")
Back to Archived Journals » Journal of Receptor, Ligand and Channel Research » Volume 7
K+ channels in biological processes: vascular K+ channels in the regulation of blood pressure
Received 18 April 2014
Accepted for publication 1 July 2014
Published 30 September 2014 Volume 2014:7 Pages 51—60
DOI https://doi.org/10.2147/JRLCR.S36062
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Matthias E Werner,1 Jonathan Ledoux2–4
1Institute of Cardiovascular Sciences, Faculty of Medical and Human Sciences, University of Manchester, Manchester, UK; 2Research Center, Montreal Heart Institute; 3Department of Molecular and Integrative Physiology, 4Department of Medicine, Université de Montréal, Montreal, QC, Canada
Abstract: Appropriate supply of blood to organs and tissues is highly dependent on arterial blood pressure and therein of the peripheral blood vessel resistance. Of the two main components of vessel resistance, the active resistance results from the modulation of the contractility level of vascular smooth muscle cells (VSMCs). The intracellular level of Ca2+ in VSMCs is an essential component of muscle contraction and is tightly regulated through modulation of the membrane potential. Since resting membrane potential of vascular cells is mainly dependent on K+ ions, ion channels permeable to K+ ions have a significant impact on contractility of smooth muscle cells and therefore on vascular diameter and blood pressure. Activation of K+ channels on both endothelial cells and VSMCs is generally associated to hyperpolarization and relaxation of vascular smooth muscle. Several types of K+ channels are expressed in VSMCs and endothelial cells, and they are classified based on their pharmacological and biophysical properties. Voltage-dependent K+ channels are activated by depolarization and are mainly involved in negative-feedback mechanisms. Ca2+-activated K+ channels can be divided into three groups, with BKCa being activated by both intracellular Ca2+ and depolarization. On the other hand, KCa2.x and KCa3.1 channels (small and intermediate Ca2+-activated K+ channels, respectively) are almost strictly dependent on rises in intracellular Ca2+ levels to increase their open probability. Kir and adenosine triphosphate (ATP)-sensitive K+ (KATP) channels, members of the same family, have a significant impact on VSMC membrane potential. The more recently studied two-pore K+ channels are thought to be metabolic sensors (like KATP channels) and would be involved in acute regulation of local blood flow. This review will summarize the main K+ channels expressed in vascular cells and their relevance in the control of vascular tone and blood pressure.
Keywords: membrane potential, Ca2+, vascular tone
Blood pressure
Regulation of blood pressure by vascular tone
Blood pressure can be defined as the pressure applied by circulating blood on blood vessels walls. It is dependent on the blood volume, the blood flow, and the resistance of the blood vessels. Blood pressure and blood flow are regulated by the constriction and dilation of resistance arteries, usually small arteries with internal diameters <300 μm.1 Physiological elevations in intravascular pressure or augmented sympathetic activity promotes smooth muscle depolarization, increases myocyte intracellular Ca2+, and thus vasoconstriction. The subsequent increase in total peripheral resistance within the vasculature increases blood pressure.2 The vascular smooth muscle cells (VSMCs) within the blood vessel wall provide structural integrity to the vessel and regulate vascular tone and blood pressure by their contractile state. From a basal resting tone, the VSMCs are constantly in a semi-contracted state and can contract further, leading to a reduced blood vessel diameter and increased blood pressure (Figure 1). On the other hand, relaxation of VSMCs leads to a larger vessel diameter and hence to a reduction of blood pressure. Although the systemic outcome of this change in vessel resistance is a modification in blood pressure, generally, vascular tone – and hence VSMC contractile state – is finely tuned in order to maintain or adapt blood flow to the tissue perfused by the artery involved. Indeed, according to Poiseuille’s Law, with all other parameters constant, a reduction of the vessel radius by a factor of two would result in a decrease in blood flow by a factor of 16.
 | Figure 1 Relationship between VSMC contraction and blood vessel diameter. |
Impact of intracellular calcium on vascular tone
Contraction and relaxation of the VSMCs are tightly regulated by the intracellular calcium concentration ([Ca2+]i) emphasizing the importance of the homeostasis regulatory mechanisms in the control of vascular tone. Vasoconstrictors like epinephrine, endothelin-1, angiotensin II, or thromboxane A2 act through increasing [Ca2+]i by opening Ca2+ channels located in the cytoplasmic and sarcoplasmic membrane as well as on the apparent calcium sensitivity of the contractile apparatus in VSMCs, whereas relaxing factors have the opposite effect. Interestingly, an elevation in endothelial [Ca2+]i induces relaxation of the adjacent VSMCs through endothelium-derived relaxing factors such as nitric oxide (NO), prostacyclin (PGI2) and endothelium-derived hyperpolarizing factor (EDHF).3–6 Therefore, fine tuning of [Ca2+]i in both cell types is imperative for precise regulation of blood pressure and organ and tissue perfusion.
Role of the membrane potential on vascular cells and its impact on intracellular calcium and on blood vessel diameter
The membrane potential of vascular cells is dependent on the intracellular and extracellular concentration of ions as well as their respective permeability across the lipid bilayer. A resting membrane potential (Em) is generated by well-balanced influx and efflux of ions through ion channels along their gradients as well as by active ion transport through ion pumps.
