")
Back to Journals » International Journal of General Medicine » Volume 7
Kearns–Sayre syndrome: a case series of 35 adults and children
Authors Khambatta S, Nguyen D, Beckman T, Wittich C
Received 5 April 2014
Accepted for publication 29 April 2014
Published 3 July 2014 Volume 2014:7 Pages 325—332
DOI https://doi.org/10.2147/IJGM.S65560
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sherezade Khambatta, Douglas L Nguyen, Thomas J Beckman, Christopher M Wittich
Division of General Internal Medicine, Mayo Clinic, Rochester, MN, USA
Background: Kearns–Sayre syndrome (KSS) is a rare mitochondrial cytopathy, first described at Mayo Clinic in 1958.
Aims: We aimed to define patient and disease characteristics in a large group of adult and pediatric patients with KSS.
Methods: We retrospectively searched the Mayo Clinic medical index patient database for the records of patients with KSS between 1976 and 2009. The 35 patients identified with KSS were analyzed in terms of demographic characteristics, presenting signs and symptoms, diagnostic features, clinical evolution, and associations between disease features and the development of disability.
Results: The mean (standard [SD]) age at KSS presentation was 17 (10) years, but the mean age at diagnosis was 26 (15) years. Ophthalmologic symptoms developed in all patients, and neurologic and cardiac involvement was common. Only four patients (11%) in the series died, but all deaths were from sudden cardiac events. The development of physical disability was significantly associated with cognitive decline (P=0.004) but not with other clinical features, such as sex or sudden cardiac death.
Conclusion: We report the largest case series to date of patients with KSS from a single institution. In addition to the conduction system abnormalities identified in previous series, our cohort included patients with syncope and sudden cardiac death. This underscores the need to consider formal electrophysiologic studies and prophylactic defibrillators in patients with KSS.
Keywords: heart block, mitochondrial diseases, ophthalmoplegia, retinitis pigmentosa
Introduction
Kearns–Sayre syndrome (KSS) is a rare mitochondrial cytopathy, first described at Mayo Clinic in 1958.1 KSS belongs to a group of mitochondrial DNA (mtDNA) deletion syndromes that also includes Pearson syndrome and progressive external ophthalmoplegia (PEO).2 Classically, KSS has a triad of features, including presence of PEO, pigmentary retinopathy, and an age of onset younger than 20 years.1,3,4 Additionally, one or more of the following features must be present to make the diagnosis of KSS: 1) heart block, 2) cerebellar ataxia, or 3) increased cerebrospinal fluid (CSF) protein level (>100 mg/dL).1,3,4 Patients with PEO who meet some, but not all, of the criteria for KSS have been termed as having “KSS minus” or “PEO plus”.2
Although the diagnosis of KSS is based on clinical criteria, confirmation with muscle biopsy and genetic testing is now routine.3 Muscle biopsy reveals characteristic “ragged red fibers” on trichrome stain and hyperactive fibers with succinate dehydrogenase stain.3 In terms of genetic testing, most patients with KSS have large (1.3–10 kb) mtDNA deletions.2,3
Treatment of KSS includes monitoring for the development of ophthalmologic manifestations and, if present, consideration of their surgical management. Many patients are given supplements, such as coenzyme Q10.5 Current guidelines recommend permanent pacemaker implantation for patients with neuromuscular diseases with atrioventricular block, and this includes KSS.6
Clinically, KSS is a heterogeneous neurodegenerative syndrome involving the musculoskeletal, central nervous, cardiovascular, and endocrine systems. The current literature on KSS primarily consists of case reports or small case series that use clinical diagnostic criteria. In the case series, ptosis, ophthalmoplegia, and cricopharyngeal dysphagia are commonly recognized symptoms.7 Onset of disease usually occurs in childhood, with death commonly reported in early adulthood.2 Published case series of patients with mitochondrial diseases8,9 have included patients with KSS. The largest case series to date incorporated 136 patients with large-scale mtDNA deletions and included 33 Japanese adults and children with KSS.10 This study found an inverse association between the length of the deletion and the age at which KSS was diagnosed. Despite these existing studies, KSS remains a difficult syndrome to recognize because of the variety of clinical manifestations and the lack of any large case series to date.
