")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 13
Juvenile Hemochromatosis: Rheumatic Manifestations of 2 Sisters Responding to Deferasirox Treatment. A Case Series and Literature Review
Authors Alqanatish J , Alsowailmi B, Alfarhan H, Alhamzah A, Alharbi T
Received 12 August 2020
Accepted for publication 27 October 2020
Published 15 January 2021 Volume 2021:13 Pages 15—21
DOI https://doi.org/10.2147/OARRR.S276112
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Chuan-Ju Liu
Jubran Alqanatish,1– 3 Banan Alsowailmi,1 Haneen Alfarhan,1 Albandari Alhamzah,1 Talal Alharbi1,2,4
1King Saud Bin Abdulaziz University for Health Sciences (KSAU-HS), Riyadh 14611, Saudi Arabia; 2King Abdullah International Medical Research Center (KAIMRC), Riyadh 14611, Saudi Arabia; 3Department of Pediatrics, King Abdullah Specialist Children’s Hospital and King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia; 4Department of Pediatric Hematology/Oncology, King Abdullah Specialist Children’s Hospital and King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia
Correspondence: Jubran Alqanatish
King Saud Bin Abdulaziz University for Health Sciences (KSAU-HS), PO Box 22490, Riyadh 11426, Saudi Arabia
Tel +966 118011111 Ext 53501
Email [email protected]
Abstract: Hereditary hemochromatosis (HH) is an inherited iron overload. The most common form of HH is type 1 hereditary hemochromatosis (HFE-related), which is associated with mutation of the HFE gene located on chromosome 6 and inherited in an autosomal recessive pattern. Type 2 hereditary hemochromatosis or juvenile hemochromatosis is less frequent autosomal recessive disease that results from mutations in the HJV gene on chromosome 1 (type2a) or the HAMP gene on chromosome19 (type2b). Mutation of type 2 transferrin receptor gene and mutation of the ferroportin gene result in hemochromatosis type 3 and hemochromatosis type 4, respectively. Juvenile hemochromatosis is characterized by an early onset of excess accumulation of iron in various organs. It could affect the liver, heart, pancreas and joints, resulting in arthropathy. Most juvenile hemochromatosis cases exhibit severe symptoms due to early onset. Cardiac and hypogonadism are the dominating features of the disease. Prevalence of arthropathy in juvenile hemochromatosis is higher than classic HH. Early diagnosis and intervention of juvenile hemochromatosis may prevent irreversible organ damage. The diagnosis can be made based on laboratory testing (of increased transferrin saturation, serum iron and ferritin levels), liver biopsy, imaging or genotype. According to international guidelines, treatment of HH is indicated when serum ferritin concentrations are above the upper limit of normal. We report two sisters who presented to the rheumatology clinic with arthralgia, which was subsequently found to have a homozygous mutation variant of unknown significance in the HFE2 gene: c.497A>G;p.(His166Arg) and has been treated with deferasirox (Exjade®). Musculoskeletal symptoms completely resolved in both patients in two months and remained so for one year on treatment.
