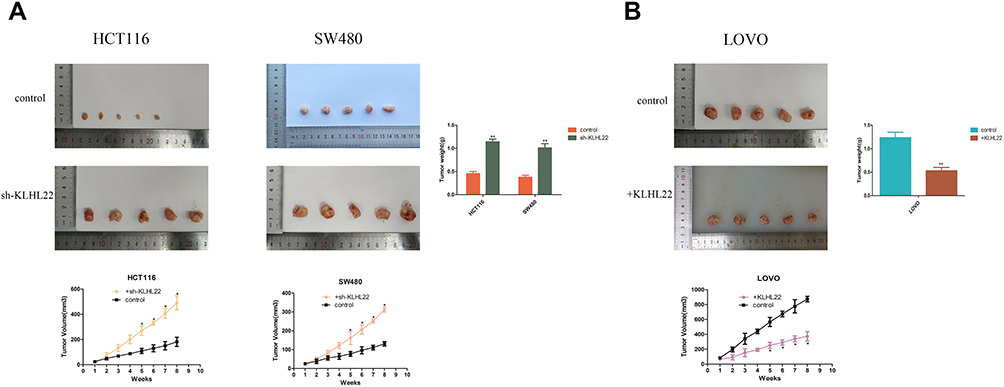 |
Figure 8 Influence of KLHL22 overexpression or silencing on tumor formation in vivo. (A) Photographs of tumors excised from mice injected with normal HCT116 and SW480 cells or KLHL22 shRNA-transfected HCT116 and SW480 cells. Pictures showed that mice were injected with normal LOVO cells or +KLHL22 LOVO cells (n = 5 per group). After 56 days, Nude mice were treated, and the weight of their tumors was weighed. *P<0.05 by Student’s t-test. (B) Tumor growth curves showed that the mice were injected with normal HCT116 and SW480 cells or KLHL22 shRNA-transfected HCT116 and SW480 cells. Tumor growth curves for mice injected with normal LOVO cells or +KLHL22 LOVO cells. (**p < 0.01 by Student’s t-test.). |
Discussion
CRC, a common human malignancy, is affecting a substantial number of people worldwide, and further study on its molecular mechanisms is urgently needed.17,18 There has been accumulating evidence showing that the difference in KLHL22 expression is closely related to cancer. However, no information is currently available on the specific role or molecular mechanism of KLHL22 in CRC patients. In this study, we aimed to test the function of the KLHL22 gene in CRC development and to identify the associated signaling mechanisms. The results of KLHL22 expression in CRC and nonmalignant tissues show that KLHL22 is downregulated in CRC and may inhibit cancer development.
EMT is a physiological process and a crucial event that triggers cancer cell metastasis, invasion and development in numerous cancers.19 In addition, it refers to the loss of original epithelial cell polarity and adhesion and the addition of a mesenchymal cell phenotype.20 During this process, the expression of E-cadherin and cytokeratin 19 (CK19) is downregulated, and cytoplasmic markers, such as vimentin, N-cadherin and S100A4, are upregulated. Many studies have shown that abnormal expression of E-cadherin and N-cadherin is significantly associated with lymph node metastasis and survival prognosis.21 Twist1, a significant mediation factor downstream of β-catenin, is involved in promoting EMT.22 Additionally, Twist1 induces a decrease in E-cadherin-mediated cell-cell adhesion to promote EMT.23–25 Our results show that decreasing KLHL22 expression was associated with gradually increasing amounts of vimentin and N-cadherin and decreasing levels of E-cadherin, indicating that KLHL22 may inhibit invasion and migration by stimulating EMT in CRC.