Because KSS is rare, an accurate description of its phenotype is needed for clinicians to recognize the syndrome. Therefore, we endeavored to report a large case series of patients with KSS. Our objective was to characterize these patients with KSS in terms of their demographic characteristics, presenting signs and symptoms, diagnostic features, clinical evolution, and associations between disease features and the development of disability.
Methods
Study design and participants
The study cohort was identified by using the previously validated medical index database at Mayo Clinic, Rochester, Minnesota.11 The database classified patients with KSS using an internal coding system based on the Hospital Adaptation of the International Classification of Diseases, Eighth Revision.11 Additional searches, using alternative coding systems such as systematized nomenclature, were completed and revealed no additional cases. This computerized medical record search of patients seen from 1976 through 2009 identified 37 patient cases. One of the identified patients had a family history of KSS but no personal diagnosis of the syndrome and was therefore excluded. A second patient did not provide consent to use his medical records for research and was also excluded. Therefore, 35 cases were used for data collection in this study. All patients included had a diagnosis of KSS documented in the medical record. This study was approved by the Mayo Clinic Institutional Review Board.
Data collection and analysis
All data were extracted from the medical record by one reviewer (SK). All diagnoses of KSS were confirmed by examining patients’ medical records for the triad of PEO, pigmentary retinopathy, and an age of onset younger than 20 years.1,3,4 Additionally, one or more of the following were required for the diagnosis of KSS: 1) heart block, 2) cerebellar ataxia, or 3) increased CSF protein level (>100 mg/dL).1,3,4
Additional abstracted data included patient demographic characteristics (sex, age at diagnosis, family history of KSS, and family history of any mitochondrial disorder); diagnostic features (presence of an mtDNA mutation, muscle biopsy results, and protein concentrations in the CSF); major presenting symptom leading to the diagnosis of KSS; laboratory studies at diagnosis (hematocrit, leukocyte count, platelet count, and levels of hemoglobin, creatinine, calcium, aspartate aminotransferase, creatinine kinase, serum lactate, and pyruvate); relevant clinical features that developed during follow up (ophthalmologic, neurologic, or endocrine); mortality; cardiovascular symptoms (syncope, dilated cardiomyopathy, cardiac arrest, pacemaker implantation, pacemaker indication, and sudden cardiac death); and electrocardiographic (ECG) abnormalities (heart block, conduction delay, axis deviation, and preexcitation). Additionally, the severity of KSS was rated by whether the patient was independently ambulatory or used a gait aid or wheelchair. This method was modified from the Kurtzke Expanded Disability Status Scale.12
Statistical analysis
Continuous data were reported as mean (standard deviation [SD]), and discrete data were reported as number of patients (%). Associations between severity of KSS (ambulatory versus [vs] use of gait aid or wheelchair) and patient characteristics (sex and confirmation of KSS diagnosis), presence of a genetic mutation, presence of a muscle defect on biopsy, presence of deafness, presence of cognitive decline, and death were determined using a two-tailed Fisher’s exact text. P<0.05 was considered statistically significant. Statistical analyses were conducted using SAS (SAS Institute Inc., Cary, NC, USA).
Results
Patient characteristics and diagnostic features
Disease characteristics for each patient are shown in Table 1. Patient characteristics and diagnostic features are shown in Table 2. Most patients (28 [80%]) had available information in the chart to confirm the treating physicians’ diagnosis of KSS, based on accepted criteria.1,3,4 Most patients were male (24 [69%]), and all patients were white except for one, who was African American. The mean (SD) age at KSS presentation was 17 (10) years, but the mean age at diagnosis was 26 (15) years. The mean duration of follow up was 10.8 (8.7) years. None of the patients had a documented family history of KSS, although one patient had a family member with mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS syndrome). Additionally, one patient had a sister with ataxia without a formal diagnosis of a mitochondrial disorder.