Keywords: hereditary hemochromatosis, HFE gene, arthritis, deferasirox
Introduction
Hemochromatosis is an overload in iron storage, classified into hereditary hemochromatosis (HH) (due to a mutation in HFE gene in 80% of cases, non-HFE hemochromatosis in 20% of HH cases) or acquired due to frequent packed red blood cell (PRBC) transfusion or high dietary iron intake. HH is an autosomal recessive and potentially lethal disorder of iron metabolism. The first description of hemochromatosis gene was by Feder et al in 1996.1 It is characterized into four types, the most frequent type is HH type, which results from HFE gene mutation (HFE gene); hemochromatosis type 2 or juvenile hemochromatosis is divided into two subtypes, mutation of hemojuvelin (HJV) results in type 2a, while mutation of the hepcidin HAMP gene results in type 2b; mutation of type 2 transferrin receptor gene results in hemochromatosis type 3; hemochromatosis type 4 is the only autosomal dominant form caused by the mutation of the ferroportin gene.2,3
Previous reports indicate that most Asian cases of HH are due non-HFE mutation.4,5
HH course in childhood is either asymptomatic or exhibits non-specific symptoms. The diagnosis is typically delayed till adulthood during investigation for the cause of organ damage such as diabetes mellitus, heart failure, liver cirrhosis, hypogonadism and arthritis. Arthropathy, osteopenia and osteoporosis are well-studied musculoskeletal features of HH.6 Arthritis may precede other symptoms, and involvement of the second and third metacarpophalangeal (MCP) joints is a characteristic feature of HH. Generally, the arthropathy of HH is non-inflammatory.7
In contrast, juvenile hemochromatosis cases exhibit severe symptoms due to early onset that may manifest with cardiac dysfunction and hypogonadotropic hypogonadism. The prevalence of arthritis was reported as high as 87.5%.8
To increase the awareness of pediatric rheumatologists and to contribute to the literature on the use of deferasirox in HH, we report the data of two sisters who presented in the rheumatology clinic with polyarthralgia. Both have homozygous variant of unknown significance in the HFE2 gene: c.497A>G;p.(His166Arg) and their musculoskeletal symptoms resolved with deferasirox treatment.
Case Series
A 9-year-old girl was referred to the rheumatology clinic due to high ferritin and transaminases for 3 years and a non-specific, infrequent bone and joint pain that increases over the wrists and ankles whenever she engages in any activity. She had no joint swelling, morning stiffness, gelling or skin rash. There was no history of fever, headache, visual symptoms or jaundice. She had hepatomegaly, and liver ultrasound showed hyperechoic liver structure with a hypodense lesion in segment 4A/B, likely representing focal fatty infiltration. A liver biopsy then revealed hemosiderosis with parenchymatous (hepatocellular) distribution, consistent with HH, grade 4 (0–4 grading system) (Figure 1). Genetic testing revealed homozygous variant of unknown significance in HFE2 gene: c.497A>G;p.(His166Arg). This variant was homozygous in the affected sisters, while heterozygous in both parents. This variant has not been described in Exome Aggregation Consortium (ExAC); gnomAD.; 1000 genome and 1410 individuals in our local Saudi genome database. Furthermore, it was predicted to be damaging by software prediction tools like PolyPhen and Mutation taster.
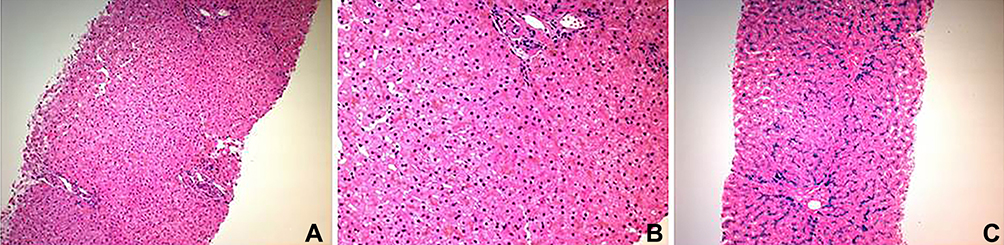 |
Figure 1 Liver biopsy is histologically unremarkable on H&E stain (A). On higher magnification, there is intra-hepatocyte brown pigments which represents iron accumulation (B).The pigments appear blue with Perls stain (C). Grade 4 hepatocyte iron overload. The biopsy is negative for significant fibrosis. |
Family history was remarkable, given a history of a similar complaint in an older sister, a 25-year-old who had been suffering from forearm and calf pain and unsettled hyperferritinemia for several years. Musculoskeletal pain was significant and disturbed her sleep with not much response to analgesia. She had occasional stiffness over her hands but no joint pain, swelling or gelling.
There was no skin rash, no headache or visual symptoms. Description of laboratory findings for both sisters is provided in (Table 1).
