")
Back to Journals » Nutrition and Dietary Supplements » Volume 6
Ketogenic dietary therapy for epilepsy and other disorders: current perspectives
Authors Neal E
Received 29 November 2013
Accepted for publication 15 January 2014
Published 3 April 2014 Volume 2014:6 Pages 25—33
DOI https://doi.org/10.2147/NDS.S41517
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Elizabeth G Neal
1Matthew's Friends Clinics, Lingfield, UK, 2UCL Institute of Child Health, London, UK
Abstract: The ketogenic diet (KD) is a high-fat, restricted-carbohydrate regime, originally designed to mimic metabolic responses to fasting and has been used since the 1920s as a treatment for epilepsy. Modified variants of the KD include the addition of medium-chain triglyceride and less-restrictive modified Atkins and low glycemic index protocols. Scientifically proven as treatment for intractable seizures in children, these ketone-generating diets are increasingly also being used in adults. They are the treatment of choice in glucose transporter type 1 deficiency syndrome and pyruvate dehydrogenase deficiency. Evidence for the potential of KD therapy to be included within the treatment options for cancer and neurodegenerative disorders is more limited, albeit an exciting area of research with future clinical potential. This review discusses the key aspects of KD therapy, including the efficacy of treatments and clinical implementation. The importance of appropriate initiation, adequate clinical supervision, regular monitoring, and assessment of nutritional needs is highlighted.
Keywords: diet, seizures, ketosis
Introduction
A dietary approach to the treatment of epilepsy can be traced back to Hippocrates’ description of a man whose seizures were cured by abstaining from all food and drink. The first scientific accounts of the benefits of fasting in epilepsy were published more than 100 years ago.1 A further study in 1921 reported that 20 out of 26 fasted patients showed seizure improvement; two of these patients remained seizure-free for more than a year.2 During fasting, hormonal and metabolic adaptation occurs to draw on energy reserves of body fat with the aim of sparing protein breakdown. Muscles and other tissues progressively switch energy source from glucose to free fatty acids that are converted to ketone bodies (acetoacetate and β-hydroxybutyrate) in liver mitochondria, via β-oxidation and acetyl-coenzyme A formation. Ketone bodies are increasingly utilized as an energy source in many tissues, notably heart, muscle, and brain. Unlike fatty acids, ketone bodies can pass across the blood–brain barrier.
The suggestion that a diet that is very high in fat and low in carbohydrate might mimic these fasting-induced metabolic changes and thus have benefit to individuals with epilepsy3 led to the development of the ketogenic diet (KD) that is still used today. Other early studies reported effective use of this treatment in children4–8 and adults9 with epilepsy. Although interest in diet therapy diminished with the development of new anticonvulsant medications toward the end of the 1930s, more recently there has been a reversal of this trend with KD research and worldwide clinical use increasing significantly over the last 20 years. This review describes the different KD therapies, evidence for efficacy, and key aspects of clinical implementation. The potential for their therapeutic use in disorders other than epilepsy is also considered.
The ketogenic diets
The classical KD described in the early studies was based on a ratio of ketone-producing foods in the diet (fat) to foods that reduced ketone production (carbohydrate and protein). Seizure control in children was found to be optimal when this ketogenic ratio was at least 3, and most of the classical KDs used today aim for a ratio of 3:1 (87% of total dietary energy derived from fat) or 4:1 (90% of total dietary energy derived from fat). The carbohydrate allowance is very limited, and protein is kept to a minimum to meet requirements for growth. A modification of this diet was first described 50 years later, using medium-chain triglycerides (MCT) to replace some of the long-chain fat sources.10 As MCT are absorbed and metabolized more efficiently, being carried directly to the liver in the portal blood, ketone body synthesis will be greater per unit of ingested fat.10 Therefore, less total fat is needed in the diet, allowing more carbohydrate and protein with MCT generally providing 30%–60% of dietary energy. Both the classical and MCT KD involve strict dietary calculation and food weighing.
In the last 10 years, alternative more relaxed protocols for implementation of KD therapy have been used successfully. The modified Atkins diet (MAD) restricts carbohydrates and encourages high-fat foods, but it does not limit protein or total dietary energy.11 Food is not weighed, but many centers use a macronutrient exchange system for implementation. The low glycemic index treatment is more generous in carbohydrates, but it only allows those with a glycemic index <50.12 Protein, fat, and calories are monitored, but not as strictly as on the KD. Food is not weighed; it is based on portion sizes.12 These four different KD therapies are compared in Table 1, which gives more detail on macronutrient compositions and their suitability for different individuals.
