")
Back to Journals » Journal of Inflammation Research » Volume 14
Iridin Prevented Against Lipopolysaccharide-Induced Inflammatory Responses of Macrophages via Inactivation of PKM2-Mediated Glycolytic Pathways
Authors Ying ZH, Li HM, Yu WY , Yu CH
Received 20 November 2020
Accepted for publication 12 January 2021
Published 5 February 2021 Volume 2021:14 Pages 341—354
DOI https://doi.org/10.2147/JIR.S292244
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Zhen-Hua Ying,1 Hui-Min Li,1 Wen-Ying Yu,2 Chen-Huan Yu2– 4
1Zhejiang Provincial People’s Hospital, Hangzhou Medical College, Hangzhou, 310006, People’s Republic of China; 2Zhejiang Key Laboratory of Experimental Animal and Safety Evaluation, Hangzhou Medical College, Hangzhou, 310013, People’s Republic of China; 3Institute of Cancer and Basic Medicine, Chinese Academy of Sciences, Hangzhou, 310018, People’s Republic of China; 4Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Hangzhou, 310022, People’s Republic of China
Correspondence: Chen-Huan Yu
Institute of Cancer and Basic Medicine, Chinese Academy of Sciences, Hangzhou, 310018, People’s Republic of China
Email [email protected]
Purpose: Abnormal glycolysis of immune cells contributed to the development of inflammatory response. Inhibition of this Warburg phenotype could be a promising strategy for preventing various inflammatory diseases. Iridin (IRD) is a natural isoflavone, and exerts anticancer, antioxidant, and anti-inflammatory effects. However, the underlying mechanism of IRD on acute inflammation remains unknown. In this study, the protective effects of IRD against lipopolysaccharide (LPS)-induced inflammation were investigated in murine macrophage RAW264.7 cells and in mice.
Methods: The inhibition of IRD on NO production in culture medium was detected by Griess assay while the levels of TNF-α, IL-1β, and MCP-1 were detected by ELISA assay. The effects of IRD on OCR and ECAR levels in LPS-treated macrophages were monitored by using Seahorse Analyzer. The apoptosis rate as well as the release of ROS and NO of RAW264.7 cells were analyzed by flow cytometric assay. The protective effects of IRD were investigated on LPS-induced inflammation in mice. The expressions of PKM2 and its downstream (p-JAK1, p-STAT1, p-STAT3, p-p65, iNOS, and COX2) in cells and in lung tissues were detected by Western blotting analysis.
Results: IRD treatment at the concentrations of 12.5– 50 μM significantly inhibited the productions of TNF-α, IL-1β, MCP-1, and ROS, and suppressed the levels of glucose uptake and lactic acid in LPS-treated RAW264.7 cells. Oral administration with IRD (20– 80 mg/kg) inhibited LPS-induced acute lung injury as well as inflammatory cytokine production in mice. Moreover, IRD targeted pyruvate kinase isozyme type M2 (PKM2) and suppressed its downstream p-JAK1, p-STAT1, p-STAT3, p-p65, iNOS, and COX2, which could be abolished by PKM2 agonist DASA-58 and antioxidant N-acetyl-L-cysteine, but partly be reversed by NF-κB activator CUT129 and JAK1 activator RO8191.
Conclusion: IRD alleviated LPS-induced inflammation through suppressing PKM2-mediated pathways, and could be a potential candidate for the prevention of inflammatory diseases.
Keywords: isoflavone, glycolysis, Warburg effect, PKM2, JAK/STATs, NF-κB
Introduction
Inflammation is a type of defensive response of the body against various antigen, stimuli, or tissue damage. After the pathogen invasion, lots of immune cells infiltrate into the infected host cells or injured endothelial cells, resulting in the moderate inflammatory response, which can promote the host to remove harmful stimuli and drive the body to initiate defensive healing.1 Among various immune cells, macrophages play an important role in the initiation and regulation of host defense (including inflammation and autoimmune response).2 The macrophages activated by varying stimuli not only repair damaged tissues via secreting a variety of inflammatory mediators (including cytokines, chemokines, and protease), but also remove infection factors and dead cells.3 However, excessive inflammatory reaction induced by over-activated macrophages can also cause various tissue damage, which happens in the pathological progress of acute inflammation such as sepsis, acute pneumonia, and pancreatitis.4,5
Emerging evidence has found that when exposed to varying stimuli, the activated macrophages could rapidly switch glucose metabolism to aerobic glycolysis, a phenomenon known as the Warburg effect, to rapidly provide ATP and metabolic intermediates for supporting immune cell proliferation and function.6,7 Furthermore, this metabolic alteration in macrophages inevitably alters cellular energy metabolism and produces mass reactive oxygen species (ROS). The excessive ROS influences the outcome of the inflammatory immune cell response and in turn aggravates abnormal metabolic events.8 Previous studies found that the activation of the pyruvate kinase M2 isoform (PKM2) signaling cascades is involved in the aerobic glycolysis of macrophages, resulting in the increased glucose uptake and lactate production, but the decreased oxygen consumption,9,10 which are closely related to the development and prognosis of inflammatory response. Therefore, PKM2 has been recently characterized as the vital enzyme of the glycolysis pathway and considered as a promising target for sepsis and acute lung damage.11,12
Recently, numerous natural products with novel structure and distinct biological activities have been discovered from the plant and animal kingdoms. These therapeutic agents provided many valuable leading compounds for new drug development, and some of them have even been in the stage of product development.13,14 To develop more effective and less toxic candidates, molecular target docking and high-throughput fluorescence-based assay for PKM2 inhibitor screening were employed in our previous study. Among 6,456 compounds derived from the Natural Product Library (Aladdin, China), Iridin (IRD) was one of the Top 10 compounds which showed the best inhibitory activities and highest affinities (data shown in Supplement Figure S1).
As well known, IRD is a main isoflavone derived from the root of the plant Belamcanda chinensis (L.) Redouté. It exists in various pharmacological activities, such as anti-inflammatory and antioxidant, cardioprotective, antitumor, hypoglycemic, and hypolipemic effects.15–17 Since isoflavones are phytoestrogens with potent estrogenic activity, IRD have been proved to be implicated in protecting against proliferative and metastatic progression of prostate cancer, breast cancer, and glioblastoma.18–20 Moreover, compared with other derivatives, IRD and its aglycone irigenin presented the strongest inhibitory effects on nitric oxide (NO) production in lipopolysaccharide (LPS)-treated RAW 264.7 macrophage cells.17 However, the underlying mechanism of IRD on acute and chronic inflammation remains unknown. In this study, we reported the novel role of IRD in LPS-induced inflammation and its underlying molecular mechanism specifically on aerobic glycolysis in murine macrophage cells.
Materials and Methods
Chemical and Reagents
IRD (purity, HPLC≥99%) was obtained from Yuanye biotechnology Co., Ltd. (Shanghai, China), and was endotoxin-free. Dexamethasone (Dex) sodium phosphate was purchased from Aladdin-e Co., Ltd. (Shanghai, China). Other chemical reagents were obtained from Huadong chemical reagent Co., Ltd (Hangzhou, China).
Cell Viability Assay
RAW264.7 cells (purchased commercially from Cell Bank/Stem Cell Bank, Chinese Academy of Sciences, Shanghai) were inoculated into a 96-well plate at 1×104/well. Each group had five duplicated wells. These cells were incubated with different concentrations of IRD (0, 6.25, 12.5, 25, 50, 100, 150, and 200 μM) for 24 hours. Then, the cell culture medium was changed and the cells were washed once with PBS. Then, the complete medium was used to dilute CCK-8. One hundred microliters of CCK-8 diluted with the complete medium (in the proportion of 1:10) was added in each well for incubation at 37°C for 2 hours. The absorbance at A450 nm wavelength was measured by a microplate reader (Bio-rad, USA).
