")
Back to Journals » International Journal of Nanomedicine » Volume 12
Ketoprofen-loaded polymer carriers in bigel formulation: an approach to enhancing drug photostability in topical application forms
Authors Andonova V , Peneva P, Georgiev GS, Toncheva VT, Apostolova E, Peychev Zh , Dimitrova S, Katsarova M, Petrova N, Kassarova M
Received 4 May 2017
Accepted for publication 6 July 2017
Published 26 August 2017 Volume 2017:12 Pages 6221—6238
DOI https://doi.org/10.2147/IJN.S140934
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Velichka Andonova,1,2 Petya Peneva,1,2 George S Georgiev,3 Vencislava T Toncheva,3 Elisaveta Apostolova,4 Zhivko Peychev,5 Stela Dimitrova,6 Mariana Katsarova,6 Nadia Petrova,7 Margarita Kassarova1,2
1Department of Pharmaceutical Sciences, Faculty of Pharmacy, Medical University-Plovdiv, 2Technological Center for Emergency Medicine (TCEMED), Plovdiv, 3Faculty of Chemistry and Pharmacy, Sofia University “St Kliment Ohridski”, Sofia, 4Department of Pharmacology and Drug Toxicology, Faculty of Pharmacy, Medical University-Plovdiv, 5Department of Medical Informatics, Biostatistics and e-learning, Faculty of Public Health, Medical University-Plovdiv, 6Department of Chemistry and Biochemistry, Faculty of Pharmacy, Medical University-Plovdiv, Plovdiv, 7Institute of Mineralogy and Crystallography, Bulgarian Academy of Sciences, Sofia, Bulgaria
Abstract: The purpose of the study was to investigate the stability and biopharmaceutical characteristics of ketoprofen, loaded in polymeric carriers, which were included into a bigel in a semisolid dosage form. The polymer carriers with in situ-included ketoprofen were obtained by emulsifier-free emulsion polymerization of the monomers in aqueous medium or a solution of the polymers used. The morphological characteristics of the carriers, the in vitro release and the photochemical stability of ketoprofen were evaluated. The model with optimal characteristics was included in a bigel formulation. The bigel was characterized in terms of pH, rheological behavior, spreadability, and in vitro drug release. Acute skin toxicity, antinociceptive activity, anti-inflammatory activity, and antihyperalgesic effects of the prepared bigel with ketoprofen-loaded polymer carrier were evaluated. The carriers of ketoprofen were characterized by a high yield and drug loading. The particle size distribution varied widely according to the polymer used, and a sustained release was provided for up to 6 hours. The polymer mixture poly(vinyl acetate) and hydroxypropyl cellulose as a drug carrier, alone or included in the bigel composition, improved the photostability of the drug compared with unprotected ketoprofen. The bigel with ketoprofen-loaded particles provided sustained release of the drug and had optimal rheological parameters. In vivo experiments on the bigel showed no skin inflammation or irritation. Four hours after its application, a well-defined analgesic, anti-inflammatory, and antihyperalgesic effect was registered. The polymer mixture of poly(vinyl acetate) and hydroxypropyl cellulose as a carrier of ketoprofen and the bigel in which it was included provided an enhanced photostability and sustained drug release.
Keywords: antihyperalgesic effect, antinociceptive activity, biphasic systems, carrageenan-induced edema, emulsifier-free radical polymerization
Introduction
Ketoprofen ([RS]2-[3-benzoylphenyl]-propionic acid) (KP) is widely used as a non-steroidal anti-inflammatory drug (NSAID). Various forms of KP are available in the pharmaceutical market: coated tablets, capsules, gels for topical application, transdermal patches, liquid spray, and solutions for injection. It is important to note that their availability is subject to change. Such is the case with the topical application gel: once an over-the-counter medicine, it is now a prescription drug in many countries around the globe. This restriction is due to the fact that it has been established that topically applied KP can induce photosensitivity. It includes both phototoxic and photoallergic activity.1 In an experimental study, it was found that benzoyl radical is the key structure that provokes photosensitivity and the photo cross-reactivity of KP, suprofen and tiaprofenic acid.2 As that radical is common in benzophenone ultraviolet (UV) filters, cross-reactions are familiar with sunscreens containing mainly oxybenzone.3 However, the instability of KP under UV/visible light and the subsequent formation of degradation products are known to cause photosensitivity after topical application.4,5 It has been found that TiO2 inclusion in fabric backing can improve photostability, reduce photodegradation and increase photo safety of the drug in the KP-loaded patch.5
It is well known that polymeric nanocarriers can improve the stability of the drug loaded, provide increased capacity for hydrophobic drug dissolution, improve bioavailability and ensure controlled release in the target biological tissues.6–10 For example, pomegranate seed oil nanoemulsions containing KP and stabilized by pullulan as a polymeric emulsifier, can improve drug photostability and have better drug solubility.7 In another study, stable KP-loaded chitosan (CH) microparticles were obtained, and the drug content was found to be more than 97.5% at the end of the first month in accelerated conditions.8
The choice of an appropriate emulsifier and its concentration is an essential step in the preparation of drug nanocarriers and can have a critical influence on their stability, bio-tolerability, and biocompatibility. It is widely agreed that the usage of emulsifiers affects drug penetration, absorption, and pharmacokinetics in general.10 On the other hand, the imprudent usage of emulsifiers may lead to severe toxic effects.11
Bigels are innovative formulations that constitute 2-phase structured systems obtained by mixing hydrogel and organogel. These systems can overcome the main disadvantages of both types of gels, namely, hydrogel’s limited ability to cross the lipophilic barriers of the skin and organogel’s low patient compliance, that is due to its stickiness and oily residues.12 At the same time, the systems have the benefits of both gel forms: 1) a high ability to accommodate both hydrophilic and lipophilic drugs; 2) an ensured topical or transdermal drug delivery; 3) an optional controlled drug delivery; and 4) an increased patient compliance.12,13 For example, Rhee et al studied an oleo-hydrogel formulation of KP, characterized by an enhanced transdermal drug permeation and skin absorption.14 In another study, carbopol-based bigels for topical delivery of metronidazole (Aarti drugs Ltd., Mumbai, India) for the treatment of bacterial vaginosis were investigated.15 A positive correlation was found between the strength of the bigels and the amount of organogel in them.
Natural (sodium alginate) and synthetic (hydroxypropyl methylcellulose) polymer was used for the preparation of polymer-fish oil bigel (hydrogel/oleogel colloidal mixture) as a transdermal drug delivery vehicle.16 The following points were noted: the bigel played a stabilizing role on the fish oil fatty acids, demonstrated excellent mechanical properties, and the drug permeation was enhanced. Olive oil, sesame oil, and fish oil are the oils most commonly used for the preparation of bigels.13,15–17 Oleogels prepared from these oils are combined with hydrogels of natural and synthetic polymers such as guar gum, gelatin, sodium alginate, hydroxypropyl methylcellulose, and carbopol.12,15–17
In a previous study, we established the stabilizing role of poly(vinyl acetate) (pVAc) nanocarriers on the included drug (indomethacin) and the possibility of a controlled release, provided by copolymers and polymeric mixtures with hydrophilic polymers.18–20 In the present study, different in situ-loaded polymer carriers of KP were obtained by one stage emulsifier-free radical homopolymerization of vinyl acetate (VAc) and copolymerization of VAc and 2-(dimethylamino) ethyl methacrylate (DMAEMC). The influence on the stability and biopharmaceutical characteristics of KP of the addition of hydrophilic polymers ([CH], carbomer [Cbp], and hydroxypropyl cellulose [HPC]) to the polymerization mixture was evaluated. Relying on the beneficial features of bigels, an in situ KP-loaded polymer carrier with optimal characteristics was included in a Cbp/almond oil bigel. All polymers used in this study are well known and with proven biocompatibility.21 Our attention was drawn to certain properties of almond oil. Fatty acid composition analysis made by Venkatachalam and Sathe indicated that oleic acid (C18:1) is the main constituent of monounsaturated lipids (62%) while linoleic acid (C18:2) is the major polyunsaturated fatty acid (29%).22 Furthermore, Hussain et al found that almond oil in various concentrations significantly enhances the penetration of KP in transdermal gels and patch across synthetic membrane/rabbit skin.23
The purpose of the study was to investigate the stability and biopharmaceutical characteristics of KP, in situ loaded into a homopolymer of VAc, a copolymer of VAc and DMAEMC and polymeric mixtures of pVAc with CH, Cbp, and HPC. The KP-loaded polymeric carrier with the optimal features was included into a Cbp/almond oil bigel as a vehicle for semisolid dosage forms, and their pharmacological effects were evaluated.
Material and methods
Materials
All materials used in the research such as KP ((2RS)-2-(3-benzoylphenyl)propanoic acid ≥99%; Sigma-Aldrich Chemie GmbH, Schnelldorf, Germany), vinyl acetate (VAc, monomers, Sigma-Aldrich Chemie GmbH), (2-dimethylaminoethyl) methacrylate (DMAEMC, monomers; Sigma-Aldrich Chemie GmbH), chitosan (CH, from shrimp shells, ≥75%, deacetylated; Sigma-Aldrich Chemie GmbH), hydroxypropyl cellulose (average Mw ~100,000; Sigma-Aldrich Chemie GmbH), Potassium persulphate (PP), methanol, carbomer (Cbp, Carbopol® 940; The Lubrizol Corporation, Wickliffe, OH, USA), propylene glycol (Sigma-Aldrich Chemie GmbH), sorbitan monostearate (SMS, Span® 60; Sigma-Aldrich Chemie GmbH), ethanol 96% (v/v), almond oil, triethanolamine, and purified water were pharmaceutical grade.
Animals
One hundred and four male Wistar rats (whose weight ranged from 180 to 400 g) were used. The rats were kept under standard laboratory conditions (temperature 22°C±1°C, humidity 45%, and 12-hour light cycle). They received food and water ad libitum.
All in vivo experiments were approved by the Bulgarian Food Safety Agency (permit number: 88/09.01.2014) and the Ethics Committee of the Medical University of Plovdiv, Bulgaria (approval number: 1/22.01.2015). The experiments were performed in accordance with the International Council for Laboratory Animal Science ethical guideline for researchers, and the relevant institutional and national rules and regulations.
