")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 11
Kawasaki disease and familial mediterranean fever gene mutations, is there any link?
Authors Salehzadeh F , Mirzarahimi M, Hosseini Asl S, Nematdoust Haghi R
Received 19 January 2019
Accepted for publication 1 May 2019
Published 21 May 2019 Volume 2019:11 Pages 127—131
DOI https://doi.org/10.2147/OARRR.S202217
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Farhad Salehzadeh,1 Mehrdad Mirzarahimi,2 Saied Hosseini Asl,3 Roghayeh Nematdoust Haghi4
1Pediatric Rheumatology, Pediatric Department, Bouali Children’s Hospital, Ardabil University of Medical Sciences (ARUMS), Ardabil, Iran; 2Pediatric, Pediatric Department, Bouali Children’s Hospital, Ardabil University of Medical Sciences (ARUMS), Ardabil, Iran; 3Molecular-Genetic, Genetic Laboratory, Imam Khomeini Hospital, Ardabil University of Medical Sciences, Ardabil, Iran; 4Pediatric Department, Bouali Children’s Hospital, Ardabil University of Medical Sciences (ARUMS), Ardabil, Iran
Background and aim: Kawasaki disease (KD) is an acute febrile, self-limiting, and systemic vasculitis of unknown etiology. MEFV gene has a major role in autoinflammatory disorders and innate immune reactions. Several reports revealed that MEFV gene mutations are associated with systemic vasculitis. The aim of this study was to determine the association between KD and MEFV gene mutations.
Methods: The peripheral blood of 30 patients who were diagnosed with KD based on ACC criteria and 224 healthy people as a control group (113 male and 111 female), were collected and the samples screened for the 12 common pathogenic variants according to manufacturer’s instructions.
Results: The mean age of patients (13 females and 17 males) was 7.7 years. Ten percent of patients showed a mutation, that was meaningfully (p<0.05%) lower than that of healthy controls (25%). E148Q was shown in two patients and compound heterozygous (E148Q-M680I) was detected in one of them with lack of FMF presentations. No significant and meaningful associations were detected between the MEFV gene variant alleles and KD.
Conclusion: Unlike in other types of pediatric vasculitis, this study did not reveal any significant association between the MEFV gene mutations and KD, moreover, because of the lower frequency of mutations in these patients, it seems that this gene has a modifier and/or protective role in KD.
Keywords: FMF, vasculitis, MEFV gene, Kawasaki disease
Introduction
Kawasaki disease (KD) is an acute febrile, self-limiting, and systemic vasculitis of unknown etiology, that occurs in infants and children, especially those in Asia (Japan has the highest incidence), but its incidence has increased worldwide,1 perhaps due to more recognition and awareness of the disease.2 An important genetic susceptibility to the disease is suggested by the higher incidence among US children of Asian/Pacific Islander descent in California and Hawaii (50.4 and 210 per 100,000 respectively).3,4
Though significant differences in epidemiological features have been observed worldwide,2 a number of factors that seem relatively constant are increased incidence in people of Asian descent, a male predominance, and marked seasonality.5,6 It manifests as high fever, mucocutaneous inflammation and cervical lymphadenopathy, bilateral nonexudative conjunctivitis, erythema of the lips and oral mucosa, changes in the extremities, and rash.3 When it was first described, the coronary artery complication was not recognized,2 but now the most serious late complication of KD – the involvement of coronary arteries, is known.7
MEFV gene, which plays a major role in autoinflammatory disorders and innate immune reactions and is predominantly expressed in monocytes and granulocytes,8 plays major roles in the pathophysiology of KD at the early phase.7 Several reports revealed that MEFV mutations are associated with different types of systemic vasculitis such as Behcet’s disease, Henoch-Schonlein purpura, and polyarteritis nodosa.9 This suggests that MEFV gene mutations result in broader spectrum of vasculitis, and might increase the baseline of inflammation and affect the clinical features of inflammatory diseases.9
To clarify the role of MEFV gene mutations in the development of the most common type of systemic vasculitis in children (KD) as one of the host genetic factors and its role in clinical preservation, courses, and prognosis of disease, we investigated the associations between KD and 12 common MEFV gene mutations in these patients.
The role of MEFV gene in inflammation induced by innate immune response may disclose the pathogenetic background of these mutations in KD.