Glucose Uptake Assay
The cells were inoculated into the 24-well plate at 1×105/well, and pre-treated with IRD for 2 hours and then with LPS for 16 hours. Each group had five duplicated wells. After being cultured with sugar-free medium for 1 hour, the cells were treated with 2-NBDG Fluorescent Dyes (the final concentration was 20 μM) in the dark at 37°C for 10 minutes. The fluorescence value was detected by a fluorescent microplate reader (excitation: 465 nm; emission: 540 nm). Cellular OCR (indicative of oxidative phosphorylation) and ECAR (indicative of glycolysis) were then measured by Seahorse Bioscience Analyzer (Seahorse Bioscience Inc., USA) according to the operation instructions.
Detection of Lactate and Cytokines in the Cell Culture Medium
The cells were inoculated into the 24-well plate at 1×105/well, and pre-treated with IRD (or activator, agonist) for 2 hours (ie, total exposed time was 18 hours) and then with LPS for 16 hours. Each group had five duplicated wells. The cell culture medium was collected and then the levels of lactate and pro-inflammatory mediators (TNF-α, IL-1β, and MCP-1) in each group were measured according to the operation instructions of the ELISA kit (Keshun Bio., China), while the levels of NO in culture medium were detected by Griess assay, as previously reported.21,22
Detection of Phagocytic Activity of RAW264.7 Macrophages
The effects of IRD on phagocytic activity of RAW 264.7 macrophages were measured by neutral red uptake assay.23 Briefly, the cells were inoculated into a 96-well plate (each well had 1×105 cells) and cultured overnight for 12 hours. Each group had five duplicated wells. The cells were pre-treated with IRD (or activator, agonist) for 2 hours and then exposed with LPS for 16 hours. After LPS stimulus, the supernatant was removed and 100 μL of 0.075% neutral red solution was added to each well and incubated for an additional 4 hours. After washing three times in PBS solution, 100 μL of lysis buffer [0.01% glacial acetic acid: ethanol=1:1, (v/v)] was added to the each well. After 2 hours of incubation at 4°C, the absorbance of each well was measured by using a microplate reader at 540 nm.
Detection of Intracellular ROS and NO
About 1×105 cells were inoculated into a 24-well plate and cultured overnight for 12 hours. After pre-treatment with IRD (or activator, agonist) for 2 hours and exposure with LPS for 16 hours, the cells were treated with DCFH2-DA (10 μM) for 30 minutes and then the intracellular ROS production was measured by flow cytometry. Similarly, the release of NO was detected by flow cytometry after the cells were incubated with DAF-FM (5 μM) for 1 hour.
Western Blot Assay
The RAW264.7 cells were inoculated into the dishes at 2×107/well, and pre-treated with IRD (or activator, agonist) for 2 hours and then with LPS for 16 hours. Then the cells were collected and added with the protein lysis solution and the protease inhibitor to obtain the lysis. After quality control, 20 μg of the lysis product was separated by SDS-PAGE electrophoresis and then transferred to PVDF membrane with locking buffer for 2 hours. The membrane was incubated with the first antibody at 4°C for 12 hours, and incubated with the second antibody for 1 hour. After washing with PBS 3-times at 3 minutes/time, the gray value of each stripe on the membrane was obtained by the imaging system (CLINX, Shanghai, China).
RNA Sequencing and Bioinformatic Analysis
Total RNAs were extracted from 5×107 cells of different groups by using a RNAiso kit (Takara, Japan). The concentration of RNA was determined by NanoDrop 1000 and Agilent Bioanalyzer before RNA sequencing. Libraries were generated with standard mRNA stranded protocols from Illumina and sequenced on Illumina HiSeq2500 by LC-bio Co. Ltd. (China). The expressed data were normalized using the median normalization. Only candidates with P-values lower than 0.05 and Fold Change filtering (Fold Change>1.5) were considered as the differentially expressed genes (DEGs). The functional annotation of DEGs was performed by online service DAVID (https://david.ncifcrf.gov/).
Target Prediction and Interaction Between IRD and PKM2
The online service tools PharmMapper and SwissDock were employed to predict the potential target of IRD as our previous study.24 Molecular docking analysis of IRD with a target protein was tested by Autodock Tools (version 1.5.6). The active sites of PKM2 were determined as: center_x=6.527, center_y=1.534, center_z=10.683; size_x=20, size_y=20, size_z=20. In order to increase the accuracy of the calculation, the parameter exhaustiveness was set as 20 and others were default values. Finally, the highest scoring conformation was selected and analyzed with the tool free Maestro.
Three serial concentrations of IRD sample and a negative control (saline buffer) were respectively added to the biotin-labeled and desalted PKM2 solutions (50 µM). The interaction between IRD and PKM2 was performed at room temperature following the ForteBio manufacturer’s protocol as per a previous report with minor revision.25
Acute Lung Inflammation Mouse Model and Drug Treatment
Male ICR (24±2 g body weights) were raised in Zhejiang animal Centre. The experiments were carried out according to the feeding standard (reared under specific pathogen-free conditions and provided with a standard rodent diet and drinking water) and ethical requirements (approval ethical number: 2019R04003) of Zhejiang animal Centre. Chinese guidelines for the welfare of the laboratory animals (GB/T 35823–2018) were followed in this study.
The mice were divided into six groups: 1) control group; 2) model group, the mice were intranasally exposed with 20 mg/kg of LPS; 3) Dexamethasone (Dex)-treated group, the LPS- challenged mice were treated with 10 mg/kg of Dex; 4–6) IRD-treated groups, the LPS- challenged mice were orally administrated with 20, 40, and 80 mg/kg of IRD once a day for 5 days, respectively. The doses of IRD used in this study were based on a previous study26 and our pre-experiment. Each group had eight mice. The drugs were given once daily for 3 days. Seven days after LPS exposure, all mice were asphyxiated by CO2 and then the blood and lung were collected. The number of total cells and neutrophils (Neu) in bronchoalveolar lavage fluid (BALF) was counted by automatic blood cell analyzer (HITACHI, Japan).
Detection of Serum Inflammatory Regulators
The serum levels of inflammatory regulators (IL-10, TNF-α, and iNOS) were detected by ELISA assay according to the manual of kits.
Histopathology Analysis
A quarter of the lung tissue was fixed in 4% paraformaldehyde, cut into slices, and prepared by H&E staining for histopathologic investigation. The histological changes were scored by using the grading system of 0 (no), 1 (slight), 2 (moderate) and 3 (severe) as a previous study reported.27 The serious degree of each slice was individually assessed by four sections (neutrophil infiltration, alveolar expansion, interstitial hyperplasia, and alveolar congestion). The sum of those four indexes was used for evaluating the degree of pathological lesions in lungs.
RNA Extraction and Real-Time PCR Analysis
Total RNAs of macrophages, which were isolated from the BALF by using lymphocyte separation medium,28 were extracted by using the Trizol reagent. PCR was conducted by using SYBRGreen method on the quantitative PCR system (Eppendorf, German). The PCR program was as follows: holding 95°C for 7 minutes; 95°C for 30 seconds, 60°C for 35 seconds, and 72°C for 35 seconds. The mRNA expression was quantified using the comparative cross threshold (CT) method. The PCR reactions were performed in duplicate and each experiment was repeated three times. The Primer sequences used in this study are shown in Table 1.
