")
Back to Journals » International Journal of Nanomedicine » Volume 13
Kidney-targeted drug delivery via rhein-loaded polyethyleneglycol-co-polycaprolactone-co-polyethyleneimine nanoparticles for diabetic nephropathy therapy
Authors Chen DF, Han SP, Zhu YQ, Hu F, Wei YH, Wang GW
Received 24 February 2018
Accepted for publication 21 April 2018
Published 19 June 2018 Volume 2018:13 Pages 3507—3527
DOI https://doi.org/10.2147/IJN.S166445
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lijie Grace Zhang
Danfei Chen,1 Shunping Han,2,3 Yongqin Zhu,1 Fang Hu,1 Yinghui Wei,3 Guowei Wang3,4
1Department of Pediatrics, The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, 310006 China; 2Department of Chemistry, Imperial College London, London, UK; 3College of Pharmaceutical Science, Zhejiang Chinese Medical University, Hangzhou, 310053 China; 4College of Biological and Chemical Engineering, Zhejiang University, Hangzhou, 310007 China
Introduction: Diabetic nephropathy (DN) is the primary root of morbidity and mortality in diabetic patients. Unfortunately, currently, no effective therapeutic strategies are available to ameliorate and reverse the progression of DN. Rhein (RH) is an anthraquinone derivative extracted from herbal medicines with various pharmacological effects on DN. However, its clinical administration is limited by its poor solubility, low bioavailability, reduced distribution into the kidney and adverse effects.
Methods and results: To improve the delivery of RH into kidney and the therapeutic effect on DN, we synthesized and utilized polyethyleneglycol-co-polycaprolactone-co-polyethylenimine triblock amphiphilic polymers to prepare RH-loaded polyethyleneglycol-co-polycaprolactone-co-polyethylenimine nanoparticles (PPP-RH-NPs). PPP-RH-NP size was optimized to 75 ± 25 nm for kidney-targeted drug delivery; the positive zeta potential allowed an effective cellular uptake and the polyethylenimine amine groups facilitate the endosomal escape quickly. The distribution and pharmacodynamics of PPP-RH-NPs were studied in a streptozocin-induced DN model, which explicitly demonstrated kidney-targeted distribution and improved the therapeutic effects of RH on DN by ameliorating several pathological indicators.
Conclusion: Therefore, this study not only stimulates further clinical research on RH but also, more importantly, proposes a promising DN therapy consisting of an effective kidney-targeted drug delivery.
Keywords: rhein, diabetic nephropathy, polyethyleneglycol-co-polycaprolactone-co-
polyethylenimine, nanoparticles, in vitro/vivo evaluation, targeting drug delivery
Introduction
Diabetic nephropathy (DN), characterized and accompanied by high blood glucose, blood pressure, proteinuria, a progressive decline in renal function and an increased risk of cardiovascular disease, has been the primary cause of morbidity and mortality in patients affected by both type I and type II diabetes mellitus.1–5 According to statistics, nearly 347 million people have diabetes, and this number is expected to increase to 430 million by 2030 in the world.6–8 DN is becoming more and more prevalent, to some extent reaching epidemic proportions. However, currently, no effective therapeutic strategies are available to counteract and reverse the progression of DN.1,9
Rhein (RH, 4,5-dihydroxyanthraquinone-2-carboxylic acid), an anthraquinone derivative extracted and separated from Rheum palmatum L., Rheum tanguticum Maxim. ex Balf. and other traditional Chinese herbal medicines, is well known for its antidiabetic effect10,11 and is considered as a remedy for DN in China, Japan and Korea.12,13 Currently, the Shenkang injection, which mostly contains RH (and other extract from R. palmatum L.) is the first-line clinical drug for the treatment of DN in China.14,15 Moreover, the use of RH capsules has already been approved in Phase II clinical trials (clinical trial approval number: 2008L03643). RH has the ability to decrease the level of fasting blood glucose (FBG), creatinine, blood urea nitrogen (BUN), urine protein, the intensity of oxidative stress and phosphorylated drosophila mothers against decapentaplegic protein expression.11,13 In addition, it can suppress TGF-β1 expression in the kidney10,16 and protect against fibrosis in streptozotocin (STZ)-induced DN model with multi-target therapy.17 However, the clinical use of RH is limited because of poor solubility, low bioavailability, reduced distribution into the kidney and adverse effects.18,19 Thus, it is of utmost importance to seek an appropriate drug delivery system or administration for improving therapeutic effect of RH on DN.
Recently, nanotechnology has generated excitement in the cancer community because of its ability to design carriers to deposit drugs more selectively in tumor tissues.20–23 Nanoparticles have been extensively investigated as drug delivery carriers into tumors and liver because of their intrinsic tendency to accumulate and retain in these tissues.24 However, the kidney during end-stage renal diseases is another site of nanoparticle accumulation.25–29 The glomerular filtration barrier consists of glomerular endothelial cells, glomerular basement membrane and podocytes. Nanoparticles with a hydrodynamic diameter of <10 nm fall below the kidney filtration threshold, pass through the glomerulus and are excreted, while nanoparticles with a size of >150 nm cannot pass through the glomerular filtration barrier to accumulate in diseased areas.29–32 Even in the sub-nanometer size regime, the glomerular filtration barrier behaves as an atomically precise band-pass filter to slow down renal clearance of few-atom gold nanoclusters with the same surface ligands but different sizes.33 What is more is nanoparticles with specific size of 5–30 nm are able to cross the glomerular filtration barrier to target podocytes in kidney glomerulus.34 Most other kidney therapy studies based on size demonstrated that the accumulation of nanoparticles is restricted to the glomerular mesangium and the kidney’s extracellular matrix, reaching the maximal glomerular deposition by using ~80 nm nanoparticles.31,35 In addition, charge selectivity is also an important criterion for kidney targeting. Nanoparticles with a surface charge of <15 mV have minimal macrophage uptake and long circulation time.30 Highly positively charged nanoparticles are rapidly cleared from the circulation by cells of the mononuclear phagocyte system, while anionic nanoparticles are subjected to glomerular filtration, hampering the accumulation and uptake efficiency.
Many research groups have already explored nanoparticles to load and deliver RH. Yuan and Gu36 described a type of RH-loaded nanoparticles that are able to solubilize RH with prolonged circulation time and sustained release, and Wei et al37 described another type of RH-loaded nanoparticles that significantly increased kidney distribution and bioavailability of RH. Generally, intracellular drug delivery of nanoparticles includes several steps including cell endocytosis, endosome/lysosome escape, polymer dissociation and drug release. The overall therapeutic index is dependent on the efficiency of each step.38 The uptake of nanoparticles (or micelles, liposomes, dendrimer polyplexes) takes place through endocytosis, which is mostly mediated by the endosome/lysosome pathway.39,40 Because of their role on controlling cells homeostatic and waste disposal system, endosomes/lysosomes possessing lower pH and several hydrolytic enzymes can devitalize and decompose exogenous substances including drug-loaded nanoparticles and consequently weaken or even inactivate the drug effect.40,41 Therefore, the next generation of nanoparticles should possess suitable stability and effective endosomal escape ability to be significantly suitable for drug delivery in clinical research.
Polyethylenimine (PEI) is a gold standard and widely used carrier for gene transfer.42 It contains 15%–20% protonatable amine groups under physiological conditions and shows kidney distribution for gene delivery, to some extent.43 More than 70% of amine groups become protonated once trapped in endosomes because of the low pH microenvironment. As a result, more protons translocate into the endosomes by the ATPase proton pumps. Subsequently, chloride ions passively enter and osmotic pressure increases, followed by water influx. Finally, endosomal rupture occurs, leading to the release of drugs and carriers from the endosome to the cytosol and targets.41,42,44
According to overall evidence, in this study, we conjugated PEI to the polyethyleneglycol-co-polycaprolactone (PEG-PCL) via Michael addition reaction and synthesized polyethyleneglycol-co-polycaprolactone-co-polyethylenimine (PEG-PCL-PEI) triblock amphiphilic polymers. PEG-PCL-PEI was used as carrier, and the nanoprecipitation method was applied to prepare RH-loaded PEG-PCL-PEI nanoparticles (PPP-RH-NPs). Moreover, their physical and chemical properties and in vitro biological characteristics were evaluated. Likewise, particle size, zeta potential and drug encapsulation efficiency (EE) and loading efficiency (LE) were optimized and screened; thus, nanoparticles of ~80 nm size and ~10 mV zeta potential were created, followed by the investigation of their pharmaceutical profile, endosomal escape in HK-2 cells and therapeutic effect in STZ-induced DN model. As anticipated, PPP-RH-NPs with the ability of endosomal escape markedly alleviated DN progression, and we also demonstrated the underlying drug delivery mechanism associated with the beneficial effects. Our data provided strong evidence for the therapeutic role of RH in DN and promoted further clinical and translational research on the use of nanoparticles in kidney diseases, which will attract more and more widespread attention especially in the field of nanomedicine and nephropathy therapy.
Materials and methods
Chemicals
RH active pharmaceutical ingredient (purity ≥98%; lot no 20130701) was purchased from Zelang Medical Technology Co., Ltd. (Nanjing, China). RH reference substance (lot no 110757200206) was purchased from National Institutes for Food and Drug Control (Hangzhou, China). Lantus (insulin glargine, a long-acting basal insulin analog) was purchased from Sanofi-Aventis Deutschland GmbH (Frankfurt, Germany). Methoxypolyethylene glycol 2000 (PEG), caprolactone, stannous octoate, acryloyl chloride, boc-alanine, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were supplied by Energy Chemical (Shanghai, China). Polyethylenimine (PEI; molecular weight 1.8 kD) and 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Hoechst 33342 and LysoTracker® Green DND99 were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Cyanine 5 NHS active ester (Cy5-NHS) was purchased from Mercury Bio-pharmaceutical Technology Co. Ltd (Hangzhou, China). Poloxamer 188 was purchased from BASF (Ludwigshafen, Germany). Anti-TGF-β1 antibody and anti-Smad2/3 antibody for Western blotting were purchased from Abcam (Cambridge, UK). Anti-fibronectin (FN) antibody for immunohistochemistry (IHC) was purchased from Proteintech Group (Chicago, IL, USA). Anti-NAPDH antibody was purchased from Bioworld Technology (Nanjing, China). Enzyme-linked immunosorbent assay (ELISA) kits for proteins, urea nitrogen and creatinine detection kit were purchased from Jiancheng Bioengineering Institute (Nanjing, China). STZ was purchased from Sigma-Aldrich Co. Other materials such as N, N-dimethylformamide (DMF), dichloromethane (DCM) and triethylamine (TEA) were of analytical grade.
Animals and cell culture
Male C57BL/6 mice (also known as C57BL/6J) were supplied by the Laboratory Animal Center of Zhejiang Chinese Medical University, Hangzhou, China, maintained at 25°C and 45% relative humidity and kept under 12 h/12 h light/dark cycles. All experiments were performed according to the guidelines for the care and use of animals established by the Zhejiang Chinese Medical University. HK-2 cell line was obtained from the Shanghai Institutes for Biological Sciences of Chinese Academy of Sciences, Shanghai, China. Culture environment was as follows: DMEM (Thermo Fisher Scientific) containing 10% fetal bovine serum, 4.5 g/L glucose, 3.7 g/L NaHCO3, 1% nonessential amino acid, 1% glutamine and 100 U/mL penicillin/streptomycin. Rabbit red blood cells (RBCs) were provided by Laboratory Animal Center of Zhejiang Chinese Medical University. The animal experiments (license no 2016063) were approved by the institutional animal ethics committee of Zhejiang Chinese Medical University.
PEG-PCL and PEG-PCL-PEI synthesis and characteristics
PEG-PCL and PEG-PCL-PEI synthesis
PEG2K (the molecular weight of PEG was 2K) and caprolactone were dried before use. PEG (2 g, 1 mmol), caprolactone (10 mL, 90 mmol) and stannous octoate (0.82 g, 2 mmol) were mixed and stirred for 24 h at 110°C under N2 condition in a round-bottom flask. Then, the products were dissolved in DCM and precipitated in cold ether thrice. The precipitate was filtered and vacuum dried to obtain a white solid (product ~6 g, yield ~75%). 1H-nuclear magnetic resonance (NMR) of PEG-PCL (CDCl3, 400 MHz, δ ppm) was as follows: 1.38 (−COCH2CH2CH2CH2CH2O−, 100.77H), 1.63 (−COCH2CH2CH2CH2CH2O−, 200.45H), 2.29 (−OCO−CH2−, 99.89H), 3.38 (CH3−O−, 3H), 3.65 (−CH2CH2−O−, 180.55H), 4.06 (−CH2−OCO−, 98.23H).