Materials and methods
This study included 224 healthy people as a control group (113 males and 111 females). All of them were over 50 years old, with negative history of FMF symptoms (including first-degree family). (This study was done to evaluate variants of MEFV mutations in normal and unaffected people).
Patient group included 30 KD patients who were treated with intravenous immunoglobulin plus oral aspirin at Bouali Children’s Hospital from 2010 through 2018 and met the appropriate diagnostic criteria for KD. Our study is complaint with the Helsinki Declaration and was approved by the domestic ethics committee of faculty of medicine ARUMS (Ardabil University of Medical Sciences), approval number: IR.ARUMS.REC.1396.254. Written informed consent was obtained from all participants (their parents).
The principle clinical features are shown in Table 1.
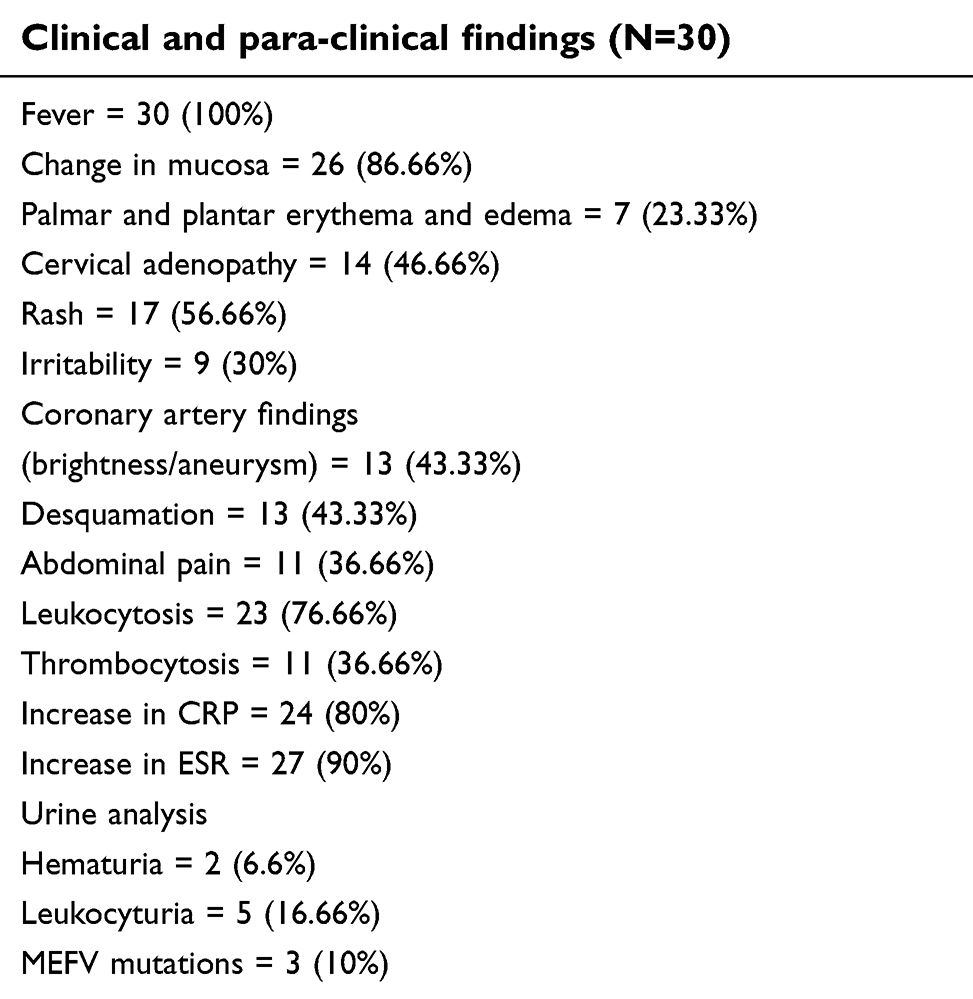 | Table 1 Patient findings |
Blood samples were collected from 30 KD patients and the samples were screened for the 12 common pathogenic variants (E148Q, F479L, P369S, I692del, M680I(G/C), M680I(G/A), M694V, V726A, A 744S, M694I, K695R, R 761H) using an RDB assay (FMF Strip Assay, Vienna lab, Vienna, Austria) according to manufacturer’s instructions.
Results
There were 13 girls and 17 boys; the median age at diagnosis was 7.7 years. The youngest patients were 1.5 years old and the oldest one was 12.5 years old. The main complaint from patients was fever in all of them (100%), and fever plus changes in mucosa in 26 (86%) patients was the second most common complaint. Table1 shows the main symptoms of patients.