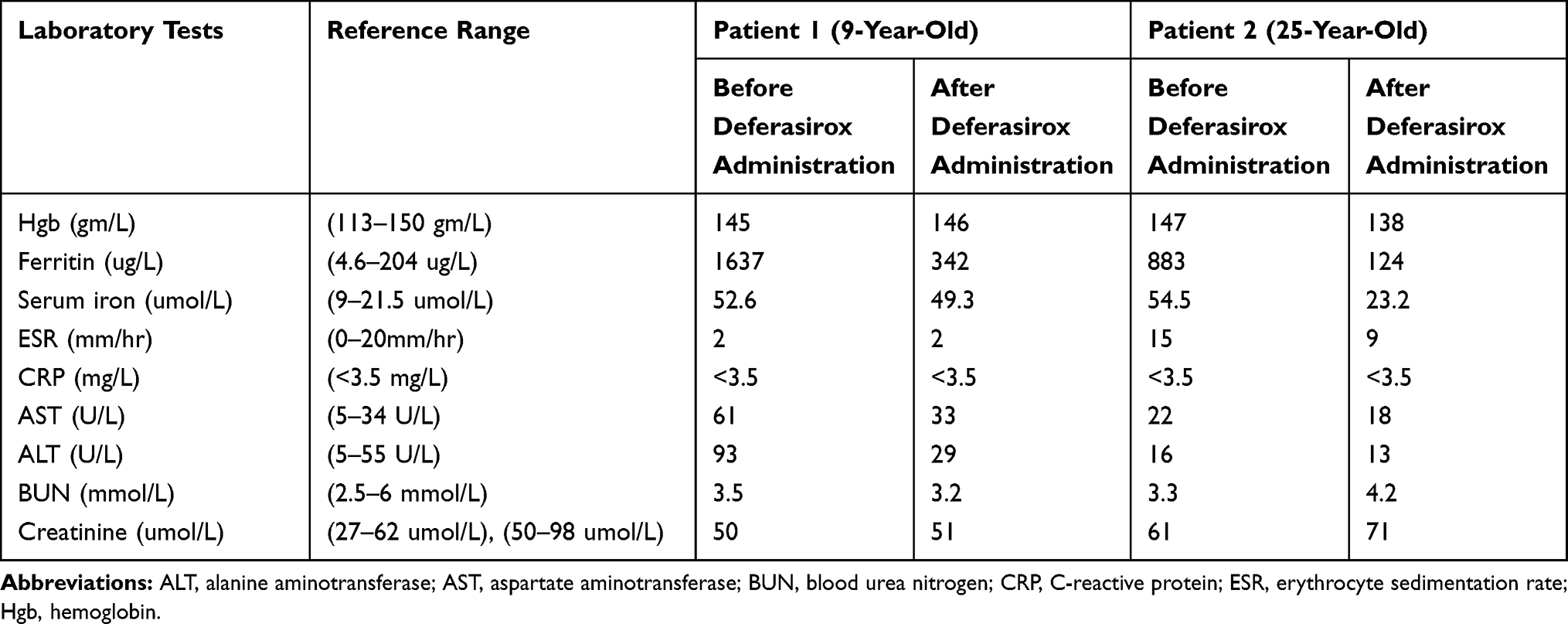 |
Table 1 Summary of Laboratory Results for Both Patients |
Liver magnetic resonance imaging (MRI) was done for the older sister and showed dark signals of the liver (L) relative to normal signals of spleen (S) due to iron deposition (Figure 2). Like the case of her sister, a diagnosis of hemochromatosis type 2A was made and genetic testing revealed, a homozygous variant of unknown significance in HFE2 gene: c.497A>G;p.(His166Arg).
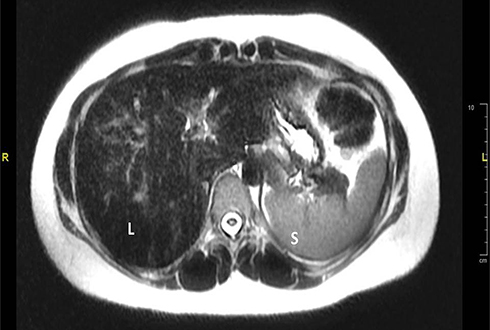 |
Figure 2 Axial T2 weighted image of the liver showed dark signals of the liver (L) relative to normal signals of spleen (S) due to iron deposition. |
In the second case, she had morbid obesity with BMI of 43 and depression and underwent gastric sleeve surgery at age of 22. It was noticed that serum ferritin level decreased after surgery but never went back to normal.