Preparation and characterization of KP-loaded polymeric nanocarriers
Development of KP-loaded polymeric nanocarriers
A previously described method of one stage emulsifier-free radical polymerization with some modifications was used to obtain KP-loaded polymer nanocarriers.18,19 In situ-loaded KP-nanoparticles (NPs) were obtained by 1) homopolymerization of VAc monomers 10% (v/v) or copolymerization of VAc 10% (v/v) and DMAEMC 20% (v/v) in an aqueous medium and 2) homopolymerization of VAc 10% (v/v) in a solution of the biocompatible and well-known polymers CH 1% (w/v), Cbp 0.5% (w/v) and HPC 1% (w/v) in the presence of KP 2% (w/v). The polymerization was conducted in a nitrogen atmosphere at 55°C for 90 min under ultrasonic impact (Ultrasonicator Siel UST7.8-200, Siel Ltd, Gabrovo, Bulgaria). PP in a concentration of 1% (w/v) was used as an initiator. The model latexes were exposed at dialysis through a membrane with molecular weight cut-off (MWCO) 12,400 Da for 2.5 hours to eliminate the low molecular weight compounds (eg, the initiator of the process, residual monomers or free KP) from the primary latex, and then the samples were freeze-dried. NP yield (%Y) was calculated using the following equation:
|
|
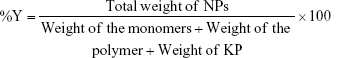
Transmission electron microscopy (TEM)
TEM images of the investigated models were produced by transmission electron microscope JEOL JEM 2100 (JEOL Ltd., Tokyo, Japan) with an accelerating voltage of 200 kV. For the phase identification of the samples, the diffraction mode of the microscope, selected area electron diffraction (SAED), was used. The following preparation procedure was applied before the observation of the samples in the microscope: micro-quantities of the studied substance were mixed with distilled water in a test tube and placed in an ultrasonic bath to homogenize for 3 min. After that, the suspension was dripped onto a carbon-coated standard Cu grid and dried under air conditions in a dust-free environment for 24 hours.
Differential thermal analysis and thermogravimetry (DTA-TG)
The apparatus Stanton Redcroft STA 780 (Stanton Redcroft Ltd, London, UK) was used for DTA-TG analysis. Samples of ~10 mg, placed in corundum crucibles, were heated from room temperature to 550°C with rate 10°C/min using Ar as purge gas (20 mL/min).
Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) analysis
ATR-FTIR analysis was carried out with a Nicolet™iS™ 10 FT-IR Spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA) equipped with a Smart iTR™ ATR sampling (ZnSe crystal) accessory (Thermo Fisher Scientific, Inc.). The spectra were recorded from 4,000 to 650 cm−1 using a DTGS detector.
Particle size, polydispersity index, and zeta potential analysis
Particle size (expressed as Z-average), particle size distribution, PSD (expressed as polydispersity index, PI), and electrical charge on the surface of the NPs (expressed as Zeta Potential, ZP) were determined using Nanotrac Wave (Microtrac, Inc., Montgomeryville, PA, USA).24 The basic specifications of the device allow PSD of tested models to be determined in the measurement range of 0.8 nm–6.54 μm (diameter) and minimum sample volume ~250 μL under measurement angle 180°.
The samples were prepared using equal quantity of NPs (1% w/w) in phosphate buffer at pH 5.5. They were subjected to ultrasonic impact for 10 min to obtain a homogeneous suspension before measuring the Z-average, PI, and ZP. Samples with a volume 3 mL were analyzed at 25°C. All measurements were performed in triplicate and the results were reported as mean values ± SD of 3 replicates (n=3) for each formulation.
Drug loading (%DL) assessment
The %DL assessment was carried out as follows: 10.0 mg of NPs were placed in a test tube and methanol was added to 100.0 mL; the samples were subjected to ultrasonication for 60 min; the quantitative determination of KP loaded in polymer carriers was made spectrophotometrically at λ=260 nm on a UV/Vis spectrophotometer Ultrospec 3300 pro (Biochrom Ltd., Cambridge, UK) after the appropriate dilution and filtration of the samples through a filter Chromafil® Xtra 1.00 μm.8–10 The measurements were made compared with the medium of examination, methanol. Control experiments were performed using NPs without KP. The quantity of KP was calculated from the standard curve with a linearity coefficient (R2) =0.9997 in a concentration range for the drug 3.44–10.32 μg/mL. The %DL and encapsulation efficiency (%EE) were calculated using the following equations:6,9,10
|
|

|
|

The measurements were done three times, and the results were represented as a mean value ± SD.
In vitro release study
In vitro release study was carried out in a thermostated vessel with equal amounts of the tested KP-loaded polymeric carriers under sink conditions; working volume for dissolution 100.0 mL phosphate buffer at pH 5.5; temperature 32°C±0.5°C; stirring speed 100 min−1. The quantitative defining of KP was made spectrophotometrically at λ=260 nm on UV/Vis spectrophotometer Ultrospec 3300 pro (Biochrom Ltd.) after filtering the samples through a filter Chromafil Xtra 1.00 μm. The measurements were made compared with the medium of examination phosphate buffer at pH 5.5. Control experiments were performed using NPs without KP. The concentrations were calculated from the standard curve with a linearity coefficient (R2) =0.999. The results were plotted after triplicate measurement (mean ± SD).
Photostability study
Photostability testing of the in situ KP-loaded was carried out under forcing conditions for 6 hours using 3 batches of every polymeric carrier. Aqueous suspensions (pH 5.5) of KP-loaded polymeric carriers were exposed to outdoor daylight. A solution of KP (pH 5.5) was used as a control. Samples for analysis were taken at the second, fourth, and sixth hour. The non-disintegrated KP was determined by a modified high-performance liquid chromatography (HPLC) analysis.25 The HPLC system was composed of a ProStar 230 solvent delivery module and PDA detector model 335, Microsorb-MV C18 column (150×4.6 mm, 5 μm particle size), all from Varian Australia Pty. Ltd., Mulgrave, Australia. A solvent system including acetonitrile (A) and H2O with pH 3.1 (B) was used in isocratic condition 45A:55B. The flow rate was 1.1 mL/min and detection at 254 nm. The compound of interest was identified according to its retention time determined using authentic KP. It was quantified using an absolute calibration curve. Star Chromatography Workstation Version 6.30 (build 5) software (Varian Australia Pty. Ltd) was used.
The measurements were performed 3 times, and the results were represented as mean ± SD.
Preparation and characterization of a bigel with a KP-loaded NPs
Preparation and characterization of the bigel
For the preparation of bigel, both phases (hydrophilic and lipophilic) were obtained separately. The hydrogel contains Cbp 1.0% (w/w), propylene glycol 5.0% (w/w), ethanol 10.0% (w/w), purified water 84.0% (w/w), and triethanolamine. The organogel contains almond oil 85.0% (w/w) and SMS 15.0% (w/w). In brief, the technology of preparation is as follows: the weighted Cbp was dispersed in the mixture of purified water, propylene glycol and ethanol (25°C, 400 rpm). A stable hydrogel was formed by adding triethanolamine (pH 5.5–6.5). SMS was dissolved in almond oil (60°C, 100 rpm). A bigel was prepared by adding the heated organogel portionwise to the hydrogel under continuous stirring (500 rpm) to obtain a homogeneous mixture and cooling to ambient temperature. KP 2.5% (w/w) and the equivalent of 2.5% (w/w) drug content KP-loaded NPs with established optimal photoprotective properties were included in the bigel after its preparation.
The prepared bigel was inspected visually for its color, homogeneity, consistency and phase separation.26,27 Optical microscopy analysis and determination of pH, viscosity, and spreadability of the bigel were done.
An optical microscope (Leica DM2000 LED; Leica Microsystems, Wetzlar, Germany), equipped with a camera (Leica DMC 2900, Leica) and software for image processing (Leica Application Suite, Leica) was used. The sample from the bigel was observed at a magnification of 40× after staining with a solution of methylene blue (methylthioninium chloride) 1.5% (w/v).
The pH value of 10% aqueous dispersion of the prepared bigel was measured by a pH meter (inoLab pH 720; WTW GmbH, Weilheim, Germany), which was calibrated with standard buffer solutions.28 The measurement was done 3 times, and the result of pH of the bigel was represented as a mean value ± SD.
Determination of the viscosity of bigel formulation was made on viscometer Selecta STS-2011 (J.P Selecta s.a, Barcelona, Spain) at 25°C±0.5°C. A spindle R6 was used at 1, 2, 3, 4, 5, 6, and 10 rpm. The results were plotted after triplicate measurement (mean ± SD).
The spreadability of the bigel was determined by pressing 1±0.01 g of a sample between two 20×20 cm horizontal plates, with the upper one weighing 125.0±1.0 g. The study temperature was maintained at 25°C±0.5°C, and the bigel was evaluated 48 hours after preparation. The spread diameter (Φ) was measured after 1 min.29–32 The measurements were done 3 times, and the results were represented as a mean value ± SD.
Determination of drug content and ATR-FTIR analysis
The drug content in bigel formulations was determined as follows: 0.100 g of bigel formulations were dispersed in freshly prepared phosphate buffer pH 5.5 in 100 mL volumetric flasks under ultrasonic impact; 5 mL of this dispersion was further filtered and diluted to 25 mL with phosphate buffer pH 5.5.33 The drug content was determined at 260 nm using UV/Vis spectrophotometer Ultrospec 3300 pro (Biochrom Ltd.). The measurements were performed 3 times, and the results were represented as a mean value ± SD.
ATR-FTIR analysis of bigels prepared with KP and KP-loaded polymeric carriers was carried out with the same apparatus described above.
In vitro dissolution testing
In vitro drug release study of KP from prepared bigel formulation was performed in Dissolution Apparatus 1 (Sotax AG, Aesch, Switzerland).34 Samples of 0.300 g bigel, containing the drug or KP-NPs equivalent of 2.5% KP, were applied onto a cellulose membrane (MWCO 12,500 Da), which had been previously soaked in distilled water for 24 hours.28 The cellulose membrane with bigel was fixed to the baskets. Three hundred mL of freshly prepared phosphate buffer pH 5.5 was used as a test medium. The experiment was conducted at 32°C±0.5°C and 50 rpm. Samples of 5 mL were taken at appropriate time intervals. The amount of active substance was determined spectrophotometrically at λ=260 nm. The results were plotted after 6-fold measurement (n=6, mean ± SD). The release data was fitted to various mathematical models in order to determine which model best fits the obtained release profile.
Stability studies
Photostability testing of bigel formulations with KP and KP-loaded NPs with the best photoprotective properties and a market product was carried out under forcing conditions for 6 hours using 3 batches of the model. The market product was used as a control. The tested models were exposed to outdoor daylight. Samples for analysis (0.500 g) were taken at the second, fourth and sixth hour. The samples were transferred to a screw-cap cuvette and diluted with 5 mL methanol. After that, they were centrifuged for 15 min at 2,500 rpm. After filtration (Chromafil Xtra 0.45 μm), the non-disintegrated KP in the supernatant was determined by HPLC analysis as described above.
In vivo experiments
Acute skin toxicity testing
Twenty-four rats weighing 250–400 g were selected for the skin toxicity test and divided into 4 groups (n=6). The animals were anesthetized with intraperitoneal injection of ketamine (90 mg/kg bw) and xylazine (10 mg/kg bw). The fur on the back of the rats was shaven, and 2 fields with dimensions 1 cm2 were marked. The sites were chosen to be as close to the rat neck as possible in order to avoid ingestion by the animal licking itself after the treatment. One field was left untreated. The tested formulation (1 mL) was rubbed gently on the skin of the other site. The rats were treated as follows: first group – bigel, containing KP-loaded polymer carrier characterized by the best photoprotective properties and optimal in vitro release; second group – bigel, containing KP; third group – blank bigel and fourth group (positive control) – a market product Fastum® gel (Ketoprofen gel [KP gel]; Berlin-Chemie AG, Berlin, Germany).