PEG-PCL (2 g, 0.25 mmol) was dissolved in 20 mL dry DCM. One-milliliter dry TEA and 80 μL acryloyl chloride were added at room temperature (RT), and the mixture was stirred for 6 h under N2. The mixture was subsequently extracted by saturated NaCl solution (20 mL × 3), concentrated and precipitated in cold ether. Finally, it was filtered and vacuum dried to obtain PEG-PCL-COCHCH2 (product ~1.2 g, yield ~60%).
PEG-PCL-COCHCH2 (1 g, 0.125 mmol) and PEI (0.36 g, 0.2 mmol) were dissolved in 10 mL DMF, and 1 mL pyridine was added. After being stirred at 60°C overnight, the mixture was dialyzed in DMF by dialysis bag (molecular weight cutoff 10 kD) for 24 h followed by dialysis in water for 24 h. The flavescent solid PEG-PCL-PEI was obtained by freeze-drying (product ~0.8 g, yield ~65%). 1H-NMR of PEG-PCL-PEI (CD3OD, 400 MHz, δ ppm) was as follows: 1.29 (−COCH2CH2CH2CH2CH2O−, 100.41H), 1.53 (−COCH2CH2CH2CH2CH2O−, 199.87H), 2.27 (−OCO−CH2−, 98.54H), 3.55 (−CH2CH2−O−, 180H), 3.96 (−CH2−OCO−, 99.39H), 2.50 (−CH2CH2−NH−, 195H).
The structure of polymers was confirmed by 1H-NMR (Bruker Avance-400 Nuclear Magnetic Resonance; Bruker Optik GmbH, Ettlingen, Germany). The molecular weight and polydispersity indexes (PDIs) were characterized by gel permeation chromatography (GPC; Waters 1515 GPC; Waters, Milford, MA, USA). PEG-PCL and PEG-PCL-PEI critical micelle concentrations (CMCs) were determined by a fluorescence method. Fluorescence intensity ratio of pyrene at 338 nm/333 nm (I338/I333) was chosen and calculated. CMCs were determined as the intersection from the I338/I333 fluorescence intensity fitting curves as a function of concentrations.45
Cy5-labeled PEG-PCL and PEG-PCL-PEI synthesis
PEG-PCL (1 g, 0.125 mmol), boc-alanine (189 mg, 1 mmol), EDC (48 mg, 0.25 mmol) and NHS (30 mg, 0.25 mmol) were mixed, dissolved in 10 mL DCM and stirred for 12 h at RT. The product was obtained by precipitation in ether, and then it was dissolved in 20 mL DCM. Two milliliters of trifluoroacetic acid were added and stirred for 1 h. Product was extracted by saturated NaCl solution (20 mL × 3), concentrated, precipitated in ether and vacuum dried. PEG-PCL-COCH2CH2NH2 (PEG-PCL-NH2) was obtained by lyophilization (product ~0.45 g, yield ~41%).
PEG-PCL-NH2 (200 mg) and PEG-PCL-PEI (200 mg), each in a flask, were dissolved in 5 mL DMF each. Cy5-NHS (1 mg and 0.75 mg) was added to flasks, respectively (grafting ratio of Cy5 was 5% theoretically). After being stirred at 45°C for 12 h in the dark, the mixture was dialyzed in DMF for 12 h and subsequently in distilled water for 24 h by dialysis bag (molecular weight cutoff 3.5 kD). The blue powder Cy5-labeled PEG-PCL-NH2 (PEG-PCLCy5) and Cy5-labeled PEG-PCL-PEI (PEG-PCL-PEICy5) were obtained by lyophilization. Polymers purified by dialysis in this section did not suffer a reduction in weight. Cy5 grafting ratios of PEG-PCLCy5 and PEG-PCL-PEICy5 were about 3.2% and 3.9%, respectively, as determined by fluorescence intensity.
Preparation of nanoparticles
PPP-RH-NPs were prepared by nanoprecipitation method, and the size of PPP-RH-NPs was optimized by high-pressure homogenizer (AH-100D; ATS Engineering Inc., Brampton, ON, Canada). Briefly, a defined amount of RH and PEG-PCL-PEI (20 mg) was dissolved in 2 mL acetone/methanol (2/1, v/v). This organic phase was added dropwise to 10 mL water containing 1% poloxamer 188 under stirring (800 rpm/min). After being stirred for 30 min, the solution was evaporated using rotary evaporators at RT for 30 min. The solution volume was adjusted to 10 mL with water, and the raw PPP-RH-NP dispersions were obtained. After high-pressure homogenization (parameter: first-stage valve to 1,000 bar, second-stage valve to 200 bar, approximate time 1 min), mature PPP-RH-NP dispersions with desirable size were successfully obtained. RH-loaded PEG-PCL nanoparticles (PP-RH-NPs), PEG-PCL-PEI nanoparticles without loading RH (PPP-NPs) and PEG-PCL nanoparticles without loading RH (PP-NPs) were prepared as controlled formulations by the same method.
The size distribution and zeta potential of the nanoparticle dispersions obtained were detected by dynamic light scattering method (Nano-ZS 90; Malvern Instruments, Worcestershire, UK). The morphology of nanoparticles was visualized by transmission electron microscopy (TEM; JEM-1200EX; JEOL, Tokyo, Japan). Briefly, one drop of PPP-RH-NP solution was placed onto a copper grid and dried, and 2% (w/v) phosphotungstic acid solution was used to stain for imaging. Drug LE and EE were quantified by high-performance liquid chromatography (HPLC; Agilent-l200 reverse-phase HPLC; Agilent Technologies, Santa Clara, CA, USA). Briefly, 1 mL of each nanoparticle dispersion was centrifuged (20,000 rpm/min, 30 min), and the supernatant was used to evaluate free RH concentration, and the total RH minus the free RH in the supernatant represented the RH loaded in nanoparticles. EE and LE were calculated according to Equations 1 and 2, respectively:
|
|
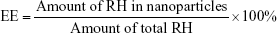
|
|

RH HPLC assay
RH standard stock solution was prepared at 0.10 mg/mL in methanol, and a series of solutions (0.1, 0.5, 1, 5, 10, 20, 40 and 80 μg/mL) were prepared by diluting the standard stock solution. HPLC was performed using a Platisil ODS column (150 × 4.6 mm, 5 μm; Dikma Technologies, Beijing, China) at 25°C. The mobile phase consisted of methanol/1% phosphoric acid (75:25, v/v) and eluted at 1.0 mL/min. RH was detected at the wavelength of 254 nm. Peak areas (A) vs RH concentrations (C) curves were obtained. Curves in the concentration range of 0.5–40 μg/mL showed an acceptable linear relationship (regression equation: A = 71.272C-12.817, R2 = 0.9998). HPLC assay conditions satisfied the methodology requirements.
In vitro drug release test
The in vitro release profiles of RH solution (RH-sol, RH dissolved in PBS containing 10% Tween 80 and 1% glycerin) and nanoparticle dispersions were tested via the dialysis method in 0.01 M PBS (pH 7.4) medium containing 2% w/v Tween 80. PPP-RH-NPs, PP-RH-NPs and RH-sol (equivalent-dose 0.5 mg RH) were placed into a dialysis bag (molecular weight cutoff 3,500 Da) and dipped into 200 mL medium in a shaker (37°C, 60 rpm/min). Samples (1 mL) were collected at 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 24 and 48 h, and an equal volume of medium was added at each time interval. HPLC analysis was used to quantify RH concentrations, and the cumulative release rate (Q) was calculated according to Equations 3 and 4:
|
|
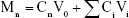
|
|
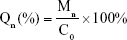
Mn, Qn, Cn, V0, Vi, Ci and C0 refer to cumulative release at each collection time, percentage of the cumulative release at each collection time, drug concentration at tn, released medium volume, sample volume removed each time, drug concentration at ti and the total drug concentration, respectively.
In vitro hemolysis assay
In vitro hemolysis assay was performed under guidance from some reported protocols.46,47 The anticoagulant rabbit RBCs were centrifuged (2,000 rpm/min, 5 min) to remove fibrinogen and washed with PBS (pH 7.4) thrice. These steps were repeated until the supernatant was clear. The RBC stock suspension (108/mL) was obtained by dilution with PBS. PP-NP and PPP-NP solutions were diluted with PBS. Each solution (900 μL) and RBC stock suspensions (100 μL) were mixed in tubes, incubated at 37°C for 2 h and centrifuged at 2,000 rpm for 5 min to collect supernatant (200 μL). Absorbance (a) at 540 nm was measured. Water was used as a positive control, while PBS was used as a negative control. The hemolysis ratio was calculated according to Equation 5:
|
|
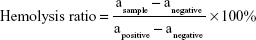
In vitro cytotoxicity assay
The in vitro cytotoxicity on HK-2 cells was determined by the MTT assay. HK-2 cells (5 × 104 cells/mL, 180 μL/well) were placed in 96-well plates for 12 h incubation. PP-NPs and PPP-NPs (20 μL each) were added at serial dilutions and incubated for 48 h. An equal amount of PBS was added to the control group. Then, 20 μL MTT (5 mg/mL in PBS) was added to each well and incubated for 4 h. Medium was removed, and 200 μL dimethyl sulfoxide (DMSO) was added to each well, followed by vibration for 1 min on a vortex mixer. The reference wavelength was set at 620 nm, and the absorbance (a) at 570 nm was read and measured using a microplate reader (Spectra Max M5 microplate reader; Molecular Devices LLC, Sunnyvale, CA, USA). Each test was repeated six times. Cell viability was determined by Equation 6:
|
|
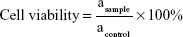
Cell uptake assay
HK-2 cells (105 cells/well in 1 mL DMEM) were seeded in 12-well plates for 12 h culture. PPPCy5-RH-NP and PPCy5-RH-NP dispersions (100 μL) were dissolved in each well and incubated for 0, 0.25, 0.5, 1, 1.5, 2 and 3 h. Then, cells were washed thrice with PBS, digested and harvested. Samples were analyzed by flow cytometry (FACS Calibur; BD, Franklin Lakes, NJ, USA), and the mean fluorescence intensity of Cy5 was obtained using its analytical software. In addition, the uptake velocity curves and efficiency were evaluated.
Subcellular distribution assay
HK-2 cells (105 cells/well in 1 mL DMEM) were cultured in glass-bottom petri dishes (35 mm) for 24 h. First, lysosomes were stained with 0.2 μL LysoTracker Green DND99 for 20 min, and then nuclei were stained with Hoechst 33342 (two drops) for 30 min. Dishes were washed with PBS. Next, PPPCy5-RH-NPs (100 μL) or PPCy5-RH-NPs (100 μL) were added and incubated for 15 min, 1 h and 3 h in serum-free medium. Subcellular distribution images of nanoparticles were observed and photographed by laser scanning confocal microscope (Nikon A1; Nikon Corporation, Tokyo, Japan) under the DAPI, 488 and 640 nm wavelength channels.
STZ-induced DN model
The induction protocol we followed was described by the Animal Models of Diabetic Complication Consortium48 and Animal Models of Disease.49,50 Eight-week-old C57BL/6 mice were treated with a single dose of STZ (200 mg/kg body weight, 100 mM, pH 4.5 citrate buffer). After treatment, mice were placed back in their cages, and they had access to normal food and 10% sucrose water. Citrate buffer was administered to other animals in place of STZ, as the healthy control (HC), and these control animals were subjected to the same treatment as the diabetic animals. On experimental day 3, the 10% sucrose water was replaced with regular water. All mice were fasted for a night before the FBG, in a tail-vein blood sample, was measured using One Touch Basic Blood Glucose Monitoring Apparatus (Aikang Technologies, Hangzhou, China) to ensure hyperglycemia, and every week body weight was recorded. The worsening of disease or the occurrence of serious complications was a sign of diabetes developing into early DN after 5 weeks from STZ treatment.49–51
In vivo real-time biodistribution in DN model mice
DN control (DNC) model and HC mice were intravenously (i.v.) treated with PPPCy5-RH-NPs (200 μL/20 g). Mice chests and abdomens were depilated by using a depilatory cream. The real-time distribution of PPPCy5-RH-NPs was observed and quantitatively analyzed via IVIS Lumina III (PerkinElmer Inc., Waltham, MA, USA) at 1, 3 and 6 h. Mice were sacrificed at 6 h, and major organs (heart, liver, spleen, lung, kidney and intestine) were excised, weighed and imaged. The ratio of total fluorescence intensity and tissue weight was recorded, and PPPCy5-RH-NP distribution in different organs and tissues was analyzed.