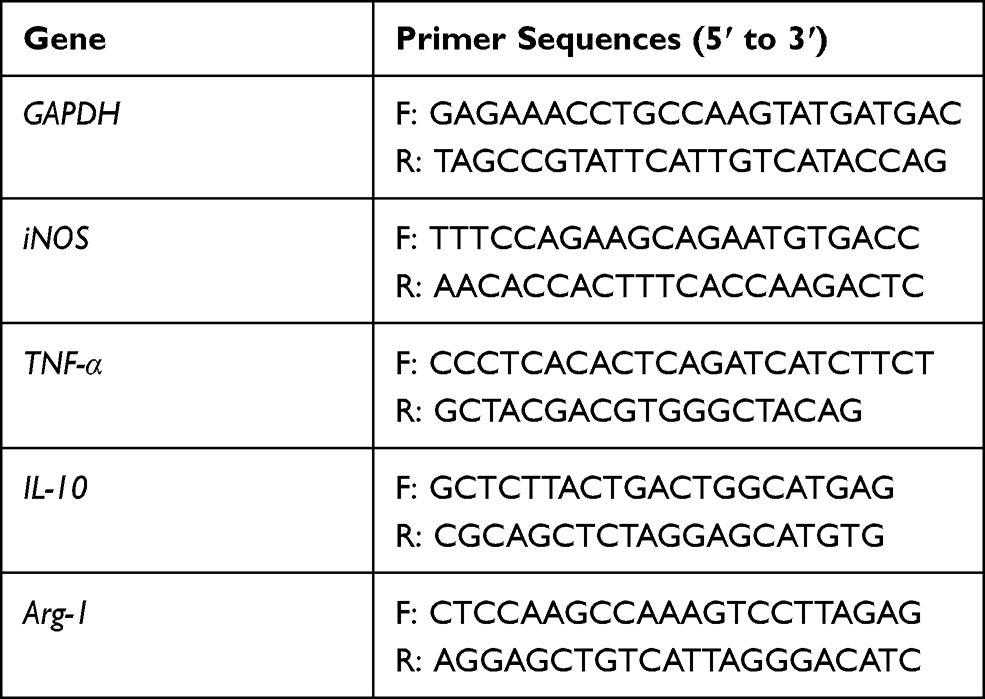 |
Table 1 Sequences of Primers Used for the RT-PCR Assay |
Statistical Analysis
The data were analyzed by SPSS software (version 22.0) and showed as means±SD.
The multiple group analysis was performed using post-hoc test and the statisticalsignificance between two groups was set up as P<0.05.
Results
Cytotoxicity of IRD on RAW264.7 Cells
To evaluate the cytotoxicity of IRD on cell proliferation, RAW264.7 cells were respectively assayed by CCK-8 after being exposed with different concentrations of IRD (0, 6.25, 12.5, 25, 50, 100, 150, and 200 μM). The results in Figure 1A showthat when treated with a concentration less than 100 μM, IRD did not affect the proliferation of RAW264.7 cells. Also, there were no significant differences on the cell viabilities among the control group and LPS+IRD-treated groups (P>0.05). Given the high price and the fact that IC50 of IRD on LPS-induced NO release was 24.8 μM, the concentrations of 12.5, 25, and 50 μM were selected for following pharmacological investigation in this study.
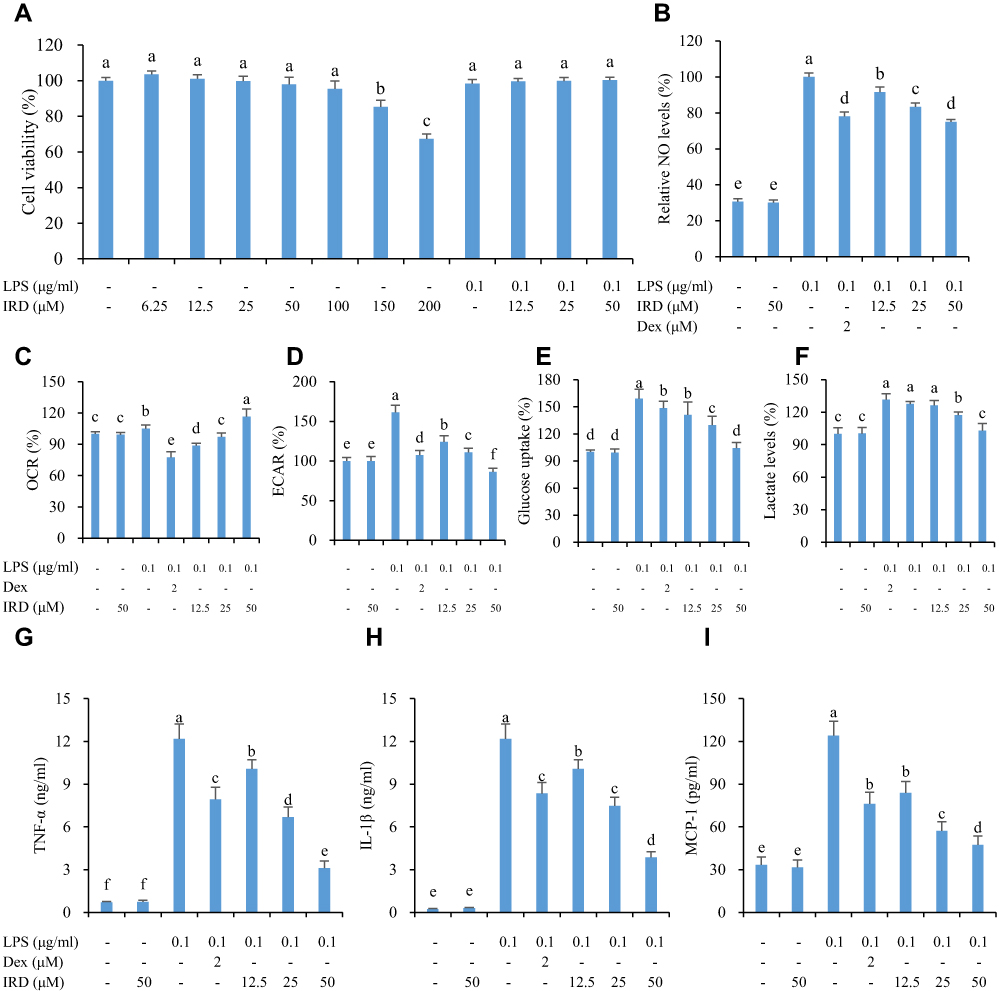 |
Figure 1 Inhibition of IRD on LPS-induced aerobic glycolysis and inflammation in RAW264.7 cells. (A) The effects of IRD and LPS on macrophage proliferation were analyzed by CCK-8 assay. (B) IRD reduced the levels of NO in culture medium detected by Griess assay. (C) IRD increased the OCR levels (indicator of oxidative phosphorylation) but (D) decreased ECAR levels (indicator of glycolysis) in LPS-treated macrophages, which were simultaneously monitored at 24 hours by using the Seahorse Analyzer. (E) IRD inhibited the aerobic glycolysis via suppressing glucose uptake and (F) lactate levels (indicators of glycolysis) in LPS-challenged RAW264.7 cells. (G) IRD inhibited the productions of inflammatory indicators TNF-α, (H) IL-1β, and (I) MCP-1 induced by LPS. Dex, dexamethasone. All data were shown as mean±SD (n=5). Different letters indicated significant differences (P<0.05) by Tukey’s test. |
IRD Reduced Warburg Effects of LPS-Activated Macrophages
As shown in Figure 1C-F, after LPS stimulation, ECAR, glucose uptake ability, and lactate secretion (three classical glycolytic indexes) of RAW264.7 cells were significantly elevated but OCR (additional glycolytic index) were decreased when compared with the control cells (P<0.05), indicating that LPS-activated macrophages exhibited severe glycolytic process (also called the Warburg effect). Treatment with IRD at concentrations of 12.5~50 μM gradually decreased the levels of ECAR, glucose uptake, and lactate, but increased the levels of OCR in the LPS-exposed macrophages, which presented a significant concentration–effect relationship. Remarkably, there were no differences of ECAR level, OCR level, glucose uptake ability, and lactate secretion between the IRD-treated group (without LPS challenge) and control group, indicating that IRD did not affect the glucose metabolism of normal macrophages. However, treatment with Dex, set as the positive control in the study, did not affect the levels of those two glycolytic indexes (Supplement Figure S1). It indicated that Dex did not reduce the Warburg effect induced by LPS.