Regarding MEFV gene analysis, three male patients (10%) showed mutations. Two patients had heterozygote E148Q and one showed compound heterozygote E148Q-M680I (Table 2). Control group showed 25% mutations, the most common variants were E148Q (18.3%), P369S (3.1%), V726A (2.2%), and A744S (1.3%), respectively.
 | Table 2 Genotype phenotype correlation |
Thirteen patients developed some degree of coronary artery findings; brightness, dilated, and aneurysm. One patient had compound heterozygous mutations (E148Q-M680I), coronary artery dilatation, and micro-aneurysm.
The time between onset of symptoms and diagnosis of KD was less than 9±2 days.
Discussion
Despite 4 decades of investigation, the etiology of KD remains unknown. Current understanding of the immune response suggests response to a classic antigen that is protective against future exposure in most patients. Expansion of the regulatory T-cell population after IVIG administration is associated with cessation of fever and clinical improvement. The self-limiting feature of the disease accompanied with a low rate of recurrence suggest emergence of T- and B-cell memory cells which are protective against future encounters with the KD agent.7
Although early studies provided evidence of an immune reaction triggered by a super antigen, subsequent studies favored a canonical response to a conventional antigen.
A study of the adaptive immune response demonstrated that both pro-inflammatory and regulatory T-cells could be found in the circulation in the first week after fever onset.9
Activation of the innate immune system, with high numbers of activated, circulating neutrophils and evidence of activation of IL-1, IL-6, and TNF signaling pathways, is an early event.7
The MEFV gene is located on the short (p) arm of chromosome 16 at position 13.3”16p 13.3”.10 MEFV gene was predominantly expressed in granulocytes and monocytes,8 which play a major role in the pathophysiology of KD at the acute phase.2 MEFV gene encodes a protein called pyrin (or marenostrin).11 Pyrin is involved in inflammation through altered apoptosis, CASP-1 activation, secretion of IL-1β, and activation of the NF-κB pathway in innate immune system.12
MEFV mutations' associations with different autoimmune and autoinflammatory diseases suggest its contribution to the development of a broader spectrum of vasculitis.
Furthermore, MEFV mutations might increase the baseline of inflammation, induce the development of rheumatic diseases, and affect the clinical course of inflammatory disorders.9
Most Japanese FMF patients showed heterozygous mutations.13,15 Among them, E148Q was the most common mutation with a weak effect on severity of disease. It seems to serve only to enhance FMF progression, that was primarily induced by another mutation.14 Tunca et al demonstrated an increased acute phase reaction in carriers of the MEFV gene.16 Rabinovich et al demonstrated that RA patients carrying MEFV mutations, particularly the E148Q mutation, present with a higher RA severity score than the non-carrier group, suggesting that MFFV mutation is a modifier of the clinical manifestation of RA.17
Migita et al's study in a Japanese population demonstrated no association between the presence of MEFV mutations and RA occurrence as well as RA-related AA amyloidosis.18
Yamaguchi et al reported that E148Q mutation was not associated with the development of KD that occurs in a higher frequency in Japanese children. It is possible that the functional effect of this mutation might be too small to stimulate inflammation for the development of vasculitis in KD.19
This study showed 10% mutations among patients with KD, whilst these mutations among healthy control group were 25%, where E148Q was the most frequent variant (18.3%). This result shows meaningfully lower mutated MEFV alleles in KD patients than healthy people. (p<0.05)
The meaningfully lower frequency of E148Q mutation in these patients reveals the possible preventive and protective role of this mutation (E148Q) in KD. Furthermore, it seems that it could modulate the severity and exacerbation of vasculitis in affected patients. This hypothesis helps us to better understand Yamaguchi`s study about the functional effect of E148Q and its incompetent effect on inflammation.19 We think that in our one patient with compound heterozygote E148Q-M680I, who developed coronary artery dilatation and micro-aneurysm, combination of pathologic mutation (M680I) resulted in vasculopathy. Moreover, the high prevalence of MEFV gene variant alleles in Mediterranean people may explain the low rate of occurrence of KD in this population in comparison with Asian people.
Different patterns of geographic prevalence of KD could have various genetic and environmental factors, and MEFV gene variant alleles could be one of several immunogenic factors.