Many signaling pathways are involved in the development of EMT; canonical Wnt/β-catenin signaling is the core protein and is conserved in various organisms ranging from worms to mammals, and β-catenin represents the key downstream factor in this pathway.26 In the cytoplasm, nascent β-catenin in the cytoplasm becomes phosphorylated and ubiquitinated when it is sequestered within a complex that consists of APC, GSK-3β and Axin-2, and GSK-3β-dependent β-catenin phosphorylation induces its proteasomal degradation. When secreted Wnt ligands bind to the Fzd and LRP coreceptors, the Dishevelled (Dvl) protein in the Wnt/β-catenin pathway is activated and phosphorylated by GSK-3β.27–29 GSK3β could phosphorylate S33/S37/T41 sites on β-catenin at centrosomes, resulting in the stabilization of β-catenin at centrosomes; stabilization is the opposite effect of the well-characterized role of GSK3β phosphorylation, which destabilizes β-catenin by targeting it for degradation.30 After stabilization, β-catenin enters the nucleus and could bind to the T-cell factor (Tcf)/lymphoid enhancer factor (Lef) family of transcription factors, thereby leading to the transcriptional activation of multiple target genes.31 From a previous study, KLHL22 could bind to several kinases with known mitotic functions, including Cdk1, casein kinases, and Nek2, to influence kinetochore-microtubule attachment and other mitotic processes at multiple levels.32 The levels of PLK1 and GUL3 influenced by KLHL22 are critical regulators of mitosis, cell division control and are also central players in maintaining genome stability during DNA replication and modulating the DNA damage response.33 In addition, some results have shown that overexpression of these factors contributes to apoptosis resistance and proliferation in cancer cells in vitro through the NF-kB and STAT signaling pathways.34 In this study, we highlighted the function of KLHL22 in CRC with overexpression and silencing of KLHL22, and the corresponding results show that KLHL22 may increase the expression of members of the complex (APC, GSK-3β and Axin-2) with subsequent downregulation of both T-β-catenin and N-β-catenin. Additionally, these changes to the cellular localization of β-catenin are consistent with the changes detected in these cells via immunofluorescence. Overall, KLHL22 may inhibit EMT and cell proliferation by decreasing Wnt/β-catenin pathway activity in CRC.
To further determine whether the effects of KLHL22 on PLK1 and EMT regulation and proliferation are partially attributable to the Wnt/β-catenin signaling pathway, we evaluated the effects of re-expressing or blocking GSK-3β in stable KLHL22-expressing or KLHL22-silenced cell lines in vitro. Furthermore, we examined the expression of relevant proteins via Western blotting and assessed the condition of the cells. The results suggest that re-expression of the upstream Wnt/β-catenin signaling gene GSK-3β promotes the cancer suppression function of mutant KLHL22 in CRC cells. Thus, we further demonstrated that the regulatory effects of KLHL22 on EMT are partially attributable to the Wnt/β-catenin signaling pathway.
In summary, our data reveal that KLHL22 is a tumor suppressor in CRC. Moreover, silencing KLHL22 induces CRC cell proliferation and stimulates invasion, migration and EMT, whereas overexpression of KLHL22 yields the opposite results. Additionally, an increase in KLHL22 leads to a reduction in xenograft tumor growth in vivo, whereas the opposite outcome occurs when KLHL22 is knocked down. Therefore, we conclude that KLHL22 regulates the activity of GSK-3β to inhibit EMT partially through the Wnt/β-catenin signaling pathways. We anticipate that this study will provide a basis for new strategic approaches to develop effective CRC therapies.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Brody H. Colorectal cancer. Nature. 2015;521(7551):S1. doi:10.1038/521S1a
2. Weinberg BA, Marshall JL, Salem ME. The growing challenge of young adults with colorectal cancer. Oncology (Williston Park). 2017;31(5):381–389.
3. Chen J, Ou Y, Yang Y, et al. KLHL22 activates amino-acid-dependent mTORC1 signalling to promote tumorigenesis and ageing. Nature. 2018;557(7706):585–589. doi:10.1038/s41586-018-0128-9
4. Beck J, Peter M. Regulating PLK1 dynamics by Cullin3/KLHL22-mediated ubiquitylation. Cell Cycle. 2013;12(16):2528–2529. doi:10.4161/cc.25839
5. Jia D, Dong R, Jing Y, et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology. 2014;60(5):1686–1696. doi:10.1002/hep.27243
6. Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling. Dev Dis J Cell Biol. 2006;172:973.