In VSMCs, Em (−45 mV) is primarily determined by K+ efflux through several plasma membrane K+ channels, including the voltage-gated K+ (KV) channels, the large-conductance Ca2+-activated K+ (BKCa) channels, inwardly-rectifying K+ (Kir) channels, ATP-sensitive K+ (KATP) channels, and two-pore domain K+ (K2P) channels.7–10 With a reversal potential for K+ around −89 mV, opening of K+ permeable channels will hyperpolarize Em. Excitation of VSMCs followed by the depolarization of the Em by influx of cations (Ca2+ and Na+) or efflux of Cl− promotes the opening of voltage-dependent Ca2+ channels (VDCCs). The opening of VDCCs allows Ca2+ influx, causing a rise in [Ca2+]i and activation of the cellular contractile machinery. Depolarization also leads to the voltage-dependent opening of KV channels, and the corresponding increase in [Ca2+]i through VDCCs in combination with the depolarization opens the voltage- and Ca2+-sensitive BKCa channels. Opening of these K+ channels causes a compensatory hyperpolarizing current that reduces open probability of VDCCs to oppose vasoconstriction.7
In vascular endothelial cells (ECs), hyperpolarization rather than depolarization is associated with raised [Ca2+]i. This is attributed to the increase in the inward electrochemical gradient for Ca2+, as ECs generally lack VDCCs. The increase in EC [Ca2+]i is of major importance for physiological function in blood vessels. It initiates and sustains vasodilation by stimulating the synthesis of NO and PGI2. It also activates small-conductance K+ (KCa2.3) and intermediate-conductance K+ (KCa3.1) channels, generating a hyperpolarization known as the endothelium-derived hyperpolarization factor (EDHF), which spreads to and relaxes the adjacent VSMCs.5,6 However, it is important to note that EDHF is not limited to KCa2.3 and KCa3.1 channels but is rather an amalgam of different mechanisms which have in common the ability to induce a hyperpolarization of the underlying smooth muscle cells. The following sections of this review will focus on the different types of K+ channels expressed in VSMCs and ECs and their role in regulating blood pressure and vascular tone.
Potassium channels
Voltage-dependent K+ channels
Voltage-dependent K+ channels (KV) consist of a large superfamily of 12 subfamilies containing several members each in addition to splice variants.11 However, vascular KV channels mainly consist of KV1, KV2, and KV3 isoforms. More recently, KV7 (KCNQ) and KV11 (HERG) family members have also been identified in VSMCs.12,13 Archetypical KV channels have cytoplasmic carboxy and amino termini and are six transmembrane domain proteins, with segment S4 being the voltage-sensor, and their quaternary structure consists of four α subunits.14 The complexity of KV currents is due to the heteromultimerization of α subunits, modifying biophysical and pharmacological properties. Additional potential convolution is provided through modulation of the KV heteromultimers by β subunits. KV channels are activated by depolarization and are therefore an optimal component of a negative-feedback mechanism in response to a contractile-prone VSMC depolarization.7 Their activity is, however, subject to regulation by protein kinases like PKA, PKC, and PKG.
In VSMCs, the main isoforms of KV channels expressed are KV1.2, KV1.5, KV1.6, KV2.1, and KV3.1, but expression pattern is variable depending on the vascular bed.15 However, studies have suggested that functional expression of KV channels can be regulated through heteromultimerization with “silent” KV5, 6, 8, and 9. Indeed, heteromultimerization with “silent” α subunits appears to modulate KV current expression, drug sensitivity, as well as biophysical properties such as inactivation.16
Blocking of KV channels with 4-aminopyridine significantly increases myogenic tone.17 KV7 are also suggested to play a prime role in hypoxia-induced vasodilation, as XE991, a KV7-specific inhibitor, abolishes dilation.18 On the other hand, in pulmonary arteries, KV1.2, KV2.1, KV3.1, and KV7 resting currents are essential to resting membrane potential and are involved in the hypoxia-induced vasoconstriction of pulmonary arteries.19,20 Interestingly, despite numerous ex vivo evidence, there is currently little in vivo evidence of a physiological role for vascular KV channels.21,22 Although expression of KV1.3 and 1.5 has been reported in ECs, a role for endothelial KV channels remains unclear, but a depolarization feedback has been proposed.23,24 Further investigation is however necessary to better understand KV channel function in endothelium.
Calcium-dependent K+ channels
The human genome holds eight genes that encode Ca2+-dependent K+ channels. Based on their genetic relationship, mechanisms of calcium sensing, pharmacology, and single-channel conductances, these channels can be divided into two distinct but only remotely related groups.25 The first group contains the large-conductance (single-channel conductance >150 pS) Ca2+-dependent BKCa (KCa1.1) channel, and the related Slack (KCa4.1), Slick (KCa4.2), and Slo3 (KCa5.1) channels. The latter three are activated by increases in intracellular Na+ and/or Cl− or by alkalization rather than by intracellular Ca2+ and will not be discussed any further in this review. The second group of Ca2+-dependent channels, the SK/IK (KCa2.x/3.1) group, contains the three small-conductance (single-channel conductance <50 pS) Ca2+-dependent K+ channels, KCa2.1 (SK1), KCa2.2 (SK2), and KCa2.3 (SK3), as well as the intermediate-conductance (single-channel conductance 50–150 pS) KCa3.1 (SK4/IK) channel. All four channels from this second group have the same calmodulin-mediated calcium sensing mechanism, which is different to the intrinsic calcium sensing mechanism of the BKCa channel (see next section). While all KCa2.x and KCa3.1 channels are expressed in neuronal tissue, only KCa2.3 and KCa3.1 are expressed in vascular tissue, where they play important roles in regulating blood pressure.26
KCa2.3 and KCa3.1 channels
Functional homomeric KCa2.3/KCa3.1 channels are formed by the association of four subunits, each of them containing six transmembrane domains with intracellularly located amino and carboxy termini. It has been suggested that different KCa2.x channel subunits (KCa2.1–KCa2.3) could form heteromeric channels, but this has not been shown in vascular cells.27 KCa2.x/KCa3.1 channels lack voltage sensitivity despite the presence of some charged amino acids in the fourth transmembrane domain, and absence of a Ca2+-binding motif would suggest insensitivity to [Ca2+]. However, KCa2.x channels are constitutively tethered with calmodulin, working as a Ca2+-sensing subunit, and induce Ca2+-dependent channel opening.28 Indeed, half maximal effective concentration (EC50) values for [Ca2+] range from 300 to 500 nM,29 which are similar to the EC50 values for calmodulin and are within the physiological range of [Ca2+]i levels in vascular cells.28,30 KCa3.1 channels are also constitutively connected with calmodulin, and their EC50 value for [Ca2+] is about 740 nM.28,30,31 The lack of voltage dependence allows these channels to remain in an open state at more negative membrane potentials than KV and BKCa channels. Consequently, these channels can hyperpolarize the membrane toward values near the K+ equilibrium potential of −89 mV. KCa2.3 and KCa3.1 channels are expressed in vascular ECs that require hyperpolarization to sustain Ca2+ influx through transient receptor potential (TRP) channels or Ca2+ release from intracellular stores during cellular activation. The transmission of hyperpolarization through gap junctions is known as a component of EDHF.22,23
The coexpression of KCa2.3 and KCa3.1 channels in ECs was initially viewed as a redundancy, but a growing body of evidence suggests that the two endothelial channels are located in spatially different microdomains and contribute in different signaling pathways.5 KCa2.3 is found at interendothelial connections and has been shown to co-localize with TRP channels (Figure 2).