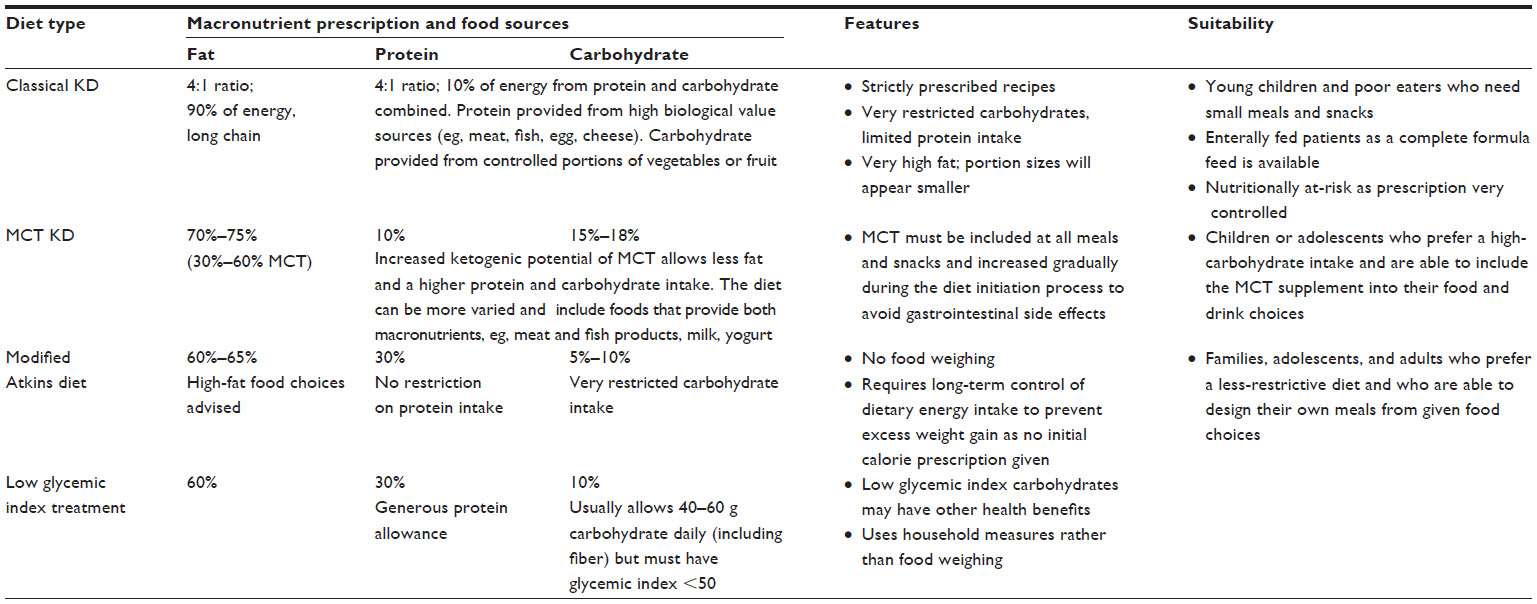 | Table 1 Characteristics of the different KD therapies |
In practice, many dietitians are adopting a more flexible, patient-tailored approach to KD therapy, especially when treating older children and adults. This may combine elements from one or more of the different types of KD, rather than sticking to a rigid diet protocol.
Efficacy of KD therapies
There have been many case series and observational studies reporting successful use of the classical and MCT KD in the treatment of intractable childhood epilepsy; efficacy has also been reported in two systematic reviews13,14 and a statistical meta-analysis of 19 studies.15 Although most studies report use of the classical KD, a randomized controlled trial (RCT), including 145 children on both classical and MCT KD, reported seizure frequency after 3 months to be significantly lower in the diet group compared to controls,16 with no difference in efficacy between these two types of KD at 3, 6, or 12 months.17 The last decade has seen a rapidly growing body of published literature on the effectiveness of the MAD. A review of data from four prospective studies found 52% of a total of 82 children had >50% seizure reduction after 3 months on this diet.18
Retrospective low glycemic index treatment data suggests similar results.19 An updated Cochrane review on the efficacy of ketogenic therapies concluded that they conveyed “short to medium term benefits in seizure control, effects which are comparable to modern antiepileptic drugs.”20 Since this review, a RCT on the use of the MAD in refractory childhood epilepsy has been published.21 Seizure frequency in 102 children after 3 months was significantly lower in the diet group compared to controls. Comparing efficacy between these different dietary protocols is challenging, due to differences in selection criteria, although there is some evidence that the stricter KD may lead to greater improvements in seizure control.22
KD therapy has been shown to be successful in treating a wide range of seizure types and syndromes,23 although it may be particularly beneficial in myoclonic epilepsies, infantile spasms, and tuberous sclerosis complex.24 It is the treatment of choice in disorders of brain energy metabolism (see Use of KD therapy in other disorders). Although success has been reported in all ages including infants, research into the dietary treatment of epilepsy in adults is more limited. A review of the available data concluded that it may be effective for almost one-half of drug-resistant adolescents and adults25 and is increasingly being requested as a treatment option in this group.
