")
Back to Archived Journals » Current Biomarker Findings » Volume 4
Ion channel messenger RNA processing defects and arrhythmia
Authors Zhou A, Dudley Jr S
Received 24 August 2014
Accepted for publication 16 October 2014
Published 24 November 2014 Volume 2014:4 Pages 151—160
DOI https://doi.org/10.2147/CBF.S37417
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Hung Khong
Anyu Zhou, Samuel C Dudley Jr
Lifespan Cardiovascular Research Center, The Warren Alpert School of Medicine, Brown University, and Providence Veterans Administration Medical Center, Providence, RI, USA
Abstract: Messenger RNA (mRNA) processing is an essential step for the expression of most eukaryote genes. Ion channels are critical for proper electrical activity in the heart, and perturbations of these channels are known to cause arrhythmia. Recently, mRNA processing defects have been shown to contribute to altered ion channel activity and arrhythmogenesis. Abnormal pre-mRNA splicing of cardiac ion channels, including the cardiac sodium channel, potassium channels, and calcium channels, because of mutations of the cis-elements within the RNA or abnormal expression of splicing factors, has been documented to contribute to arrhythmic risk. In addition to pre-mRNA splicing, other mRNA processing events, such as 3'-end formation and mRNA turnover, are also disrupted in cardiac diseases, such as congenital heart disease caused by mutation at the 3'-untranslated region of GATA4. mRNA stability is also dysregulated by altered expression of microRNAs in atrial fibrillation. In this review, we discuss our current understanding of how mRNA processing defects contribute to the risk of arrhythmias and how monitoring the products of abnormal processing may lead to diagnostic tests for arrhythmic risk.
Keywords: messenger RNA, arrhythmia, sudden death, ion channels, biomarkers
Introduction
Cardiac arrhythmias cause a significant number of deaths worldwide,1 and the risk of arrhythmias is inversely related to cardiac contractile function. Arrhythmia refers to any deviation from the normal pattern of the heartbeat, encompassing abnormalities of rate, regularity, site of impulse origin, and sequence of activation. The mechanisms of arrhythmia caused by ion channel defects are complicated and have been well reviewed in other articles.2–4 In short, normal cardiac excitation and relaxation involves a delicate balance of complex dynamic interactions between ionic currents passing through a variety of membrane channels. Cardiac excitation reflects membrane depolarization of cardiac myocytes, primarily because of the activation of voltage-dependent Na+ channels that underlie the action potential upstroke. Activation is then followed by a long depolarized plateau phase that permits Ca2+-induced Ca2+ release from the sarcoplasmic reticulum, binding of Ca2+ to contractile proteins on the sarcomeres, and coordinated contraction. Repolarization follows secondary to the time-dependent and voltage-dependent activation of repolarizing potassium currents. Relaxation of contraction is coupled to the electrical repolarization phase, which allows filling of the ventricles prior to the next excitation. Abnormal activity of cardiac ion channels can disrupt this electrical sequence and cause arrhythmia.
Abnormal activity of cardiac ion channels has many causes, including amino acid sequence changes and accompanying functional abnormalities caused by genetic defects, mutations, and polymorphisms.5–7 The expression level of ion channels can also be altered by dysregulation of transcription, post-transcriptional RNA processing, and protein degradation.8–11 Cardiac contractile dysfunction is associated with ion channel changes,11 and these ion channel changes are thought to contribute to increased arrhythmic risk.
Genetic and epigenetic alterations have been linked to arrhythmias.12–15 Now, it is being recognized that defects of messenger RNA (mRNA) processing can cause arrhythmogenesis (Table 1). This review focuses on mRNA processing, especially pre-mRNA splicing and mRNA stability and its impact on cardiac arrhythmias. Elucidation of mRNA processing defects is providing insights into the fundamental mechanisms of cardiac arrhythmias as well as the identification of possible targets for developing novel antiarrhythmic therapeutics to correct the electrical remodeling associated with heart disease.