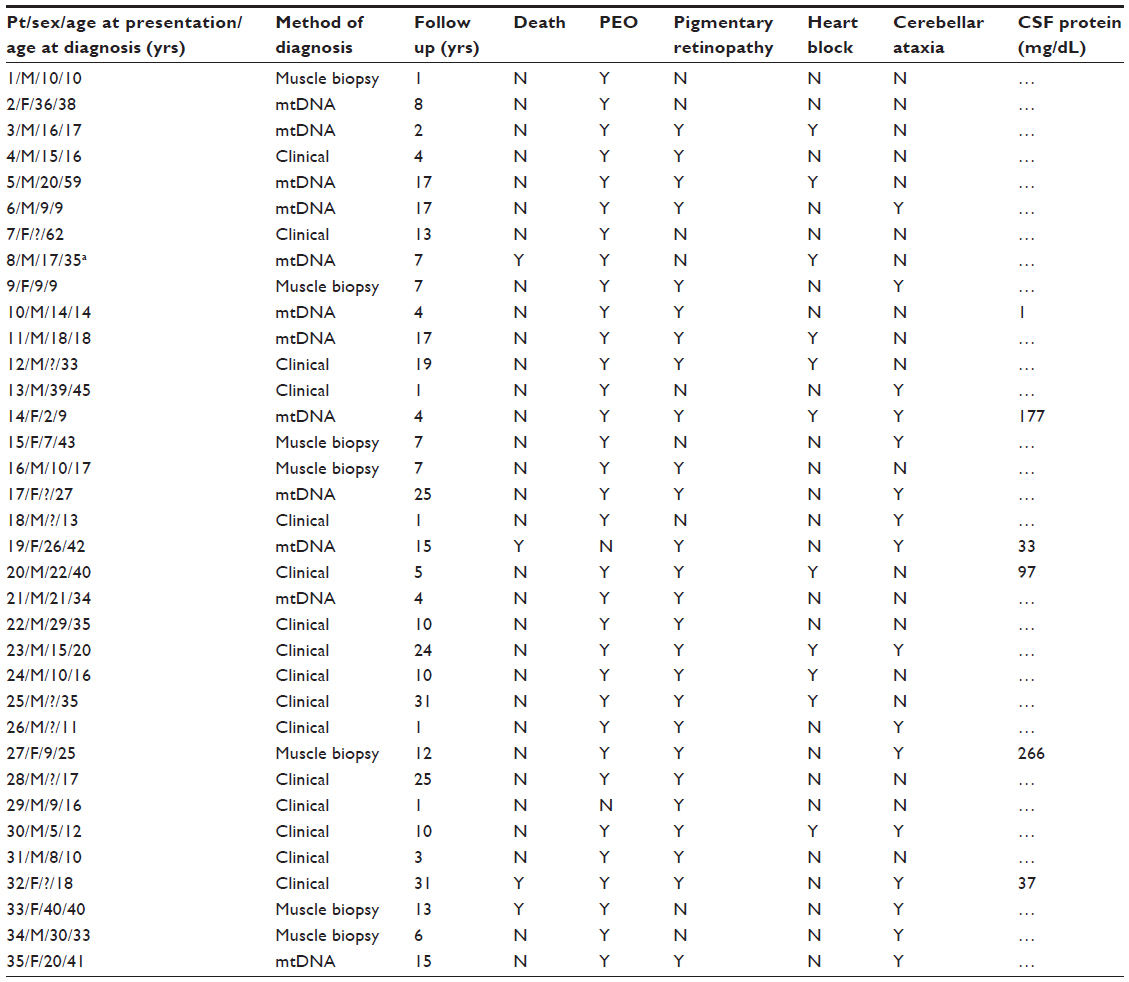 | Table 1 Patient disease characteristics |
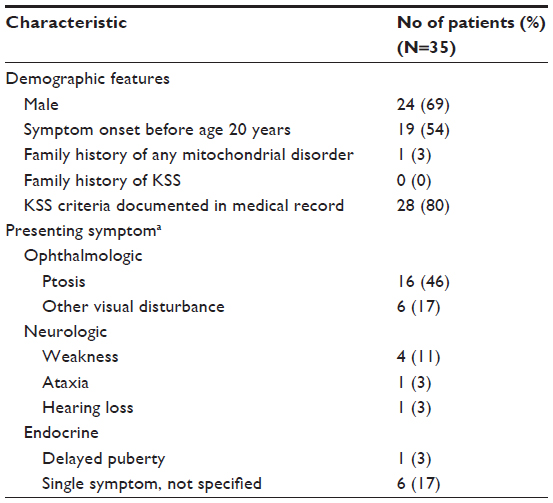 | Table 2 Patient characteristics and presenting symptoms |
Fifteen patients (43%) underwent genetic testing for an mtDNA mutation consistent with KSS, and abnormalities were found in 12 of these (80%). Muscle biopsy was performed in 17 patients (49%), 15 (88%) of whom had results consistent with a mitochondrial myopathy. CSF was tested in only five patients (14%), which showed an increased protein value in only two. In the 16 patients who had no genetic or muscle testing, the diagnosis of KSS was made on a clinical basis only; in general, these patients were older.