The reported benefits of KD therapy frequently extend beyond a reduction in the number and the intensity of seizures and include more rapid recovery from seizures, reduced dependence on emergency medication, and increased alertness, awareness, responsiveness, and energy levels. Anecdotal data suggest these improvements in the quality of life can be independent of seizure control and may be partly due to the reduced anticonvulsant drug load that can often be facilitated by successful dietary treatment. The effects of the diet can be long lasting, and some individuals who have seen benefit can return to a normal diet without resumption of seizure activity.
Mechanism of action
Determining the mechanism by which KD therapy acts to reduce seizure activity in the brain is ongoing and remains the focus of many international research groups. The restricted carbohydrate and high-fat intake will result in reduced glucose utilization and increased ketone body production from fatty acid oxidation, with a shift away from glycolysis toward increased Krebs cycle activity and direct mitochondrial metabolism of ketone bodies. Although a single underlying mechanism remains unknown, it is likely these metabolic changes will decrease neuronal excitability via multiple coexisting mechanistic pathways beyond that of a simple, direct anticonvulsant effect of ketone bodies. The increased mitochondrial adenosine triphosphate (ATP) production and reduced glycolytic ATP production will selectively activate ATP-sensitive potassium channels that will, in turn, have a stabilizing effect on the neuronal cell membrane.26–29
The KD may also have a role in altering neurotransmitter function by disrupting excitatory glutamate synaptic transmission and facilitating an increased production of inhibitory γ-aminobutyric acid (GABA).26–29 Other hypothesized mechanisms of KD action include the inhibition of mammalian target of rapamycin activity,27,29 the refilling of Krebs cycle substrates by anaplerosis,27 an increase in extracellular adenosine,27,28 increased leptin levels,27 and reduced intracellular reactive oxygen species, due to increased nicotinamide adenine dinucleotide phosphate production.26,28,29 Increased polyunsaturated acid levels have been reported on the KD,26,27 which may confer anticonvulsant benefit via modifications in cell membrane composition, stimulation of nuclear receptor proteins (eg, peroxisome proliferator-activated receptor α), and decreased production of reactive oxygen species by upregulation of mitochondrial uncoupling protein activity and expression.26,27
The direct anticonvulsant action of structurally specific medium-chain fatty acids has also been reported.30 There is growing recognition of additional neuroprotective, antiepileptogenic benefit of the KD-induced alterations in cellular fuel supply and possible dietary influence on gene expression. Further detail on mechanistic hypotheses can be found in a number of excellent articles on this subject.26–30
Clinical implementation
Ketogenic dietary therapy should only be initiated under the supervision of a hospital team, including medical support from a neurologist or physician who has experience in treating epilepsy and a registered dietitian who is confident in the calculation and fine-tuning of the chosen dietary protocol. Although computer programs are now available to aid recipe development, the dietitian will assess individual nutritional requirements and develop appropriate dietary prescriptions.
KD therapy should not be used in individuals who have disorders of fatty acid metabolism or who require treatment with a high-dietary carbohydrate intake;24 other medical contraindications are included in Table 2. Compliance of parent and/or caregiver with the dietary restrictions is paramount to success; this should be taken into consideration prior to commencing treatment. The KD can easily be administered to enterally-fed children via nasogastric tube or gastrostomy, and ketogenic formulas are available for convenience (KetoCal, Nutricia North America, Inc., Gaithersburg, MD, USA; Nutricia Advanced Medical Nutrition, Schiphol, The Netherlands). Although not an absolute contraindication, concurrent dietary restrictions due to physical feeding difficulties or food intolerances will make KD therapy more difficult to implement, especially if there are associated behavioral eating issues.
 | Table 2 Medical contraindications to use of KD therapy |
For many years, the KD was only initiated during a hospital admission and after a closely monitored fast of up to 48 hours with controlled fluid intake. The advantages of this type of protocol have not been proved,31–33 and many diets are now started at home without an initial fast or fluid restriction. However, some centers will choose to hospitalize children for initiation – especially the very young – in view of the restrictive nature of the KD therapy and the importance of adequate monitoring of metabolic changes during the early days. Diets should only be started on an outpatient basis if there has been a thorough teaching program prior to commencement, including instructions on the daily home monitoring of blood or urine ketones and the management of possible early side effects, such as excess ketosis and hypoglycemia.