IRD Inhibited LPS-Induced Release of Inflammation-Related Mediators
Compared with the control group, the levels of TNF-α, IL-1β, MCP-1, and NO in the cell culture medium of the LPS-challenged group were significantly increased (P<0.05). Both IRD (Figure 1B, G-I) and Dex (Supplement Figure S1) significantly decreased the levels of those four inflammation-related mediators (P<0.05). These results indicated that both IRD and Dex reduced the secretions of TNF-α, IL-1β, MCP-1, and NO in LPS-activated macrophages.
IRD Inhibited LPS-Induced ROS Production and NO Release
The results in Figure 2 showthat the fluorescence intensity (which reflected the levels of intracellular ROS) of the LPS-stimulated group was significantly increased (P<0.05), while low expression was observed in the control group. It indicated that LPS caused the accumulation of intracellular ROS. However, the fluorescence intensities as well as the levels of ROS and NO were significantly decreased in the IRD-treated group in a concentration-dependent manner. Thus, it demonstrated that IRD effectively eliminated LPS-induced accumulation of endogenous ROS and NO in RAW264.7 cells.
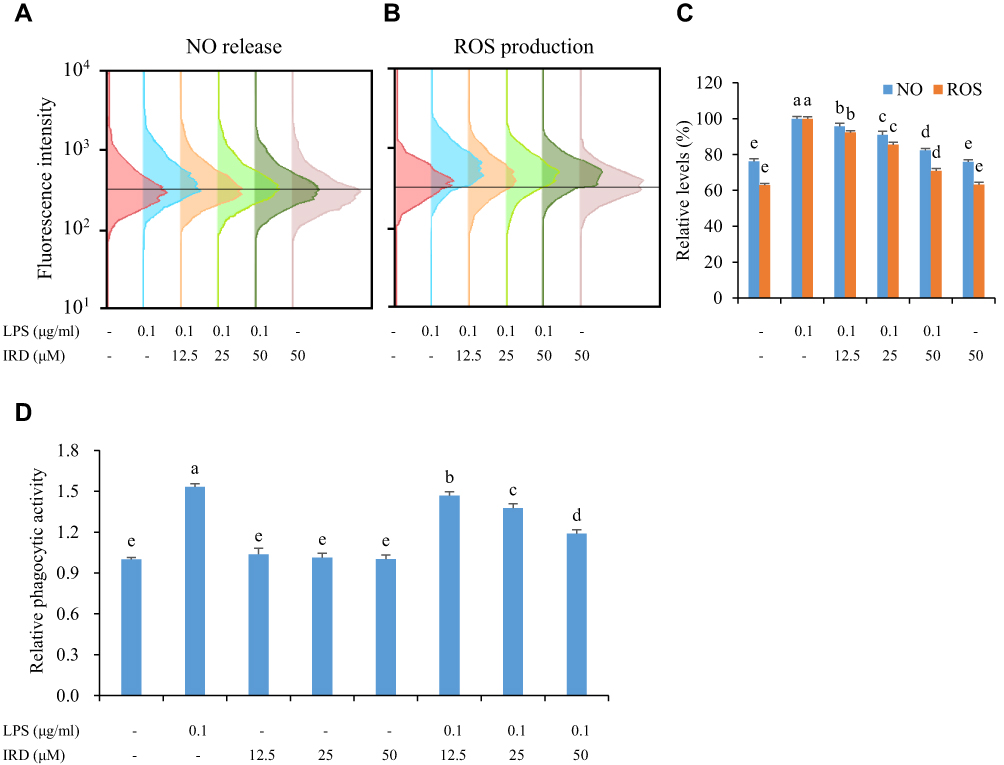 |
Figure 2 IRD inhibited the release of ROS and NO as well as phagocytic activity of macrophages stimulated by LPS. (A) The cells were treated with DAF-FM probe and the release of NO in each group was detected by flow cytometry assay. (B) The cells were treated with DCFH-DA probe and the production of ROS in each group was detected by flow cytometry assay. (C) The relative levels of NO and ROS in each group. (D) IRD inhibited excessive phagocytic activity of RAW264.7 macrophages induced by LPS. All data were shown as mean±SD (n=5). Different letters indicated significant differences (P<0.05) by Tukey’s test. |
IRD Ameliorated LPS-Induced Acute Lung Inflammation in Mice
Seventy-two hours after LPS challenge, the severe neutrophil infiltration and edema were seen in the lung tissues of the mice, resulting in the highest lung indexes and pathologic scores among the groups (Figure 3). In addition, the number of total cells and neutrophils in the BALF of LPS-treated mice was also significantly increased compared with the normal mice (P<0.05). However, treatment with IRD obviously inhibited the accumulation of neutrophils and reduced the levels of lung edema. Thus, the number of total cells and neutrophils as well as the lung indexes and pathologic scores in the BALF of IRD-treated groups were also significantly decreased in a dose-dependent manner compared with those in the model group (P<0.05), indicating the protective effects of IRD on LPS-induced acute lung injury and inflammatory response.
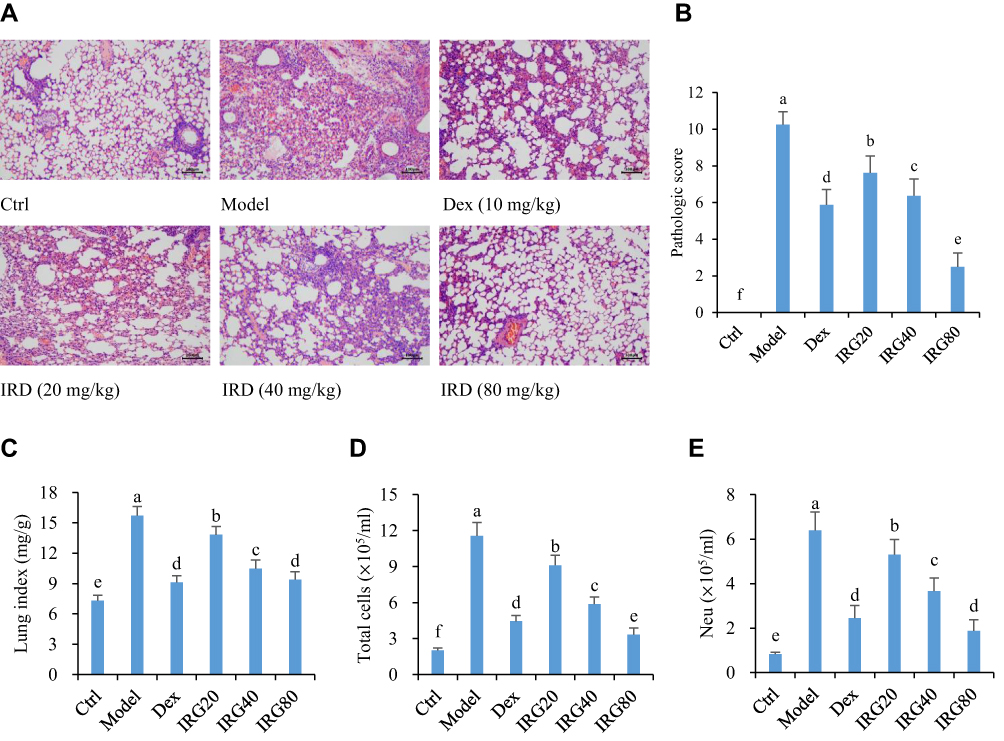 |
Figure 3 IRD ameliorated LPS-induced lung injury. (A) The histological sections of lung were dyed by HE staining (Scale bar, 100 μm). In the normal group, the pulmonary alveoli presented continuous structure and intact wall without any exudation, while the severe neutrophil infiltration and edema as well as the damaged alveolar structures could be found in the lung tissues of the model group. The levels of neutrophil infiltration and edema were significantly reduced in the Dex-treated group and IRD-treated groups. (B) IRD reduced the pathologic scores of LPS-exposed mouse lung in a dose-dependent manner. (C) IRD decreased the lung indexes as well as (D) the number of total cells and (E) neutrophils (Neu) in bronchoalveolar lavage fluid (BALF). All data were shown as mean±SD (n=8). Different letters indicated significant differences (P<0.05) by Tukey’s test. |
IRD Inhibited LPS-Induced Macrophage Polarization and Inflammation in Mice
As shown in Figure 4, the levels of iNOS and TNF-α in the serum of LPS-treated mice were significantly increased compared with those in the normal mice (P<0.05), but the serum IL-10 levels were decreased (P<0.05). Similarly, the mRNA expressions of the M1 polarization markers iNOS and TNF-α in the macrophages of LPS-exposed mice were significantly increased, but the M2 polarization markers IL-10 and Arg-1 were significantly decreased compared with those in the normal mice (P<0.05). However, treatment with IRD significantly reduced both the levels of iNOS and TNF-α in the serum and the expressions of iNOS and TNF-α mRNA in the macrophages of LPS-challenged mice, but increased the serum levels of IL-10 as well as the IL-10 and Arg-1 mRNA expressions in the macrophages. These results indicated that IRD reprogramed macrophages from inflammatory M1 polarization phenotype to an anti-inflammatory M2 phenotype and thereby inhibited the production of the proinflammatory cytokines.