Major presenting signs or symptoms
Analysis of the major presenting signs and symptoms leading to the diagnosis of KSS (Table 2) showed that most patients (22 [63%]) initially had an abnormality of the ophthalmologic system. Of these ophthalmologic presenting symptoms, ptosis was the most common (16 [46%]); the other six patients had retinal pigment in the characteristic “salt and pepper”13 distribution (three patients), diplopia (two patients), and decreased vision (one patient). Neurologic symptoms were the next most common (six [17%]), which included nonspecific weakness (four), ataxia (one), and hearing loss (one). In one patient (3%), delayed puberty was the initial symptom of KSS.
Laboratory studies at diagnosis
At the time of KSS diagnosis, no significant abnormalities were found in the hemoglobin, hematocrit, leukocyte count, platelet count, or creatinine, calcium, or aspartate aminotransferase levels in any of the patients tested. However, four of the 20 patients with data available had small increases in creatinine kinase value. Serum lactate levels were increased (normal <2.3 mmol/L) in six of 18 patients (33%) at diagnosis, with only one patient having a level higher than 5 mmol/L. Six of 13 patients (46%) had increased pyruvate levels.
Clinical features developing during follow up
Clinical features developing during follow up are described in Table 3. Only four patients (11%) died during the study period (average duration of follow-up of 10.8 years). The mean (SD) age at death was 46 (23) years, and all patients died of sudden cardiac death.
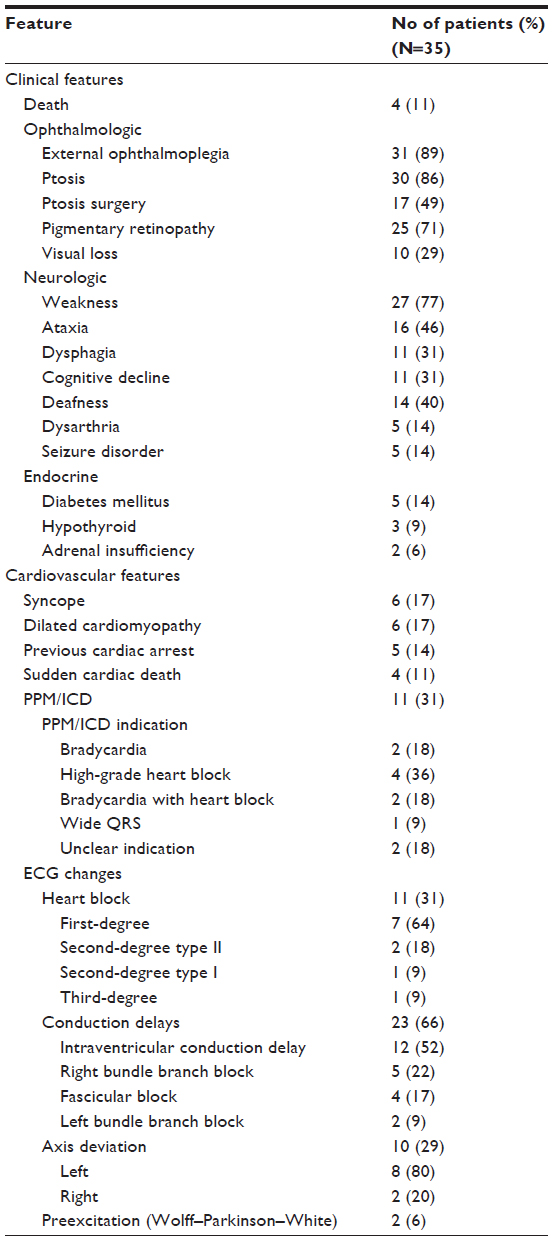 | Table 3 Clinical and cardiovascular features and ECG abnormalities developing during follow up |
An ophthalmologic abnormality developed in all patients as a part of their KSS. PEO was present in 31 patients (89%), and ptosis developed in 30 (86%), 17 of whom underwent surgery for ptosis at least once. Pigmentary retinopathy (25 [71%]) and visual loss (ten [29%]) were also noted.