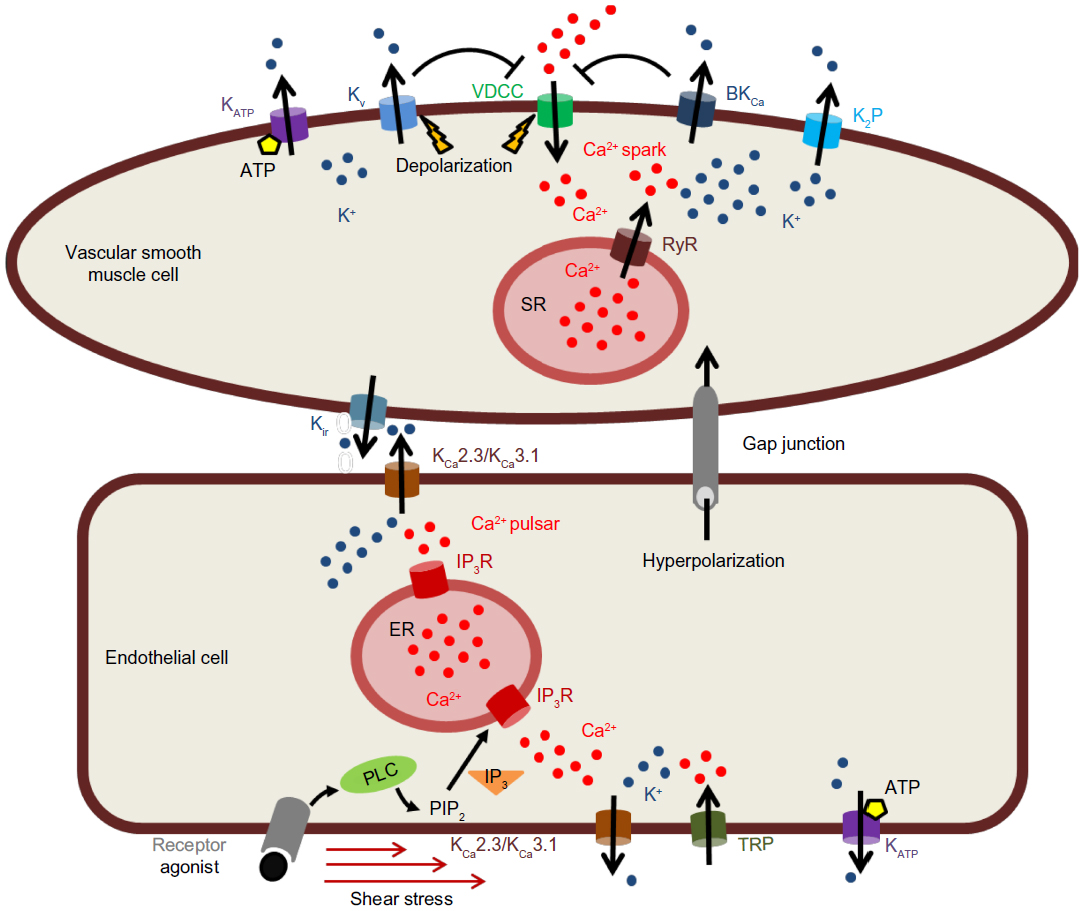 | Figure 2 Modulation of membrane potential by K+ channels in vascular cells. |
In this position, KCa2.3 has been suggested to detect local Ca2+ increases in response to shear stress stimulation.32 A very recent work by Sonkusare et al33 shows that activation of as few as three vanilloid (TRPV) family member TRPV4 channels per cell can cause activation of EC KCa2.3 and KCa3.1 channels followed by maximal dilation of pressurized resistance arteries. However, in this study both channels seemed to be involved by TRPV4 channel opening.
KCa3.1 channels are predominantly localized in EC–VSMC connections, where Ca2+ release through inositol 1,4,5-trisphosphate receptors (IP3Rs) regulates the function of these channels (Figure 2). Ledoux et al34 have identified local Ca2+ release events (“Ca2+ pulsars”) in vascular ECs, which are mediated by IP3Rs in the endothelial endoplasmic reticulum (ER) in cellular projection through the internal elastic lamina and making contact to adjacent VSMCs to pass on EDHF through gap junctions. The authors showed that the endothelium-dependent vasodilator acetylcholine elevated IP3 followed by increased frequency of Ca2+ pulsars, whereas blunting IP3 signaling or depleting ER Ca2+ inhibited these events. KCa3.1 channels also co-localized to the endothelial projections, and blocking these channels caused cellular depolarization. Inhibition of Ca2+ pulsars also depolarized ECs, but additional block of KCa3.1 channels had no further effect in the absence of pulsars.34
Despite different intracellular localization of KCa2.3 and KCa3.1 channels and their different signal transduction pathways, activation of either channel initiates hyperpolarization and subsequent vasodilatory responses through EC hyperpolarization spreading to the underlying VSMCs, promoting closure of VDCCs, and ensuring relaxation and vasodilation.5,35 The significance of this KCa2.3/KCa3.1-mediated EDHF for the systemic regulation of blood pressure is demonstrated by the observation of an elevated blood pressure in KCa3.1- and/or KCa2.3-deficient mice.32 Particularly, deficiency of either channel increases mean arterial blood pressure (MAP) by 7–9 mmHg. Lack of KCa2.3 increases MAP by increasing systolic pressure (SP) as well as diastolic pressure (DP) but without a change in pulse pressure (PP), whereas lack of KCa3.1 seems to increase MAP because of a higher SP as well as a higher PP, but at a normal DP.36 Interestingly, lack of both channels increased SP, DP, and PP, but did not increase MAP further, suggesting other compensatory mechanisms. These findings suggest that the blood pressure changes caused by the deficiency of either channel are not additive and that the loss of one channel cannot be compensated by the other.