The animals were returned to their cages and observed for symptoms of local and systemic toxicity. The skin toxicity was evaluated at the first, twenty-fourth and forty-eighth hour after the application. The irritation score was determined, using a modified Draize scoring system.35
Antinociceptive activity (Hot plate test)
Forty rats (body weight of 180–250 g) were divided into 5 groups (n=8). Groups 1 to 4 were treated as described in the previous section (Acute skin toxicity test), and the fifth group (control) was treated with saline (0.9% NaCl). The same amount (1 mL/rat) of the formulation was applied topically on the plantar surface of all paws. The antinociceptive effect was evaluated immediately before the application and at 30, 90, and 180 min after the treatment. Hot plate test was conducted as described previously.36 In brief, the following steps were followed: the animals were placed on a metal surface heated to a temperature of 55°C±0.5°C and observed for any behavioral sign of nociception (licking a hind paw, vocalization, or an escape response). The latency was recorded and reported as mean ± SEM.
Anti-inflammatory activity (Carrageenan-induced edema)
Forty rats (body weight of 180–250 g) were divided into 5 groups (n=8) and treated with the formulations, reported in the above section (Antinociceptive activity [Hot plate test]).
The same amount (1 mL/rat) of the formulations was applied topically on the plantar surface of the right hind paw of the rat. The skin penetration of the formulation was assured by gentle massage. Immediately after the treatment, the rats received a subplantar injection of 0.1 mL carrageenan (1% [w/v] suspension in saline) into the right hind paw.37
Subsequent measurements of the right hind paw of the animals were taken immediately before the treatment with the formulations and at the first, second, and fourth hour after the subplantar injection. The paw was submerged up to the tibiotarsal joint in the water column of a plethysmometer (UgoBasile, Gemonio, Italy). The paw volume was recorded, and the change in the volume was calculated according to the formula:38
|
|
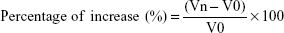
Vn = the volume registered at the nth hour;
V0 = the volume registered for the same animal before treatment.
Antihyperalgesic effect
The same animals used in the previous experiment (Anti-inflammatory activity [Carrageenan-induced edema]) were tested. Immediately after the evaluation of paw volume, the pain threshold was assessed using Analgesimeter (UgoBasile). The apparatus applied mechanical pressure on the rat hind paw. Linearly increasing force (16 g/s) was applied between the third and fourth metatarsal. Nociceptive threshold was measured as the strength of the pressure at which the rat withdrew the testing paw (PPT-units). The maximal possible pressure (cut-off limit) is 250 g. The results were shown as mean value ± SEM.
Statistical analysis
Data was analyzed using SPSS 19.0 (IBM Corp., Armonk, NY, USA). A one-sample Kolmogorov–Smirnov test was used for the evaluation of the normal distribution. One-way analysis of variance with Tuckey post hoc test was performed in the case of a normal distribution; non-parametric Wilcoxon signed rank test, and Mann–Whitney U-test was conducted in the other case. Results were considered significant at P<0.05.
Results and discussion
Preparation and characterization of KP-loaded polymeric nanocarriers
Preparation and yield
Polymeric carriers of KP were obtained by a one-stage emulsifier-free radical homopolymerization of VAc and copolymerization of VAc and DMAEMC. The drug was included in situ during the polymerization. Under the action of a free-radical initiator (PP), VAc monomers (or VAc and DMAEMC, respectively) can polymerize while dispersed in water to form a milky-white emulsion. The main advantage of this method is the formation of a polymer chain without the usage of an emulsifier. It is possible because during the polymerization the forming macromolecule acts as an emulsifier: a polar head ([SO4]−) that remains on the surface of the emulsion droplet and a growing long hydrophobic chain, which is located inside the emulsion droplet. The preparation of the copolymer of VAc and DMAEMC with in situ-loaded KP (KP-p[VAc-co-DMAEMC]) is carried out in an analogous manner.
The homopolymerization of VAc in aqueous solutions of hydrophilic biopolymers such as CH and Cbp was studied in a previous work.19,20 It was found that the viscosity of polymeric solutions affects the process of homopolymerization, the yield of NPs and their morphology, as well as the %DL. During the polymerization, the polymer chains of pVAc were formed, and they were mixed with those of the hydrophilic polymer in the solution, including drug molecules. In this way, the models KP-pVAc+CH, KP-pVAc+Cbp and KP-pVAc+HPC were obtained. KP-pVAc NPs stood out with the highest %Y (95.4±1.2), followed by KP-pVAc+CH (93.9±1.1), KP-pVAc+HPC (93.0±1.9), KP-pVAc+Cbp (91.0±1.7), and KP-p(VAc-co-DMAEMC) (89.7±2.3).
TEM
TEM was used to observe the polymer particle morphology. All models were characterized by a nano-dimensional structure and irregular shape (Figure 1). The micrographs show a different contrast inside the polymer carriers, which is evidence for a different particle density. It seems that the drug is preferably incorporated into the polymer matrix (the darker part), while the hydrophilic polymer forms a shell around the particles (the lighter part). Such a structure leads to a sustained drug release, as it can be seen from the in vitro release study. A different shape and size of the polymeric NPs was observed in a previous work where the same polymer carriers (excluding the polymer mixture of pVAc and HPC and copolymer p[VAc-co-DMAEMC]) loaded with indomethacin were studied.19,20 It seems that the drug itself and its concentration influenced the particle morphology. The irregular shape can be explained by the state of polymers in a solution and after freeze-drying. All micrographs show the tendency of NPs to form agglomerates, probably due to the lower ZP of the NPs, as described below.
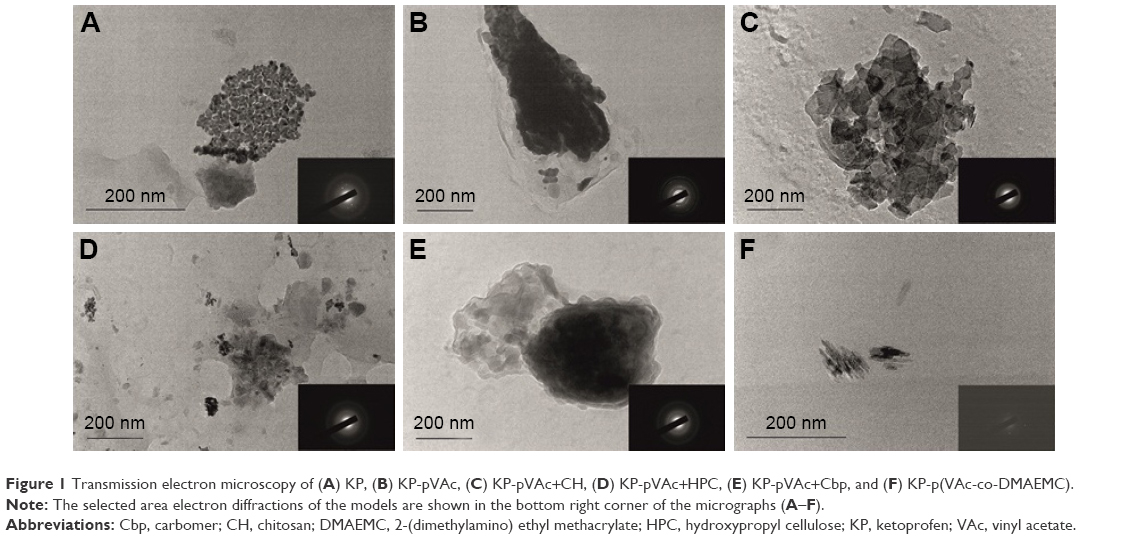 | Figure 1 Transmission electron microscopy of (A) KP, (B) KP-pVAc, (C) KP-pVAc+CH, (D) KP-pVAc+HPC, (E) KP-pVAc+Cbp, and (F) KP-p(VAc-co-DMAEMC). |
SAED, is a crystallographic experimental technique that can be performed along with TEM analysis. The electron diffraction can identify micro-quantities of the crystalline phase distributed in the amorphous matrix. The electron diffraction of KP demonstrates its crystalline structure (Figure 1A, the micrograph in the bottom right corner). All the polymer carriers have a characteristic conformation, resulting in a crystalline structure, demonstrated on the SAED patterns (in the bottom right corner of each photomicrography on Figure 1). This effect can be explained by the crystallization of KP itself, included in the polymer carriers. It is expected that, together, the KP crystalline structure and the polymer carrier itself, will provide a sustained drug release.
DTA-TG analysis
Thermal analysis (DTA-TG) gives information about the degree of miscibility and interaction between polymers and the drug used. While TG only measures changes caused by mass loss (ΔM), DTA also registers changes in the material where no mass loss occurs, for example, crystal structure changes, melting, glass transition, etc. Figure 2 presents DTA and TG curves of the different KP-loaded polymer carriers (solid line) compared with the drug (dotted line) and plain polymer (dashed line).