7. Bates RC, Mercurio A. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther. 2005;4(4):365–370. doi:10.4161/cbt.4.4.1655
8. Zha L, Zhang J, Tang W, et al. HMGA2 elicits EMT by activating the Wnt/β-catenin pathway in gastric cancer. Dig Dis Sci. 2013;58(3):724. doi:10.1007/s10620-012-2399-6
9. Ahmad A, Sarkar SH, Bitar B, et al. Garcinol regulates EMT and Wnt signaling pathways in vitro and in vivo, leading to anticancer activity against breast cancer cells. Mol Cancer Ther. 2012;11(10):2193–2201. doi:10.1158/1535-7163.MCT-12-0232-T
10. Liang J, Liang L, Ouyang K, Li Z, Yi X. MALAT1 induces tongue cancer cells’ EMT and inhibits apoptosis through Wnt/β-catenin signaling pathway. J Oral Pathol Med. 2017;46(2):98. doi:10.1111/jop.12466
11. Fu Y, Zheng S, An N, et al. β-catenin as a potential key target for tumor suppression. Int J Cancer. 2011;129(7):1541–1551. doi:10.1002/ijc.26102
12. Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31(12):2714–2736. doi:10.1038/emboj.2012.150
13. Anson M, Craindenoyelle AM, Baud V, et al. Oncogenic β-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J Clin Inves. 2012;122(2):586–599. doi:10.1172/JCI43937
14. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11(24):3286–3305. doi:10.1101/gad.11.24.3286
15. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871. doi:10.1016/j.cell.2009.11.007
16. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. doi:10.1038/nrc2620
17. Luo H, Hao E, Tan D, et al. Apoptosis effect of Aegiceras corniculatum on human colorectal cancer via activation of FoxO signaling pathway. Food and Chem Toxicol. 2019;134:110861. doi:10.1016/j.fct.2019.110861
18. Yang J, McDowell A, Kim EK, et al. Development of a colorectal cancer diagnostic model and dietary risk assessment through gut microbiome analysis. Exp Mol Med. 2019;51(10):117. doi:10.1038/s12276-019-0313-4
19. Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253(5020):661–665. doi:10.1126/science.1651562
20. Larue L, Delmas V. The WNT/Beta-catenin pathway in melanoma. Fron Biosci J Virtual Lib. 2006;11(1):733. doi:10.2741/1831
21. Kang HJ, Eun-Jeong K, Byoung-Gwon K, et al. Quantitative analysis of cancer-associated gene methylation connected to risk factors in korean colorectal cancer patients. J Preventive Med Public Health. 2012;45(4):251–258. doi:10.3961/jpmph.2012.45.4.251
22. Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993;262(5140):1734–1737. doi:10.1126/science.8259519
23. Macdonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi:10.1016/j.devcel.2009.06.016
24. Liu X, Rubin JS, Kimmel AR. Rapid, Wnt-induced changes in GSK3beta associations that regulate beta-catenin stabilization are mediated by Galpha proteins. Curr Biol Cb. 2005;15(22):1989–1997. doi:10.1016/j.cub.2005.10.050
25. Metcalfe C, Mendozatopaz C, Mieszczanek J, Bienz M. Stability elements in the LRP6 cytoplasmic tail confer efficient signalling upon DIX-dependent polymerization. J Cell Sci. 2010;123(9):1588–1599. doi:10.1242/jcs.067546
26. Lien WH, Fuchs E. Wnt some lose some: transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 2014;28(14):1517. doi:10.1101/gad.244772.114
27. Huelsken J, Behrens J. The Wnt signalling pathway. J Cell Sci. 2002;115(21):3977–3978. doi:10.1242/jcs.00089
28. Jamieson C, Sharma M, Henderson BR. Wnt signaling from membrane to nucleus: Î2-catenin caught in a loop. Int J Biochem Cell Biol. 2012;44(6):847–850. doi:10.1016/j.biocel.2012.03.001
29. Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409–414. doi:10.1038/nature01593
30. Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi:10.1016/j.cell.2004.06.006
31. Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432(7015):324–331. doi:10.1038/nature03100
32. Gawden-Bone C, Zhou Z, King E, Prescott A, Watts C, Lucocq J. Dendritic cell podosomes are protrusive and invade the extracellular matrix using metalloproteinase MMP-14. J Cell Sci. 2010;123(9):1427–1437. doi:10.1242/jcs.056515
33. Zarrabi K, Dufour A, Li J, et al. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J Biol Chem. 2011;286(38):33167–33177. doi:10.1074/jbc.M111.256644
34. Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther. 2002;1(3):226–231. doi:10.4161/cbt.72
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.