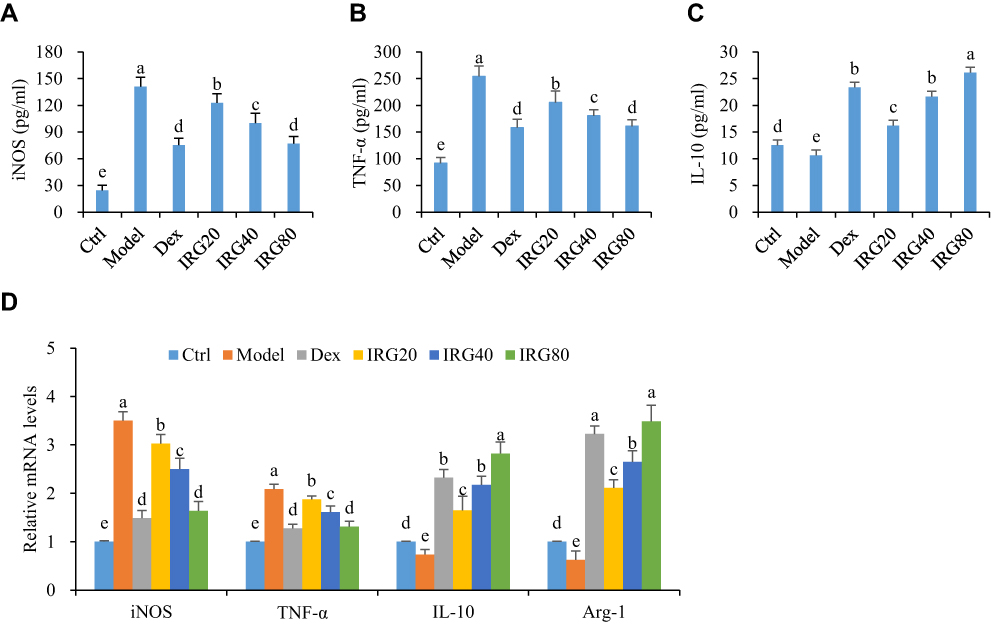 |
Figure 4 IRD inhibited LPS-induced macrophage polarization and inflammation in mice. (A) IRD decreased the levels of iNOS and (B) TNF-α, but (C) increased IL-10 in the BALF of LPS-exposed mice. (D) IRD promoted macrophage polarization towards M2 phenotype through reducing mRNA expressions of M1 phenotype markers (iNOS and TNF-α) and increasing M2 markers (IL-10 and Arg-1) in the lung tissues of LPS-exposed mice. All data were shown as mean±SD (n=8). Different letters indicated significant differences (P<0.05) by Tukey’s test. |
PKM2 Was a Potential Target of IRD
After heatmap analysis (Figure 5A), 97 DEGs were found, including 65 upregulated and 32 downregulated DEGs in the IRD-treated group. By the KEGG and interactive relationship analysis (Figure 5B), those DEGs were mainly enriched in the AGE-RAGE pathway, glycolysis/gluconeogenesis, cytokine-cytokine receptor interaction, Th17 cell differentiation, TLR signaling pathway and JAK-STAT signaling pathway. More importantly, glycolysis as one of significantly enriched term contained 17 DEG in the network, indicating that glycolysis contributed to the inhibitory effects of IRD on LPS-induced inflammatory response.
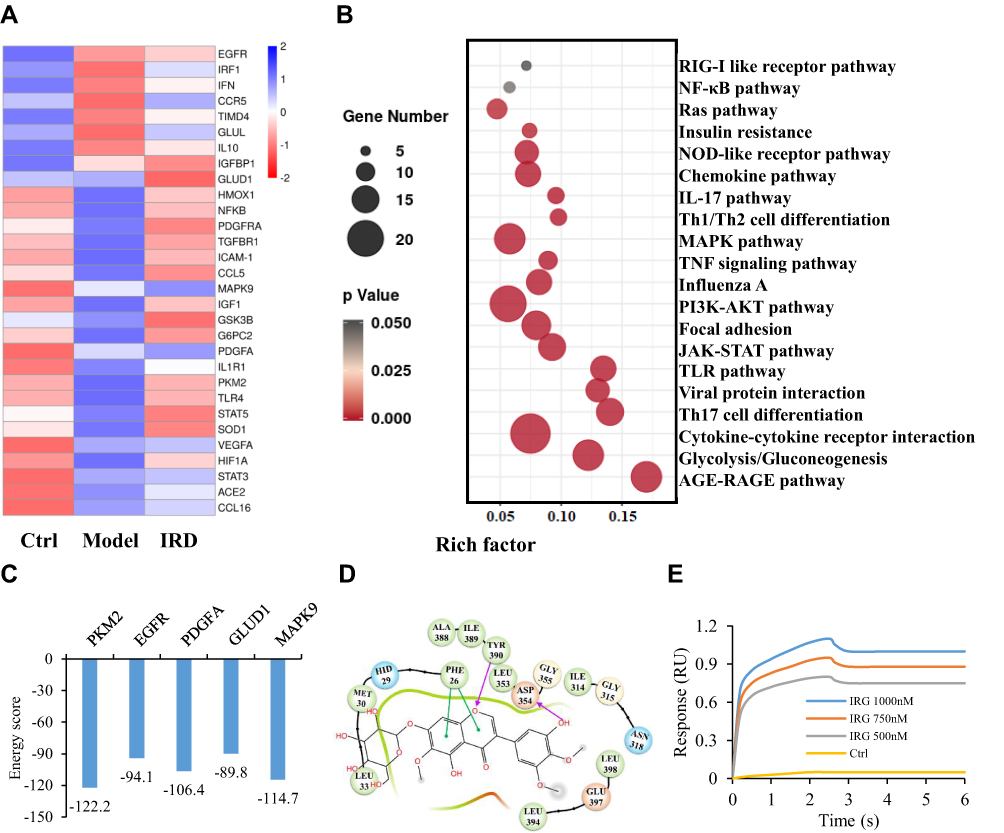 |
Figure 5 PKM2 was one of the main targets of IRD. (A) The heatmap analysis of the top 30 differentially expressed mRNAs (DEM). Statistical significance was defined as adjusted P-value<0.05 and biological significance was defined as FC>2.0 according to |log2FC|>1.0. (B) The high transcripts of DEM involved into top-20 hits of different metabolism pathways. (C) The top five potential targets of IRD predicted by reverse molecular docking technology. (D) The structure of PKM2 domain bound to IRD with the active sites. (E) The molecule interactions between PKM2 and IRD analyzed by surface plasmon resonance technology. |
Testing on web docking tools PharmMapper and SwissDock revealed that PKM2 (PDB ID: 5X1V) was one of the potential targets with the lowest energy scores among the proteins from the RCSB Protein Data Bank (Figure 5C). The binding affinity of IRD to the active pocket of PKM2 was -6.9 kcal/mol. The docking mode is shown in Figure 5D. It could be found that the IRD were closely bound in the active pocket of PKM2. Furthermore, IRD could form hydrogen bonding interaction with PKM2 at the TYP 390 and ASP 354 sites, and have Pi–Pi interaction at PHE 26. These interactions were the most important forces between PKM2 and IRD, leading to the formation of stable complexes. To further verify the interaction between IRD and PKM2, the binding affinity was determined by biomolecular assays. As shown in Figure 5E, compared with the control, affinity curves rose more steeply with the increase of IRD concentration until it reached an inflection point. It demonstrated that IRD was directly bounded to the PKM2 protein.