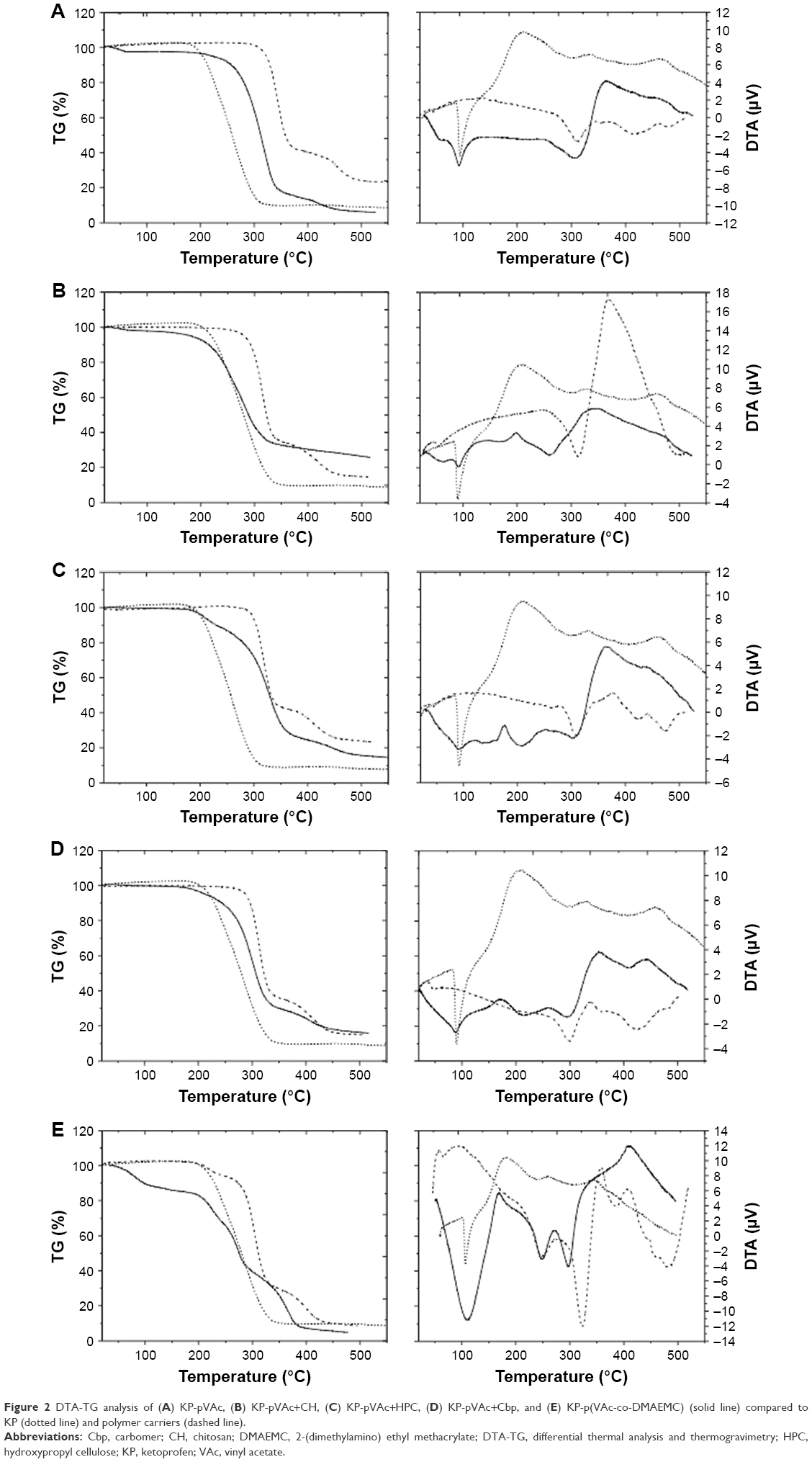 | Figure 2 DTA-TG analysis of (A) KP-pVAc, (B) KP-pVAc+CH, (C) KP-pVAc+HPC, (D) KP-pVAc+Cbp, and (E) KP-p(VAc-co-DMAEMC) (solid line) compared to KP (dotted line) and polymer carriers (dashed line). |
The DTA curve of KP shows a sharp endothermic peak between 87°C and 150°C. This peak is associated with the melting point of the drug and the value (Tmp=96.6°C) corresponds to the values found in the literature (94°C–97°C).39 The TG-curve does not show mass loss at this temperature. The process that occurs in the temperature range 235°C–350°C is also an endothermic one but it corresponds to the thermal decomposition of the KP (ΔM~80%). The TG-curve of KP shows mass losses in one step. Both curves reveal that the drug is thermally stable up to 235°C.
The TG curves of the KP-NPs exhibit mass losses in one (2b), two (2a, 2d), and three (2c, 2e) steps and thermal events (DTA) corresponding to these losses are presented as well. The first endothermic process for KP-pVAc and KP-pVAc+CH (near 60°C) is attributed to dehydration.40 The DTA curves show that the endothermic peaks which correspond to the melting point of the drug were shifted to 91°C for KP-pVAc+CH, KP-pVAc+Cbp, and KP-p(VAc-co-DMAEMC); 90°C for KP-pVAc; and 89°C for KP-pVAc+HPC. These results suggest that, first, there are no changes in the KP structure after its incorporation into the polymer, and, second, that the nature of the interaction between KP and the polymer carrier is not chemical. The TG curves do not show mass loss excluding the model KP-p(VAc-co-DMAEMC) (ΔM~13%) up to 150°C. In the DTA curves of the models, the endothermic processes occurred in the temperature range 200°C–400°C corresponding to the thermal decomposition of the KP-NPs (ΔM: 50%÷80% for all the models). Both curves reveal that the KP-pVAc is thermally stable up to 265°C. Thermal stability up to 207°C was found for KP-pVAc+CH. For the composites KP-pVAc+Cbp and KP-pVAc+HPC, this stability is shifted toward lower 180°C. As can be seen, the presence of KP in all the models is distinct, and the thermal stability of the models decreases as follows: KP-pVAc>KP-pVAc+CH>KP-pVAc+Cbp>KP-pVAc+HPC>KP-p(VAc-co-DMAEMC).
ATR-FTIR analysis
ATR-FTIR analysis was used to explain whether there is a chemical or physical interaction between the drug used and the different polymer carriers. Infrared spectroscopy data of KP and KP-NPs is shown in Figure 3. ATR-FTIR spectrum of KP indicates 2 main bands centered at 1,695 and 1,654 cm−1, which corresponds to the C=O stretching of the carbonyl group.40–42 The fingerprint region shows a triplet in the region of 703 cm−1. Both findings speak for crystalline nature of KP used in the study.42 The medium-weak, multiple bands located in region 1,400–1,600 cm−1 correspond to the presence of an aromatic core.
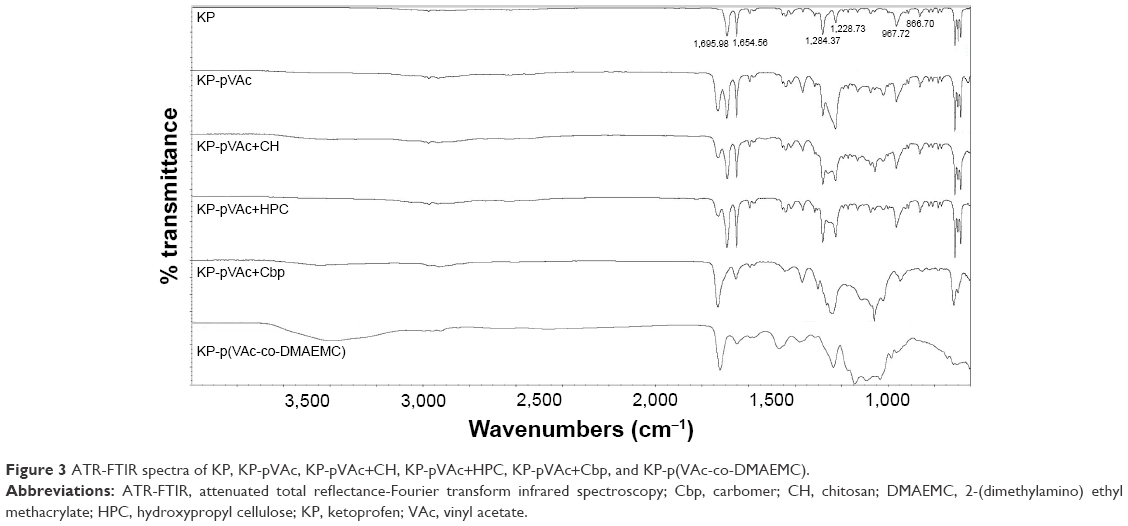 | Figure 3 ATR-FTIR spectra of KP, KP-pVAc, KP-pVAc+CH, KP-pVAc+HPC, KP-pVAc+Cbp, and KP-p(VAc-co-DMAEMC). |
The comparison of IR spectra of KP and of all the KP-loaded polymer carriers suggests that there is no change in the chemical structure of the drug after its incorporation into the polymer.41–43 It indicates the presence of a physical mixture of KP and the polymer carriers with the existence of a weak H-bonding between OH-group (from –COOH). Disappearance, displacement and low intensity of KP characteristic absorption peaks in the spectra of KP-pVAc+HPC and KP-p(VAc-co-DMAEMC) assumes a lower drug content in these carriers compared with KP-pVAc, KP-pVAc+CH, and KP-pVAc+Cbp.
Examination of the spectra of KP-loaded polymer carriers showed that the greatest number of hydrogen bonds had been formed in KP-pVAc+HPC and KP-p(VAc-co-DMAEMC). This finding could be helpful for ensuring a sustained drug release.
Particle size, PI, and ZP analysis
PSD plays a vital role in the determination of the critical physicochemical properties of the particulate system.44 It gives information about the reliability of the process of preparation of NPs.
Particle size (expressed as Z-average), PSD (expressed as PI), and the electrical charge on the surface of the NPs (expressed as ZP) are shown in Table 1. It is evident that all the models that represent polymer mixtures and copolymer, dispersed in the water medium (pH 5.5), possess micrometer-scaled mean diameters. Possible reasons for the larger values of this characteristic of the particles in water in comparison with their TEM analysis could be particle aggregation and a swelling of the hydrophilic polymers. The greater amount of hydrophilic polymer on the surface of the particle, the greater the swelling, and, respectively the particle mean diameter.
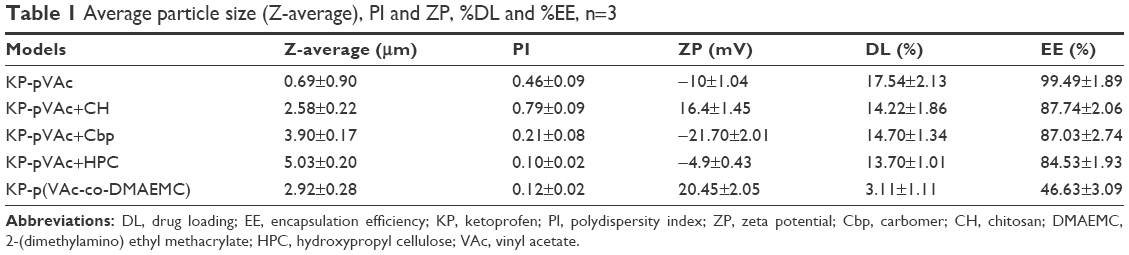 | Table 1 Average particle size (Z-average), PI and ZP, %DL and %EE, n=3 |
The PI of the models varies from 0.1 to 0.79 as follows: KP-pVAc+HPC<KP-p(VAc-co-DMAEMC)<KP-pVAc+Cbp<KP-pVAc<KP-pVAc+CH. The KP-loaded homopolymer carrier (KP-pVAc) was characterized by a high PI value of 0.46, which reveals a broad PSD as compared with the same carrier, but loaded with indometacin.19 The highest PI value was registered for KP-pVAc+CH, which shows a much broader PSD in comparison with other KP-loaded carriers and to the same carrier loaded with indometacin.20
The absolute values of ZP were registered in the range from 4.9 to 21. The low ZP values (bellow 25) are an indicator for the instability of the aqueous suspensions of these models and can explain the tendency of NPs to form agglomerates, as it was observed by their TEM.45
The results obtained by PSD, PI, and ZP analysis show the need for further improvement of the polymerization process in order to ensure carriers with optimal size, narrow PSD, and a higher ZP values.