KD therapy can be used alongside other anticonvulsant drugs, although a more cautious initiation may be needed in patients taking medications that increase the risk of acidosis. Ongoing community support from the diet team is essential, with regular monitoring throughout the duration of dietary treatment including full blood hematology and biochemistry. There have been reports of a number of side effects, most commonly constipation and other gastrointestinal problems, short-term acidosis, hyperlipidemia, growth faltering, and renal calculi.34–37
A 3-month trial of dietary treatment is recommended; if there is no improvement by then – despite appropriate dietary fine tuning – it should be discontinued. If KD therapy is successful, it will usually be continued for at least 2 years, but this period of time is individualized based on patient response, not on specific guidelines. Discontinuation must be done cautiously; the longer it has been used and the more successful it has been in seizure treatment, the more gradual should be the change back to a normal diet. This will employ a stepwise process that may take weeks or months.
Worldwide use of KD therapies has increased dramatically since the early 1990s. The past few years have seen the development of parent and professional support groups, notably Matthew’s Friends based in the UK (matthewsfriends.org) and The Charlie Foundation based in the USA (charliefoundation.org). Not only have these organizations facilitated global and local scientific conferences and parent information events, they are an invaluable resource to aid dietary implementation with family support and sharing of recipes and practical ideas.
Nutritional status and supplementation
To prevent nutritional deficiencies, full vitamin, mineral, and trace element supplementation should be given while on KD therapy; requirements will differ depending on which dietary protocol is used and should be assessed on an individual basis by the dietitian. Children with drug-resistant epilepsy are at risk of insufficient vitamin D status prior to starting a KD,38 and although levels can be normalized on diet therapy with vitamin D supplementation, a decline in both whole body and spine bone mineral content while on the KD has been reported,39 despite reduction in anticonvulsant medication. Selenium deficiency can occur in children on the KD40 with the risk of impaired myocardial function.41 Hypomagnesemia has also been seen34 and may be a particular problem in children on the classical KD, despite micronutrient supplementation.42 There is one report of vitamin C deficiency in a child on the KD who presented with scurvy and oral complications.43 Risk of these micronutrient deficiencies highlights the importance of regular dietary review of the adequacy of supplementation and measurement of blood levels wherever possible. Conversely, plasma levels of vitamin A and vitamin E can often be raised42 as a consequence of high dietary intakes associated with a high-fat diet; these should also be regularly monitored.
Carnitine is an amino-acid derivative that is needed for the esterification and transport of long-chain fatty acids into the mitochondria for subsequent β-oxidation. Carnitine deficiency has been reported in epilepsy patients treated with anticonvulsant medications,44,45 and the KD may further deplete stores as carnitine requirements will be increased if a diet high in long-chain fat is followed. A fall in the carnitine status of children and young adults during the first few months of KD treatment has been seen,46 with some cases requiring supplementation, although levels tended to normalize with time on the diet therapy. Although carnitine deficiency is unlikely if high ketone levels are being maintained, free carnitine levels should be regularly monitored while on KD therapies, and additional supplementation may be needed if deficiency is suspected.
In view of the potential atherogenic risk of KD-induced alterations in lipid levels,35 advice should be given as part of the dietary prescription to ensure this high-fat diet provides a healthy balance of fatty acid intakes. The omega 3:omega 6 fatty acid ratio within plasma phospholipids will have an influence on cardiovascular risk, with one study suggesting that an omega 3-enriched KD may have benefit in ensuring this ratio is kept within the normal range.47 The possibility of controlling lipid levels by KD modification is also supported by one study of 12 children with preexisting hyperlipidemia; a very low saturated fat and low cholesterol diet with optimized polyunsaturated fatty acid intake resulted in a significant reduction in both total and low-density lipoprotein cholesterol levels.48
KD therapy in neurometabolic disease
KD therapy is the recommended first-line treatment24 for two rare neurometabolic diseases that affect the energy metabolism of the brain, glucose transporter type 1 (GLUT1) deficiency syndrome, and pyruvate dehydrogenase (PDH) deficiency. GLUT1 deficiency syndrome is caused by a defect in the protein responsible for glucose transfer across the blood–brain barrier. Frequently presenting with seizures early in life, brain growth may be impaired with developmental delay and other neurological problems, including a complex movement disorder. It is characterized by low glucose concentration in the cerebrospinal fluid in the absence of hypoglycemia, alongside low-to-normal cerebrospinal fluid lactate.49 Ketone bodies can be used as an alternative energy source for the brain and – in most cases – the KD will significantly reduce seizure frequency,50,51 with improvements in neurological function and movement disorders also reported.50,52,53 The classical KD has been most widely used to treat GLUT1, although case reports suggest MAD may also be used successfully.54,55 Diets should be continued well into puberty; the benefit in adults is unclear.