Unlike in the other common types of pediatric vasculitis, this study did not reveal any significant association between MEFV mutations and KD, moreover it seems to have a modifier and/or protective role in KD.
Conclusion
This study demonstrated no association between the presence of MEFV variant alleles and KD, and emphasizes its possible protective role in KD.
Availability of Data and Materials
The author can be contacted for data requests.
Consent for publication
Written informed consent was obtained from the parents of patients for publication of this study.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Burns JC, Glod´E MP. Kawasaki syndrome. Lancet. 2004;364(9433):533–544. doi:10.1016/S0140-6736(04)16814-1
2. Luca NJ, Yeung RS. Epidemiology and management of Kawasaki disease. Drugs. 2012;72(8):1029–1038. doi:10.2165/11631440-000000000-00000
3. Uehara R, Belay ED. Epidemiology of Kawasaki disease in Asia, Europe, and the United States. J Epidemiol. 2012;22(2):79–85. doi:10.2188/jea.JE20110131
4. Holman RC, Christensen KY, Belay ED, et al. Racial/ethnic differences in the incidence of Kawasaki syndrome among children in Hawaii. Hawaii Med J. 2010;69(8):194–197.
5. Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. 2005;9(4):185–194. doi:10.1016/j.ijid.2005.03.002
6. Burns JC, Newburger JW. Genetics insights into the pathogenesis of Kawasaki disease. Circ Cardiovasc Genet. 2012;5(3):277–278. doi:10.1161/CIRCGENETICS.111.960831
7. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the committee on rheumatic fever, endocarditis and kawasaki disease, council on cardiovascular disease in the young, american heart association. Circulation. 2004;110(17):2747–2771. CIR.0000145143.19711.78. doi: 10.1161/01
8. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial mediterranean fever. The International FMF consortium. Cell. 1997;90(4):797–807. doi:10.1016/S0092-8674(00)80539-5
9. Ozen S, Bakkaloglu A, Yilmaz E, et al. Mutations in the gene for familial mediterranean fever: do they predispose to inflammation. J Rheumatol. 2003;30(9):2014–2018.
10. MEFV – Mediterrianean Fever. US National Library of medicine,National Institutes of Health Department of Health & Human Services.
11. Bernot A, Clepet C, Dasilva C, et al The French FMF consortium: a candidate gene for familial mediterranean fever. The French FMF consortium. Nat Genet. 1997;17:25–31. doi:10.1038/ng0997-25
12. Ting JP, Kastner DL, Hoffman HM. CAT- ERPILLERs, pyrin and hereditary immune - logical disorders. Nat Rev Immunol. 2006;6(3):183–195. doi:10.1038/nri1788
13. Nakamura A, Yazaki M, Tokuda T, Hattori T, Ikeda S. A Japanese patient with familial Mediterranean fever associated with compound heterozygosity for pyrin variant E148Q/M694I. Intern Med. 2005;44(3):261–265. doi:10.2169/internalmedicine.44.261
14. Kotone-Miyahara Y, Takaori -Kondo A, Fukunaga K, et al. E148Q/M694I mutation in 3 Japanese patients with familial mediterranean fever. Int J Hematol. 2004;79(3):235–237. doi:10.1532/IJH97.03119
15. Komatsu M, Takahashi T, Uemura N, Takada G. Familial mediterranean fever medicated with an herbal medicine in Japan. Pediatr Int. 2004;46(1):81–84. doi:10.1111/j.1442-200x.2004.01892.x
16. Tunca M, Kirkali G, Soyturk M, Akar S, Pepys MB, Hawkins PN. Acute phase response and evolution of familial mediterranean fever. Lancet. 1999;353(9162):1415. doi:10.1016/S0140-6736(98)09449-5
17. Rabinovich E, Livneh A, Langevitz P, et al. Severe disease in patients with rheumatoid arthritis carrying a mutation in the mediterranean fever gene. Ann Rheum Dis. 2005;64(7):1009–1014. doi:10.1136/ard.2004.029447
18. Migita K, Nakamura T, Maeda Y, et al. MEFV mutations in Japanese rheumatoid arthritis patients. Clin Exp Rheumatol. 2008;26(6):1091–1094.
19. Yamaguchi K, Ikeda K, Ihara K, et al. Lack of association between E148Q MEFV variant and Kawasaki disease. Hum Immunol. 2009;70(6):468–471. doi:10.1016/j.humimm.2008.10.017
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.