%DL assessment
A high %DL and %EE was found for all the polymer carriers excluding the copolymer KP-p(VAc-co-DMAEMC) (Table 1). The highest values for both parameters were established for KP-pVAc. The polymer mixtures of pVAc and CH and Cbp were characterized by very similar %DL and %EE. These values were slightly lower for the polymer mixture KP-pVAc+HPC and the smallest %DL and %EE were detected for KP-p(VAc-co-DMAEMC). It should be noted that the theoretical values of %DL were 17.64%, 16.20%, 16.89%, 16.20%, and 6.67% for KP-pVAc, KP-pVAc+CH, KP-pVAc+Cbp, KP-pVAc+HPC, and KP-p(VAc-co-DMAEMC), respectively. It is evident that the %DL obtained for both the homopolymer and polymer mixture carriers comes close to the theoretical %DL. Probably during the process of polymerization of VAc and DMAEMC, the drug was retained in the liquid medium and after that it was dialyzed, thus forming a copolymer with a low content of in situ-loaded KP (KP-p[VAc-co-DMAEMC]).
In vitro release study
In vitro, the release study of KP-NPs was conducted in a phosphate buffer at pH 5.5 for 6 hours. The results from the cumulative drug release from the polymer carriers are shown in Figure 4. The 5 models released the drug at different rates and extents.
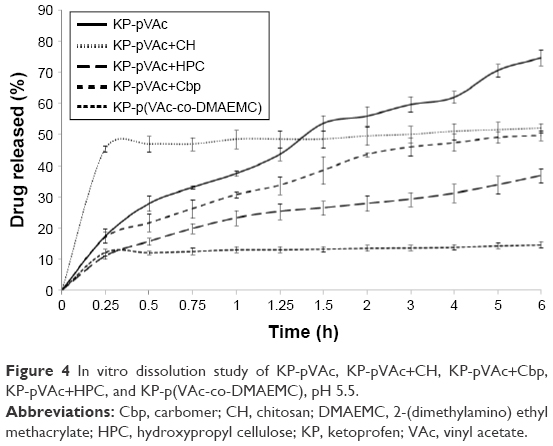 | Figure 4 In vitro dissolution study of KP-pVAc, KP-pVAc+CH, KP-pVAc+Cbp, KP-pVAc+HPC, and KP-p(VAc-co-DMAEMC), pH 5.5. |
A burst effect (up to 15 min) was found for KP-pVAc+CH (~46%) and KP-p(VAc-co-DMAEMC) (~12%). After that, a sustained drug release was observed following zero order kinetics with coefficients of determination (R2) 0.9687 and 0.9661 for KP-pVAc+CH and KP-p(VAc-co-DMAEMC), respectively. The biphasic sustained release of KP-pVAc+CH could be explained by the distribution of KP into KP-pVAc+CH. The initial burst release was due to the drug that was localized on the particle surface and into the outer hydrophilic shell, while the drug incorporated into the particle core was released in a prolonged manner. Similarly, the drug attached to the particle surface as well as higher number of hydrogen bonds detected by ATR-FTIR analysis, are possible causes of the type of dissolution profile of KP-p(VAc-co-DMAEMC).
At the end of the in vitro release study, the cumulative amount of KP released from KP-pVAc, KP-pVAc+Cbp, and KP-pVAc+HPC reached 75%, 50%, and 37%, respectively. The Higuchi square root model showed higher coefficients of determination values (R2) for the 3 models: 0.9900, 0.9835, and 0.9746, respectively. The different rates and extents of drug release of KP-pVAc, KP-pVAc+Cbp and KP-pVAc+HPC could be explained by 1) the particle mean diameter: the smaller particles, the greater surface area, are associated with a higher rate and extent of drug release (KP-pVAc), and, conversely: the bigger particles, the slower the drug release (KP-pVAc+HPC) and 2) the presence of weak hydrogen bonds between KP and polymer carriers in varying degrees. In a previous work exploring the in vitro dissolution of indomethacin from a homopolymer of VAc, copolymers of VAc and 3-dimethyl (methacryloyloxyethyl) ammonium propane sulfonate, as well as polymer mixtures of pVAc with CH and Cbp, it was found that the drug release follows first order kinetics.19,20 It is evident that the processes of diffusion controlled the drug release from different pVAc carriers such as a homopolymer, copolymers, or polymer mixtures with hydrophilic CH, Cbp, and HPC.
Photostability study
Forced KP degradation in aqueous media (pH 5.5) under outdoor daylight was carried out to test the photoprotection of different polymeric carriers. The study was conducted for 6 hours. A solution of KP was used as a control. Samples for analysis were taken on the second, fourth and sixth hour. KP was analyzed by a validated HPLC method with limit of detection 30 ng/mL, limit of quantification 110 ng/mL, a correlation coefficient (r2) 0.9999 and coefficient of variation 4.21%. The recovery of the method was 99.1%.
The results showed that the drug concentration decreased during the experiment and at the second hour, KP determined was ~1% for KP-pVAc, ~2% for KP solution and KP-pVAc+Cbp, and ~4% for KP-pVAc+HPC (Figure 5). Only traces of KP were found in the aqueous suspension of KP-pVAc+CH, and no quantity of the drug was found in KP-p(VAc-co-DMAEMC) suspension at that time interval.
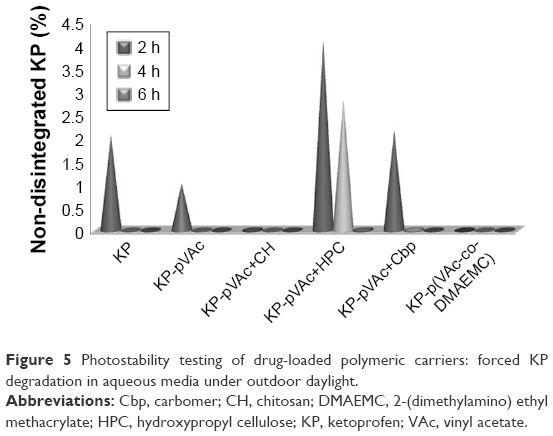 | Figure 5 Photostability testing of drug-loaded polymeric carriers: forced KP degradation in aqueous media under outdoor daylight. |
The drug was detected at the fourth hour (~2.8%) in the suspension of KP-pVAc+HPC only. At that time, traces of KP were found in KP-pVAc+Cbp.
Drug traces were found in the aqueous suspension of KP-pVAc+HPC at the sixth hour.
It is evident that the pVAc carrier alone, the mixture of pVAc and CH, as well as the copolymer p(VAc-co-DMAEMC) did not protect the drug used from photo-degradation. Moreover, the drug degradation was more rapid compared with the KP solution. The photostability study provided results for a photoprotection of polymeric carriers used on KP that were not similar to the results obtained by the thermal stability study (DTA-TG analysis presented above). The photostability is a consequence of the size, shape, and structure of the drug carriers.46 The enhanced photoprotection of the pVAc+HPC carrier on in situ KP loaded could be explained by the swollen shell from HPC and the greatest mean particle size of this model.
Preparation and characterization of a bigel with KP-NPs
Preparation and characterization of a bigel as semisolid formulation
The carbopol hydrogel (aqueous phase) and almond oil organogel (apolar phase) were prepared separately. SMS in the concentration of 15% (w/w) was used as a gelling agent for the preparation of almond oil organogel.47,48 The bigel (BG30) was obtained with a hydrogel/organogel ratio of 70/30 (w/w). The gel formation was confirmed by a tube inversion test (Figure 6A). BG30 possessed white color with a creamy appearance and pleasant scent of almond oil. It was not greasy to the touch. Figure 6B shows the optical micrographs of BG30 regarding the mutual disposition of phases. It is evident that the almond oil organogel is dispersed in the form of small gel droplets with regular or oval shape into the carbopol hydrogel. This microstructure of BG30 can provide higher stability during storage. The value of pH of BG30 was 6.67±0.04 and it is a prerequisite for physiological tolerance, and a possible lack of skin irritation in topical application.49 Figure 6C shows the effect of shearing on the viscosity of bigel BG30. The formulation shows a pseudoplastic flow; it was found that the increase in the shearing rate leads to a reduced viscosity of the bigel. The spreadability determines the way of application (easy or difficult), the accurate dosing, and patient preference. The spread diameter of the resulting circle (Φ) for BG30 was 37.3±0.6 mm. In the study on the spreadability of various oil-in-water creams, Arvouet-Grand et al and later Lardy et al have graded the investigated formulations from fluid to very stiff by the value of Φ obtained.26,27 According to both classifications, BG30 is a semi-stiff formulation.
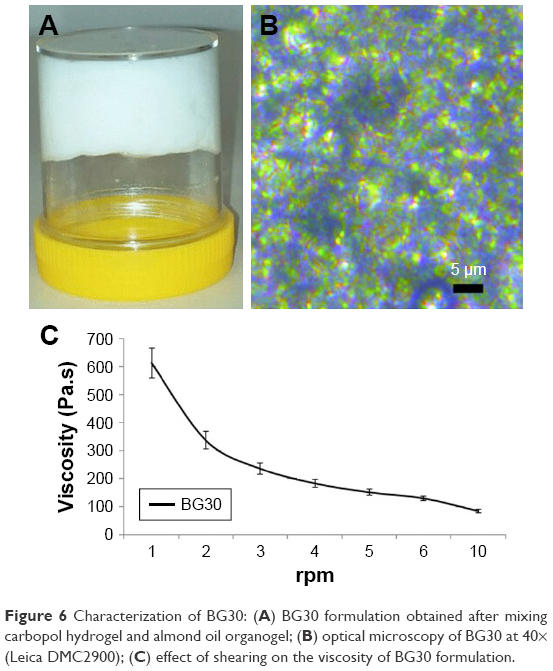 | Figure 6 Characterization of BG30: (A) BG30 formulation obtained after mixing carbopol hydrogel and almond oil organogel; (B) optical microscopy of BG30 at 40× (Leica DMC2900); (C) effect of shearing on the viscosity of BG30 formulation. |
Drug content and ATR-FTIR analysis
The polymer mixture of pVAc and HPC was found to be a promising carrier of KP (KP-pVAc+HPC), which could provide drug photoprotection and a sustained release. In this regard, the model KP-pVAc+HPC was used in the experiments below.
The drug content of bigel formulations was (98.33%±0.99%) and (97.04%±1.06%) for BG30-KP and BG-KP-pVAc+HPC, respectively. It was found that KP and KP-loaded NPs were uniformly distributed in all the bigel formulation.33,50–52
ATR-FTIR analysis was done on BG30-KP and BG30-KP-pVAc+HPC to indicate the compatibility of the drug or drug-loaded polymer carrier and bigel formulation. Figure 7 shows the IR-spectra of BG30-KP and BG30-KP-pVAc+HPC. Both spectra were compared with the spectra of the plain bigel formulation (BG30), drug and drug-loaded polymer carrier (KP-pVAc+HPC). The spectra of bigel formulations BG30-KP and BG30-KP-pVAc+HPC contain all the characteristic peaks of the bigel (BG30) and some of those that indicate the presence of the drug and KP-loaded polymer carrier. The apparent disappearance of the KP- characteristic absorption bands in the spectrum of BG30-KP could be explained by the low concentration of the drug used (2.5%, w/w). There are no preconditions or signs of chemical interaction between KP and bigel or the KP-loaded polymer carrier and bigel. However, both models (BG30-KP and BG30-KP-pVAc+HPC) represent a physical mixture with the existence of a high number of hydrogen bonds that could affect the biopharmaceutical properties of the bigel formulations.