PDH is a severe degenerative mitochondrial disorder that presents with lactic acidosis and variable degrees of neurological degeneration during infancy and childhood. Caused by an enzyme deficiency within the pyruvate dehydrogenase complex that prevents the metabolism of pyruvate to acetyl-coA, pyruvate is instead metabolized to lactate, and ATP production is compromised. The KD will bypass the metabolic block by providing ketones as an alternative fuel to glucose. Although it will not fully reverse clinical symptoms, the progressive loss of neurological function can be slowed by the dietary treatment, especially if it is initiated early in life.56,57 The classical KD is usually recommended as stricter carbohydrate restriction has been associated with greater improvement in clinical outcome,57 although one recent case report suggests a modified diet can also be used with success.58
Use of KD therapy in other disorders
There is a growing evidence base to support the use of KD therapy within the established treatment options for a range of other disorders,59,60 most notably cancer of the brain and other organs. The metabolic switch toward glucose as a preferred energy source that occurs in malignant cancer cells was first reported in the 1920s,61 facilitated by the upregulation of key enzymes involved in glucose metabolism and transport.62 While normal tissues can adapt to glucose depletion by using ketones for energy, this may not be possible in the glucose-dependent malignant cell. The use of a low-carbohydrate, high-fat KD could be a way to specifically target tumors based on these metabolic differences.63 Although positive reports are mainly from animal studies,64–69 this is an exciting area of research with potentially important clinical applications especially for neurological cancers. The MCT KD was successfully used to treat two children with advanced stage malignant astrocytomas; glucose uptake decreased at the tumor sites, with one patient showing significant clinical improvement and remaining free of disease progression during 12 months on the diet.70 In a group of 16 adults with advanced metastatic cancer of various sites treated with an omega-3 supplemented, carbohydrate-restricted diet, improvements were seen in blood parameters, emotional functioning, and sleep in the five patients who were able to complete the 3-month study period.71
A novel therapeutic strategy for management of brain cancers has been proposed, involving the initial use of KD therapy to lower circulating glucose and to raise ketone bodies prior to considering surgery or other treatments;61 further clinical trials involving the use of diet alongside conventional cancer treatments are currently in development.62
Evidence from animal studies suggests that KD therapy may have a protective role in neurotrauma and ischemia, with the injured brain preferring to use ketones as a fuel.72 The metabolic changes induced by KD therapy may also be beneficial in certain neurodegenerative disorders.72 Parkinson’s disease is characterized by progressive death of brain dopaminergic neurons; ketone bodies could provide a fuel source for these at-risk neurons, and the ketone-induced enhancement of mitochondrial function may protect cells against insults that demand a high energy supply. This is supported by animal data73 and a small feasibility study that reported a decreased Parkinson’s disease rating score in five patients on the KD.74
Studies in mice suggest KD therapy may protect against the formation of the amyloid protein-containing extracellular plaques that are characteristically deposited in Alzheimer’s disease. In a randomized, double-blind study of 152 patients with mild-to-moderate Alzheimer’s disease, the group treated with MCT ketogenic compounds showed significant improvement in cognitive function compared with controls,75 although the benefit of the diet was only seen in patients lacking the apolipoprotein ε4 genotype.
Animal studies also suggest that KD therapy may help slow the progressive loss of motor neurons seen in amyotrophic lateral sclerosis,76 although there are no published clinical data.
Conclusion
The traditional KD and its newer modified variants are well-established and scientifically proven treatments for intractable pediatric epilepsy; increasingly, they are also being used in adults. They are a first-line treatment for GLUT1 deficiency and PDH. Although there is clearly future potential for this type of diet within the management of cancer and neurodegenerative disorders, further studies are needed before KD therapy can be incorporated routinely into clinical practice to clarify who would benefit from this type of treatment and how best it should be implemented. KD therapies should always be under the supervision of a dietitian and physician, with consideration given to nutritional supplementation, monitoring, and the risk of side effects. Although use is increasing worldwide, in many countries, the availability of this type of diet therapy is both limited and variable.