After ophthalmologic findings, neurologic symptoms were the next most common. Weakness was found in 27 patients (77%), which varied from mild weakness to extreme debility. Other symptoms included ataxia, deafness, cognitive decline, dysphagia, dysarthria, and seizure disorder. Endocrinopathies included diabetes mellitus, hypothyroidism, and adrenal insufficiency. Additional clinical features in patients with KSS were short stature (ten [9%]), developmental delay (four [11%]), and encephalopathy (two [6%]).
Cardiovascular features developing during follow up
Cardiovascular features that developed during follow up (Table 3) included syncope and dilated cardiomyopathy as the most frequently described symptoms (six patients each [17%]). One patient died of heart failure, and five (14%) had survived a previous cardiac arrest. One patient was known to have congenital heart disease.
Permanent pacemaker/implantable cardioverter-defibrillator (PPM/ICD) devices were implanted in eleven patients (31%). The indications for PPM/ICD placement included high-grade heart block, bradycardia, a combination of heart block and bradycardia, and widening of the QRS complex. The indication was unclear for two patients (18%). Importantly, the four patients who died all died of sudden cardiac death, and only one of these patients had a PPM/ICD in place. The other three patients had no prior ECG changes to indicate PPM/ICD placement. Nonetheless, one patient had prior syncope, and one patient had a previous cardiac arrest and ventricular arrhythmias.
ECG changes developing over the patients’ lifetimes are shown in Table 3. Heart blocks (eleven [31%]) included first degree, second degree type I, second degree type II, and third degree. Conduction delays occurred in 23 patients (66%), and ten patients (29%) had an axis deviation. Preexcitation in the form of Wolff–Parkinson–White syndrome was seen in two patients (6%), and two patients (6%) had a history of ventricular arrhythmias.
Associations between disability and clinical features
At their last visit to Mayo Clinic, six patients (17%) used a wheelchair, and four (11%) noted some gait instability requiring the use of a gait assist device. Two young adults noted that they were unable to work because of disease-related issues, including heart failure and fatigue. However, 25 of the patients in this study (71%) had no disability from the mitochondrial myopathy.
Statistical analysis indicated that the presence of disability, defined as the use of a gait aid or wheelchair, was associated only with cognitive decline (P=0.004). There were no statistically significant associations between disability and patient sex, diagnosis, genetic defect, muscle defect, deafness, or sudden cardiac death (Table 4).