IRD Downregulated the Expressions of Glycolysis-Related Proteins
The results in Figures 6, 7 and Supplement Figure S2 showed that the expressions of PKM2 and its downstream (p-JAK1, p-STAT1, p-STAT3, p-p65, iNOS, and COX2) proteins in the control group were almost undetectable or very weak, whereas expressions of those proteins were significantly increased in LPS-treated group. IRD could significantly reduce the expressions of PKM2 and its downstream (p-JAK1, p-STAT1, p-STAT3, p-p65, iNOS, and COX2) proteins in both LPS-exposed RAW264.7 cells and lung tissues of LPS-treated mice, indicating that IRD could bind to PKM2 and thereby downregulate the JAK/STAT and NF-κB pathways. However, those decreased protein expressions induced by IRD in LPS-exposed macrophages could be reversed by PKM2 activator DASA-58 or antioxidant NAC, but partly be reversed by NF-κB activator CUT129 and JAK1 activator RO8191.
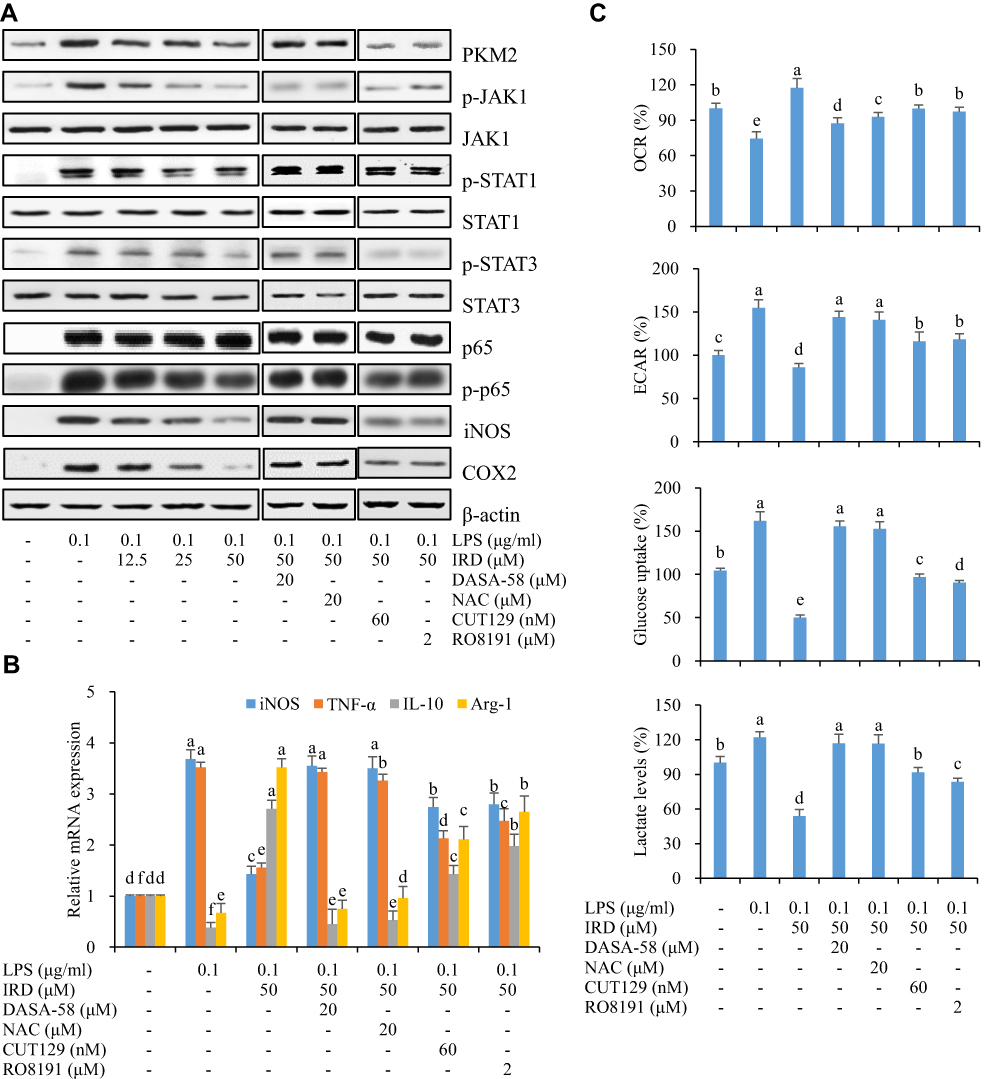 |
Figure 6 IRD reduced LPS-induced aerobic glycolysis in RAW264.7 cells via PKM2-mediated JAK/STAT and NF-κB pathways. (A) IRD downregulated the expressions of PKM2 and its downstream (p-JAK1, p-STAT1, p-STA3, p-p65, iNOS, and COX2), which were reversed by PKM2 agonist DASA-58 and antioxidant N-acetyl-L-cysteine (NAC), but partly reversed by NF-κB activator CUT129 and JAK1 activator RO8191. The relative expressions of those proteins are shown in Supplement Figure S2. (B) DASA-58 and NAC reversed M2 macrophage phenotype induced by IRD while CUT129 and RO8191 partly did it. (C) DASA-58 and NAC weakened the inhibition of IRD on the aerobic glycolysis while CUT129 and RO8191 partly reduced it. All data were shown as mean±SD (n=5). Different letters indicated significant differences (P<0.05) by Tukey’s test. |
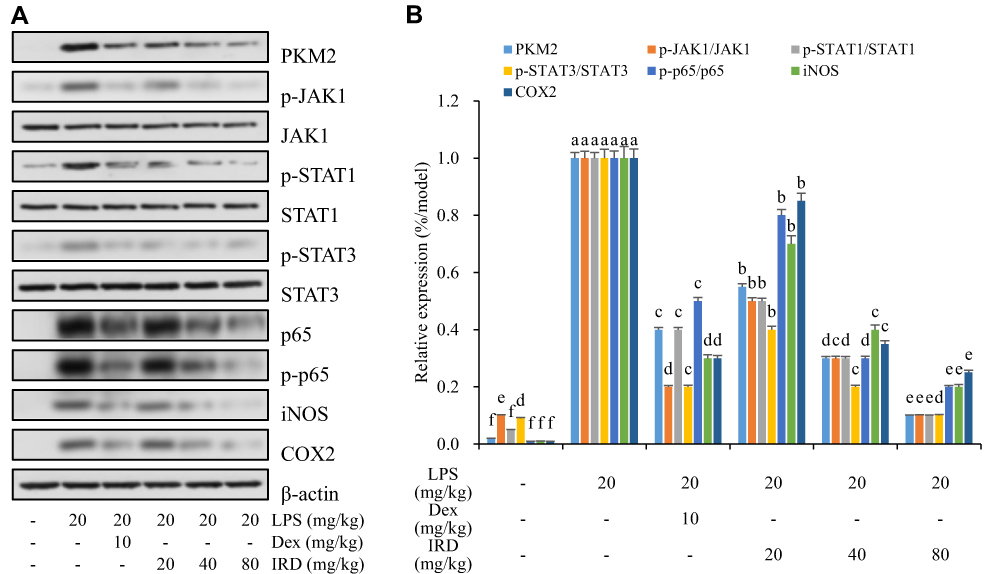 |
Figure 7 IRD downregulated the protein expressions of PKM2-mediated JAK/STAT and NF-κB pathways in lung tissues of LPS-treated mice. (A) Representative bands and (B) quantitative results of the protein expressions of PKM2 and its downstream (p-JAK1, p-STAT1, p-STA3, p-p65, iNOS, and COX2) in mouse lung tissues. All data are shown as mean±SD (n=5). Different letters indicated significant differences (P<0.05) by Tukey’s test. |
Similarly, after DASA-58 or NAC treatment, 2-NDBG uptake abilities of RAW264.7 cells were enhanced, and the contents of lactic acid in cell culture medium also were increased compared with those in IRD-treated cells. Also, both DASA-58 and NAC revered M2 macrophage phenotype induced by IRD. However, co-treatment with IRD and CUT129 (or RO8191) in LPS-exposed cells showed weaker effects on the above-mentioned indexes. The findings presented that both DASA-58 and NAC significantly weakened the inhibition of IRD on excessive glycolysis of macrophages while CUT129 and RO8191 had partial reversal effects, indicating that IRD prevented against LPS-induced inflammatory responses in macrophages via suppressing PKM2-mediated JAK/STAT and NF-κB pathways.