Normal (quiescent) contractile VSMCs do not express KCa2.3 or KCa3.1 channels.36 Instead, the majority of Ca2+-dependent K+ currents in these cells is carried by BKCa channels. However, the expression pattern of Ca2+-dependent K+ channels changes drastically if VSMCs undergo phenotypic changes. For instance, proliferation of smooth muscle cells is accompanied by an upregulation of KCa3.1 as determined by mRNA and protein expression of this channel.37–39 This phenotypic change can trigger restenosis after angioplasty and atherosclerosis as stimulation of KCa3.1 expression was also detected in proliferating neointimal smooth muscle cells in balloon-catheterized coronary arteries of swine and carotid arteries of rats.36–39 Furthermore, induction of KCa3.1 expression was also observed in human neointima smooth muscle cells from coronary bypass vessels.40 These findings suggest that induction of KCa3.1 expression in atypical smooth muscle growth is a common feature and an indicator of a phenotypic alteration in a number of animal species as well as in humans.41
BKCa channel
Functional BKCa channels are heteromultimers consisting of four α-subunits and four β-subunits.42 The α-subunit contains eleven hydrophobic domains (S0–S10), with an extracellular amino terminus and an intracellular carboxy terminus. S0–S6 are located in the cytoplasmic membrane, with a pore domain between S5 and S6.42 Like other voltage-gated channels (see section on KV channels), the S4 most likely functions as the voltage sensor of the channel. Several studies have suggested that the α-subunit has also an intrinsic Ca2+ sensor, a “Ca2+ bowl” located in the tail region of the protein (for a detailed review of these studies see Knaus et al, 1994).42 As mentioned previously, only one gene encodes for the α-subunit, but many splice variants with different properties were detected. The existence of splice variants could explain different BKCa channel properties like voltage sensitivity or phosphorylation by different kinases.43,44 In blood vessels, BKCa channels are primarily expressed in smooth muscle cells. Only a few studies have shown expression of the channel in the endothelium of pig (summarized in Köhler and Ruth, 2010).36 However, the BKCa channel conducts most of the Ca2+-sensitive K+ currents in healthy contractile VSMCs. The β-subunit of the BKCa channel contains two transmembrane domains with a long extracellular linker, while the amino and the carboxy termini are located in the cytoplasm. Four β-subunit genes have been identified, but the predominant subunit in VSMCs is the β1 isoform.45 The β1-subunit increases the voltage and calcium sensitivity of the channel by interacting with the α-subunit.46
In VSMCs, BKCa channels regulate the membrane potential by producing spontaneous transient outward currents (STOCs) in response to highly localized releases of Ca2+ (Ca2+ sparks) through ryanodine receptors (RyR) from the sarcoplasmic reticulum (SR) (Figure 2).47 This spark-to-STOC coupling is only possible when BKCa channels, located in the cell membrane, and the RyR, located in the SR membrane, are in very close proximity.48 One single Ca2+ spark, which measures roughly 2 μm in diameter, generates a spatially restricted (1% of the cell) large increase in [Ca2+]i (>10 μM), while the global Ca2+ is raised by only 2 nM.47,48 At negative membrane potentials, BKCa channels are normally less sensitive to [Ca2+]i than KCa2.3/KCa3.1 channels and thus are less active around resting membrane potential of VSMCs.49,50 But BKCa channels are also activated by membrane potential depolarization. The voltage-sensitivity is variable, as it is modulated by [Ca2+]i.51 Increase of [Ca2+]i shifts the voltage-dependent parameters to more negative voltages and allows the channel to open under a physiological range of membrane potentials.52,53
The hyperpolarization resulting from the BKCa channel activation provides an important negative feedback on Ca2+ influx by closing the VDCCs and thus counteracting vasoconstriction. Therefore, BKCa channels in VSMCs are deeply involved in blood pressure control and have been associated with other EDHF-dilator responses, as BKCa channels are the anticipated targets of diffusible EDHFs (eg, EETs, H2O2, and NO).54,55 However, it is currently not clear whether lack of BKCa channels results in a loss of blood vessel dilation mediated by these diffusible EDHFs.