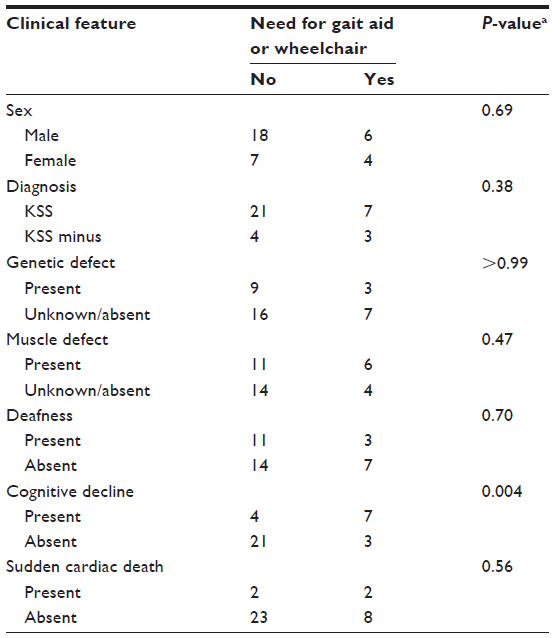 | Table 4 Associations between clinical features and development of disability |
Discussion
To our knowledge, this is the largest case series of patients with KSS. We found that patients with KSS all had ophthalmologic symptoms and commonly had neurologic and cardiac involvement. This finding is consistent with current diagnostic criteria for KSS.1,3,4 However, the findings of this study, may dispute some commonly held beliefs regarding KSS, including lack of certain diseases or laboratory associations, and longevity beyond middle adulthood. Only four of 35 patients in our series died (11%), but all deaths were from sudden cardiac events. Although most of these patients had no ECG changes to indicate the need for PPM/ICD placement, our findings support previous recommendations to carefully consider the placement of such devices in patients with KSS. Finally, we found that the development of physical disability (need for a gait aid or wheelchair) was significantly associated with cognitive decline but not with other clinical features, such as sex or sudden cardiac death.
All the patients in our study had eye involvement. Extraocular muscles have a high energy requirement, and mitochondria make up approximately 60% of the cell volume in eye muscles.8 Not surprisingly, the most common visual abnormality identified in our study was ophthalmoplegia, followed by ptosis and pigmentary retinopathy. Additionally, the characteristic “salt and pepper” retinopathy was detected in a majority of patients. On the basis of these findings, we believe that the diagnosis of KSS should be considered in all young patients with these ocular and visual symptoms.
Our study also demonstrated frequent cardiovascular involvement in adults with KSS. The extent of cardiac abnormalities varied from ECG changes to high-grade heart block, cardiomyopathy, and cardiac arrest. Previous studies have noted the presence of cardiac conduction system abnormalities in patients with KSS.14,15 Moreover, exercise intolerance has been described in patients with mitochondrial myopathies, including KSS.16 Less than half the patients (eleven [31%]) received a PPM/ICD as prophylactic treatment for the indications of bradycardia or heart block. All of the patients who died in this series succumbed to sudden cardiac death, and only one of these four patients had received a prophylactic PPM/ICD. Nonetheless, the other three patients had no ECG changes or symptoms indicating a need for PPM/ICD. In the general adult population, the overall incidence of sudden cardiac death has been estimated at 0.1% to 0.2% per year.17 Findings from the current study confirm the results from previous studies regarding progressive heart block in patients with KSS,9,14,15 support previous calls to consider routine prophylactic PPM/ICD placement in patients with KSS, and possibly suggest a low threshold for formal electrophysiologic testing in these patients.
Previously published case series have included patients with KSS. Yamashita et al10 described 136 patients with mtDNA deletions. Using the diagnostic definition of Rowland et al,4 they found that 33 (24%) of these patients met the criteria for KSS. Deafness, short stature, and insulin-dependent diabetes mellitus were all frequently observed. All patients were younger than 20 years at diagnosis and had PEO. They also noted that patients with KSS had significantly longer DNA deletions and more deleted mitochondrial transfer (t)RNA than other patients in the series. In contrast to that case series, our study used the treating physician’s diagnosis rather than a retrospective application of criteria in patients with a positive genetic test. Therefore, the patients we describe with the clinical diagnosis of KSS were more heterogeneous, including some with disease presentation after age 20 years and without PEO or pigmentary retinopathy.
Our findings dispute several commonly held beliefs about patients with KSS. First, the existing diagnostic criteria require that patients have symptom onset before age 20 years, and it is commonly believed that patients with KSS die in early adulthood. However, seven patients (20%) were older than 20 years at disease presentation, and only four (11%) died, at a mean age of 46 years, during the study time frame (mean follow up of 10.8 years). Potential explanations for the finding of diagnosis after age 20 years include recall bias, insidious onset of symptoms, or that milder disease may present at an older age. Second, anemia has previously been noted as a feature of KSS but was not a predominant feature of patients in our study.18 Third, KSS has been associated with several endocrine disorders.19 In this series, we identified patients with short stature, delayed puberty, diabetes mellitus, and hypothyroidism. However, hyperaldosteronism and hypoparathyroidism, which have been reported in previous studies,19 were not observed in our cohort of patients.