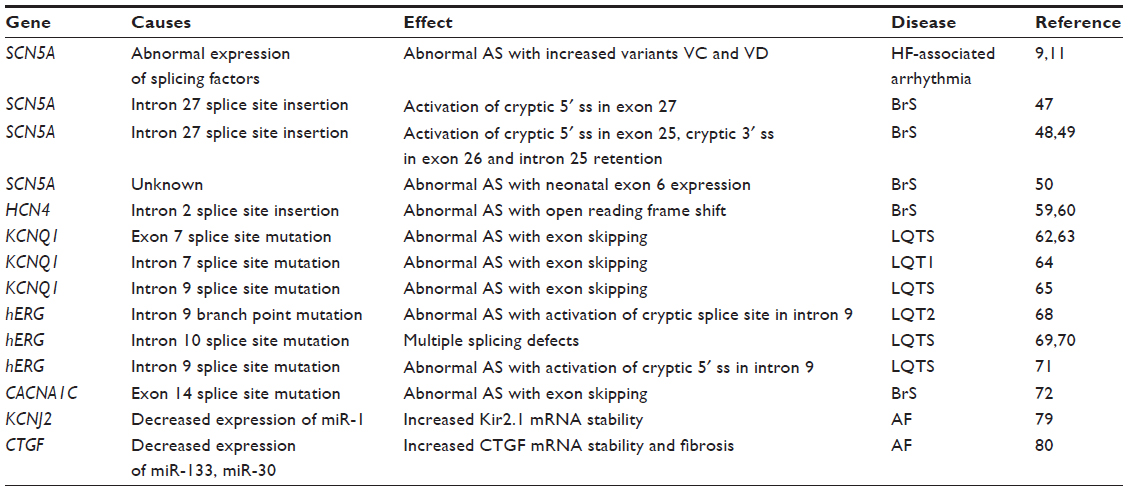 | Table 1 Abnormal mRNA processing associated with arrhythmia |
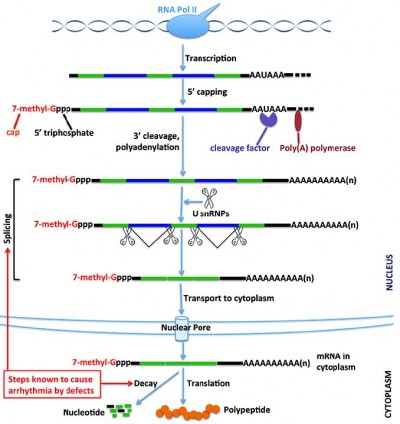
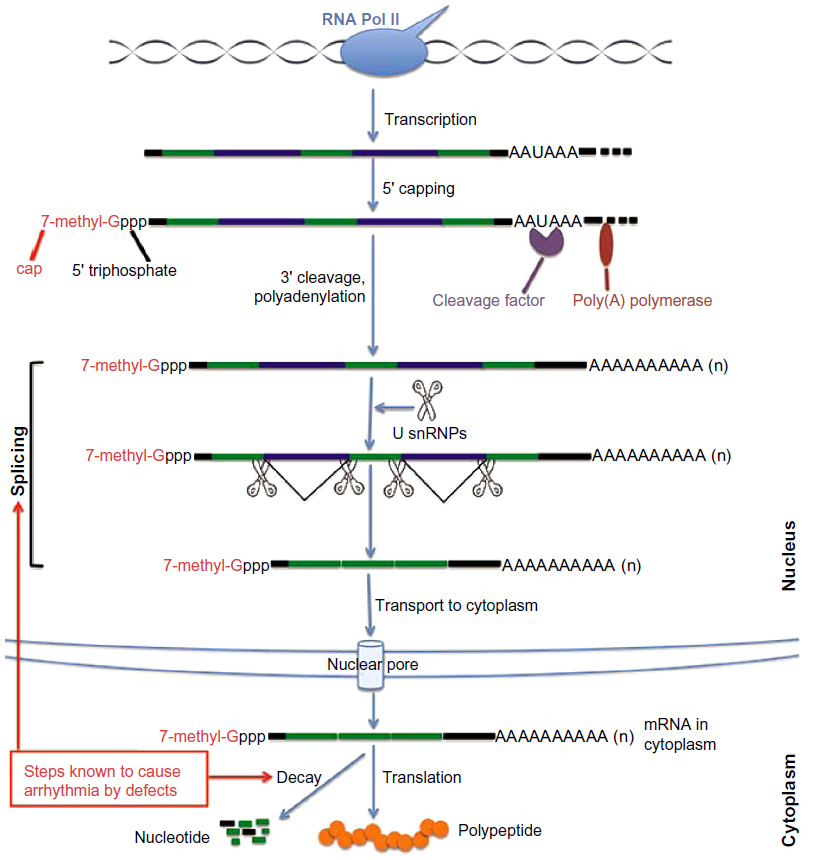 | Figure 1 A diagram of the mRNA processing pathway. The initial primary RNA transcript (pre-mRNA) synthesized by RNA polymerase II needs to be extensively processed into mature mRNAs before it can be exported out of the nucleus and into the cytoplasm for translation. Processing steps include 5′ capping, pre-mRNA splicing, 3′-end cleavage, and polyadenylation. Once RNA nuclear processing is complete, the capped, spliced, and polyadenylated mRNA is exported from the nucleus to the cytoplasm through nuclear pore complexes. The mature mRNA is translated into protein or is degraded in the cytoplasm. Altered steps known to cause arrhythmia are labeled. |
mRNA processing
When a eukaryotic gene is transcribed, the initial primary transcript synthesized by RNA polymerase II must be extensively modified before it can leave the nucleus and be translated into protein. This process includes 5′ capping, 3′-end polyadenylation, editing, and splicing (Figure 1). These nuclear processing steps, which largely determine the fate of the resulting transcript, require a large set of proteins, adding a layer of potential regulation that can affect export, localization, translation, and stability of the mature RNA.16 This processing allows the cell to fine-tune gene expression in a fast, precise, and cost-effective manner.17
RNA processing is tightly coupled with transcription. It begins while RNA polymerase II is in the process of transcribing the gene into RNA. This processing affects not only protein-coding RNA but also small nuclear RNAs, microRNAs (miRNAs), and other noncoding RNAs.18–20 The C-terminal domain of RNA polymerase II provides the basis for the coupling between transcription and RNA processing.18 The first step of RNA processing is the addition of an inverted guanosine to the 5′ end, and methylation of this guanosine to create a “cap” that marks the beginning of the mRNA. Capping helps protect the transcript degradation from 5′-to-3′ exonucleases present in both the nucleus and cytoplasm. It is also important in mediating mRNA recruitment to ribosomes and maintaining quality, since mRNAs without a cap are degraded.21 Protein complex binding to the cap structure coordinates subsequent steps in pre-mRNA splicing, nuclear export, translation, and decay.22
In humans and other eukaryotes, the vast majority of protein-coding genes contain many segments (introns) that are part of the primary transcript (pre-mRNA) but are not included in the mature RNA. The removal of introns and the splicing together of the sequences included in the mRNA (exons), which contain the protein-coding open reading frame and the 3′ and 5′ untranslated regions, is accomplished by a process called pre-mRNA splicing. The definition of exons and the execution of the splicing reaction are carried out by the nuclear spliceosome,23 a large and dynamic molecular machinery composed of small nuclear RNAs and splicing factors, including RNA binding proteins that bind in a sequence-specific manner to RNA and either enhance or silence splicing at nearby splice sites.24,25 Pre-mRNA splicing is a highly regulated, stepwise process. In addition to the consensus sequences, 5′ and 3′ splice sites at exon-intron boundaries, the branch point sequence, polypyrimidine tract, cis-elements within the pre-mRNA, and trans-regulatory factors are all necessary for proper splice site recognition. Any splicing errors will result in a disconnection between the coding gene and its encoded protein product.