Nevertheless, the relevance of BKCa channels in regulating blood pressure is demonstrated by the following studies using BKCa channel knockout mice and a variety of hypertensive animal models. BKCa channel deficiency causes mild hypertension, and accordingly, hypertensive disease models show a reduced BKCa channel function or expression. Mice deficient of the pore-forming α-subunit have increased systemic blood pressure as a consequence of the loss of the VSMCs hyperpolarizing K+ current, and by primary hyperaldosteronism.56 Furthermore, BKCa channel–deficient mice show substantial erectile dysfunction and reduced sensitivity to the phosphodiesterase-5 inhibitor sildenafil.57,58 VSMCs from mice lacking the auxiliary β1-subunit of the BKCa channel also show reduced numbers of STOCs and thus have a more depolarized resting membrane potential. Consequently, vascular smooth muscle contractility is augmented and the systemic arterial blood pressure is elevated in these mice.46,59 In contrast, decreased BKCa channel function in VSMCs has been described in numerous models of hypertension, and hence, upregulation of BKCa α-subunits could represent a mechanism to lessen hypertension.55 In angiotensin II–hypertensive rats and in spontaneous hypertensive rats, reduced BKCa channel activity in VSMCs has been observed and has been related to a downregulation of the expression of the β1-subunit.60,61 In L-nitro-arginine-treated hypertensive rats and in rats with pulmonary hypertension, a downregulation of the expression of the pore-forming α-subunit was demonstrated.62,63 In aged rats, lower expression levels of both α- and β-subunits have been reported.64
Moreover, two single nucleotide-polymorphisms in the human BKCa channel α-subunit gene (IVS17 + 37T>C and C864T) and one in the β1-subunit gene (E65K) have been identified.65,66 The genetic changes in the α-subunit gene are associated with increased severity of systolic and general hypertension as well as increased risk of myocardial infarction. Contrarily, the nucleotide-polymorphism in the β1-subunit gene causes enhanced channel activity (gain-of-function mutation) and is protective against diastolic hypertension in post-menopausal women. Consequently, these discoveries undoubtedly demonstrate a substantial role of BKCa channel α- and β-subunits in controlling blood pressure and prove that both subunits represent possible drug targets for a blood pressure–lowering therapy in hypertension.
Other K+ channels
Kir and KATP channels
The pore-forming proteins of both Kir and KATP channels belong to the same gene family, the inwardly rectifying potassium channels (Kir). Six subfamilies are currently recognized, designated Kir1.0 to Kir6.0. Their name originates from the steep inward rectification of their current-voltage relation, hence they conduct inward K+ current much more readily than outward current.67 This inward rectification is in fact the consequence of intracellular polyamines and Mg2+ ions blocking Kir pore in response to depolarizing membrane potential. Kir channel subunits comprise only two transmembrane domains, and functional channels are built by tetramers.67 Expression studies suggest that one channel of the Kir2.0 family (Kir2.1, but not Kir2.2 or Kir2.3), is expressed in VSMCs, and the channel is activated by moderate elevations of extracellular K+ (from 6 to 15 mM).67 The elevation of external K+ causes a graded shift of the voltage-dependency of Kir2.1 conductance, which leads to hyperpolarization and vasodilation. This outcome might appear unanticipated, since an increase in extracellular K+ levels would predict a decrease in the electrochemical gradient for K+, therefore reducing the overall influence of K+ channels on the VSMC membrane potential and consequently on the vascular tone. However, activation of the Kir channels exceeds this effect and the overall result of a somehow modest increase in extracellular K+. The expression of Kir channels is inversely proportional to the artery diameter. Moreover, their K+ sensitivity makes Kir a powerful metabolic sensor as they can trigger an increase in blood flow in response to K+ accumulation following neuronal activity (ie, neurovascular coupling), for example.68,69 Lack of Kir2.1 in mice completely abrogated the Kir currents, and arteries from these animals did not dilate in response to elevated K+.70 However, whether blood pressure is changed in these animals is not known. Endothelial Kir channels are thought to be involved in the flow-mediated dilation as they are also activated by shear stress, and they can be found in several but not all ECs.34 Wu et al proposed that cerebral VSMCs also express Kir2.2, and Kir2.4 expression has been detected in cultured human pulmonary smooth muscle cells.71,72 However, unequivocal evidence that Kir channels play a relevant role in the regulation of systemic blood pressure is still lacking.
KATP channels are hetero-octameric complexes comprising four pore-forming subunits (Kir6.1 or Kir6.2) and four regulatory sulphonylurea receptors (SUR2A and SUR2B), which are proteins from the ATP-binding cassette family.67 KATP channels in VSMCs most likely result from the association of SUR2B with Kir6.1/Kir6.2 subunits (1:1 ratio). Showing a weaker inward rectification than their Kir2 family channel counterpart, substantial interest has been directed toward these channels as they exhibit activity under normal physiological conditions and significantly contribute to the control of membrane potential, arterial diameter, and blood pressure regulation.67,73 Similarly to Kir2 channels, KATP channels are physiologically important through their capacity to couple cellular metabolism and electrical excitability, although different mechanisms are involved. Hence, change in intracellular ATP/ADP ratio is the main stimulus to KATP channel opening, membrane potential hyperpolarization and vasodilation in conditions of high metabolic activity. Binding of ATP to the Kir6 subunits blocks the channel, but this is compensated by the activating influence of the ADP/SUR interaction.73 Therefore, a decrease in ATP intracellular levels or an increase in ADP cytoplasmic concentration would both result in an increase in KATP open probability. Furthermore, recent findings also indicate that the ATP/ADP ratio may not be the only physiological regulator of KATP channels, as the nucleotide sensitivity of the channels is modulated by changes in membrane levels of phosphatidyl-inositol-bisphosphate (PIP2) and fatty acyl CoA esters, phosphorylation by protein kinases as well as by caveolin-1.74,75 Alternatively, KATP channels are also sensitive to shear stress, pH, and hyperosmolarity and induce a subsequent vasodilation. Altogether, these studies suggest that KATP channels can be finely tuned to significantly modulate blood flow in health and disease.