Pharmacodynamics study on DN mice model
DN mice were treated with Lantus (0.5 U/mouse, subcutaneous injection), RH-sol and PPP-RH-NPs (dose equivalent to RH 5 mg/kg, i.v. injection). HC and DNC mice were i.v. treated with saline (six mice in each group). Administration was performed every other day for 20 days. Body weight and FBG were monitored every 2 days before administration.
Mice were kept in metabolic cages, and urine was collected for 24 h, at the end of the treatment. The indexes such as 24 h urinary volume (24 h UV), 24 h urinary protein excretion (24 h UPE) and urinary creatinine (UCr) were measured. Blood samples were collected with EDTA as anticoagulant and centrifuged to analyze BUN and serum creatinine (SCr). Creatinine clearance rate (CCr) was calculated according to Equation 7:
|
|
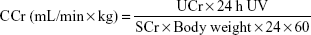
Mice were sacrificed, and kidneys were removed and weighed. Kidney index was defined as the ratio between kidney weight and body weight. One kidney was fixed in 4% paraformaldehyde and embedded in paraffin for sectioning. Hematoxylin and eosin (H&E) staining and periodic acid–Schiff (PAS) staining were performed. Sections were examined for histopathological changes by light microscope. FN expression in kidneys was analyzed by IHC. The other kidney was stored at −80°C and homogenized for extraction of proteins. Western blotting was performed to evaluate the expression of fibrosis proteins such as TGF-β1 and Smad2/3 in kidneys.
Statistical analyses
Data are expressed as mean ± SD. Significant differences in the mean values were evaluated by one-way analysis of variance (ANOVA). A statistical test with a P < 0.05 was considered statistically significant, and extreme significance was set at P < 0.01.
Results
Synthesis and characteristics of the polymer
PEG-PCL-PEI synthesis is depicted in Figure 1A. The structure of polymers was confirmed by 1H-NMR. Based on three protons in methoxy polyethylene glycol, the molecular weight of PEG-PCL was 7.7 kD (2,000 + 100/2 × 114 = 7,700). 1H-NMR of PEG-PCL-COCHCH2 is shown in Figure 1C. Peak shifts were according to PEG-PCL (Figure 1B). The peaks at 5.5–6.5 ppm (−OCO−CHCH2, 3.59H) were attributed to three protons in the acryloyl-substituted group. The proton numbers were more than theoretical values, indicating the presence of a residual acryloyl chloride in the product, although it had no effect on the next reaction. The molecular weight of PEI was 1.8 kD according to the 1H-NMR (Figure 1D). The peaks at 2.56 ppm (−CH2CH2−NH−) were assigned as 170 protons. Since peaks of the methoxyl group and solvent overlapped, the peak at 3.55 (−CH2CH2−O−, 180H) was set as calibration (Figure 1E). The molecular weight of PEG-PCL-PEI was approximately 9.8 kD (7.7 K + 195/4 × 43 = 9,796). The comparison of the peaks’ actual and theoretical values indicated that PEG-PCL-PEI purity was high enough (approximately 90%) for drug delivery.
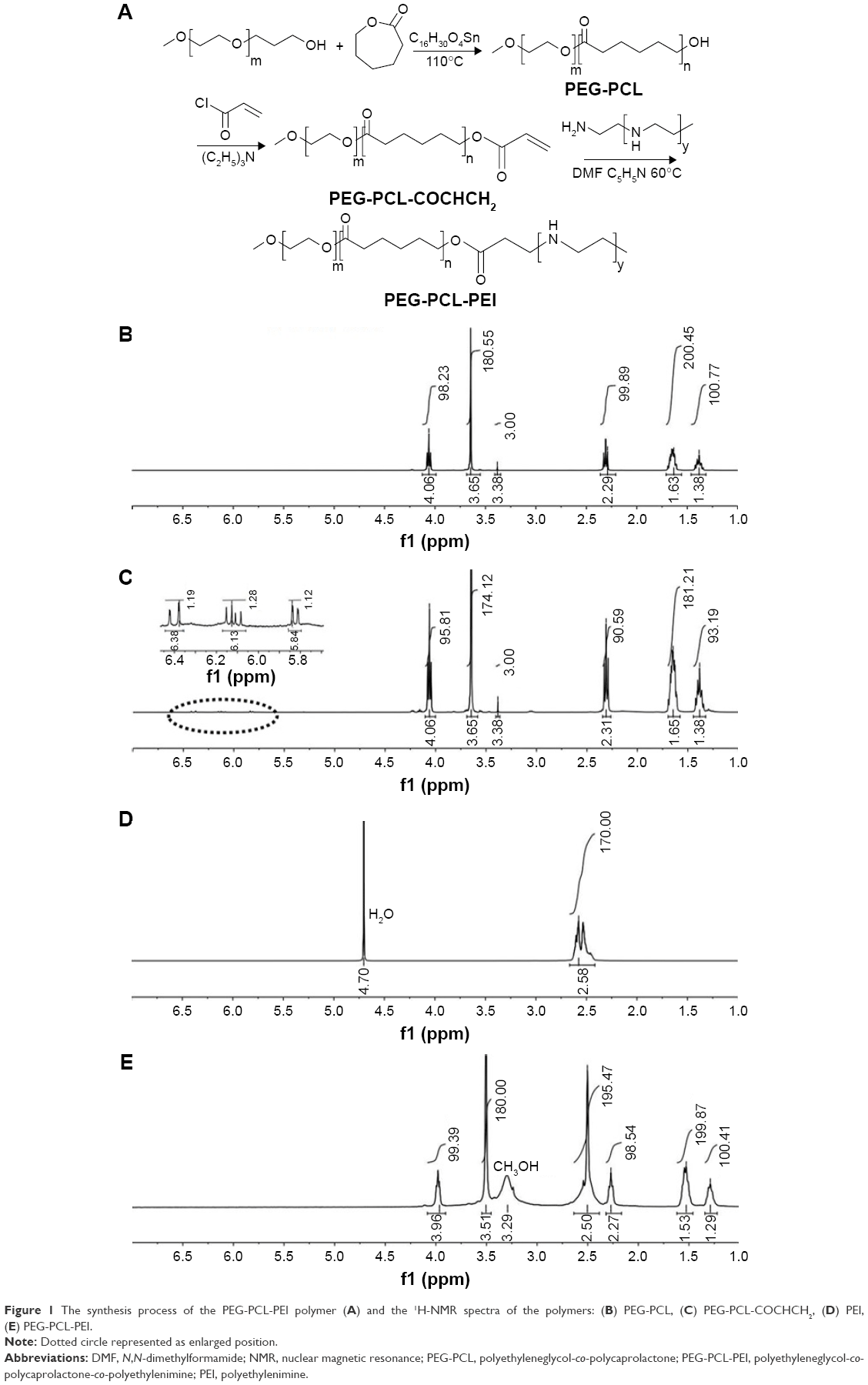 | Figure 1 The synthesis process of the PEG-PCL-PEI polymer (A) and the 1H-NMR spectra of the polymers: (B) PEG-PCL, (C) PEG-PCL-COCHCH2, (D) PEI, (E) PEG-PCL-PEI. |
The molecular weight of each polymer and PDI was assessed by GPC (Figure 2A). PEG-PCL-PEI PDI was 1.08, confirming a narrow distribution, whereas PEG-PCL and PEI showed wide distributions. The possible reason was that PEG-PCL-PEI was dialyzed by the dialysis bag with a molecular weight cutoff of 10 kD which might remove low molecular weight polymers. A calibration curve was obtained through by using polyethylene as the standard reference. The weight-average molecular weight of PEG-PCL-PEI was 9.5 kD, which was consistent with the 1H-NMR result. Fluorescent probe Cy5 was chosen to label PEG-PCL and PEG-PCL-PEI for the detection of cellular uptake. According to the fluorescence intensity, Cy5 grafting ratios were about 3.2% and 3.9% for PEG-PCLCy5 and PEG-PCL-PEICy5, respectively (Figure 2B).
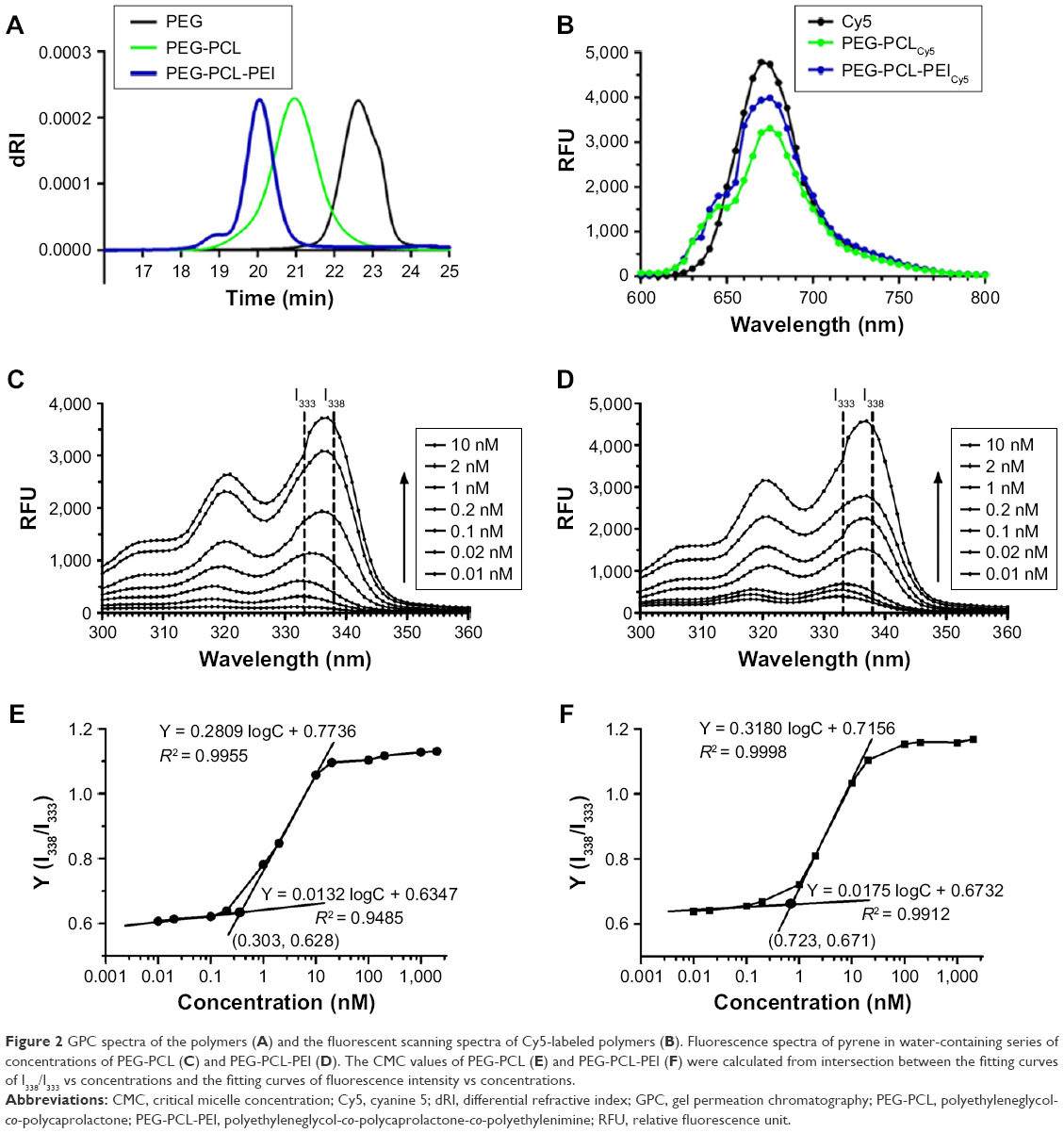 | Figure 2 GPC spectra of the polymers (A) and the fluorescent scanning spectra of Cy5-labeled polymers (B). Fluorescence spectra of pyrene in water-containing series of concentrations of PEG-PCL (C) and PEG-PCL-PEI (D). The CMC values of PEG-PCL (E) and PEG-PCL-PEI (F) were calculated from intersection between the fitting curves of I338/I333 vs concentrations and the fitting curves of fluorescence intensity vs concentrations. |
Amphiphilic polymers form micelles once reaching a sufficient concentration called the critical micelle concentration (CMC). CMC values of PEG-PCL and PEG-PCL-PEI were determined by a fluorescence method. Pyrene exhibited different fluorescent peaks, which were very sensitive to solvent polarity levels. As shown in Figure 2C and D, fluorescence intensity from 338 to 333 nm was an indicator of the local environment. The excitation spectra were recorded in the 300–360 nm wavelength range with an emission wavelength of 390 nm. Pyrene was easily buried in the inner core of micelles because of hydrophobic interactions. Pyrene fluorescent intensity in water was weak, whereas a red-shift excitation wavelength and enhanced intensity could be observed in the hydrophobic cores of micelles. Curves were nearly horizontal at low concentrations as shown in Figure 2E and F. As PEG-PCL and PEG-PCL-PEI concentrations increased, a red-shift excitation wavelength from 333 to 338 nm was observed. PEG-PCL and PEG-PCL-PEI fluorescence intensity rapidly changed at 0.2 and 1 nmol/L. CMC values were considered as the intersection between the fitting curves of I338/I333 vs concentrations and the fitting curves of fluorescence intensity vs concentrations. As shown in Figure 2E and F, PEG-PCL-PEI has a higher CMC value than PEG-PCL (0.723 and 0.303 nmol/L, respectively). Since the nature of PEI block was hydrophilic, a higher PEG-PCL-PEI concentration was needed to form the hydrophobic cores in micelles.