Discussion
IRD is the strongest anti-inflammatory component derived from some species of the family Iridaceae.17 However, the recent research mainly focused on the anti-tumor activities and structure-activity relationship between IRD and its derivatives.15,16 The underlying inflammatory mechanism of IRD was still unclear. In this study, we found that IRD not only significantly reduced glycolysis and inflammatory response of macrophages activated by LPS, but also inhibited the expressions of PKM2, p-JAK1, p-STAT1, p-STAT3, p-p65, iNOS, and COX2.
LPS as the major active component of endotoxins is a well-known TLR4 activator, which can activate macrophages to release a variety of inflammatory substances (including cytokines) via the activating NF-κB pathway leading to inflammation. The activated NF-κB accelerated accumulation of intracellular ROS and the transcription of downstream iNOS and COX2, which in turn promoted the activation of NF-κB.29,30 On the other hand, LPS impacted the phenotype of macrophage polarization which was implicated in the development and progression of the airway inflammation.31 During infectious diseases, macrophages act as the highly plastic and pluripotent immune cells, which can be divided into M1 type classically activated macrophages and M2 type alternatively activated macrophages. M1 macrophages secrete pro-inflammatory cytokines and chemokines to present antigens, while M2 macrophages secrete inhibitory cytokines, such as IL-10 and TGF-β1, to down-regulate the immune response and play an anti-inflammatory role in the body.32,33 Several preclinical investigations have indeed demonstrated the importance of macrophages in the pathogenesis of acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) and the effectiveness of macrophage phenotype modulation, thereby supporting its scientific rationale.34–36 In this study, IRD reduced the mRNA expressions of M1 markers iNOS and TNF-α but elevated the expressions of IL-10 and Arg-1 in the macrophages of LPS-treated mice. Correspondingly, after IRD pre-treatment, the levels of iNOS and TNF-α released by LPS-exposed cells were significantly decreased while IL-10 were increased. These results demonstrated that IRD promoted the M2 macrophage phenotype, resulting in amelioration of LPS-induced lung inflammation.
After LPS challenge, a series of inflammatory cells such as macrophages was persistently activated and recruited to the exposed position to remove the stimulus. However, the excessive activation of these cells ultimately triggered uncontrolled systemic inflammation and sepsis. To satisfy energy demands of cell proliferation and biomacromolecule synthesis, the cells had to alter the cellular metabolic pathway that converts glucose into pyruvate. This process required upregulated PKM2 expression that catalyzes the final and rate-limiting reaction of the glycolytic pathway.7,37,38 Recently, increasing evidence indicated that besides tumor metabolism and growth, PKM2 took great part in the progress of various acute and chronic inflammation.39–41 Aberrant activation of PKM2 promotes its dimerization and subsequent nuclear translocation, which in turn acts as a protein kinase and phosphorylates the various transcription factors, including STAT3, MAPK, NF-κB, and NLRP3, thus boosting the release of pro-inflammatory cytokines (such as MCP-1 and TNF-α). Some clinical and experimental researches have shown that the levels of PKM2 positively correlates with the severity of inflammation and macrophage activation in vitro and in vivo.42–44 Treatment with PKM2 inhibitor shikonin or siRNA suppressed macrophage activation, and consequently reduced the production of IL-1β and IL-18.9,44,45 Therefore, blocking PKM2, or modulating aerobic glycolysis, represented a promising approach for preventing inflammation-related diseases. In this study, the results showed that 6.25–50 μM of IRD can effectively reduce the levels of glucose uptake and lactate production, and inhibit the production of intracellular ROS and the release of MCP-1 and TNF-α in a dose-dependent manner. Furthermore, IRD binds to the active sites of PKM2, thereby downregulating PKM2 and its downstream (p-JAK1, p-STAT1, p-STAT3, p-p65, iNOS, and COX2) in LPS-treated RAW264.7 cells. After the PKM2 activator DASA-58 was used to enhance the glycolysis of macrophages or the antioxidant NAC to remove the production of endogenous ROS in cells, the inhibition of IRD on the glycolysis, and inflammation could be significantly weakened. However, NF-κB activator CUT129 and JAK1 activator RO8191 partly reversed those effects. Therefore, IRD inhibited PKM2 expression and thereby downregulated its downstream JAK/STATs and NF-κB signaling pathways.
Conclusion
The present study demonstrated that IRD reduced glycolysis and the release of inflammatory cytokines in LPS-activated macrophages via inhibiting PKM2-mediated JAK1/STAT NF-κB and pathways. Thus, IRD could be a potential candidate for prevention and treatment of inflammatory diseases, and the findings suggested an innovative pharmacotherapy for acute inflammation via regulating Warburg effect of immune cells.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Science Foundation of China (No. 81603368 and 81673583), Zhejiang Provincial Science and Technology Project (No. 2015C33164), Zhejiang Provincial Medicine and Technology Project (No. 2020KY527) and Zhejiang Innovation Discipline Project of Laboratory Animal Genetic Engineering (No. 201604).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Pelka K, De Nardo D. Emerging concepts in innate immunity. Methods Mol Biol. 2018;1714:1–18.
2. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci. 2014;10:520–529. doi:10.7150/ijbs.8879
3. Patel U, Rajasingh S, Samanta S, Cao T, Dawn B, Rajasingh J. Macrophage polarization in response to epigenetic modifiers during infection and inflammation. Drug Discov Today. 2017;22:186–193. doi:10.1016/j.drudis.2016.08.006
4. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4–5):383–396. doi:10.1016/j.imbio.2017.11.001
5. Habtezion A, Gukovskaya AS, Pandol SJ. Acute pancreatitis: a multifaceted set of organelle and cellular interactions. Gastroenterology. 2019;156(7):1941–1950. doi:10.1053/j.gastro.2018.11.082
6. Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. 2019;19(5):291–304. doi:10.1038/s41577-019-0124-9
7. Saha S, Shalova IN, Biswas SK. Metabolic regulation of macrophage phenotype and function. Immunol Rev. 2017;280(1):102–111. doi:10.1111/imr.12603
8. Franchina DG, Dostert C, Brenner D. Reactive oxygen species: involvement in T cell signaling and metabolism. Trends Immunol. 2018;39(6):489–502. doi:10.1016/j.it.2018.01.005
9. Xie M, Yu Y, Kang R, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280. doi:10.1038/ncomms13280
10. Yang L, Xie M, Yang M, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5:4436. doi:10.1038/ncomms5436
11. Mills EL, O’Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol. 2016;46(1):13–21. doi:10.1002/eji.201445427
12. Palsson-McDermott EM, O’Neill LA. The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays. 2013;35(11):965–973. doi:10.1002/bies.201300084
13. Jia Z, Anandh Babu PV, Chen W, Sun X. Natural products targeting on oxidative stress and inflammation: mechanisms, therapies, and safety assessment. Oxid Med Cell Longev. 2018;2018:6576093. doi:10.1155/2018/6576093
14. Tabolacci C, Forni C, Jadeja RN, Facchiano F. Natural compounds against cancer, inflammation, and oxidative stress. Biomed Res Int. 2019;2019:9495628. doi:10.1155/2019/9495628
15. Zhang L, Wei K, Xu J, et al. Belamcanda chinensis (L.) DC-An ethnopharmacological, phytochemical and pharmacological review. J Ethnopharmacol. 2016;186:1–13. doi:10.1016/j.jep.2016.03.046
16. Liu M, Yang S, Jin L, Hu D, Wu Z, Yang S. Chemical constituents of the ethyl acetate extract of Belamcanda chinensis (L.) DC roots and their antitumor activities. Molecules. 2012;17(5):6156–6169. doi:10.3390/molecules17056156
17. Lee JW, Lee C, Jin Q, et al. Chemical constituents from Belamcanda chinensis and their inhibitory effects on nitric oxide production in RAW 264.7 macrophage cells. Arch Pharm Res. 2015;38(6):991–997. doi:10.1007/s12272-014-0529-8
18. Morrissey C, Bektic J, Spengler B, et al. Phytoestrogens derived from Belamcanda chinensis have an antiproliferative effect on prostate cancer cells in vitro. J Urol. 2004;172(6):2426–2633. doi:10.1097/01.ju.0000143537.86596.66
19. Sengupta R, Barone A, Marasa J, et al. Novel chemical library screen identifies naturally occurring plant products that specifically disrupt glioblastoma-endothelial cell interactions. Oncotarget. 2015;6(21):18282–18292. doi:10.18632/oncotarget.4957
20. Monthakantirat O, De-Eknamkul W, Umehara K, et al. Phenolic constituents of the rhizomes of the Thai medicinal plant Belamcanda chinensis with proliferative activity for two breast cancer cell lines. J Nat Prod. 2005;68(3):361–364. doi:10.1021/np040175c
21. Tang J, Diao P, Shu X, Li L, Xiong L. Quercetin and quercitrin attenuates the inflammatory response and oxidative stress in LPS-Induced RAW264.7 cells: in vitro assessment and a theoretical model. Biomed Res Int. 2019;2019:7039802. doi:10.1155/2019/7039802
22. Yeo J, Lee YM, Lee J, et al. Nitric Oxide-scavenging nanogel for treating rheumatoid arthritis. Nano Lett. 2019;19(10):6716–6724. doi:10.1021/acs.nanolett.9b00496
23. Lee JH, Ahn DU, Paik HD. In vitro immune-enhancing activity of ovotransferrin from egg white via MAPK signaling pathways in RAW 264.7 macrophages. Korean J Food Sci Anim Resour. 2018;38(6):1226–1236. doi:10.5851/kosfa.2018.e56
24. Yu WY, Li L, Wu F, et al. Moslea Herba flavonoids alleviated influenza A virus-induced pulmonary endothelial barrier disruption via suppressing NOX4/NF-κB/MLCK pathway. J Ethnopharmacol. 2020;253:112641. doi:10.1016/j.jep.2020.112641
25. Li R, Tian JZ, Wang MR, Zhu LN, Sun JS. EsGLUT4 and CHHBP are involved in the regulation of glucose homeostasis in the crustacean Eriocheir sinensis. Biol Open. 2017;6(9):1279–1289. doi:10.1242/bio.027532
26. Wang N, Geng C, Sun H, Wang X, Li F, Liu X. Hesperetin ameliorates lipopolysaccharide-induced acute lung injury in mice through regulating the TLR4-MyD88-NF-kappaB signaling pathway. Arch Pharm Res. 2019;42(12):1063–1070. doi:10.1007/s12272-019-01200-6
27. Tang SE, Wu SY, Chu SJ, et al. Pre-treatment with ten-minute carbon dioxide inhalation prevents lipopolysaccharide-induced lung injury in mice via down-regulation of toll-like receptor 4 expression. Int J Mol Sci. 2019;20(24):6293. doi:10.3390/ijms20246293
28. Shi Q, Zhao L, Xu C, Zhang L, Zhao H. High molecular weight hyaluronan suppresses macrophage M1 polarization and enhances IL-10 production in PM(2.5)-induced lung inflammation. Molecules. 2019;24(9):1766. doi:10.3390/molecules24091766
29. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103–115. doi:10.1038/cr.2010.178
30. Zhang J, Wang X, Vikash V, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. 2016;2016:4350965. doi:10.1155/2016/4350965
31. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol. 2019;106(2):345–358. doi:10.1002/JLB.3RU1018-378RR
32. Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci. 2018;19(6):1801. doi:10.3390/ijms19061801
33. Bosco MC. Macrophage polarization: reaching across the aisle? J Allergy Clin Immunol. 2019;143(4):1348–1350. doi:10.1016/j.jaci.2018.12.995
34. Song C, Li H, Li Y, et al. NETs promote ALI/ARDS inflammation by regulating alveolar macrophage polarization. Exp Cell Res. 2019;382(2):111486. doi:10.1016/j.yexcr.2019.06.031
35. Wang Y, Xu Y, Zhang P, et al. Smiglaside A ameliorates LPS-induced acute lung injury by modulating macrophage polarization via AMPK-PPARγ pathway. Biochem Pharmacol. 2018;156:385–395. doi:10.1016/j.bcp.2018.09.002
36. Verdeguer F, Aouadi M. Macrophage heterogeneity and energy metabolism. Exp Cell Res. 2017;360(1):35–40. doi:10.1016/j.yexcr.2017.03.043
37. Chiba S, Hisamatsu T, Suzuki H, et al. Glycolysis regulates LPS-induced cytokine production in M2 polarized human macrophages. Immunol Lett. 2017;183:17–23. doi:10.1016/j.imlet.2017.01.012
38. Thapa B, Lee K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. 2019;52(6):360–372. doi:10.5483/BMBRep.2019.52.6.140
39. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50. doi:10.1016/j.cmet.2019.06.001
40. Alquraishi M, Puckett DL, Alani DS, et al. Pyruvate kinase M2: a simple molecule with complex functions. Free Radic Biol Med. 2019;143:176–192. doi:10.1016/j.freeradbiomed.2019.08.007
41. Zhu L, Zhao Q, Yang T, Ding W, Zhao Y. Cellular metabolism and macrophage functional polarization. Int Rev Immunol. 2015;34(1):82–100. doi:10.3109/08830185.2014.969421
42. Zhang R, Ji J, Blaženović I, et al. Investigation into cellular glycolysis for the mechanism study of energy metabolism disorder triggered by lipopolysaccharide. Toxins (Basel). 2018;10(11):441. doi:10.3390/toxins10110441
43. Almousa AA, Morris M, Fowler S, Jones J, Alcorn J. Elevation of serum pyruvate kinase M2 (PKM2) in IBD and its relationship to IBD indices. Clin Biochem. 2018;53:19–24. doi:10.1016/j.clinbiochem.2017.12.007
44. Zhong WJ, Yang HH, Guan XX, et al. Inhibition of glycolysis alleviates lipopolysaccharide-induced acute lung injury in a mouse model. J Cell Physiol. 2019;234(4):4641–4654. doi:10.1002/jcp.27261
45. Palsson-McDermott EM, Curtis AM, Goel G, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21(1):65–80. doi:10.1016/j.cmet.2014.12.005
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.