Disclosure
The author reports no conflicts of interest in this work.
References
Guelpa G, Marie A. La lutte contre l’epilepsie par la desintoxication et par la reeducation alimentaire. Revue de Therapie Medico-Chirurgicale. 1911;78:8–13. | |
Geyelin HR. Fasting as a method for treating epilepsy. Medical Recordings. 1921;99:1037–1039. | |
Wilder RM. The effect of ketonemia on the course of epilepsy. Mayo Clinic Bulletin. 1921;2:307. | |
Peterman MG. The ketogenic diet in the treatment of epilepsy: a preliminary report. Archives of Pediatrics and Adolescent Medicine. 1924;28(1):28–33. | |
Helmholz HF. The treatment of epilepsy in childhood: five years’ experience with the ketogenic diet. JAMA. 1927;88(26):2028–2032. | |
Talbot FB, Metcalf KM, Moriarty ME. A clinical study of epileptic children treated by the ketogenic diet. The Boston Medical and Surgical Journal. 1927;196(3):89–96. | |
McQuarrie I, Keith HM. Epilepsy in children: relationship of variations in the degree of ketonuria to occurrence of convulsions in epileptic children on ketogenic diets. Archives of Pediatrics and Adolescent Medicine. 1927;34:1013–1029. | |
Lennox WG. Ketogenic diet in the treatment of epilepsy. N Engl J Med. 1928;199(2):74–75. | |
Barborka CJ. Epilepsy in adults: results of treatment by ketogenic diet in one hundred cases. Archives of Neurology and Psychiatry. 1930;23(5):904–914. | |
Huttenlocher PR, Wilbourn AJ, Signore JM. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology. 1971;21(11):1097–1103. | |
Kossoff EH, Krauss GL, McGrogan JR, Freeman JM. Efficacy of the Atkins diet as therapy for intractable epilepsy. Neurology. 2003;61(12):1789–1791. | |
Pfeifer HH, Thiele EA. Low-glycemic-index treatment: a liberalized ketogenic diet for treatment of intractable epilepsy. Neurology. 2005;65(11):1810–1812. | |
Lefevre F, Aronson N. Ketogenic diet for the treatment of refractory epilepsy in children: A systematic review of efficacy. Pediatrics. 2000;105(4):E46. | |
Keene DL. A systematic review of the use of the ketogenic diet in childhood epilepsy. Pediatr Neurol. 2006;35(1):1–5. | |
Henderson CB, Filloux FM, Alder SC, Lyon JL, Caplin DA. Efficacy of the ketogenic diet as a treatment option for epilepsy: meta-analysis. J Child Neurol. 2006;21(3):193–198. | |
Neal EG, Chaffe H, Schwartz RH, et al. The ketogenic diet in the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7(6):500–506. | |
Neal EG, Chaffe H, Schwartz RH, et al. A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia. 2009;50(5):1109–1117. | |
Auvin S. Should we routinely use modified Atkins diet instead of regular ketogenic diet to treat children with epilepsy? Seizure. 2012;21(4):237–240. | |
Muzykewicz DA, Lyczkowski DA, Memon N, Conant KD, Pfeifer HH, Thiele EA. Efficacy, safety, and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia. 2009;50(5):1118–1126. | |
Levy RG, Cooper PN, Giri P. Ketogenic diet and other dietary treatments for epilepsy [review]. Cochrane Database Syst Rev. 2012;3:CD001903. | |
Sharma S, Sankhyan N, Gulati S, Agarwala A. Use of the modified Atkins diet for treatment of refractory childhood epilepsy: a randomized controlled trial. Epilepsia. 2013;54(3):481–486. | |
El-Rashidy OF, Nassar MF, Abdel-Hamid IA, et al. Modified Atkins diet vs classic ketogenic formula in intractable epilepsy. Acta Neurol Scand. 2013;128(6):402–408. | |
Nangia S, Caraballo RH, Kang HC, Nordli DR, Scheffer IE. Is the ketogenic diet effective in specific epilepsy syndromes? Epilepsy Res. 2012;100(3):252–257. | |
Kossoff EH, Zupec-Kania BA, Amark PE, et al. Charlie Foundation, Practice Committee of the Child Neurology Society; Practice Committee of the Child Neurology Society; International Ketogenic Diet Study Group. Optimal clinical management of children receiving the ketogenic diet: recommendations of the international ketogenic diet study group. Epilepsia. 2009;50(2):304–317. | |