Deferasirox (Exjade®) was introduced for both patients at a dose of 500 mg daily, 8 mg/kg/day for the older sister and 15mg/kg/day for the younger sister, respectively and serum ferritin was monitored. Deferasirox dose was calculated to the nearest whole tablet for easy administration. Their musculoskeletal complaints improved gradually and faded away in two months with improvement in serum ferritin level. Aspartate aminotransferase and alanine aminotransferase levels improved in the young girl and remained normal in the young woman following the administration of deferasirox. Renal function continued normally in both.
The mother was also diagnosed with rheumatoid arthritis and was treated with methotrexate and penicillamine. She had normal serum ferritin and her response to DMARDS was acceptable.
Discussion
Gottschalk et al report that, among approximately 5000 patients referred to the rheumatology outpatient clinics in two German centers with different rheumatologic diagnoses, 11 patients with typical signs of hemochromatosis arthropathy were identified.9
In a large cohort which evaluated 93 patients with HH looking specifically to musculoskeletal manifestations, 24% of patients have arthropathy.10
In some cases musculoskeletal symptoms alone might lead to the diagnosis of HH.11 Common joints to be involved are metacarpophalangeal joints (MCPs), the wrist, hip, knee and ankle. The classical joint involvement of HH is the second and third metacarpophalangeal joints. HH arthropathy tends to be symmetrical in most cases.12 In a survey by McDonnell et al, arthralgias were present in 44% of patients with hemochromatosis and were the most common and troublesome complaints.13 In our patient, arthralgia appeared at the age of 6 in the first case and age of 14 in the older sister. It involved MCP joints, wrist and knees in the younger girl and ankle and hips in the older one.
It is difficult to differentiate HH arthropathy from inflammatory arthropathy due to autoimmune problems but HH arthropathy does not usually include night time pain or extended morning stiffness.14 Other clinical features that may help to differentiate HH arthropathy from other rheumatic diseases are younger age at onset, location, and male predominance.15 Doppler ultrasound showed increased signals consistent with inflammatory synovitis in a 40-year-old and 58-year-old male patients who were diagnosed with HH.16 Coexistence of calcium pyrophosphate dihydrate crystal deposition disease (CPPD) with hemochromatosis is a possible cause of the increased signals in Doppler ultrasound.17
Arthritis symptoms in patients with hemochromatosis could be related to iron deposition as demonstrated by articular cartilage biopsies, which confirmed the presence of iron in chondrocytes, as seen by electron microscopy.18 However, this matter in the pathogenesis of HH remains controversial as there is a lack of correlation between the iron deposits and the radiologic findings. Furthermore, the progression of disease is not halted by iron depletion therapy.18
The other musculoskeletal symptom that has been described in patients with HH is osteoporosis. Low bone mineral density is caused by different mechanisms in patients with HH, iron overload, liver disease, and hypogonadism.19,20 Osteoporosis has been described more in adults due to obvious reasons, including aging, hypogonadism, and putative alcoholism. In children, osteoporosis due to juvenile hemochromatosis has been documented.21 International guidelines for hemochromatosis do not recommend bone assessment in HH, and most of the guidelines do not include osteoporosis assessment, except in France.22 Therefore, we did not do it for our patients.
To exclude common causes of secondary osteoporosis, assessment of parathyroid hormone will help to exclude hyperparathyroidism; vitamin D level to exclude vitamin D deficiency; paucity of liver cirrhosis in liver histopathology will help to exclude liver disease.