We found that the development of disability in patients with KSS is statistically associated with cognitive decline but not with other clinical features, such as sex or sudden cardiac death. It is probably expected that cognitive decline and disability would coexist. This study may not have been adequately powered to detect additional associations. Nevertheless, the lack of association between disability and other clinical features may be reassuring to patients with KSS.
Defects in mtDNA have been described in patients with KSS. A single mtDNA mutation can result in the expression of multiple phenotypes.20 Indeed, we found that multiple phenotypes (ranging from mild to severe disease) were present with the same mtDNA mutation. Additionally, mtDNA deletions generally occur de novo and thus, usually cause disease in only one family member.20,21 This observation was supported by our study – no patient in this series had a family history of KSS.
Ophthalmoplegia caused by mitochondrial abnormalities represents a heterogeneous group of illnesses, which has given rise to the concept of “ophthalmoplegia plus”.22 Moreover, the original diagnostic criteria for KSS were based on a limited number of cases.1,3,4 Phenotypic designations such as KSS remain useful tools for clinicians to recognize disease. Categorization of patients by signs and symptoms may allow treating clinicians to better predict prognosis and response to treatment on the basis of similar cases in the literature. Keeping the designation of KSS may help to alert the physician that cardiac conduction defects may occur and lead to earlier intervention.
The link between the phenotypic presentation and underlying pathophysiology in KSS is advancing. For example, it is now known that ragged-red fibers seen on muscle biopsy indicate a combined defect of respiratory complexes I and IV.23 Moreover, mitochondrial disorders of other complexes or of only I or IV do not generally produce ragged-red fibers. In addition, the brain development Engrailed genes are important in protecting neurons against mitochondrial dysfunction.24 Novel mtDNA deletions are being discovered and suggest that these deletions play a role in the pathogenesis of KSS.25,26 Constant advances in biochemical markers and expanding genotypes of mitochondrial disorders make diagnosis a challenge.27
This study had limitations. First, it was conducted at a single institution, which may limit external validity; further, only 35 patients were identified; with a larger sample size, potentially more associations between patient disability and clinical features may have been discovered. Second, in retrospective review, not all patients in the cohort had documented evidence in the chart to fulfill all diagnostic criteria for KSS. However, this may have been due to incomplete information in the chart or the patient having KSS minus or PEO plus.2 We were able to confirm a clinical diagnosis of KSS in 27 of the 35 patients based on information in the chart. This may explain why only ten patients had short stature and why some patients were older than 40 years. We included all cases (35) in which the treating physician documented their clinical diagnosis of KSS in the medical record. Finally, since some of the cases included in the series are decades old, some may not have had the genetic testing, metabolic studies, or muscle biopsy that are now standard because the tests were not available at the time.
Conclusion
We report, to our knowledge, the largest case series of patients with KSS from a single institution. We found that consistent with the major criteria for diagnosis, ophthalmic involvement presents in most patients at a young age. However, many patients in this series (20%) were older than the suggested cutoff of 20 years at diagnosis and, contrary to common belief, most lived beyond middle adulthood. Additionally, we failed to confirm the prevalence of anemia, hypoadrenalism, and hypoparathyroidism in patients with KSS, as had been suggested by previous research. In addition to the conduction system abnormalities that were identified in previous series, our cohort included patients with syncope and sudden cardiac death, which underscores the need to consider formal electrophysiologic studies and prophylactic defibrillators in these patients.
Acknowledgment
We thank Barbara Abbott for help with the medical index database search.
Disclosure
The authors report no conflicts of interest in this work.