Payne NE, Cross JH, Sander JW, Sisodiya SM. The ketogenic and related diets in adolescents and adults – a review. Epilepsia. 2011;52(11):1941–1948. | |
Bough KJ, Rho JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia. 2007;48(1):43–58. | |
Hartman AL, Rho JM. The biochemical basis of dietary therapies for neurological disorders. In: Neal EG, editor. Dietary Treatment of Epilepsy: Practical Implementation of Ketogenic Therapy. Oxford, UK: Wiley-Blackwell; 2012:34–44. | |
Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36(1):32–40. | |
Danial NN, Hartman AL, Stafstrom CE, Thio LL. How does the ketogenic diet work? Four potential mechanisms. J Child Neurol. 2013;28(8):1027–1033. | |
Chang P, Terbach N, Plant N, Chen PE, Walker MC, Williams RS. Seizure control by ketogenic diet-associated medium chain fatty acids. Neuropharmacology. 2013;69:105–114. | |
Bergqvist AG, Schall JI, Gallagher PR, Cnaan A, Stallings VA. Fasting versus gradual initiation of the ketogenic diet: a prospective, randomized clinical trial of efficacy. Epilepsia. 2005;46(11):1810–1819. | |
Vaisleib II, Buchhalter JR, Zupanc ML. Ketogenic diet: outpatient initiation, without fluid, or caloric restrictions. Pediatr Neurol. 2004;31(3):198–202. | |
Rizzutti S, Ramos AM, Muszkat M, Gabbai AA. Is hospitalization really necessary during the introduction of the ketogenic diet? J Child Neurol. 2007;22(2):33–37. | |
Kang HC, Chung DE, Kim DW, Kim HD. Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia. 2004;45(9):1116–1123. | |
Kwiterovich PO Jr, Vining EP, Pyzik P, Skolasky R Jr, Freeman JM. Effect of a high-fat ketogenic diet on plasma levels of lipids, lipoproteins, and apolipoproteins in children. JAMA. 2003;290(7):912–919. | |
Vining EPG, Pyzik P, McGrogan J, et al. Growth of children on the ketogenic diet. Dev Med Child Neurol. 2002;44(12):796–802. | |
Sampath A, Kossoff EH, Furth SL, Pyzik PL, Vining EP. Kidney stones and the ketogenic diet: risk factors and prevention. J Child Neurol. 2007;22(4):375–378. | |
Bergqvist AG, Schall JI, Stallings VA. Vitamin D status in children with intractable epilepsy, and impact of the ketogenic diet. Epilepsia. 2007;48(1):66–71. | |
Bergqvist AG, Schall JI, Stallings VA, Zemel BS. Progressive bone mineral content loss in children with intractable epilepsy treated with the ketogenic diet. Am J Clin Nutr. 2008;88(6):1678–1684. | |
Bergqvist AG, Chee CM, Lutchka L, Rychik J, Stallings VA. Selenium deficiency associated with cardiomyopathy: a complication of the ketogenic diet. Epilepsia. 2003;44(4):618–620. | |
Bank IM, Shemie SD, Rosenblatt B, Bernard C, Mackie AS. Sudden cardiac death in association with the ketogenic diet. Pediatr Neurol. 2008;39(6):429–431. | |
Christodoulides SS, Neal EG, Fitzsimmons G, et al. The effect of the classical and medium chain triglyceride ketogenic diet on vitamin and mineral levels. J Hum Nutr Diet. 2012;25(1):16–26. | |
Willmott NS, Bryan RA. Case report: scurvy in an epileptic child on a ketogenic diet with oral complications. Eur Arch Paediatr Dent. 2008;9(3):148–152. | |
Anil M, Helvaci M, Ozbal E, Kalenderer O, Anil AB, Dilek M. Serum and muscle carnitine levels in children receiving sodium valproate. J Child Neurol. 2009;24(1):80–86. | |
Hamed SA, Abdella MM. The risk of asymptomatic hyperammonemia in children with idiopathic epilepsy treated with valproate: relationship to blood carnitine status. Epilepsy Res. 2009;86(1):32–41. | |
Berry-Kravis E, Booth G, Sanchez AC, Woodbury-Kolb J. Carnitine levels and the ketogenic diet. Epilepsia. 2001;42(11):1445–1451. | |
Dahlin M, Hjelte L, Nilsson S, Amark P. Plasma phospholipid fatty acids are influenced by a ketogenic diet enriched with n-3 fatty acids in children with epilepsy. Epilepsy Res. 2007;73(2):199–207. | |
Liu YM, Lowe H, Zak MM, Kobayashi J, Chan VW, Donner EJ. Can children with hyperlipidemia receive ketogenic diet for medication-resistant epilepsy? J Child Neurol. 2013;28(4):479–483. | |