More recent studies are pointing to direct iron toxicity on bone metabolism.23 In vitro, iron is capable of inhibiting bone crystal growth with significant changes in crystallinity and carbonate substitution.24
Diagnosis of HH is now usually done at an early stage of the disease before even the symptoms start.25 The most frequent symptom in routine health checks is lethargy.26 Family screening also helps in early diagnosis of HH, in particular, patient’s first degree relatives with established diagnosis of HH.27 Early detection of HH is essential to prevent long term end organ damage. Abnormal iron study results or abnormal liver function should alert clinician to consider HH.28
Radiological findings, as mentioned previously, such as the specific location on the second and third MCP joints is characteristic of HH. In plain radiography; a joint space narrowing of the MCP joint, a hook-shaped osteophyte of the metacarpal head, and an early subchondral cyst formation are common findings.14,29,30
The x-ray of both hands in patient 1 did not show the typical features of chronic HH arthritis, which could be due to her younger age. Those patients with HH may show early secondary osteoarthritis and require joint replacement surgery.31
MRI has been shown to be reliable for detecting and quantifying visceral iron excess, especially in the liver and spleen.32 In patient 2 MRI was enough to show the evidence of iron overload (Figure 2).
The role of liver biopsy started to be limited in the diagnosis of HH due to the availability of genetic testing and accuracy of MRI in the assessment of iron load.33 The importance of liver biopsy is in prognostic implications as it might help in detecting advanced liver disease and cirrhosis. A synovial biopsy might be done in cases with atypical arthritis, and it would guide to the diagnosis if it shows hemosiderin deposits in the synoviocytes.
Laboratory findings in HH are increased serum iron concentration, raised transferrin concentration (plasma total iron binding capacity), and increased plasma ferritin.
Both patients that we described here, were treated with deferasirox. iron and ferritin levels were on a downward trend; ferritin level 2 months after the start of deferasirox therapy was (342 ug/L, iron levels were 49.3 umol/l), (124 ug/L, iron levels were 23.2 umol/l) for both girls, respectively.
The international guidelines published by both the American and European Liver Associations advocate that therapy should be commenced when the serum ferritin concentrations are above the upper limit of normal.26,34
There is no robust evidence for the best therapeutic option in HH. Morbidity and mortality are definitely reduced when removal of iron overload is done early in the disease course, before the development of complications.35,36 Venesection is a common modality of treatment for patients with HH.26,34
There are variable recommendations with respect to the level of iron burden at which therapy should be initiated, the endpoint of initial venesection, and the optimum maintenance therapy. These recommendations have been summarized by Powell et al37 (Table 2)
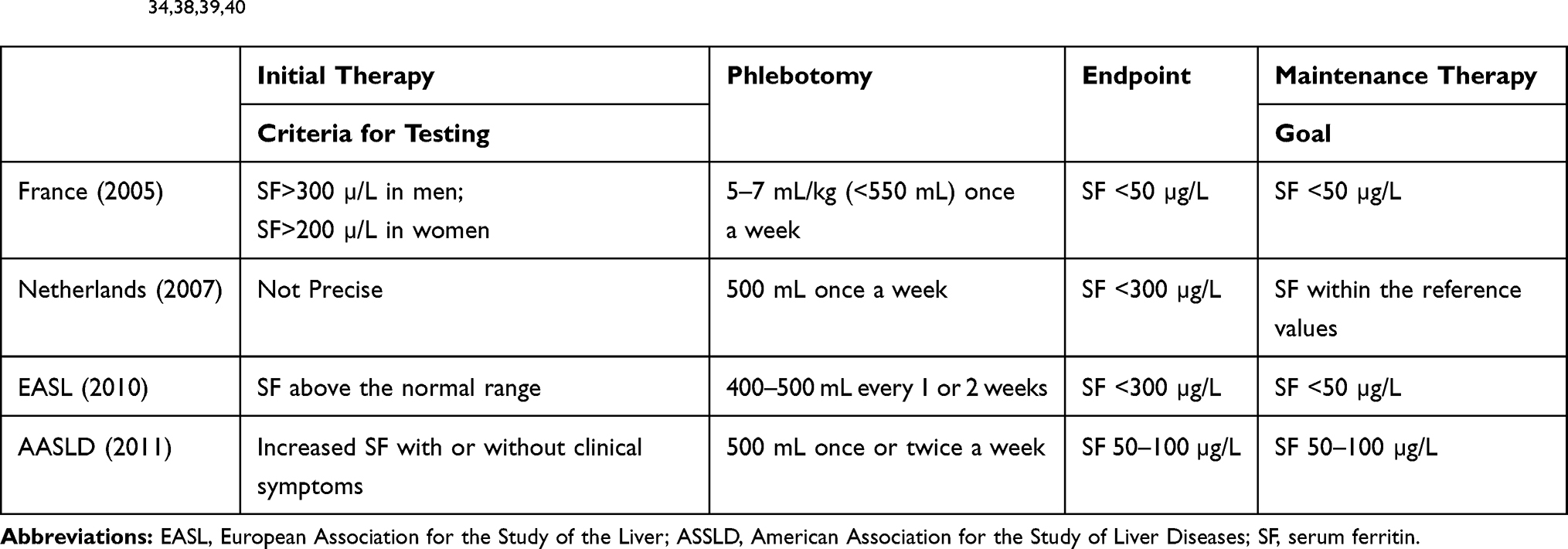 |
Table 2 Main International Recommendations on Venesection Therapy in HFE Haemochromatosis (with Permission from Powell,2016)34,38,39,40 |