PPP-RH-NP preparation and characteristics
The nanoprecipitation method and high-pressure homogenization method were applied to make the optimal PPP-RH-NPs with a nanoparticle size of ~80 nm. Without high-pressure homogenization, the raw PPP-RH-NP dispersions were composed of the bigger nanoparticle size of 118.3 ± 3.6 nm (Figure 3B and D), which would not be efficient for kidney drug delivery. After high-pressure homogenization was applied, the mature PPP-RH-NP dispersions were formed with nanoparticle size of 75.4 ± 2.9 nm, PDI of 0.11 ± 0.05 and zeta potential of 6.4 ± 2.5 mV (Figure 3A, C and E). Morphological appearance of PPP-RH-NP dispersions showed yellow and opalescent color (Figure 3A[e]) and PPPCy5-RH-NP dispersions appeared slightly chartreuse (Figure 3A[d]). The morphology of the raw and mature nanoparticles was visualized by TEM (Figure 3B and C). The raw nanoparticles formed large size agglomeration to some degree, but the mature nanoparticles showed a monodisperse spherical structure with a desirable size without aggregations and adhesions. Size and PDI of other nanoparticles are described in Table 1. Their sizes were approximately 80 nm, and PDI ranged from 0.1 to 0.2. The use of PEG-PCL resulted in negatively charged nanoparticles, while the use of PEG-PCL-PEI caused the exact opposite, because protonated amine groups in PEI contributed to the positive charges. RH concentrations were quantified by HPLC after high-speed centrifuging. Drug LEs of PP-RH-NPs and PPP-RH-NPs were 2.31% ± 0.38% and 8.57% ± 0.53%, respectively. EEs of PP-RH-NPs and PPP-RH-NPs were 47.36 ± 3.81% and 93.64% ± 5.28%, respectively. Hence, RH LE could be remarkably enhanced by the conjugation of PEI on PEG-PCL.
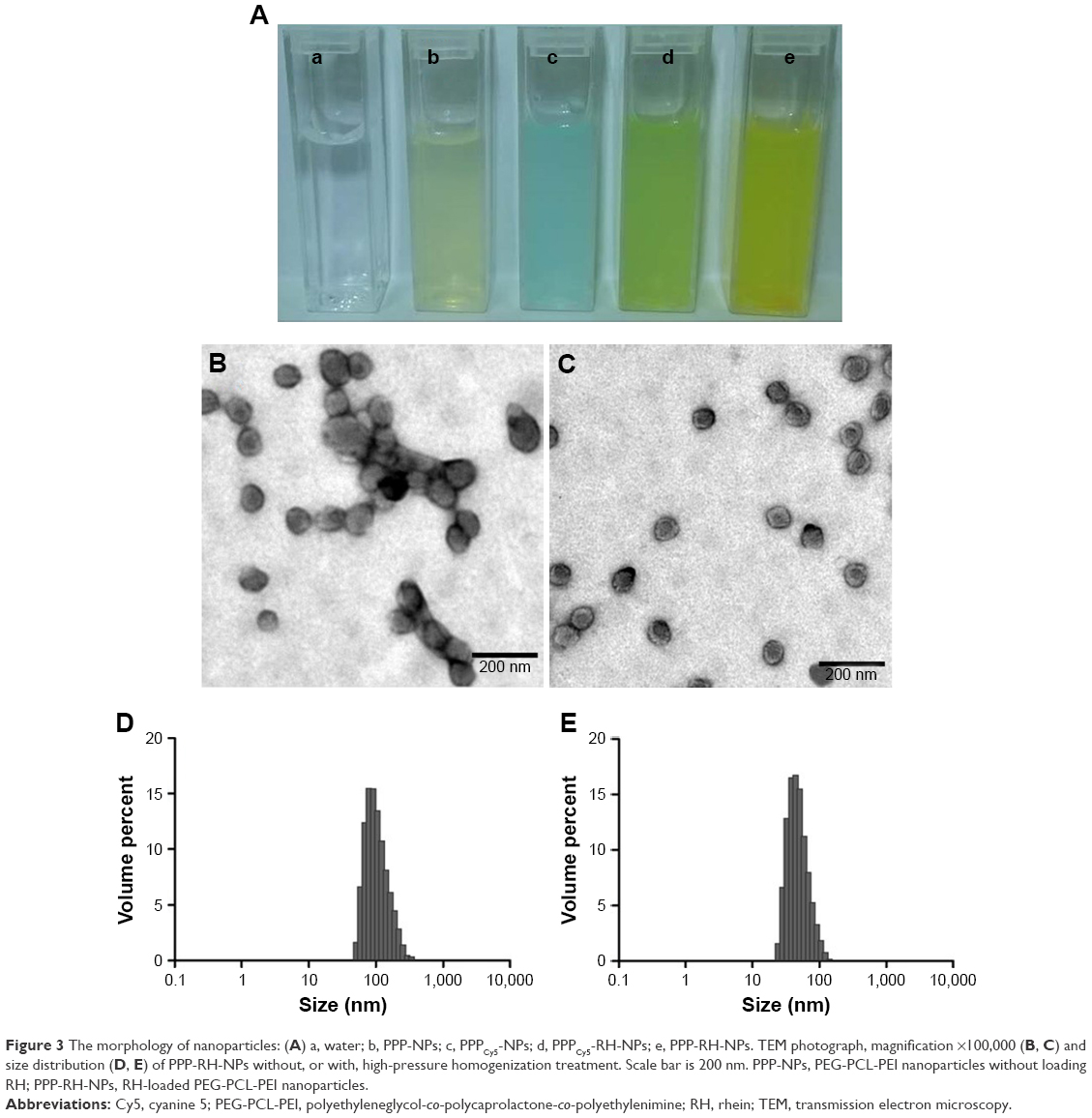 | Figure 3 The morphology of nanoparticles: (A) a, water; b, PPP-NPs; c, PPPCy5-NPs; d, PPPCy5-RH-NPs; e, PPP-RH-NPs. TEM photograph, magnification ×100,000 (B, C) and size distribution (D, E) of PPP-RH-NPs without, or with, high-pressure homogenization treatment. Scale bar is 200 nm. PPP-NPs, PEG-PCL-PEI nanoparticles without loading RH; PPP-RH-NPs, RH-loaded PEG-PCL-PEI nanoparticles. |
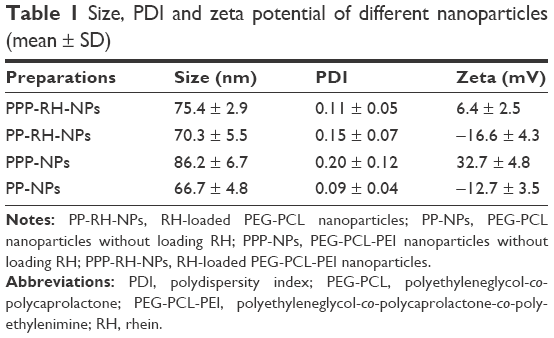 | Table 1 Size, PDI and zeta potential of different nanoparticles (mean ± SD) |
Characteristics of in vitro drug release
The in vitro release profiles of PP-RH-NPs, PPP-RH-NPs and RH-sol were tested as shown in Figure 4. With regard to RH-sol, nearly 90% was released from the solvent within 4 h, while PP-RH-NPs exhibited a biphasic release behavior, such as initial burst release for 3 h with an accumulative release rate (Q) of 55.14%. Then, the release rate became gradually stable, and the accumulative release rate was approximately 75.92% after 48 h. Specifically, PPP-RH-NPs showed a sustained-release profile without apparent burst release. The accumulative release rate of PPP-RH-NPs was approximately 31.26% and 75.92% after 3 and 48 h, respectively. The faster burst release of PP-RH-NPs was probably caused by the fact that the formulation of PP-RH-NPs was produced by hydrophobic interactions. In this case, nanoparticles were not sufficiently stable. With regard to PPP-RH-NPs, ionic interaction existed between carboxylic groups or phenol hydroxyl groups in RH and the repeated amine group units in PEI. Besides hydrophobic interactions, ionic interactions strengthened the carrier–drug interactions and contributed to self-assemble nanoparticles, forming a more stable core with a sustained-release manner.
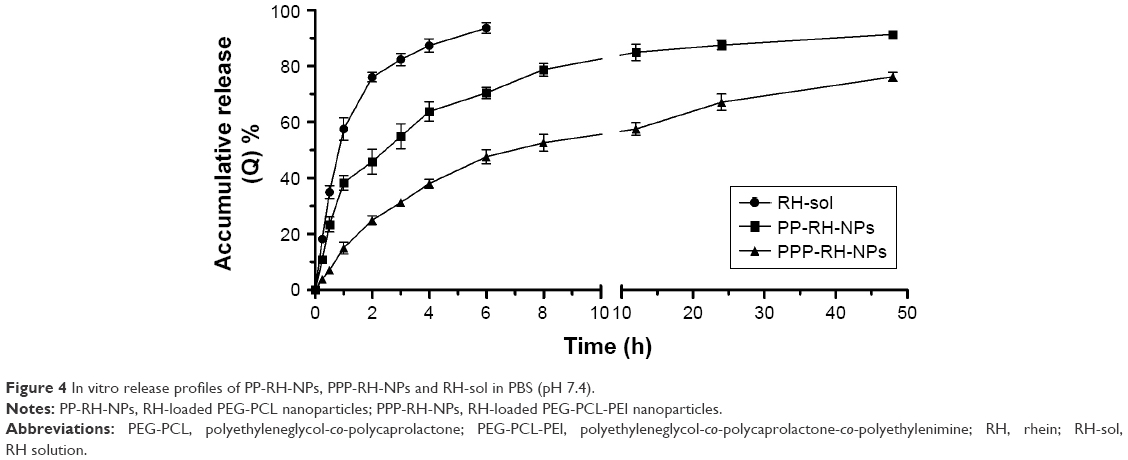 | Figure 4 In vitro release profiles of PP-RH-NPs, PPP-RH-NPs and RH-sol in PBS (pH 7.4). |
The release kinetics of PPP-RH-NPs was predicted via the zero-order, first-order and Higuchi models.52 The kinetic parameters are summarized in Table 2. From our results, it could be concluded that the release profile of PPP-RH-NPs in PBS (pH 7.4) fitted well to the Higuchi model (accumulative release rate = 0.1216t1/2 + 0.0695, R2 = 0.8874) consistent with the sustained-release property.
 | Table 2 Release kinetics of PPP-RH-NPs in PBS (pH 7.4) |
In vitro hemolysis and cytotoxicity assay
The hemolytic potential was evaluated using rabbit RBCs (Figure 5A). Both PP-NPs and PPP-NPs caused hemolysis at high concentrations. The hemolytic activity was negligible (<5%) at a nanoparticle concentration of <0.05 mmol/L, indicating the value below which the hemolysis was avoided. In vitro cytotoxicity on HK-2 cells was determined by the MTT assay (Figure 5B). Cell viability was higher than 90% after PP-NP and PPP-NP treatment for 48 h even at the concentration of 0.1 mmol/L. PPP-NPs possessed higher cell growth inhibition and hemolysis ratio compared to PP-NPs at the same concentration. PEI contained kinds of amino groups, which is an important feature of cationic polymers and cationic liposomes, as well as a disadvantage because of toxicity and hemolysis.53,54 Therapeutic dose could be achieved at a relatively low carrier concentration due to high drug LE and EE. PPP-NPs had good biocompatibility with negligible toxicity and hemolysis at the concentration of 0–0.05 mmol/L.