Although KATP channels are expressed in both VSMCs and ECs, the current understanding of the endothelial function of KATP channels remains incomplete. Indeed, transgenic mice expressing an endothelial-specific dominant negative Kir6.1 subunit show a significant increase in coronary resistance as evidenced by an elevated basal coronary perfusion pressure.76 This observation suggests that endothelial Kir6.1 channel is more important for coronary than systemic circulation. In addition, endothelial Kir6.2 channels are suggested to be important in the mechanotransduction of shear stress in the pulmonary microcirculation, since lack of endothelial Kir6.2 channel function may cause incorrect adjustment to interrupted blood flow in the lung.77
K2P channels
Structurally distinct from other vascular K+ channels, K2P channels are characterized by their two pore regions (P1 and P2; hence their name) per functional unit. Each subunit consists of homo- or heterodimers of four transmembrane domain (M1–M4) subunits. Studies have identified 10 out of the 15 members of the K2P channel family in vascular cells.78 K2P channels are classified into six separate subfamilies based on their structural and functional properties: TWIK, TASK, TREK, THIK, TALK, and TRESK subfamilies.78 Numerous research studies have shown that K+ channels of the K2P channel family are expressed in several vascular beds, including cerebral, mesenteric, and pulmonary VSMCs.79–83 K2P channels are considered responsible for background leak currents and are therefore involved in controlling and stabilizing the membrane potential.10 Although their gating is independent of membrane potential, K2P channel activity is modulated by a wide variety of factors (eg, chemical stimuli like pH, mechanical stimuli, phospholipids, and polyunsaturated fatty acids).10 Moreover, the sensitivity of the K2P channels to the different stimuli varies according to the K2P channel class. Interestingly, K2P channels have been suggested as important players in hypoxic vasoconstriction of pulmonary vasculature.82 Indeed, while hypoxia generally induces vasodilation in most vascular beds, low pO2 (partial pressure of oxygen) will be associated with a vasoconstriction in pulmonary arteries. It has also been suggested that TASK-1 inhibition could be involved in the pathogenicity of ET-1, leading to pulmonary arterial hypertension.83 TREK-1 appears to be highly expressed in ECs and involved in endothelium-dependent vasodilation. Hence, recent studies using TREK-1 deficient mice showed a reduced NO production and vasodilatory response to bradykinin in cutaneous and cerebral arteries, indicators generally associated with an endothelial dysfunction.84 Surprisingly, despite this endothelial dysfunction, arterial blood pressure in TREK-1 deficient mice remains unaltered. The authors explained this discrepancy by a more substantial role of TREK-1 in the cerebral circulation, with only a minor role in the regulation of systemic blood pressure. However, opposing results have been reported where no changes have been observed in cerebral vasculature from TREK-1 knockout mice, supporting further studies to better understand the role of K2P channels in VSMCs.85
Future research directions
The key factor in the active regulation of blood pressure is the contractile state of VSMCs, making membrane potential a brilliant tool to manipulate vascular tone in a fast, efficient, and precise manner. K+ channels are major components in this regulatory process (Figure 2), in both endothelium and VSMCs. The multitude of distinct K+ channels expressed in the vasculature (Figure 2) allows fine-tuning of the vascular response to the direct environment as well as to ensure a constant blood flow. Moreover, the impressive complexity of the numerous combinations of K+ channels expressed represents a fascinating toolbox given by Mother Nature to compensate pathological conditions or to respond to various physiological conditions.
Opportunities for future investigations on the regulation of vascular tone, especially through K+ channels, are numerous. Recent developments of experimental tools will allow us to further push the limit of our understanding of ion channel regulation. We should also better understand the interaction of the proteins with molecules inhibiting or activating the channels, leading to the development of new therapeutic avenues. However, better pharmacological tools may not necessarily be the consequence of more specific compounds, due to possible heteromultimer formation (ie, KV channels). The microenvironment, as briefly alluded to in this review, is also a critical component of ion channel activity modulation. Indeed, multiprotein complexes including – but not limited to – anchoring proteins will most likely be a target of future studies, as they could result in a better understanding of the different regulatory proteins. Ion channel microenvironment also includes lipid environment and intracellular microdomains (eg, Ca2+ and cyclic nucleotides). Evidently, increasing interests focus on dynamic regulation of ion channel function through intracellular microdomains such as Ca2+ pulsars, wavelets, sparks, and sparklets.
Finally, in our current pharmacogenomic era, it appears essential to identify genetic regulators of K+ channel expression to better understand the intrinsic variability in the human genome and its impact on pathophysiology. Again, such knowledge will be fertile in future developments and will undeniably have a significant clinical impact on improving human conditions and treatments of vascular diseases.
Disclosure
The authors have nothing to disclose.
References
Christensen KL, Mulvany MJ. Location of resistance arteries. J Vasc Res. 2001;38(1):1–12. | |
Stergiopulos N, Westerhof N. Role of total arterial compliance and peripheral resistance in the determination of systolic and diastolic aortic pressure. Pathol Biol (Paris). 1999;47(6):641–647. | |
Fleming I, Busse R. NO: the primary EDRF. J Mol Cell Cardiol. 1999;31(1):5–14. | |
Bogatcheva NV, Sergeeva MG, Dudek SM, Verin AD. Arachidonic acid cascade in endothelial pathobiology. Microvasc Res. 2005;69(3):107–127. | |
Edwards G, Félétou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflüg Arch Eur J Physiol. 2010;459(6):863–879. | |