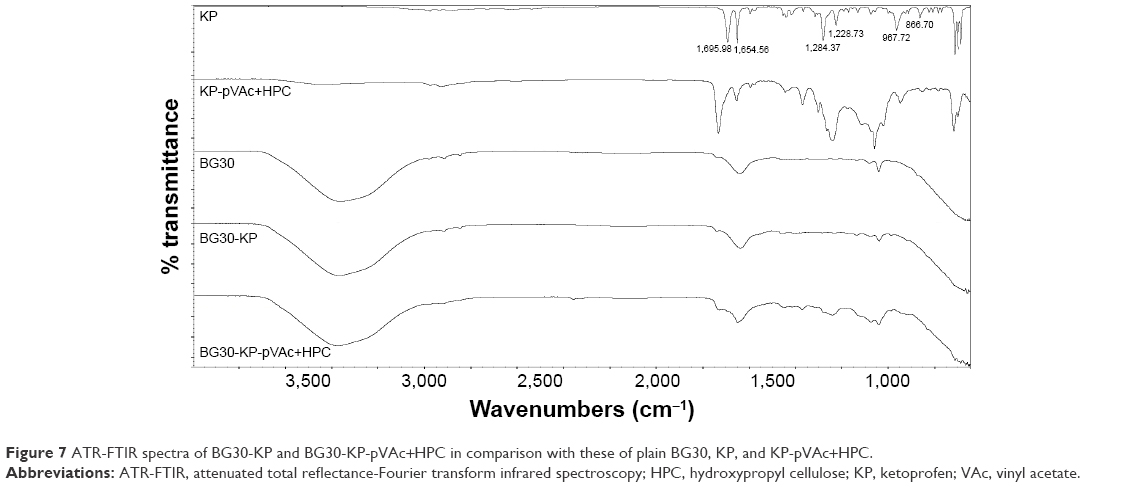 | Figure 7 ATR-FTIR spectra of BG30-KP and BG30-KP-pVAc+HPC in comparison with these of plain BG30, KP, and KP-pVAc+HPC. |
In vitro dissolution study
The in vitro drug release of BG30-KP and BG30-KP-pVAc+HPC was compared with the market product KP gel. Figure 8 shows the KP-dissolution profiles through a cellulose membrane in a 300 mL phosphate buffer with pH 5.5 for 5 hours. The market product released ~90% of the drug for 15 min. The bigel formulations were characterized by lower rate and extent of drug release compared with the KP gel. The cumulative amount of drug released after 5 hours from the bigel formulations BG30-KP and BG30-KP-pVAc+HPC was (94.7%±2.3%) and (86.6%±6.8%), respectively. This result was to be expected considering the type of semisolid drug formulation. Generally, hydrogels, followed by emulgels and bigels, are characterized by the highest rate and extent of drug release.27 Surprisingly, BG30-KP-pVAc+HPC was characterized by a faster drug release at the fifteenth minute (38.0%±2.1%) compared with BG30-KP (34.6%±1.7%), both following a prolonged release. The data showed that at the second hour, KP released was (59.6%±2.7%) for BG30-KP-pVAc+HPC and (85.0%±1.9%) for BG30-KP, respectively. Although the drug release was retarded in BG30-KP-pVAc+HPC compared with BG30-KP, it reached drug release levels higher than 85% at the end of the dissolution study.
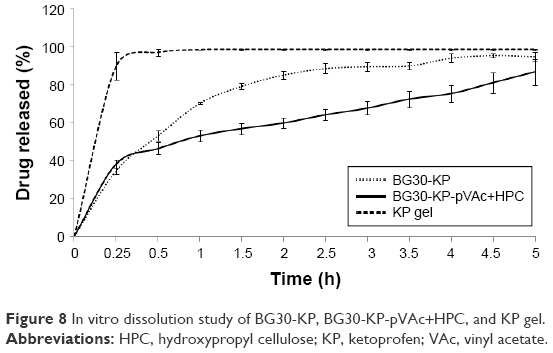 | Figure 8 In vitro dissolution study of BG30-KP, BG30-KP-pVAc+HPC, and KP gel. |
In order to describe the kinetics and the drug release mechanism of the prepared bigels, the data obtained from the in vitro the release study was fitted to different kinetic models such as zero order, first order, Higuchi, Hixon–Crowell, and Korsmeyer–Peppas models. The goodness of fit was evaluated using the determination coefficient (R2) values (Table 2). According to (R2) values, the in vitro release data was in favor of first order kinetics. The “n” value (Korsmeyer–Peppas model) is above 0.5 for BG30-KP, which determines an anomalous transport. For BG30-KP-pVAc+HPC, “n” value was below 0.5, which demonstrates that the mechanism controlling the drug release was the quasi-Fickian diffusion. First order release kinetics was obtained for KP through a series of simple gels containing HPC and Carbosil that varied in solvent composition.53 In another study, Solinís et al demonstrated that the release of salbutamol and KP enantiomers from hydroxypropyl methylcellulose K100M matrices containing 2 types of cellulose derivatives follow a quasi-Fickian diffusion mechanism as a stereoselective process.54 The same diffusion mechanism is possible for BG30-KP-pVAc+HPC model due to its complex composition, and a greater number of hydrogen bonds, confirmed by ATR-FTIR analysis.
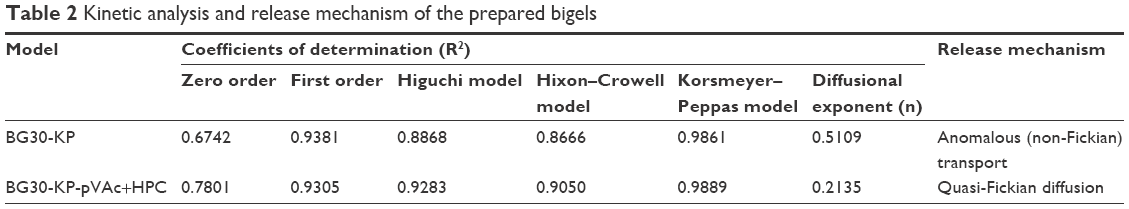 | Table 2 Kinetic analysis and release mechanism of the prepared bigels |
Photostability testing of bigel formulation with KP-pVAc+HPC
Bigel formulations BG30-KP and BG30-KP-pVAc+HPC were tested under stress conditions in an accelerated photostability testing, and the results were compared with those of the market product. All models were exposed to outdoor daylight for 6 hours. The results of the photostability testing are shown in Figure 9. Apparently, model BG30-KP-pVAc+HPC provides better drug stability compared with BG30-KP and the market product. This result was particularly prominent for both bigel formulations at the fourth hour of the study, while non-degraded KP was 62% in BG30-KP and 96% in BG30-KP-pVAc+HPC. At the end of the study, the drug determined in BG30-KP, BG30-KP-pVAc+HPC and the market product was about 54%, 70%, and 40%, respectively. These results strongly suggest that a polymer mixture of pVAc and HPC as a drug carrier included in the bigel formulation will enhance the photostability of KP.
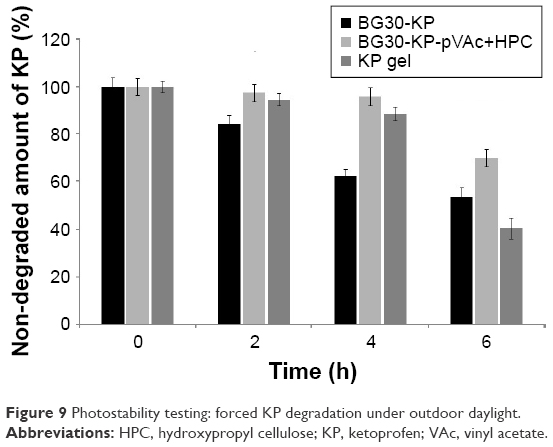 | Figure 9 Photostability testing: forced KP degradation under outdoor daylight. |
Results from in vivo experiments
Acute skin toxicity test
No symptoms of skin toxicity (redness, edema), irritation or inflammation were observed during the first 48 hours after the treatment. These results showed that all tested formulations could be considered safe for dermal use. Our results are consistent with the ones of Elkomy et al.55 The authors tested the skin toxicity of solid lipid NPs as carriers for the topical delivery of KP on rabbits and similarly registered no edema or erythema after the treatment.
Antinociceptive activity (Hot plate test)
Data showed normal distribution. We recorded a significant increase in the latency of rats treated with BG30-KP-pVAc+HPC in comparison with the BG30-KP group (15.9±0.7 vs 10.4±1.4 s; P<0.005), the blank BG30 group (15.9±0.7 vs 8.9±0.9 s; P<0.001), the KP gel group (15.9±0.7 vs 10.4±0.9 s; P<0.005) and controls (15.9±0.7 vs 7.6±1.1 s; P<0.001) at the third hour after treatment with the formulations (Figure 10). BG30-KP-pVAc+HPC showed a well-defined antinociceptive effect. Moreover, BG30-KP-pVAc+HPC was more effective even than the positive control group (KP gel). These findings support the hypothesis for a sustained KP release from BG30-KP-pVAc+HPC compared with the market product and BG30-KP.
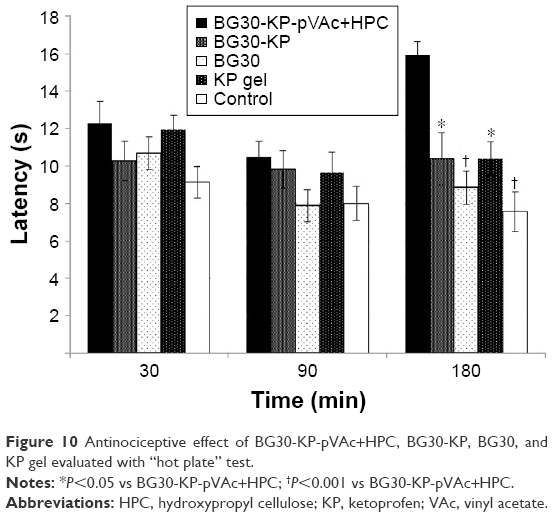 | Figure 10 Antinociceptive effect of BG30-KP-pVAc+HPC, BG30-KP, BG30, and KP gel evaluated with “hot plate” test. |
Anti-inflammatory activity (carrageenan-induced edema)
As shown in Figure 11, BG30-KP-pVAc+HPC reduced the edema, and the effect was most prominent at the fourth hour after the application in comparison with controls (32.1%±5.5% vs 52.5%±4.4%; U=10; P<0.05; Mann–Whitney U-test). A tendency of a decrease in the paw edema was also registered after administration of BG30-KP and KP gel when compared with the control group, but it did not quite reach the conventional levels of statistical significance. A well-defined anti-inflammatory activity was observed at the first hour after BG30-KP application in comparison with the control group (16.2%±4.2% vs 29.8%±2.7%; U=11; P<0.05; Mann–Whitney U-test). The paw edema in rats, treated with BG30-KP and KP gel was quite different at the first hour and similar at the second and fourth hour. The blank BG30 did not influence the paw edema, and the results were analogous to the control group. The results in the carrageenan-induced paw edema could also be explained by the sustained KP release from the bigel formulations. BG30-KP and KP gel deliver KP to the inflamed tissues faster than the tested formulation BG30-KP-pVAc+HPC.