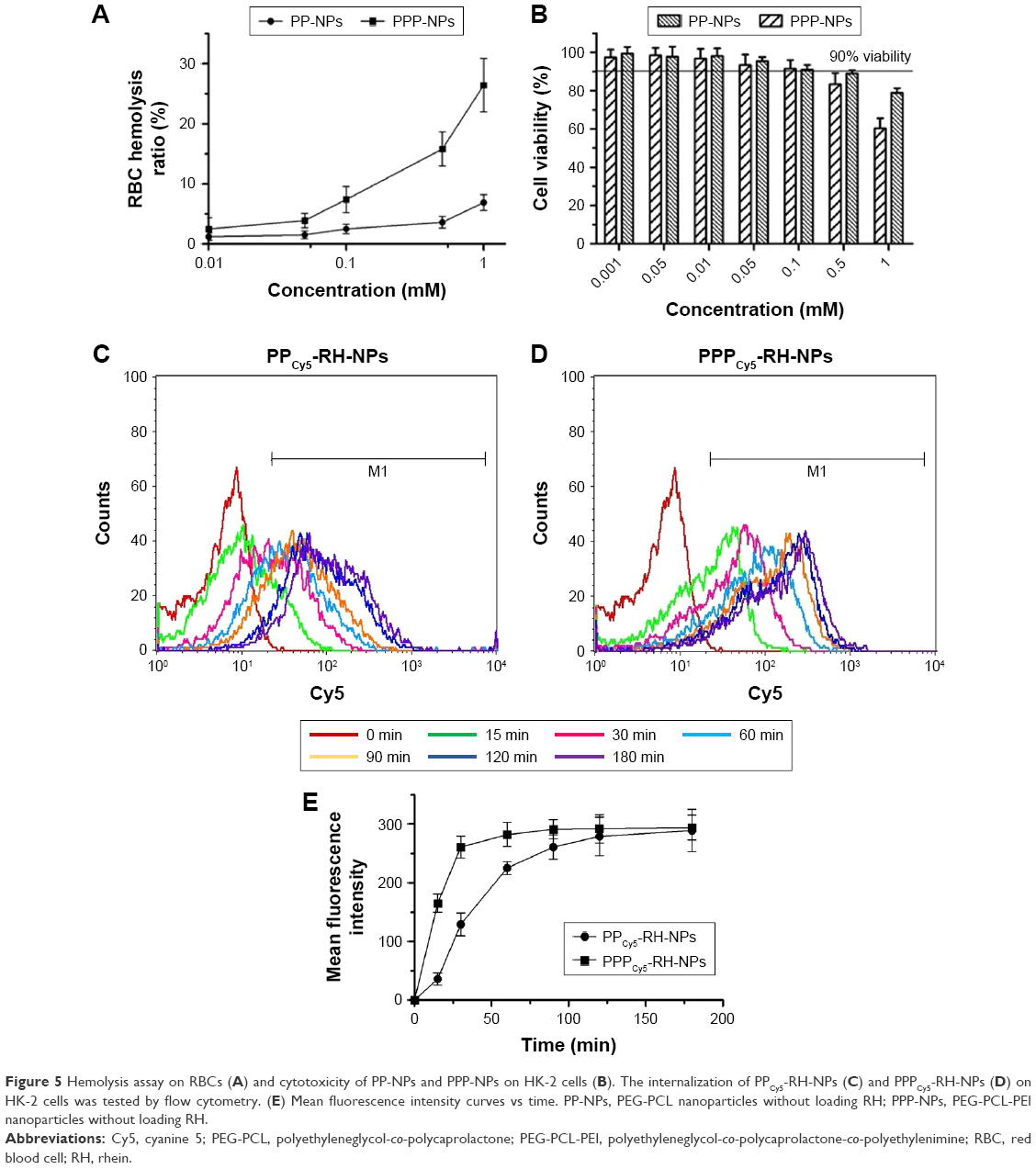 | Figure 5 Hemolysis assay on RBCs (A) and cytotoxicity of PP-NPs and PPP-NPs on HK-2 cells (B). The internalization of PPCy5-RH-NPs (C) and PPPCy5-RH-NPs (D) on HK-2 cells was tested by flow cytometry. (E) Mean fluorescence intensity curves vs time. PP-NPs, PEG-PCL nanoparticles without loading RH; PPP-NPs, PEG-PCL-PEI nanoparticles without loading RH. |
Cell uptake by flow cytometry
Cell uptake of PPCy5-RH-NPs and PPPCy5-RH-NPs was tested by flow cytometry (Figure 5C and D). Both nanoparticles internalized in a time-dependent manner, since the mean fluorescence intensity and Cy5-positive cells increased over time (Figure 5E). Cell uptake of PPPCy5-RH-NPs was up to 60% within 15 min. PPPCy5-RH-NPs and PPCy5-RH-NPs were completely internalized within 30 and 90 min, respectively. Cellular uptake of PPPCy5-RH-NPs was faster than that of PPCy5-RH-NPs. Zeta potential of PPPCy5-RH-NPs was positive approximately 6.4 ± 2.5 mV, while cell membranes were negatively charged due to the high level content of phospholipids and proteins. Internalization of PPPCy5-RH-NPs was probably facilitated by the interaction between positive PPPCy5-RH-NPs and positive-charged or negative-charged membrane.
Subcellular distribution study by confocal laser scanning microscopy
Internalization of nanoparticles and subcellular distribution were visualized by confocal laser scanning microscopy. Lysosomes (green) and Cy5-labeled nanoparticles (red) were merged to form yellow dots. As shown in Figure 6A, only a few PPCy5-RH-NPs could be observed at 15 min, and PPCy5-RH-NPs were located in lysosomes after 1 h of incubation. The yellow dots at 3 h suggested that nanoparticles were not released from endosomes. PPCy5-RH-NPs failed to escape from endosomes quickly, which could result in the loss of pharmacological activity. On the contrary, the cellular uptake and internalization of PPPCy5-RH-NPs were very fast, and the majority of nanoparticles adhered to the cell membrane within 15 min as shown in Figure 6B. The number and intensity of red dots increased with time lapse; PPPCy5-RH-NPs were internalized into lysosomes after 1 h of incubation. Many red dots were not merged with the green ones at 3 h, demonstrating the quick escape from endosomes into the cytosol and showing resistance to the metabolism of endosomes. Protonable residues in PEI had effect on the swelling and disruption of endosomes via the proton sponge effect.42,55 The endosomal escape property ability could promote drug release in the cytosol and targets, thus increasing intracellular pharmacological effects.
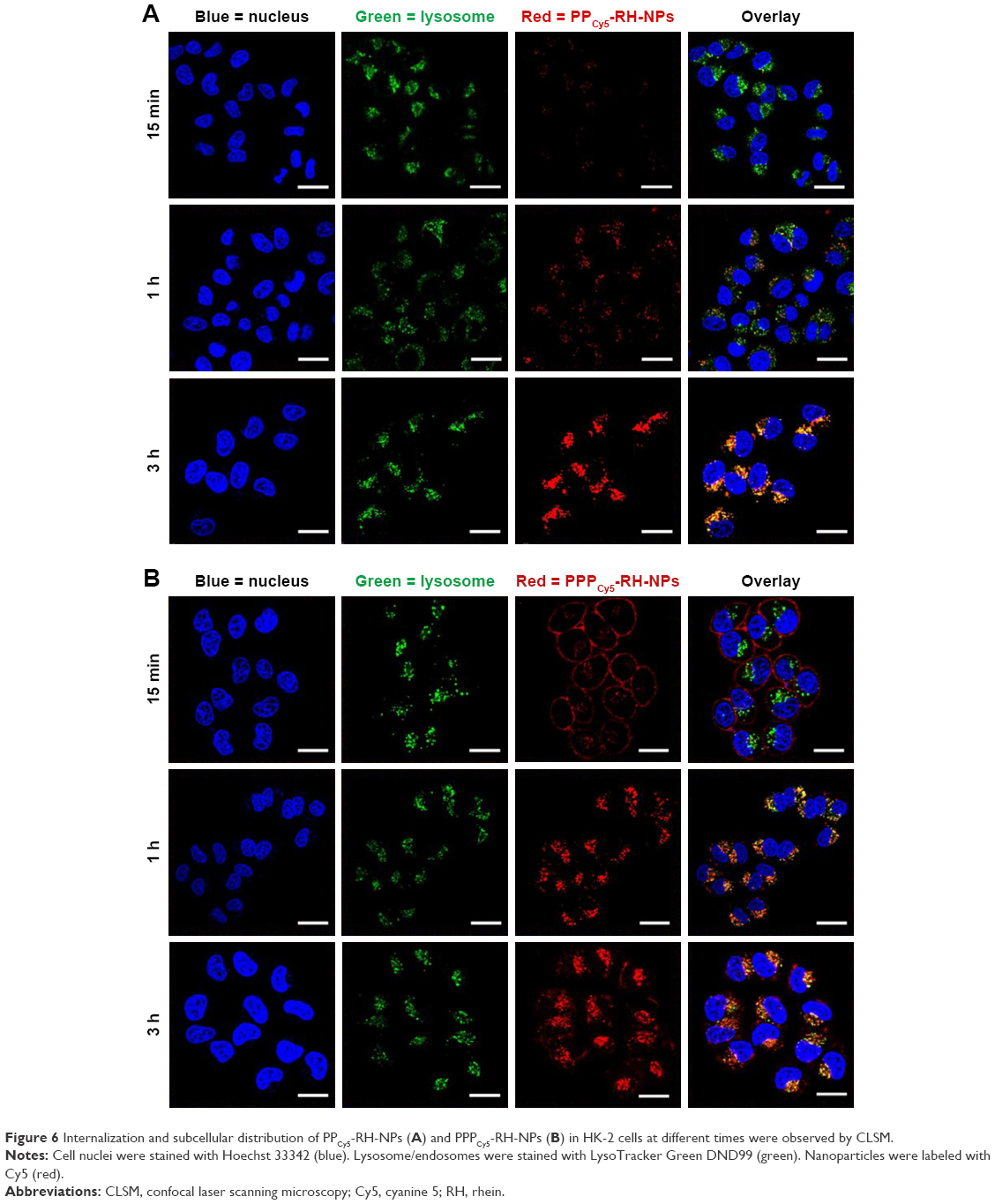 | Figure 6 Internalization and subcellular distribution of PPCy5-RH-NPs (A) and PPPCy5-RH-NPs (B) in HK-2 cells at different times were observed by CLSM. |
DN model establishment
A single high dose intraperitoneal injection of STZ is the classic method to destroy pancreas cells to establish the type I diabetes model. FBG and body weight in mice were monitored to identify whether the model was successfully established. As shown in Figure 7A, FBG in HC mice was maintained at 6.3 ± 0.9 mmol/L. At 3 days after STZ injection, FBG rose to 18.5 ± 2.2 mmol/L. If the FBG was >15 mmol/L, the mice can be considered as diabetes model. With the aggravation of the disease, FBG in model mice continued to increase, reaching and maintaining a value of 23.9 ± 1.7 mmol/L, while body weight (Figure 7B) decreased sharply to 17.7 ± 1.2 g compared to the weight of HC mice of 24.5 ± 1.6 g, due to the loss of glucose and proteins. Therefore, at this point, the diabetic mice were considered to have early DN.