Garland CJ, Hiley CR, Dora KA. EDHF: spreading the influence of the endothelium. Br J Pharmacol. 2011;164(3):839–852. | |
Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268(4 Pt 1):C799–C822. | |
Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77(4):1165–1232. | |
Quayle JM, Standen NB. KATP channels in vascular smooth muscle. Cardiovasc Res. 1994;28(6):797–804. | |
Gurney A, Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J EBJ. 2009;38(3):305–318. | |
Gutman GA, Chandy KG, Grissmer S, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57(4):473–508. | |
Ohya S, Sergeant GP, Greenwood IA, Horowitz B. Molecular variants of KCNQ channels expressed in murine portal vein myocytes: a role in delayed rectifier current. Circ Res. 2003;92(9):1016–1023. | |
Greenwood IA, Yeung SY, Tribe RM, Ohya S. Loss of functional K+ channels encoded by ether-à-go-go-related genes in mouse myometrium prior to labour onset. J Physiol. 2009;587(Pt 10):2313–2326. | |
Korovkina VP, England SK. Molecular diversity of vascular potassium channel isoforms. Clin Exp Pharmacol Physiol. 2002;29(4):317–323. | |
Cox RH. Molecular determinants of voltage-gated potassium currents in vascular smooth muscle. Cell Biochem Biophys. 2005;42(2):167–195. | |
Castle NA. Pharmacological modulation of voltage-gated potassium channels as a therapeutic strategy. Expert Opin Ther Pat. 2010;20(11):1471–1503. | |
Plane F, Johnson R, Kerr P, et al. Heteromultimeric Kv1 channels contribute to myogenic control of arterial diameter. Circ Res. 2005;96(2):216–224. | |
Hedegaard ER, Nielsen BD, Kun A, et al. KV 7 channels are involved in hypoxia-induced vasodilatation of porcine coronary arteries. Br J Pharmacol. 2014;171(1):69–82. | |
Gurney AM. Multiple sites of oxygen sensing and their contributions to hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol. 2002;132(1):43–53. | |
Joshi S, Balan P, Gurney AM. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res. 2006;7:31. | |
Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12(1):113–127. | |
Mackie AR, Brueggemann LI, Henderson KK, et al. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325(2):475–483. | |
Fountain SJ, Cheong A, Flemming R, Mair L, Sivaprasadarao A, Beech DJ. Functional up-regulation of KCNA gene family expression in murine mesenteric resistance artery smooth muscle. J Physiol. 2004;556(Pt 1):29–42. | |
Hogg DS, McMurray G, Kozlowski RZ. Endothelial cells freshly isolated from small pulmonary arteries of the rat possess multiple distinct K+ current profiles. Lung. 2002;180(4):203–214. | |
Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev. 2005;57(4):463–472. | |
Wulff H, Köhler R. Endothelial small-conductance and intermediate-conductance KCa channels: an update on their pharmacology and usefulness as cardiovascular targets. J Cardiovasc Pharmacol. 2013;61(2):102–112. | |
Monaghan AS, Benton DCH, Bahia PK, et al. The SK3 subunit of small conductance Ca2+-activated K+ channels interacts with both SK1 and SK2 subunits in a heterologous expression system. J Biol Chem. 2004;279(2):1003–1009. | |
Fanger CM, Ghanshani S, Logsdon NJ, et al. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J Biol Chem. 1999;274(9):5746–5754. | |
Bond CT, Maylie J, Adelman JP. Small-conductance calcium-activated potassium channels. Ann N Y Acad Sci. 1999;868:370–378. | |
Xia XM, Fakler B, Rivard A, et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395(6701):503–507. | |
Ahn SC, Seol GH, Kim JA, Suh SH. Characteristics and a functional implication of Ca(2+)-activated K(+) current in mouse aortic endothelial cells. Pflugers Arch. 2004;447(4):426–435. | |
Brähler S, Kaistha A, Schmidt VJ, et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. 2009;119(17):2323–2332. | |
Sonkusare SK, Bonev AD, Ledoux J, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336(6081):597–601. | |
Ledoux J, Taylor MS, Bonev AD, et al. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105(28):9627–9632. | |
Grgic I, Kaistha BP, Hoyer J, Köhler R. Endothelial Ca+-activated K+ channels in normal and impaired EDHF-dilator responses – relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol. 2009;157(4):509–526. | |
Köhler R, Ruth P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflugers Arch. 2010;459(6):969–976. | |
Neylon CB, Lang RJ, Fu Y, Bobik A, Reinhart PH. Molecular cloning and characterization of the intermediate-conductance Ca(2+)-activated K(+) channel in vascular smooth muscle: relationship between K(Ca) channel diversity and smooth muscle cell function. Circ Res. 1999;85(9):e33–e43. | |
Köhler R, Wulff H, Eichler I, et al. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 2003;108(9):1119–1125. | |
Köhler R, Kaistha BP, Wulff H. Vascular KCa-channels as therapeutic targets in hypertension and restenosis disease. Expert Opin Ther Targets. 2010;14(2):143–155. | |
Feng J, Liu Y, Clements RT, et al. Calcium-activated potassium channels contribute to human coronary microvascular dysfunction after cardioplegic arrest. Circulation. 2008;118(Suppl 14):S46–S51. | |
Toyama K, Wulff H, Chandy KG, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118(9):3025–3037. | |
Knaus HG, Eberhart A, Glossmann H, Munujos P, Kaczorowski GJ, Garcia ML. Pharmacology and structure of high conductance calcium-activated potassium channels. Cell Signal. 1994;6(8):861–870. | |
Chen L, Tian L, MacDonald SH-F, et al. Functionally diverse complement of large conductance calcium- and voltage-activated potassium channel (BK) alpha-subunits generated from a single site of splicing. J Biol Chem. 2005;280(39):33599–33609. | |
Tian L, Duncan RR, Hammond MS, et al. Alternative splicing switches potassium channel sensitivity to protein phosphorylation. J Biol Chem. 2001;276(11):7717–7720. | |
Tanaka Y, Meera P, Song M, Knaus HG, Toro L. Molecular constituents of maxi KCa channels in human coronary smooth muscle: predominant alpha + beta subunit complexes. J Physiol. 1997;502(Pt 3):545–557. | |
Brenner R, Peréz GJ, Bonev AD, et al. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature. 2000;407(6806):870–876. | |
Nelson MT, Cheng H, Rubart M, et al. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270(5236):633–637. | |
Pérez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol. 1999;113(2):229–238. | |