References
Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases. AMA Arch Ophthalmol. 1958;60(2):280–289. | |
DiMauro S, Hirano M. Mitochondrial DNA deletion syndromes; 2003 [updated May 3, 2011]. In: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1203/. Accessed May 25, 2014. | |
Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. 2010;10(2):118–126. | |
Rowland LP, Hays AP, DiMauro S, De Vivo DC, Behrens M. Diverse clinical disorders associated with morphological abnormalities in mitochondria. In: Scarlato G, Cerri C, editors. Mitochondrial Pathology in Muscle Diseases: Proceedings of the Satellite Symposium of the 5th International Congress on Neuromuscular Diseases. Padua: Piccin Medical Books; 1983:141–158. | |
Ogasahara S, Nishikawa Y, Yorifuji S, et al. Treatment of Kearns–Sayre syndrome with coenzyme Q10. Neurology. 1986;36(1):45–53. | |
Epstein AE, DiMarco JP, Ellenbogen KA, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices); American Association for Thoracic Surgery; Society of Thoracic Surgeons. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008;117(21):e350–e408. | |
Kornblum C, Broicher R, Walther E, et al. Cricopharyngeal achalasia is a common cause of dysphagia in patients with mtDNA deletions. Neurology. 2001;56(10):1409–1412. | |
Grönlund MA, Honarvar AK, Andersson S, et al. Ophthalmological findings in children and young adults with genetically verified mitochondrial disease. Br J Ophthalmol. 2010;94(1):121–127. | |
Limongelli G, Tome-Esteban M, Dejthevaporn C, Rahman S, Hanna MG, Elliott PM. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur J Heart Fail. 2010;12(2):114–121. | |
Yamashita S, Nishino I, Nonaka I, Goto Y. Genotype and phenotype analyses in 136 patients with single large-scale mitochondrial DNA deletions. J Hum Genet. 2008;53(7):598–606. | |
Pakomov SV, Buntrock JD, Chute CG. Automating the assignment of diagnosis codes to patient encounters using example-based and machine learning techniques. J Am Med Inform Assoc. 2006;13(5):516–525. | |
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–1452. | |
Isashiki Y, Nakagawa M, Ohba N, et al. Retinal manifestations in mitochondrial diseases associated with mitochondrial DNA mutation. Acta Ophthalmol Scand. 1998;76:6–13. | |
Charles R, Holt S, Kay JM, Epstein EJ, Rees JR. Myocardial ultrastructure and the development of atrioventricular block in Kearns–Sayre syndrome. Circulation. 1981;63(1):214–219. | |
Hirano M, Davidson M, DiMauro S. Mitochondria and heart disease. Current Opinion in Cardiology. 2001;16(3):201–210. | |
Andreu AL, Hanna MG, Reichmann H, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341(14):1037–1044. | |
Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98(21):2334–2351. | |
Finsterer J. Hematological manifestations of primary mitochondrial disorders. Acta Haematol. 2007;118(2):88–98. | |
Harvey JN, Barnett D. Endocrine dysfunction in Kearns–Sayre syndrome. Clin Endocrinol (Oxf). 1992;37(1):97–103. | |
Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mitochondrial DNA in Kearns–Sayre syndrome. Neurology. 1988;38(9):1339–1346. | |
Rowland LP, Hausmanowa-Petrusewicz I, Bardurska B, et al. Kearns–Sayre syndrome in twins: lethal dominant mutation or acquired disease? Neurology. 1988;38(9):1399–1402. | |
Drachman DA. Ophthalmoplegia plus. The neurodegenerative disorders associated with progressive external opthalmoplegia. Arch Neurol. 1968;18(6):654–674. | |
Sarnat HB, Marín-Garcia J. Pathology of mitochondrial encephalomyopathies. Can J Neurol Sci. 2005;32(2):152–166. | |
Alvarez-Fischer D, Fuchs J, Castagner F, et al. Engrailed protects mouse midbrain dopaminergic neurons against mitochondrial complex I insults. Nat Neurosci. 2011;14(10):1260–1266. | |
Marin-Garcia J, Goldenthal MJ, Sarnat HB. Kearns–Sayre syndrome with a novel mitochondrial DNA deletion. J Child Neurol. 2000;15(8):555–558. | |
Marín-García J, Goldenthal MJ, Flores-Sarnat L, Sarnat HB. Severe mitochondrial cytopathy with complete A-V block, PEO, and mtDNA deletions. Pediatr Neurol. 2002;27(3):213–216. | |
Rodenburg RJ. Biochemical diagnosis of mitochondrial disorders. J Inherit Metab Dis. 2011;34(2):283–292. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.