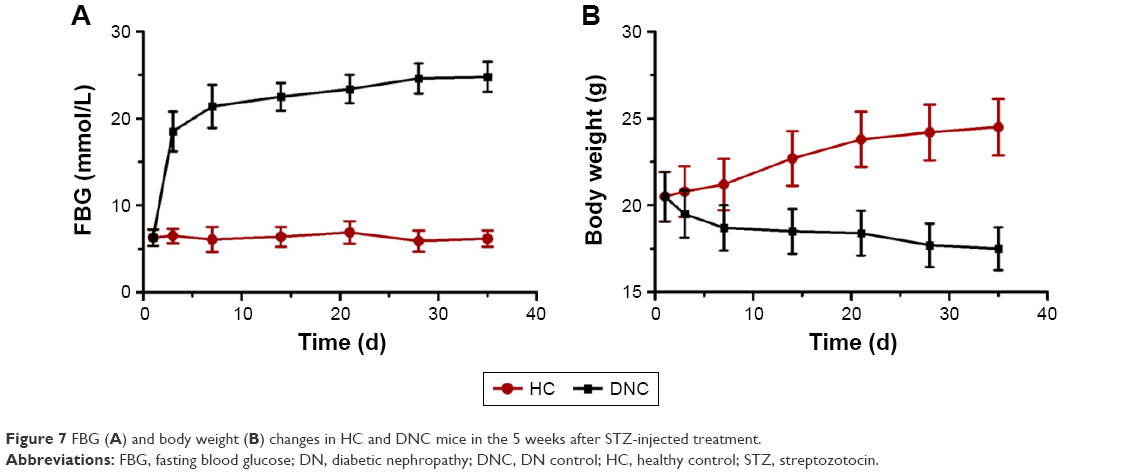 | Figure 7 FBG (A) and body weight (B) changes in HC and DNC mice in the 5 weeks after STZ-injected treatment. |
In vivo fluorescence imaging in DN model mice
Soon after the i.v. injection of PPPCy5-RH-NPs, the mice body surface, without hair, showed strong fluorescence, as shown in Figure 8A (hair has a strong influence on fluorescence, thus it should be removed). As time goes on, PPPCy5-RH-NP distribution into various organs was observed, being metabolized in both HC and DNC mice; thus, the fluorescence intensity gradually weakened. At 6 h, PPPCy5-RH-NPs were mainly accumulated in the chest and epigastrium. To accurately observe the fluorescence distributions in various organs, mice were anatomized and fluorescence was measured by a quantitative analysis instrument. The kidney fluorescence intensity was the highest in DNC mice, and lungs and liver, whereas the highest fluorescence intensity in HC mice was in the liver (Figure 8B). These results suggested that PPPCy5-RH-NPs could target the diseased kidney reaching the highest concentration, providing solid proof on the delivery of drug specifically to the diseased kidney, and consequently improving the therapeutic effect on DN.
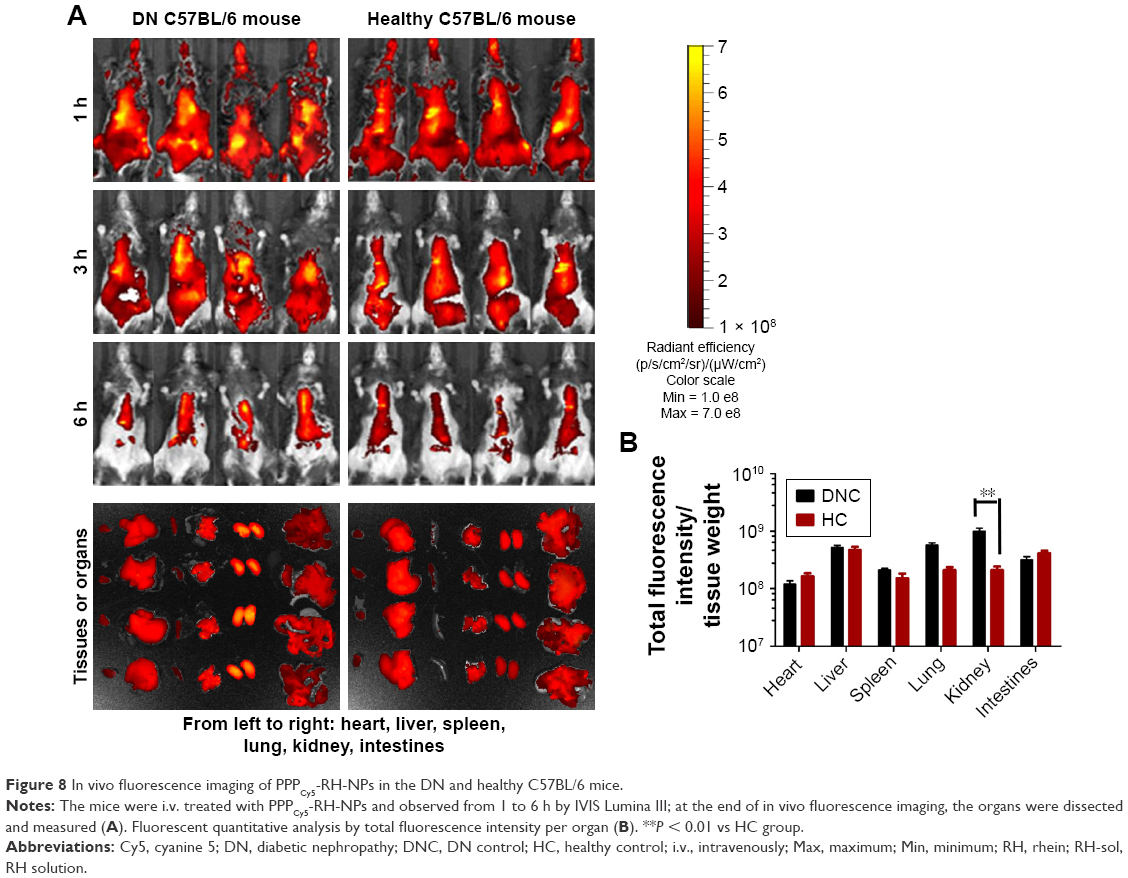 | Figure 8 In vivo fluorescence imaging of PPPCy5-RH-NPs in the DN and healthy C57BL/6 mice. |
Pharmacodynamics study on DN model mice
The change in body weight and FBG is shown in Figure 9A and B. FBG value in the Lantus group mice decreased rapidly and was sustained at 9–12 mmol/L during the treatment period. The body weight of the mice was gradually increased in this group. Mice in the PPP-RH-NP group showed a significant decrease in FBG as compared to the DNC group (17.5 vs 23.9 mmol/L). A body weight increase in the PPP-RH-NP group compared to the DNC group was observed. The FBG and body weight in the RH-sol group remained the same and then changed, only after 10 days. Blood and urine biochemistry parameters were examined at the end of the treatment. As shown in Figure 9C–H, the administration of Lantus, RH-sol and PPP-RH-NPs could decrease certain parameters (24 h UV, 24 h UPE, serum urea nitrogen (SUN), SCr and kidney index) and increase UCr and CCr, but these parameters were still not as good as in HC mice. RH-sol showed the same results as Lantus regarding some similar biochemical parameters, but both were much less than for PPP-RH-NPs. PPP-RH-NPs could protect DN mice from renal dysfunctions, significantly. UCr and CCr of mice in the PPP-RH-NP group were increased to 2.05 ± 0.19 mmol/L and 1.88 ± 0.13 mL/min·kg, respectively, while in the RH-sol group, UCr and CCr were 1.66 ± 0.26 mmol/L and 1.61 ± 0.19 mL/min·kg, respectively. The values of 24 h UV, 24 h UPE, serum urea nitrogen (SUN) and SCr in the PPP-RH-NP group were 1.34 ± 0.28 mL/24 h, 1.98 ± 0.13 mg/24 h, 9.70 ± 1.49 mmol/L and 50.6 ± 8.4 μmol/L, respectively, while in the DNC group, the values were 2.13 ± 0.35 mL/24 h, 3.45 ± 0.46 mg/24 h, 17.6 ± 3.24 mmol/L and 108.7 ± 12.5 μmol/L, respectively.
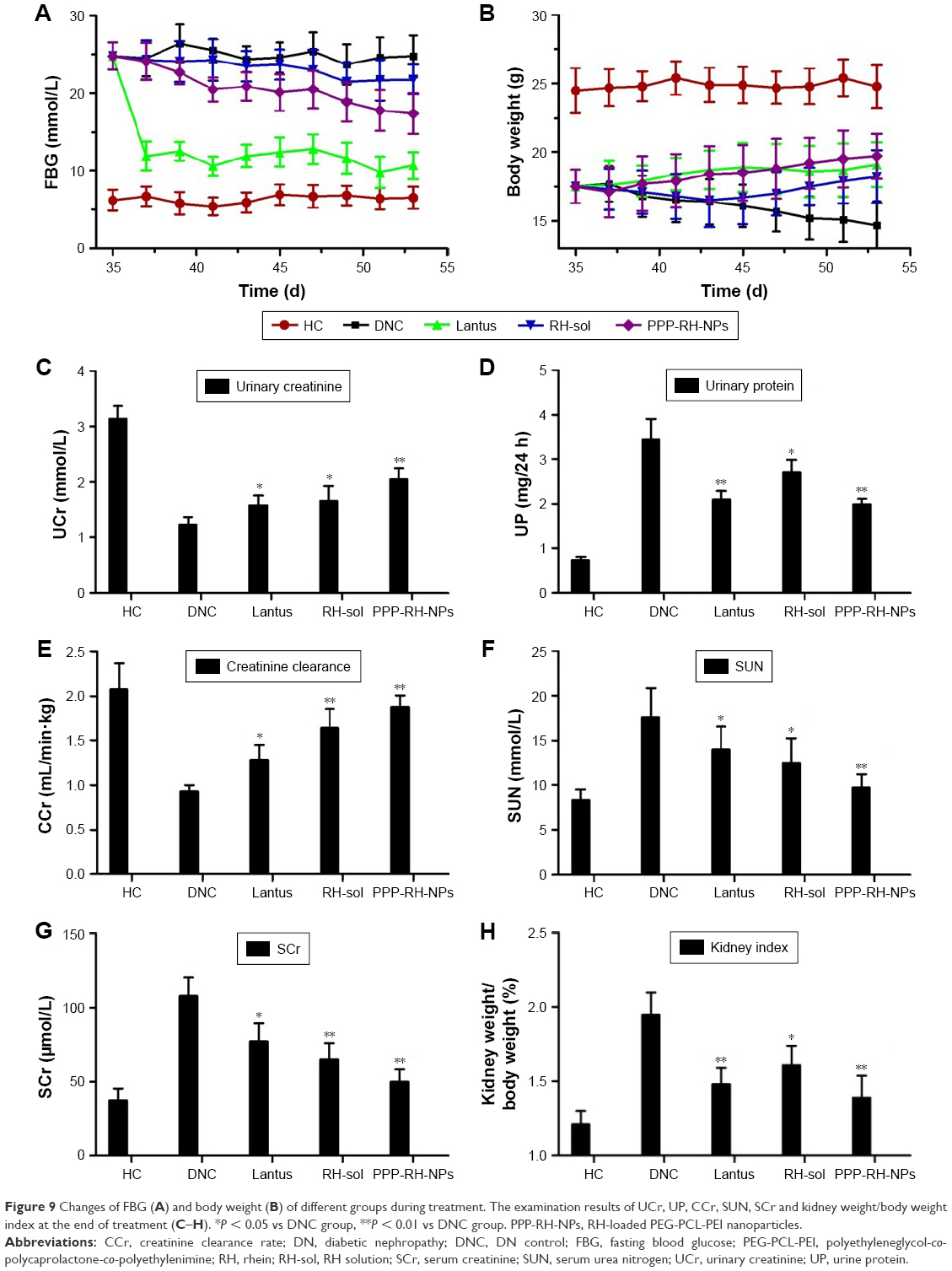 | Figure 9 Changes of FBG (A) and body weight (B) of different groups during treatment. The examination results of UCr, UP, CCr, SUN, SCr and kidney weight/body weight index at the end of treatment (C–H). *P < 0.05 vs DNC group, **P < 0.01 vs DNC group. PPP-RH-NPs, RH-loaded PEG-PCL-PEI nanoparticles. |
H&E staining, PAS staining and IHC of kidneys were carried out to confirm DN pathological features (Figure 10A). In the DNC group, characteristic glomerulus enlargement, glomerulosclerosis, expansion of glomerular mesangial matrix and mesangium, thickened glomerular basement membrane, kidney tubules and capillary vessel lesions and vessel lumen enlargement could be observed. PAS staining on DN tissue shows dark violet stained glomerular basement membrane and mesangium matrix infiltrated and expanded in the capsular space, confirming the proliferation of mesangial cells and matrix, which were in line with the thickened glomerular basement membrane in the H&E staining. FN is one of the representative extracellular matrix proteins in DN process, playing a crucial role in renal fibrosis and glomerulosclerosis.3,56 IHC staining showed much more FN expression in DNC, resulting in its increased secretion by the glomerulus during the DN development. Glomerular basement membrane and mesangial matrix gradually thickened with the accumulation of FN in glomerulus. After treatment with Lantus, RH-sol and PPP-RH-NPs, most of the pathological changes could be improved and resolved to some extent. Lantus and PPP-RH-NPs markedly controlled the glomerular volume, improved the vascular circulation and contributed to the amelioration of lumen enlargement as shown by H&E staining images. Although Lantus lowered FBG, similar results by PAS staining were noticed between the Lantus and DNC groups, suggesting no proliferation inhibition of mesangial matrix and basement membrane, while RH-sol and PPP-RH-NPs exerted a significant inhibitory effect on mesangial matrix proliferation, indicating that lowering blood glucose was not the only efficient therapeutic option in DN treatment. IHC staining showed that Lantus and RH-sol had a slight effect on FN compared to DNC, while PPP-RH-NPs significantly prevented the FN secretion and accumulation and DN deterioration.