Blatz AL, Magleby KL. Single apamin-blocked Ca-activated K+ channels of small conductance in cultured rat skeletal muscle. Nature. 1986;323(6090):718–720. | |
Lang DG, Ritchie AK. Large and small conductance calcium-activated potassium channels in the GH3 anterior pituitary cell line. Pflüg Arch Eur J Physiol. 1987;410(6):614–622. | |
McManus OB. Calcium-activated potassium channels: regulation by calcium. J Bioenerg Biomembr. 1991;23(4):537–560. | |
Barrett JN, Magleby KL, Pallotta BS. Properties of single calcium-activated potassium channels in cultured rat muscle. J Physiol. 1982;331:211–230. | |
Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol. 1997;110(3):257–281. | |
Campbell WB, Falck JR. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension. 2007;49(3):590–596. | |
Félétou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol. 2009;156(4):545–562. | |
Sausbier M, Arntz C, Bucurenciu I, et al. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation. 2005;112(1):60–68. | |
Werner ME, Meredith AL, Aldrich RW, Nelson MT. Hypercontractility and impaired sildenafil relaxations in the BKCa channel deletion model of erectile dysfunction. Am J Physiol Regul Integr Comp Physiol. 2008;295(1):R181–R188. | |
Werner ME, Zvara P, Meredith AL, Aldrich RW, Nelson MT. Erectile dysfunction in mice lacking the large-conductance calcium-activated potassium (BK) channel. J Physiol. 2005;567(Pt 2):545–556. | |
Plüger S, Faulhaber J, Fürstenau M, et al. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res. 2000;87(11):E53–E60. | |
Amberg GC, Bonev AD, Rossow CF, Nelson MT, Santana LF. Modulation of the molecular composition of large conductance, Ca(2+) activated K(+) channels in vascular smooth muscle during hypertension. J Clin Invest. 2003;112(5):717–724. | |
Amberg GC, Santana LF. Downregulation of the BK channel beta1 subunit in genetic hypertension. Circ Res. 2003;93(10):965–971. | |
Bonnet S, Savineau J-P, Barillot W, Dubuis E, Vandier C, Bonnet P. Role of Ca(2+)-sensitive K(+) channels in the remission phase of pulmonary hypertension in chronic obstructive pulmonary diseases. Cardiovasc Res. 2003;60(2):326–336. | |
Bratz IN, Dick GM, Partridge LD, Kanagy NL. Reduced molecular expression of K(+) channel proteins in vascular smooth muscle from rats made hypertensive with N{omega}-nitro-L-arginine. Am J Physiol Heart Circ Physiol. 2005;289(3):H1277–H1283. | |
Nishimaru K, Eghbali M, Lu R, Marijic J, Stefani E, Toro L. Functional and molecular evidence of MaxiK channel beta1 subunit decrease with coronary artery ageing in the rat. J Physiol. 2004;559(Pt 3):849–862. | |
Tomás M, Vázquez E, Fernández-Fernández JM, et al. Genetic variation in the KCNMA1 potassium channel alpha subunit as risk factor for severe essential hypertension and myocardial infarction. J Hypertens. 2008;26(11):2147–2153. | |
Sentí M, Fernández-Fernández JM, Tomás M, et al. Protective effect of the KCNMB1 E65K genetic polymorphism against diastolic hypertension in aging women and its relevance to cardiovascular risk. Circ Res. 2005;97(12):1360–1365. | |
Standen NB, Quayle JM. K+ channel modulation in arterial smooth muscle. Acta Physiol Scand. 1998;164(4):549–557. | |
Filosa JA, Bonev AD, Straub SV, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9(11):1397–1403. | |
Longden TA, Dabertrand F, Hill-Eubanks DC, Hammack SE, Nelson MT. Stress-induced glucocorticoid signaling remodels neurovascular coupling through impairment of cerebrovascular inwardly rectifying K+ channel function. Proc Natl Acad Sci U S A. 2014;111(20):7462–7467. | |
Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res. 2000;87(2):160–166. | |
Wu B-N, Luykenaar KD, Brayden JE, et al. Hyposmotic challenge inhibits inward rectifying K+ channels in cerebral arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292(2):H1085–H1094. | |
Tennant BP, Cui Y, Tinker A, Clapp LH. Functional expression of inward rectifier potassium channels in cultured human pulmonary smooth muscle cells: evidence for a major role of Kir2.4 subunits. J Membr Biol. 2006;213(1):19–29. | |
Teramoto N. Physiological roles of ATP-sensitive K+ channels in smooth muscle. J Physiol. 2006;572(Pt 3):617–624. | |
Zhang H, Flagg TP, Nichols CG. Cardiac sarcolemmal K(ATP) channels: Latest twists in a questing tale! J Mol Cell Cardiol. 2010;48(1):71–75. | |
Davies LM, Purves GI, Barrett-Jolley R, Dart C. Interaction with caveolin-1 modulates vascular ATP-sensitive potassium (KATP) channel activity. J Physiol. 2010;588(Pt 17):3255–3266. | |
Malester B, Tong X, Ghiu I, et al. Transgenic expression of a dominant negative K(ATP) channel subunit in the mouse endothelium: effects on coronary flow and endothelin-1 secretion. FASEB J. 2007;21(9):2162–2172. | |
Chatterjee S, Levitan I, Wei Z, Fisher AB. KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation. 2006;13(8):633–644. | |
Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90(2):559–605. | |
Bryan RM Jr, You J, Phillips SC, et al. Evidence for two-pore domain potassium channels in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2006;291(2):H770–H780. | |
Gardener MJ, Johnson IT, Burnham MP, Edwards G, Heagerty AM, Weston AH. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br J Pharmacol. 2004;142(1):192–202. | |
Gönczi M, Szentandrássy N, Johnson IT, Heagerty AM, Weston AH. Investigation of the role of TASK-2 channels in rat pulmonary arteries; pharmacological and functional studies following RNA interference procedures. Br J Pharmacol. 2006;147(5):496–505. | |
Gurney AM. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ Res. 2003;93(10):957–964. | |
Tang B, Li Y, Nagaraj C, et al. Endothelin-1 inhibits background two-pore domain channel TASK-1 in primary human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2009;41(4):476–483. | |
Garry A, Fromy B, Blondeau N, et al. Altered acetylcholine, bradykinin and cutaneous pressure-induced vasodilation in mice lacking the TREK1 potassium channel: the endothelial link. EMBO Rep. 2007;8(4):354–359. | |
Namiranian K, Lloyd EE, Crossland RF, et al. Cerebrovascular responses in mice deficient in the potassium channel, TREK-1. Am J Physiol Regul Integr Comp Physiol. 2010;299(2):R461–R469. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.