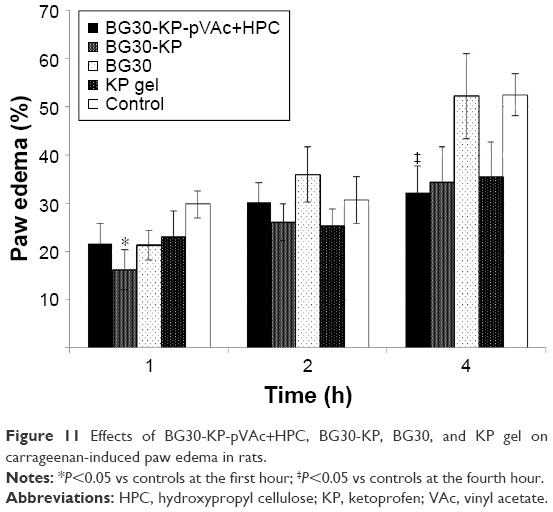 | Figure 11 Effects of BG30-KP-pVAc+HPC, BG30-KP, BG30, and KP gel on carrageenan-induced paw edema in rats. |
Sakeena et al compared the effects of KP in nanoemulsion to KP gel.38 The authors reported anti-inflammatory properties of the formulation at second and fourth hour after the application and the peak of the effect of the nanoemulsion was at the second hour due to the rapid absorption of the NSAID. In our study, BG30-KP was found to be more effective at the first hour, and the peak of the anti-inflammatory effect of BG30-KP-pVAc+HPC was registered at the fourth hour after the application.
Antihyperalgesic effect
No statistically significant difference was registered at the pre-treatment point (Mann-Whitney U-test; P>0.05). We registered a significant increase in the PPT units evaluated in the BG30-KP group when compared with the BG30 group (15.2±2.4 vs 7.8±0.6; U=9.0; P=0.015; Mann–Whitney U-test) at the first hour after treatment (Figure 12). No statistical significance was found between the marketed product and the BG30-KP group (14.1±1.9 vs 15.2±2.4; U=30.5; P=0.878). The hyperalgesic effect was diminished by treatment with KP gel in comparison with blank BG30 (14.1±1.9 vs 7.8±0.6; U=7.5; P=0.007).
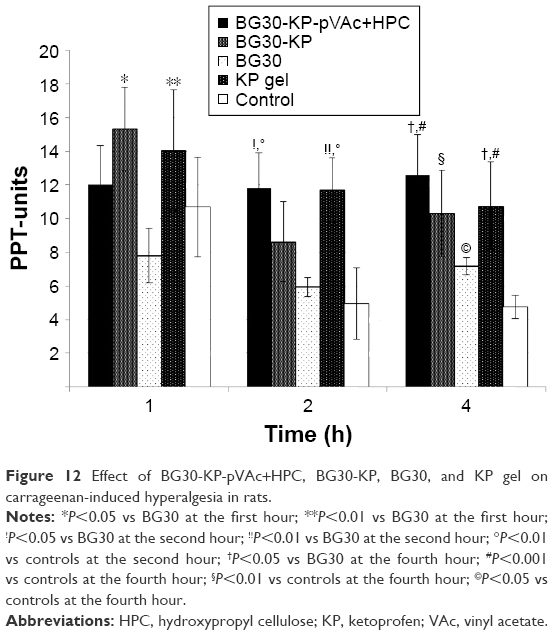 | Figure 12 Effect of BG30-KP-pVAc+HPC, BG30-KP, BG30, and KP gel on carrageenan-induced hyperalgesia in rats. |
The treatment with BG30-KP-pVAc+HPC significantly increased the reaction time of the rats when compared with the blank BG30 and the control group (11.8±2.4 vs 5.9±0.5; U=12.0; P=0.038; Mann–Whitney U-test and 11.8±2.4 vs 4.9±0.7; U=6.0; P=0.005, respectively) at the second hour of the test. Similar results were registered in the KP gel group. The PPT units measured in the positive control (KP gel) group were 10.7±2.6 – a value significantly higher than the one found in blank bigel group (5.9±0.5; U=7.0; P=0.007). The same results were observed in the KP gel group and the control group (10.7±2.6 vs 4.9±0.7; U=6.5; P=0.005).
At the fourth hour of the experiment, we registered a significant increase in the latency of BG30-KP-pVAc+HPC rats in comparison with the BG30 and control groups (12.6±2.2 vs 7.2±0.9; U=11.5; P=0.028 and 12.6±2.2 vs 4.8±0.5; U=1.5; P<0.001, respectively). The treatment with BG30-KP and KP gel evoked a well-defined antihyperalgesic effect at the fourth hour of the test. The registered PPT units were 10.3±2.0 in the BG30-KP and 4.8±0.5 in the control group (U=7.0; P=0.007). The respective measurements in KP gel and control groups were 10.8±1.2 and 4.8±0.5 (U=0.5; P<0.001). The positive control group showed an increased latency in comparison with the bigel group (10.8±1.2 vs 7.2±0.9; U=11.0; P=0.028). Surprisingly, a statistical difference was also found between the control group and the bigel group (4.8±0.5 vs 7.2±0.9; U=11.0; P=0.028; Mann–Whitney U-test).
KP gel reversed the mechanical hyperalgesia, induced by carrageenan injection. No significant difference was registered in the PPT score at the first hour after BG30-KP-pVAc+HPC treatment. The sustained release of KP from BG30-KP-pVAc+HPC determined the profile of its antihyperalgesic effect. The rapid release of KP from BG30-KP formulation led to a reduction in the mechanical hypersensitivity at the first hour, but such an effect was not registered at the second hour after treatment. The slow release of KP from BG30-KP-pVAc+HPC led to a reduced hyperalgesia. The results were very similar to those obtained for KP gel at the second and fourth hour after treatment. Comparing the PPT-units measured in the BG30-KP-pVAc+HPC group, a tendency of an increase could be registered, while the PPT-units in KP gel group showed a downward tendency over the 4 hours of the experiment.
Conclusion
The method of one-stage emulsifier-free emulsion polymerization was successfully applied for the preparation of in situ KP-loaded polymer carriers (KP-pVAc and KP-pVAc-co-DMAEMC). Polymer mixtures of pVAc and hydrophilic and biocompatible Cbp, CH and HPC were studied. The obtained polymer carriers were characterized by a nano-dimensional structure and an irregular shape. There was no change in the chemical structure of KP after its incorporation into polymer carriers. A decreasing thermal stability was observed as follows KP-pVAc>KP-pVAc+CH>KP-pVAc+Cbp>KP-pVAc+HPC>KP-p(VAc-co-DMAEMC). All the tested drug carriers were characterized by a high yield, %DL, %EE, and a sustained KP release. It was found that the processes of diffusion controlled the drug release from different pVAc carriers such as a homopolymer, copolymers, or polymer mixtures with hydrophilic CH, Cbp, and HPC. The polymer mixture of pVAc and HPC is a suitable carrier of KP: the polymer mixture provides the best drug photostability and sustained in vitro release when included in a bigel formulation. The obtained Carbopol hydrogel/almond oil bigel with a 30% apolar phase was a suitable base for topical drug formulations with KP and the KP-loaded polymer carrier. The data from the in vitro release study showed that the drug release was sustained from BG30-KP-pVAc+HPC compared with BG30-KP. The combination of polymer carriers of KP and bigel provided a more prominent drug photostability and a sustained release.
Also, the bigel did not induce skin inflammation or irritation. The formulation BG30-KP-pVAc+HPC showed a well-defined antinociceptive effect at the third hour after the application and was more effective even than the positive control group (KP gel). In the study of anti-inflammatory activity, BG30-KP was found to be more effective at the first hour, and the peak of the anti-inflammatory effect of BG30-KP-pVAc+HPC was registered at the fourth hour after application. The reduction of the mechanical hypersensitivity at the second and fourth hour after treatment was similar to the effect of the marketed product (KP gel).
In conclusion, the inclusion of KP in a pVAc+HPC polymer carrier and bigel base leads to enhanced drug photostability and the development of a safe and effective semisolid dosage form for dermal application.
Disclosure
The authors report no conflicts of interest in this work.