Leen WG, Klepper J, Verbeek MM, et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. 2010;133(Pt 3):655–670. | |
Klepper J. Impaired glucose transport into the brain: the expanding spectrum of glucose transporter type 1 deficiency syndrome. Curr Opin Neurol. 2004;17(2):193–196. | |
Klepper J, Scheffer H, Leiendecker B, et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics. 2005;36(5):302–308. | |
Klepper J, Diefenbach S, Kohlschütter A, Voit T. Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot Essent Fatty Acids. 2004;70(3):321–327. | |
Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009;31(7):545–552. | |
Ito S, Oguni H, Ito Y, Ishigaki K, Ohinata J, Osawa M. Modified Atkins diet therapy for a case with glucose transporter type 1 deficiency syndrome. Brain Dev. 2008;30(3):226–228. | |
Slaughter L, Vartzelis G, Arthur T. New GLUT-1 mutation in a child with treatment-resistant epilepsy. Epilepsy Res. 2009;84(2–3):254–256. | |
Prasad C, Rupar T, Prasad AN. Pyruvate dehydrogenase deficiency and epilepsy. Brain Dev. 2011;33(10):856–865. | |
Wexler ID, Hemalatha SG, McConnell J, et al. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology. 1997;49(6):1655–1661. | |
El-Gharbawy AH, Boney A, Young SP, Kishnani PS. Follow-up of a child with pyruvate dehydrogenase deficiency on a less restrictive ketogenic diet. Mol Genet Metab. 2011;102(2):214–215. | |
Stafstrom CE, Rho JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. 2012;3:59. | |
Paoli A, Rubini A, Volek JS, Grimaldi KA. Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur J Clin Nutr. 2013;67(8):789–796. | |
Warburg O, Posener K, Negelein E. Über den Stoffwechsel der Carcinomzelle. Biochem Zeitschr. 1924:309–344. | |
Klement RJ, Kämmerer U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr Metab (Lond). 2011; 8:75. | |
Seyfried TN, Mukherjee P. Targeting energy metabolism in brain cancer: review and hypothesis. Nutr Metab (Lond). 2005;2:30. | |
Tisdale MJ, Brennan RA, Fearon KC. Reduction of weight loss and tumour size in a cachexia model by a high fat diet. Br J Cancer. 1987;56(1):39–43. | |
Tisdale MJ, Brennan RA. A comparison of long-chain triglycerides and medium-chain triglycerides on weight loss and tumour size in a cachexia model. Br J Cancer. 1988;58(5):580–583. | |
Freedland SJ, Mavropoulos J, Wang A, et al. Carbohydrate restriction, prostate cancer growth, and the insulin-like growth factor axis. Prostate. 2008;68(1):11–19. | |
Otto C, Kaemmerer U, Illert B, et al. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omega-3 fatty acids and medium-chain triglycerides. BMC Cancer. 2008;8:122. | |
Zhou W, Mukherjee P, Kiebish MA, Markis WT, Mantis JG, Seyfried TN. The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr Metab (Lond). 2007;4:5. | |
Stafford P, Abdelwahab MG, Kim do Y, Preul MC, Rho JM, Scheck AC. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr Metab (Lond). 2010;7:74. | |
Nebeling LC, Miraldi F, Shurin SB, Lerner E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr. 1995;14(2):202–208. | |
Schmidt M, Pfetzer N, Schwab M, Strauss I, Kämmerer U. Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: A pilot trial. Nutr Metab (Lond). 2011;8(1):54. | |
Barañano KW, Hartman AL. The ketogenic diet: uses in epilepsy and other neurologic illnesses. Curr Treat Options Neurol. 2008;10(6):410–419. | |
Tieu K, Perier C, Caspersen C, et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003;112(6):892–901. | |
Vanitallie TB, Nonas C, Di Rocco A, Boyar K, Hyams K, Heymsfield SB. Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology. 2005;64(4):728–730. | |
Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond). 2009;6:31. | |
Zhao Z, Lange DJ, Voustianiouk A, et al. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006;7:29. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.