Alternative therapies like deferasirox (Exjade®), an oral iron chelator approved by the FDA in November 2005, has been shown to eliminate iron excess effectively in patients with HFE hemochromatosis.
Our patients were treated with deferasirox, inclined on the experience that did not support the effect of venesection in arthralgia and MSK symptoms.15 Patients were provided the options of deferasirox therapy or venesection, and they chose the former. Among many advantages of deferasirox is the once daily administration, oral use availability, and high affinity to bind with iron in a 2:1 ratio.41
It is well-known that certain precautions should be assumed when administering the dose of deferasirox, and it was clear to us that the younger girl’s response is different from the older sister, which has been attributed to the failure of compliance with these precautions. The common precaution that the patient failed to comply with is administration on empty stomach, use of citrus juice, and avoidance of metal containing appliances during handling.
A Phase II study by Cancado et al confirms the safety and efficacy of deferasirox, an oral iron chelator, in patients with HH. Ten patients with confirmed HH mutation were included and all completed the 12-month treatment period with a significant reduction in median serum ferritin (P < 0.001), mean transferrin saturation (P < 0.05), median liver iron concentration (P < 0.001), and mean alanine aminotransferase (P < 0.05). The two minor side effects of deferasirox encountered in that study were transient diarrhea and nausea, so the author has concluded that it is a safe alternative to phlebotomy in selected patients with HH.42
The other study which supports the use of deferasirox as an alternative therapy for iron overload in patients with HH was by Phatak et al, the first clinical trial to assess the safety and efficacy of deferasirox therapy using four dose levels (5, 10, 15, and 20 mg/kg/day). The study concludes that a starting dose of 10 mg/kg/day appears to be the most appropriate in this population; close monitoring of renal and hepatic function is required, especially given that six of the 49 patients involved in the study had transaminase increases exceeding three times normal values.43
Neglar et al reported two patients who had chronic anemia that prevented the initiation of venesection but were treated successfully with deferasirox.44
In conclusion, presentation of HH to a pediatric rheumatologist is not frequent and most of the cases are either asymptomatic or exhibit non-specific musculoskeletal symptoms. In the two cases we reported here deferasirox was very effective in treating the musculoskeletal symptoms and lowering serum ferritin level. Effect of deferasirox on serum iron was more obvious in the older sister; this was attributed to her compliance with the instructions and precaution pertinent to the drug administration.
Until more robust evidence is available, it seems that deferasirox is a safe alternative to venesection for the treatment of selected patients with HH.
Ethical Considerations
Institutional approval was not required to publish the case details. However all patients’ clinical data were regarded as confidential and written consents were obtained from the adult patient and the parents of the minor.