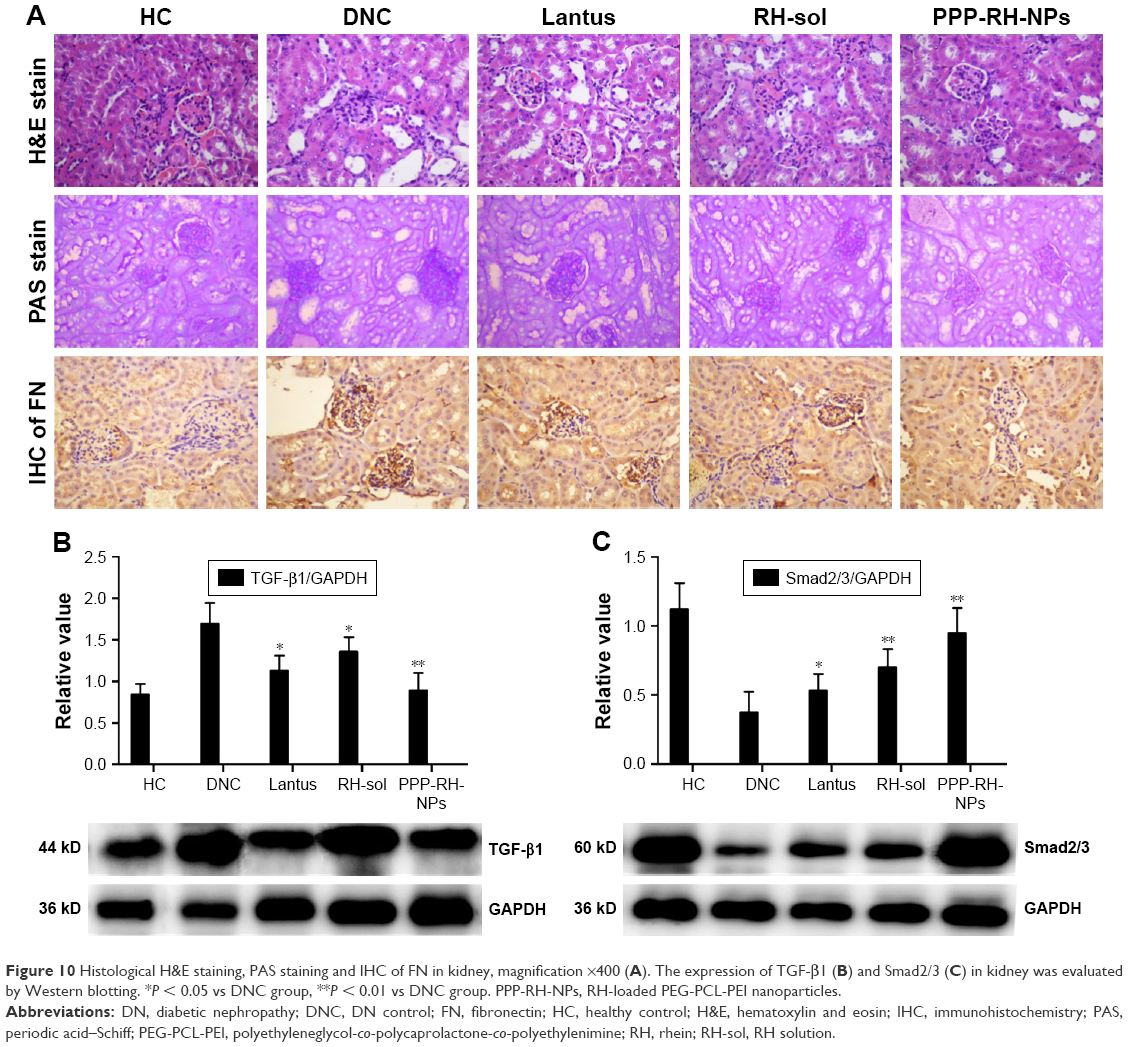 | Figure 10 Histological H&E staining, PAS staining and IHC of FN in kidney, magnification ×400 (A). The expression of TGF-β1 (B) and Smad2/3 (C) in kidney was evaluated by Western blotting. *P < 0.05 vs DNC group, **P < 0.01 vs DNC group. PPP-RH-NPs, RH-loaded PEG-PCL-PEI nanoparticles. |
TGF-β1 and Smad2/3 were representative biomarkers involved in DN pathological processes.2,5,57–59 TGF-β1 and Smad2/3 signaling pathways are crucial intermediary that are involved in increasing blood sugar and release of cytokines and other inflammatory biochemical factors, inducing kidney damage, which is closely related to diabetes renal cell proliferation, glomerular hypertrophy and renal extracellular matrix protein accumulation. Moreover, these signaling pathways are also directly involved in both renal fibrosis and injury of the glomerular filtration barrier.60 TGF-β1 and Smad2/3, Western blotting in kidneys is shown in Figure 10B and C. TGF-β1 expression and Smad2/3 phosphorylation were suppressed to some degree after Lantus, RH-sol and PPP-RH-NP treatment compared to the DNC group. More specifically, PPP-RH-NPs had more distinct effects on inhibiting the aberrant expression of TGF-β1 and preventing the signal transduction by interfering in the phosphorylation of Smad2/3, than other groups. Lantus had a greater inhibitory effect on TGF-β1 expression than RH-sol, while RH-sol showed enhanced dephosphorylation of Smad2/3 than Lantus, indicating the different therapeutic mechanisms. In other words, Lantus helped with lowering blood glucose levels to reduce the burden on the kidney, whereas RH could regulate the abnormal expression of proteins in kidney to improve renal functions.
Discussion
Nanotechnology creates vehicles that improve drug-solubilizing ability and release properties as well as prolonging their circulation time, which are necessary to improve the drug curative effect.22,38 With regard to amphiphilic polymers, hydrophobic drugs are mostly encapsulated into the micellar cores and a small number of drugs are localized at the micellar outer layers or surfaces, leading to an insufficient drug-loading efficacy.61 Thus, a high drug LE is one of the most important factors for a micelle delivery system. Although high EE can be achieved using mesostructured materials and inorganic nanoparticles, they suffer from difficulty in biodegradation and poor biocompatibility when used in clinical practice.62 Currently, polymer–drug conjugates, supramolecular assemblies and aggregations, metal complex systems and ionic pairs are effective approaches for obtaining high drug LE. In the current study, RH had a slightly acidic property because of carboxylic groups and phenol hydroxyl groups. The repeated amine groups in PEI block were protonated under physiological conditions. Besides hydrophobic interactions, ionic interaction between RH and PEI strengthened the carrier–drug interactions, resulting in a relatively high entrapment efficiency and drug LE.
Pharmacodynamic action depends on the final concentration of drug released in the targets, but after circulation, distribution and penetration, most nanoparticles are directly endocytosed in endosomes/lysosomes of cells, which are not the pharmacological targets.38,41,63 Hydrolytic enzymes in endosomes/lysosomes would break down any type of exogenous and endogenous material. If the nanoparticles are unable to quickly escape from endosomes, the low pH and enzymes can contribute to the metabolism and degradation of the drug. Therefore, the next nanoparticle generation should possess suitable stability and high endosome escape potential for an effective drug delivery and use in clinical practice. PEI has been widely used for gene transfer because of the proton sponge mechanism of various types of amine groups over a wide pH range. Amine groups are protonated when they are trapped in endosomes, resulting in more protons pumping into endosomes. Chloride ions enter passively with increasing osmotic pressure and endosomes are finally disrupted.41,42,44,55 In the current study, PEI was conjugated to PEG-PCL polymer to synthesize triblock amphiphilic PEG-PCL-PEI polymer. With the aid of PEI, PEG-PCL-PEI could possess the ability of endosomal escape and enhance cell uptake, thus resulting in an improved pharmacological function in transporting drugs.
Currently, most of the main nanomedical studies focus on the tumor diagnosis and treatment because of the advantage of enhanced permeability and retention (EPR) effect in tumors,20,64 which can facilitate the intercept and uptake of nanoparticles. Despite that, the overall delivery efficiency of drugs accumulated into tumor is less than 1%.24 In addition, targeting drug delivery to other organs and tissues is more difficult and poorer because of the existence of physical barriers (eg, blood–brain barrier) and chemical barriers (eg, liver metabolic enzymes), thus many researchers applied receptor-mediated, antibody-guided or peptide-homing functional nanoparticles to increase accumulation in the target lesions.22,38 Compared to other organs, kidney is the most important filtering and clearing organ of the body. Renal blood flow is high, and nanoparticles can easily reach the kidney. Kidney has a specific transport system, which can redistribute the drug from the carrier to glomerulus and renal tubules. In addition, it has some proprietary enzymes that can metabolize and dispose of many endogenous and exogenous substances. Furthermore, it is a target organ for many hormones, providing a favorable condition for the treatment and control of renal diseases by renal targeting.
One of the most noticeable kidney properties is the complex filtering system composed of glomerular basement membrane, mesangial matrix, podocyte and mesangial cells.26,30 In normal renal tissue, the channel filter system can filter and drain off nanoparticles of size <10 nm, while nanoparticles >75 nm cannot be filtered.29,31 In addition, the channel size of the filtration system increases in different stages or in different time periods of kidney disease. For example, in DN, the canal pores can continuously increase and be injured when filtering macromolecular proteins.30 Moreover, a type of albumin nanoparticle with defined sizes of ~95 nm to deliver celastrol selectively to mesangial cells has been recently reported.35 Hence, as shown in the real-time biodistribution in DN model mice, controlling particle size is an effective approach to realize renal target delivery. However, accumulation in peritubular capillaries does not seem to correlate with the size of nanoparticles, showing conspicuous accumulation characteristics in different portions of the kidney. Besides, the podocytes, mesangial, endothelial and epithelial cells of the glomerulus are involved in most kidney pathological processes, because of their different functions in kidney disease duration and progression; also needed is to seek for a new strategy for cell-targeted delivery, such as protein- and peptide-based carrier systems.65 The charge is another important factor affecting the effect of nanoparticles. Negative charge can prolong blood circulation time of nanoparticles and reduce the retention and clearance by the reticular endothelial system, but the negative charge does not trigger endocytosis to allow the uptake by cells. On the contrary, positive charges do not remain long in blood circulation, but this disadvantage can be amended by the aid of polymer PEG chains. Hydrophilic brushes can markedly reduce recognition by the reticular endothelial system, and positive charge can promote cellular uptake efficiently and improve drug pharmacodynamic action. Cell uptake and internalization of PPPCy5-RH-NPs were completely finished within 30 min, whereas those of PPCy5-RH-NPs were inefficient. Therefore, in this study, the nanoparticles with particle size of 75.4 ± 2.9 nm and a potential of 6.4 ± 2.5 mV were used to deliver RH specifically to the kidney, showing better therapeutic effect on DN.
DN is a severe diabetic microvascular complication, and it is related to metabolic disorders (including sugar metabolism and creatinine metabolism), hemodynamics (renin angiotensin system disorder), oxidative stress (increased reactive oxygen species [ROS]), inflammation (including chemokine CCL2, CX3CL1 and CCL5, adhesion molecule and NF-κB) and genetic factors, involving several cytokines and signaling pathways and their respective functions and cross-functions.1,4 The main strategies for the clinical DN treatment include the control of blood glucose, blood pressure, blood fat and administration of corticoid. Although current treatments have certain therapeutic effect on DN patients, the long-term outcome is still undesirable, and deterioration of kidney and related complications still appear, eventually leading to kidney failure and death. In terms of the molecular mechanism, DN development is associated with signaling pathways such as TGF-β1 and Smad2/3 that serve as intermediaries inducing kidney damage. In addition, these signaling pathways are also directly involved in both renal fibrosis and injury of the glomerular filtration barrier.5,57–59 Abnormal expression of TGF-β1 could activate and initiate the signaling pathways and then promote Smad2/3 phosphorylation to transmit signals related to diabetes renal cell proliferation, glomerular hypertrophy and renal extracellular matrix protein accumulation.60 Therefore, TGF-β1 and Smad2/3 signaling pathways represent important therapeutic molecular targets. The latest strategies in DN treatment are mainly focused on (anti-) microRNA gene drugs (eg, miR200b, miR29b, miR93, miR192 and miR146a).9,56,66–68 The microRNA can directly interfere with the pathogenic factors and mRNA transcription and expression to prevent the worsening of kidney disease. However, the extremely easy degradation by enzymes and uncontrollable off-target phenomenons of genetic drugs make their use difficult in clinical trials. RH could not only decrease the levels of FBG, creatinine, BUN, urine protein and the intensity of oxidative stress, but could also interfere with TGF-β1 and Smad2/3 signaling pathways and protect against DN through a multi-target therapy. By loading and delivering it in PEG-PCL-PEI carriers, PPP-RH-NPs could deliver RH specifically to kidney, further improving the pharmacological effects on DN.