References
Bagheri H, Lhiaubet V, Montastruc JL, Chouini-Lalanne N. Photosensitivity to ketoprofen: mechanisms and pharmacoepidemiological data. Drug Saf. 2000;22(5):339–349. | ||
Sugiura M, Hayakawa R, Xie Z, Sugiura K, Hiramoto K, Shamoto M. Experimental study on phototoxicity and the photosensitization potential of ketoprofen, suprofen, tiaprofenic acid and benzophenone and the photocross-reactivity in guinea pigs. Photodermatol Photoimmunol Photomed. 2002;18(2):82–89. | ||
Gonçalo M. Phototoxic and photoallergic reactions. In: Johansen JD, Frosch PJ, Lepoittevin J-P, editors. Contact Dermatitis. Berlin: Springer; 2010:361–374. | ||
Matamoros V, Duhec A, Albaigés J, Bayona JM. Photodegradation of carbamazepine, ibuprofen, ketoprofen and 17 α-ethinylestradiol in fresh and seawater. Water Air soil Pollut. 2009;196(1–4):161–168. | ||
Choi YG, Lee JH, Bae IH, et al. Titanium dioxide inclusion in backing reduces the photoallergenicity of ketoprofen transdermal patch. Arch Toxicol. 2011;85(3):219–226. | ||
Khadka P, Ro J, Kim H, et al. Pharmaceutical particle technologies: an approach to improve drug solubility, dissolution and bioavailability. AJPS. 2014;9(6):304–316. | ||
Ferreira LM, Cervi VF, Gehrcke M, et al. Ketoprofen-loaded pomegranate seed oil nanoemulsion stabilized by pullulan: selective antiglioma formulation for intravenous administration. Colloids Surf B Biointerfaces. 2015;130:272–277. | ||
Amit K, Mahalaxmi R, Srinivas P, Deepak K. Enhancement of solubility and dissolution of poorly soluble drug: ketoprofen as a model drug. J Chem Pharm Res. 2011;3(1):268–276. | ||
Cirri M, Bragagni M, Mennini N, Mura P. Development of a new delivery system consisting in “drug–in cyclodextrin–in nanostructured lipid carriers” for ketoprofen topical delivery. Eur J Pharm Biopharm. 2012;80(1):46–53. | ||
Kheradmandnia S, Vasheghani-Farahani E, Nosrati M, Atyabi F. Preparation and characterization of ketoprofen-loaded solid lipid nanoparticles made from beeswax and carnauba wax. Nanomedicine. 2010;6(6):753–775. | ||
Leonardi A, Bucolo C, Romano GL, et al. Influence of different surfactants on the technological properties and in vivoocular tolerability of lipid nanoparticles. Int J Pharm. 2014;470(1–2):133–140. | ||
Sagiri SS, Singh VK, Kulanthaivel S, et al. Stearate organogel-gelatin hydrogel based bigels: physicochemical, thermal, mechanical characterizations and in vitro drug delivery applications. J Mech Behav Biomed Mater. 2015;43:1–17. | ||
Lupi FR, Gentile L, Gabriele D, Mazzulla S, Baldino N, de Cindio B. Olive oil and hyperthermal water bigels for cosmetic uses. J Colloid Interface Sci. 2015;459:70–78. | ||
Rhee GJ, Woo SJ, Hwang SJ, Lee YW, Lee CH. Topical oleo-hydrogel preparation of ketoprofen with enhanced skin permeability. Drug Dev Ind Pharm. 1999;25(6):717–726. | ||
Singh VK, Anis A, Banerjee I, Pramanik K, Bhattacharya MK, Pal K. Preparation and characterization of novel carbopol based bigels for topical delivery of metronidazole for the treatment of bacterial vaginosis. Mater Sci Eng C Mater Biol Appl. 2014;44:151–158. | ||
Rehman K, Mohd Amin MC, Zulfakar MH. Development and physical characterization of polymer-fish oil bigel (hydrogel/oleogel) system as a transdermal drug delivery vehicle. J Oleo Sci. 2014;63(10):961–970. | ||
Singh VK, Banerjee I, Agarwal T, Pramanik K, Bhattacharya MK, Pal K. Guar gum and sesame oil based novel bigels for controlled drug delivery. Colloids Surf B Biointerfaces. 2014;123:582–592. | ||
Andonova V, Georgiev G, Toncheva V, Kassarova M. Preparation and study of Poly(vinyl acetate) and Poly(styrene) nanosized latex with Indometacin. Pharmazie. 2012;67(7):601–604. | ||
Andonova V, Georgiev G, Toncheva V, et al. Indomethacin loading and in vitro release properties from vinyl acetate homo- and co-polymer nanoparticles, coated with polyzwitterion and carbopol® shells. Int J Pharm Pharm Sci. 2014;6(1):691–699. | ||
Andonova V, Georgiev G, Toncheva V, Karashanova D, Katsarov P, Kassarova M. Carbopol® and chitosan coated nanoparticles with in-situ loaded indomethacin. AJPTR. 2014;4(1):664–678. | ||
de Moraes Porto ICC. (2012). Polymer biocompatibility. In: De Souza Gomes A, editor. Polymerization. Croatia: InTech; 2012:47–63. | ||
Venkatachalam M, Sathe SK. Chemical Composition of Selected Edible Nut Seeds. J Agric Food Chem. 2006;54(13):4705–4714. | ||
Hussain A, Khan GM, Shah SU, et al. Development of a novel ketoprofen transdermal patch: effect of almond oil as penetration enhancers on in-vitro and ex-vivo penetration of ketoprofen through rabbit skin. Pak J Pharm Sci. 2012;25(1):227–232. | ||
Almeida H, Lobão P, Frigerio C, et al. Preparation, characterization and biocompatibility studies of thermoresponsive eye drops based on the combination of nanostructured lipid carriers (NLC) and the polymer Pluronic F-127 for controlled delivery of ibuprofen. Pharm Dev Technol. 2017;22(3):336–349. | ||
Dvorak J, Hajkova R, Matysova L, Novakova L, Koupparis MA, Solich P. Simultaneous HPLC determination of ketoprofen and its degradation products in the presence of preservatives in pharmaceuticals. J Pharm Biomed Anal. 2004;36(3):625–629. | ||
Jain A, Gautam SP, Gupta Y, Khambete H, Jain S. Development and characterization of ketoconazole emulgel for topical drug delivery. Pharm Sin. 2010;1(3):221–231. | ||
Ibrachim MM, Hafez SA, Mahdy MM. Organogels, hydrogels and bigels as transdermal delivery systems for diltiazem hydrochloride. AJPS. 2013;8(1):48–57. | ||
Nikumbh K, Sevankar S, Patil M. Formulation development, in vitro and in vivo evaluation of microemulsion-based gel loaded with ketoprofen. Drug Deliv. 2015;22(4):509–515. | ||
Arvouet-Grand A, Vennat B, Lejeune B, Pourrat A. Formulation of propolis extract emulsions part I: o/w creams based on nonionic surfactants and various consistency agents. Drug Dev Ind Pharm. 1995;21(16):1907–1915. | ||
Lardy F, Vennat B, Pouget MP, Pourrat A. Functionalization of hydrocolloids: principal component analysis applied to the study of correlations between parameters describing the consistency of hydrogels. Drug Dev Ind Pharm. 2000;26(7):715–721. | ||
Manna S, Lakshmi US, Racharla M, Sinha P, Kanthal LK, Kumar SPN. Bioadhesive HPMC gel containing gelatin nanoparticles for intravaginal delivery of tenofovir. J App Pharm Sci. 2016;6(8):22–29. | ||
Dantas MGB, Reis SAGB, Damasceno CMD, et al. Development and evaluation of stability of a gel formulation containing the monoterpene borneol. Sci World J. 2016;2016:7394685. | ||
Ueda CU, Shah VP, Derdzinski K, et al. Topical and transdermal drug products. Pharmacopeial Forum. 2009;35(3):750–764. | ||
Hosny KM, Rambo SM, Al-Zahrani MM, Al-Subhi SM, Fahmy UA. Ketoprofen emulgel: preparation, characterization, and pharmacodynamic evaluation. Int J Pharm Sci Rev Res. 2013;20(2):306–310. | ||
Widera G, Johnson J, Kim L, et al. Effect of delivery parameters on immunization to ovalbumin following intracutaneous administration by a coated microneedle array patch system. Vaccine. 2006;24(10):1653–1664. | ||
Apostolova EG, Kokova VY, Peychev LP. Experimental study on the antinociceptive effect of retigabine in rats. IJBAMR. 2015;5(1):568–574. | ||
Porzio S, Caselli G, Pellegrini L, et al. Efficacy of a new topical gel-spray formulation of ketoprofen lysine salt in the rat: percutaneous permeation in vitro and in vivo and pharmacological activity. Pharmacol Res. 1998;37(1):41–47. | ||
Sakeena MH, Yam MF, Elrashid SM, Munavvar AS, Azmin MN. Anti-inflammatory and analgesic effects of ketoprofen in palm oil esters nanoemulsion. J Oleo Sci. 2010;59(12):667–671. | ||
Tita B, Marian E, Rusu G, Bandur G, Tita D. Effects of experimental conditions on the thermal behavior of some non-steroidal anti-inflammatory deugs. Rev Chim (Bucharest). 2013;64(12):1390–1394. | ||
Gálico DA, Holanda BB, Perpétuo GL, Schnitzler E, Filho-Treo O, Bannach G. Thermal and spectroscopy studies on solid ketoprofen of lighter trivalent lanthanides. J Therm Anal Calorim. 2011;108(1):371–379. | ||
Silverstein RM, Bassler GC, Kiemle DJ, Bryce DR. Spectrometric Identification of Organic Compounds. 9th ed. New York: John Wiley and Sons; 2014. | ||
Chan S-Y, Chung Y-Y, Cheah X-Z, Tan EY-L, Quah J. The characterization and dissolution performances of spray dried solid dispersion of ketoprofen in hydrophilic carriers. AJPS. 2015;10(5):372–385. | ||
Zlatkov A, Peikov P, Obreshkova D, Pencheva I. Spectral analyses methods for chemical compounds. 1st ed. Plovdiv: Macros; 2010. | ||
Tapi.com [homepage on the Internet]. Israel: Guy Samburski. Physical Quality Specifications Lead to Success; 2017. Available from: https://www.tapi.com/useful-information/articles/physical_quality/. Accessed April 15, 2017. | ||
Honary S, Zahir F. Effect of zeta potential on the properties of nano-drug delivery systems – a review (part 2). Trop J Pharm Res. 2013;12(2):265–273. | ||
Pohlmann AR, Detoni CB, Paese K, Coradini K, Beck RCR, Guterres SS. Polymeric nanocapsules for topical delivery. In: Dragicevic N, Maibach HI, editors. Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement: Nanocarriers. Berlin: Springer; 2016:201–203. | ||
Singh VK, Anis A, Banerjee I, Pramanik K, Bhattacharya MK, Pal K. Preparation and characterization of novel carbopol based bigels for topical delivery of metronidazole for the treatment of bacterial vaginosis. Mater Sci Eng C Mater Biol Appl. 2014;44:151–158. | ||
Behera B, Singh VK, Kulantaivel S, Bhattacharya MK, Paramanik K. Physical and mechanical properties of sunflower oil and synthetic polymers based bigels for the delivery of nitroimidazole antibiotic – a therapeutic approach for controlled drug delivery. Eur Polym J. 2015;64:253–264. | ||
Worth AP, Cronin MTD. The use of pH measurements to predict the potential of chemicals to cause acute dermal and ocular toxicity. Toxicology. 2001;169(2):119–131. | ||
Varma VN, Maheshwari PV, Navya M, Reddy SC, Shivakumar HG, Gowda DV. Calcipotriol delivery into the skin as emulgel for effective permeation. Saudi Pharm J. 2014;22(6):591–599. | ||
Raju YP, Haritha K, Satyanarayana RP, et al. Formulation and characterization of aceclofenac – Aloe vera transemulgel. Curr Drug Deliv. 2015;12(6):703–708. | ||
Karri VVSNR, Raman SK, Kuppusamy G, Mulukutla S, Ramaswamy S, Malayandi R. Terbinafine hydrochloride loaded nanoemulsion based gel for topical application. Int J Pharm Investig. 2015;45(1):79–89. | ||
Gallagher S, Trottet L, Heard CM. Ketoprofen: release from, permeation across and rheology of simple gel formulation that simulate increasing dryness. Int J Pharm. 2003;268(1–2):37–45. | ||
Solinís MA, de la Cruz Y, Calvo B, et al. Release of salbutamol sulphate and ketoprofen enantiomers from matrices containing HPMC and cellulose derivatives. Chirality. 2002;14(10):806–813. | ||
Elkomy MH, Elmenshawe SF, Eid HM, Ali AMA. Topical ketoprofen nanogel: artificial neural network optimization, clustered bootstrap validation, and in vivo activity evaluation based on longitudinal dose response modeling. Drug Deliv. 2016;23(9):3294–3306. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.