Consent for Publishing Patient Data
We confirm that written informed consents were obtained from the adult patient and the parents of the minor, to have the description of their cases published.
Disclosure
Principle investigator and co-authors report no conflicts of interest in this work.
References
1. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Gen. 1996;13:339–408.
2. Santacoloma M, Londoño HG, Limas LM. Hereditary hemochromatosis: presentation of 2 cases and literature review. Rev Col Gastroenterol. 2010;25(2):188–193.
3. Pietrangelo A. Juvenile hemochromatosis. J Hepatol. 2006;45(6):892–894. doi:10.1016/j.jhep.2006.09.003
4. Lok CY, Merryweather-Clarke AT, Viprakasit V, et al. Iron overload in the Asian community. Blood. 2009;114(1):20–25. doi:10.1182/blood-2009-01-199109
5. McDonald CJ, Wallace DF, Crawford DH, Subramaniam VN. Iron storage disease in Asia‐Pacific populations: the importance of non‐HFE mutations. J Gastroenterol Hepatol. 2013;28(7):1087–1094.
6. Schumacher HR
7. Carroll GJ, Breidahl WH, Olynyk JK. Characteristics of the arthropathy described in hereditary hemochromatosis. Arthritis Care Res (Hoboken). 2011;64(1):9–14.
8. Vaiopoulos G, Papanikolaou G, Politou M, Jibreel I, Sakellaropoulos N, Loukopoulos D. Arthropathy in juvenile hemochromatosis. Arthritis Rheum. 2003;48(1):227–230.
9. Gottschalk R, Neeck G, Wigand R, Vogtherr B, Kaltwasser JP. Hemochromatosis arthropathy–an early manifestation of genetic hemochromatosis. Z Rheumatol. 1997;56(3):156–162.
10. Nguyen CD, Morel V, Pierache A, et al. Bone and joint complications in patients with hereditary hemochromatosis: a cross-sectional study of 93 patients. Ther Adv Musculoskelet Dis. 2020;12:1759720X20939405. doi:10.1177/1759720X20939405
11. Wimalawansa SM, Alsamkari R. Unusual presentation of hemochromatosis as isolated metacarpophalangeal joint osteoarthritis: A case report. Hand (N Y). 2011;6:329–332. doi:10.1007/s11552-011-9335-7
12. Sahinbegovic E, Dallos T, Aigner E, et al. Musculoskeletal disease burden of hereditary hemochromatosis. Arthritis Rheum. 2010. 62:3792–3798.
13. McDonnell SM, Preston BL, Jewell SA, et al. A survey of 2,851 patients with hemochromatosis: symptoms and response to treatment. Am J Med. 1999;106:619–624. doi:10.1016/S0002-9343(99)00120-5
14. Guggenbuhl P, Brissot P, Loréal O. Haemochromatosis: the bone and the joint. Best Pract Res Clin Rheumatol. 2011;25(5):649–664. doi:10.1016/j.berh.2011.10.014
15. Husar-Memmer E, Stadlmayr A, Datz C, Zwerina J. HFE-related hemochromatosis: an update for the rheumatologist. Curr Rheumatol Rep. 2014;16(1):393. doi:10.1007/s11926-013-0393-4
16. Oke AR, Wong E, McCrae F, Young-Min S. Hereditary hemochromatosis arthropathy and Doppler ultrasound findings of synovitis. Rheumatology. 2017;56(7):1240–1241. doi:10.1093/rheumatology/kex069
17. Parperis K. Comment on: hereditary hemochromatosis arthropathy and Doppler ultrasound findings of synovitis. Rheumatology. 2018;57(4):765–766. doi:10.1093/rheumatology/kex436
18. Schumacher HR. Articular cartilage in the degenerative arthropathy of hemochromatosis. Arthritis Rheum. 1982;25:1460–1468. doi:10.1002/art.1780251212
19. Guggenbuhl P, Deugnier Y, Boisdet JF, et al. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos Int. 2005;16(12):1809–1814. doi:10.1007/s00198-005-1934-0
20. Valenti L, Varenna M, Fracanzani AL, et al. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos Int. 2009;20:549–555. doi:10.1007/s00198-008-0701-4