Conclusion
We synthesized a novel nontoxic PEG-PCL-PEI polymer and PPP-RH-NPs. In the STZ-induced DN model, PPP-RH-NPs showed a good kidney distribution, consequently ameliorating a number of pathological indicators and reversing the worsening DN. Therefore, these nanoparticles improved pharmacological effects of RH on DN, opening new horizons in the use of RH. More importantly, our data provided a strong platform for targeting drug delivery against nephropathy including DN, promoting further clinical and translational research on nanoparticles in kidney disease.
Acknowledgment
This study was financially supported by the Science and Technology Program on Traditional Chinese Medicine of Zhejiang Province (No 2017ZB031), Science and Technology Program on Medicine and Health of Zhejiang Province (No 2017KY511) and National Science Foundation of China (No 81373982).
Disclosure
The authors report no conflicts of interest in this work.
References
Fernandez-Fernandez B, Ortiz A, Gomez-Guerrero C, Egido J. Therapeutic approaches to diabetic nephropathy-beyond the RAS. Nat Rev Nephrol. 2014;10(6):325–346. | ||
Kato M, Natarajan R. Diabetic nephropathy-emerging epigenetic mechanisms. Nat Rev Nephrol. 2014;10(9):517–530. | ||
Hu C, Sun L, Xiao L, et al. Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr Med Chem. 2015;22(24):2858–2870. | ||
Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7(6):327–340. | ||
Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395–423. | ||
Martínez-Castelao A, Navarro-González JF, Górriz JL, de Alvaro F. The concept and the epidemiology of diabetic nephropathy have changed in recent years. J Clin Med. 2015;4(6):1207–1216. | ||
Reddy MA, Natarajan R. Recent developments in epigenetics of acute and chronic kidney diseases. Kidney Int. 2015;88(2):250–261. | ||
Saran R, Robinson B, Abbott KC, et al. US Renal Data System 2016 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis. 2017;69(3 suppl 1):A7–A8. | ||
Lorenzen JM, Haller H, Thum T. MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat Rev Nephrol. 2011;7(5):286–294. | ||
Guo XH, Liu ZH, Dai CS, Li H, Liu D, Li LS. Rhein inhibits renal tubular epithelial cell hypertrophy and extracellular matrix accumulation induced by transforming growth factor beta1. Acta Pharmacol Sin. 2001;22(10):934–938. | ||
Peng L, Yang J, Ning C, et al. Rhein inhibits integrin-linked kinase expression and regulates matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio in high glucose-induced epithelial-mesenchymal transition of renal tubular cell. Biol Pharm Bull. 2012;35(10):1676–1685. | ||
Gao Q, Qin WS, Jia ZH, et al. Rhein improves renal lesion and ameliorates dyslipidemia in db/db mice with diabetic nephropathy. Planta Med. 2010;76(1):27–33. | ||
Zheng JM, Zhu JM, Li LS, Liu ZH. Rhein reverses the diabetic phenotype of mesangial cells over-expressing the glucose transporter (GLUT1) by inhibiting the hexosamine pathway. Br J Pharmacol. 2008;153(7):1456–1464. | ||
Xu S, Lv Y, Zhao J, Wang J, Zhao X, Wang S. Inhibitory effects of Shenkang injection and its main component emodin on the proliferation of high glucose-induced renal mesangial cells through cell cycle regulation and induction of apoptosis. Mol Med Rep. 2016;14(4):3381–3388. | ||
Shi S, Cai Y, Cai X, et al. A network pharmacology approach to understanding the mechanisms of action of traditional medicine: Bushenhuoxue formula for treatment of chronic kidney disease. PLoS One. 2014;9(3):e89123. | ||
He D, Lee L, Yang J, Wang X. Preventive effects and mechanisms of rhein on renal interstitial fibrosis in obstructive nephropathy. Biol Pharm Bull. 2011;34(8):1219–1226. | ||
Zeng CC, Liu X, Chen GR, et al. The molecular mechanism of rhein in diabetic nephropathy. Evid Based Complement Alternat Med. 2014;2014:487097. | ||
Wang J, Zhao Y, Xiao X, et al. Assessment of the renal protection and hepatotoxicity of rhubarb extract in rats. J Ethnopharmacol. 2009;124(1):18–25. | ||
He LN, Yang AH, Cui TY, et al. Reactive metabolite activation by CYP2C19-mediated rhein hepatotoxicity. Xenobiotica. 2015;45(4):361–372. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602–2663. | ||
Pelaz B, Alexiou C, Alvarez-Puebla RA, et al. Diverse applications of nanomedicine. ACS Nano. 2017;11(3):2313–2381. | ||
Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13(11):813–827. | ||
Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1(5):16014. | ||
Kim YK, Kwon JT, Jiang HL, Choi YJ, Cho MH, Cho CS. Kidney-specific peptide-conjugated poly (ester amine) for the treatment of kidney fibrosis. J Nanosci Nanotechnol. 2012;12(7):5149–5154. | ||
Lin Y, Li Y, Wang X, Gong T, Zhang L, Sun X. Targeted drug delivery to renal proximal tubule epithelial cells mediated by 2-glucosamine. J Control Release. 2013;167(2):148–156. | ||
Zhang Z, Zheng Q, Han J, et al. The targeting of 14-succinate triptolide-lysozyme conjugate to proximal renal tubular epithelial cells. Biomaterials. 2009;30(7):1372–1381. | ||
Zuckerman JE, Gale A, Wu P, Ma R, Davis ME. siRNA delivery to the glomerular mesangium using polycationic cyclodextrin nanoparticles containing siRNA. Nucleic Acid Ther. 2015;25(2):53–64. | ||
Kamaly N, He JC, Ausiello DA, Farokhzad OC. Nanomedicines for renal disease: current status and future applications. Nat Rev Nephrol. 2016;12(12):738–753. | ||
Wang J, Masehi-Lano JJ, Chung EJ. Peptide and antibody ligands for renal targeting: nanomedicine strategies for kidney disease. Biomater Sci. 2017;5(8):1450–1459. | ||
Choi CH, Zuckerman JE, Webster P, Davis ME. Targeting kidney mesangium by nanoparticles of defined size. Proc Natl Acad Sci U S A. 2011;108(16):6656–6661. | ||
Zuckerman JE, Davis ME. Targeting therapeutics to the glomerulus with nanoparticles. Adv Chronic Kidney Dis. 2013;20(6):500–507. | ||
Du B, Jiang X, Das A, et al. Glomerular barrier behaves as an atomically precise bandpass filter in a sub-nanometre regime. Nat Nanotechnol. 2017;12(11):1096–1104. | ||
Bruni R, Possenti P, Bordignon C, et al. Ultrasmall polymeric nanocarriers for drug delivery to podocytes in kidney glomerulus. J Control Release. 2017;255:94–107. | ||
Guo L, Luo S, Du Z, et al. Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis. Nat Commun. 2017;8(1):878. | ||
Yuan Z, Gu X. Preparation, characterization, and in vivo study of rhein-loaded poly (lactic-co-glycolic acid) nanoparticles for oral delivery. Drug Des Devel Ther. 2015;9:2301–2309. | ||
Wei Y, Luo X, Guan J, et al. Biodegradable nanoparticles for improved kidney bioavailability of rhein: preparation, characterization, plasma, and kidney pharmacokinetics. Drug Dev Ind Pharm. 2017;43(11):1885–1891. | ||
Sun Q, Zhou Z, Qiu N, Shen Y. Rational design of cancer nanomedicine: nanoproperty integration and synchronization. Adv Mater. 2017;29(14). | ||
Seo SJ, Chen M, Wang H, Kang MS, Leong KW, Kim H-W. Extra-and intra-cellular fate of nanocarriers under dynamic interactions with biology. Nano Today. 2017;14:84–99. | ||
Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. J Control Release. 2014;190:485–499. | ||
Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151(3):220–228. | ||
Akinc A, Thomas M, Klibanov AM, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J Gene Med. 2005;7(5):657–663. | ||
Boletta A, Benigni A, Lutz J, Remuzzi G, Soria MR, Monaco L. Nonviral gene delivery to the rat kidney with polyethylenimine. Hum Gene Ther. 1997;8(10):1243–1251. | ||
Godbey WT, Wu KK, Mikos AG. Tracking the intracellular path of poly (ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci U S A. 1999;96(9):5177–5181. | ||
Astafieva I, Zhong XF, Eisenberg A. Critical micellization phenomena in block polyelectrolyte solutions. Macromolecules. 1993;26(26):7339–7352. | ||
Dobrovolskaia MA, Clogston JD, Neun BW, Hall JB, Patri AK, McNeil SE. Method for analysis of nanoparticle hemolytic properties in vitro. Nano Lett. 2008;8(8):2180–2187. | ||
Amin K, Dannenfelser RM. In vitro hemolysis: guidance for the pharmaceutical scientist. J Pharm Sci. 2006;95(6):1173–1176. | ||
Breyer MD, Böttinger E, Brosius FC III, et al; AMDCC. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16(1):27–45. | ||
Furman BL. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc Pharmacol. 2015;70:5.47.1–20. | ||
Sigaudo-Roussel D, Fromy B, Saumet JL. Diabetic neuropathy in animal models. Drug Discov Today Dis Models. 2007;4(1):39–44. | ||
Tesch GH, Allen TJ. Rodent models of streptozotocin-induced diabetic nephropathy (methods in renal research). Nephrology. 2007;12(3):261–266. | ||
Grassi M, Grassi G. Application of mathematical modeling in sustained release delivery systems. Expert Opin Drug Deliv. 2014;11(8):1299–1321. | ||
Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24(7):1121–1131. | ||
Mayr J, Bachl J, Schlossmann J, Díaz DD. Antimicrobial and hemolytic studies of a series of polycations bearing quaternary ammonium moieties: structural and topological effects. Int J Mol Sci. 2017;18(2):303. | ||
Benjaminsen RV, Mattebjerg MA, Henriksen JR, et al. The possible “proton sponge” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol Ther. 2013;21(1):149–157. | ||
Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2012;23(3):458–469. | ||
Meng XM, Tang PMK, Li J, et al. TGF-β/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82. | ||
Loeffler I, Wolf G. Transforming growth factor-β and the progression of renal disease. Nephrol Dial Transplant. 2013;29(Suppl 1):i37–i45. | ||
Meng XM, Chung ACK, Lan HY. Role of the TGF-β/BMP-7/Smad pathways in renal diseases. Clin Sci. 2013;124(4):243–254. | ||
Meng X, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–338. | ||
Reddy B, Yadav HKS, Nagesha DK, Raizaday A, Karim A. Polymeric micelles as novel carriers for poorly soluble drugs. J Nanosci Nanotechnol. 2015;15(6):4009–4018. | ||
Shi Y, Miller ML, Di Pasqua AJ. Biocompatibility of mesoporous silica nanoparticles? Comments Inorg Chem. 2016;36(2):61–80. | ||
Martens TF, Remaut K, Demeester J, De Smedt S, Braeckmans K. Intracellular delivery of nanomaterials: how to catch endosomal escape in the act. Nano Today. 2014;9(3):344–364. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Dolman ME, Harmsen S, Storm G, Hennink WE, Kok RJ. Drug targeting to the kidney: advances in the active targeting of therapeutics to proximal tubular cells. Adv Drug Deliv Rev. 2010;62(14):1344–1357. | ||
Bowen T, Jenkins RH, Fraser DJ. MicroRNAs, transforming growth factor beta-1, and tissue fibrosis. J Pathol. 2013;229(2):274–285. | ||
Gomez IG, MacKenna DA, Johnson BG, et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. 2015;125(1):141–156. | ||
Trionfini P, Benigni A, Remuzzi G. MicroRNAs in kidney physiology and disease. Nat Rev Nephrol. 2015;11(1):23–33. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.