21. Angelopoulos NG, Goula AK, Papanikolaou G, Tolis G. Osteoporosis in HFE2 juvenile hemochromatosis. A case report and review of the literature. Osteoporosis Int. 2006;17(1):150–155. doi:10.1007/s00198-005-1920-6
22. Anonymous. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3–22. doi:10.1016/j.jhep.2010.03.001
23. Weinberg ED. Iron loading: a risk factor for osteoporosis. Biometals. 2006;19(6):633–635. doi:10.1007/s10534-006-9000-8
24. Guggenbuhl P, Filmon R, Mabilleau G, Basle MF, Chappard D. Iron inhibits hydroxyapatite crystal growth in vitro. Metabolism. 2008;57:903–910. doi:10.1016/j.metabol.2008.02.004
25. Allen KJ, Gurrin LCP, Constantine CCP, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221–230. doi:10.1056/NEJMoa073286
26. Adams P, Brissot P, Powell LWEASL. International consensus conference on haemochromatosis. J Hepatol. 2000;33:487–496. doi:10.1016/S0168-8278(01)80874-6
27. Powell LW, Dixon JL, Ramm GA, Purdie DM. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med. 2006;166:294–301. doi:10.1001/archinte.166.3.294
28. Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology (Baltimore, Md). 2011;54(1):328. doi:10.1002/hep.24330
29. Schumacher HR
30. Schumacher HR
31. Carroll G, Breidahl W, Olynyk J. Characteristics of the arthropathy described in hereditary hemochromatosis. Arthritis Care Res (Hoboken). 2011. 64:9–14.
32. Gandon Y, Olivie D, Guyader D, et al. Non-invasive assessment of hepatic iron stores by MRI. Lancet. 2004;363:357–362. doi:10.1016/S0140-6736(04)15436-6
33. Hernando D, Levin YS, Sirlin CB, Reeder SB. Quantification of liver iron with MRI: state of the art and remaining challenges. J Magn Resonance Imaging. 2014;40(5):1003–1021. doi:10.1002/jmri.24584
34. Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328–343.
35. Adams PC, Speechley M, Kertesz AE. Long-term survival analysis in hereditary hemochromatosis. Gastroenterology. 1991;101(2):368. doi:10.1016/0016-5085(91)90013-B
36. Niederau C, Fischer R, Purschel A, Stremmel W. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology. 1996;110:1107–1119. doi:10.1053/gast.1996.v110.pm8613000
37. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016;388(10045):706–716.
38. European Association For The Study Of The Liver. EASL clinical practice guidelines for HFE hemochromatosis. Journal of hepatology. 2010;53(1):3–22.
39. Brissot P, De Bels F. Prise en charge de l'hémochromatose liée au gène HFE. La Presse Médicale. 2007;36(9):1295–300.
40. Swinkels DW, Jorna AT, Raymakers RA. Synopsis of the Dutch multidisciplinary guideline for the diagnosis and treatment of hereditary haemochromatosis.
41. Tanaka C. Clinical pharmacology of deferasirox. Clin Pharmacokinet. 2014;53:679–694. doi:10.1007/s40262-014-0151-4
42. Cançado R, Melo MR, de Moraes Bastos R, et al. Deferasirox in patients with iron overload secondary to hereditary hemochromatosis: results of a 1-yr Phase 2 study. Eur J Haematol. 2015;95(6):545–550. doi:10.1111/ejh.12530
43. Phatak P, Brissot P, Wurster M, et al. A Phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology. 2010;52(5):1671–1779. doi:10.1002/hep.23879
44. Nagler M, Gregor M, Wuillemin WA. Iron chelation with deferasirox in two patients with HFE hemochromatosis and chronic anemia. Acta Haematol. 2011;126:119–121. doi:10.1159/000328039
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.