Most pre-mRNAs can be spliced in different ways. Exons are either retained in the mRNA or removed from it in different combinations to create a diverse array of mRNAs from the same pre-mRNA. This process is known as alternative RNA splicing. The inclusion or exclusion of selected exonic sequences in specific cells, at specific developmental stages, and in response to specific extracellular stimuli, generates multiple mature RNA transcripts from a single pre-mRNA sequence via alternative splicing pathways. Alternative splicing is an important mechanism to generate multiple proteins from a single gene in eukaryotes and thus it expands gene expression complexity without an increase in the overall number of genes.26–28 In human cells, approximately 95% of the multi-exon genes yield alternatively spliced transcripts.29 The outcome of alternative splicing is decided by cis-elements, known as enhancers and silencers within exons and/or introns, trans-acting factors, such as serine/arginine-rich proteins or heterogeneous nuclear ribonucleoproteins, and is influenced by transcription.30–32
Endonucleolytic cleavage occurs 10–30 nucleotides downstream of a signal sequence (conserved AAUAAA sequence in mammals or an AU-rich sequence in yeast). Following cleavage, most eukaryotic mRNAs, with the exception of replication-dependent histone transcripts in some organisms, add a poly(A) tract at their 3′-ends.33–35 This process requires the assembly of a large protein complex guided by the conserved AAUAAA sequence and GU-rich sequences downstream of the processing site. This multiprotein complex contains an endonuclease that cleaves the pre-mRNA to create the 3′ end and poly(A) polymerase, which adds the poly(A) tail, a homopolymer of 250 adenosine residues. These latter steps are coordinated by interaction between a splicing factor bound to the last intron of the pre-mRNA and poly(A) polymerase.36 The formation of the 3′-end promotes transcription termination, facilitates nuclear RNA transport, and enhances the translation and stability of mRNA. On the other hand, the importance of 3′-end formation on gene expression regulation is underscored by the fact that it may stimulate further rounds of transcription initiation.37
At this point, the capped, spliced, and polyadenylated mRNA is assembled into an mRNP complex that is competent for export from the nucleus to the cytoplasm through nuclear pore complexes. After being transported to the cytoplasmic side of the nuclear pore complex, the newly synthesized mRNA must undergo a surveillance program in which it is scanned by a “pioneer round” of translation that determines whether the transcript will encode a full-length protein.38 This process is important for quality control. Following the surveillance process, the mRNA can be translated, silenced, stored, or degraded. All these steps are highly regulated, and the fate of a particular mRNA is determined by the interplay between cis-elements (such as AU-rich elements) within the mRNA and the trans-acting factors (including proteins and miRNAs) that bind to them.
AU-rich elements are the largest and most studied group of cis-acting mRNA elements which involve regulation of mRNA stability. Approximately 9% of cellular mRNAs contains canonical AU-rich elements,39 and their differential binding by a number of AU-rich element-binding proteins (such as Hu family proteins and AU-rich binding factor-1) determine their overall accumulation, translation, and decay. Besides AU-rich elements, GU-rich elements also have been shown to be involved in mRNA stability regulation by binding to CUG-repeat binding proteins.40 Both translation and decay are targets of numerous signal transduction pathways.
Aberrant pre-mRNA splicing and arrhythmias
Cardiac Na+ channel pre-mRNA splicing and arrhythmias
Voltage-gated Na+ channels are responsible for the action potential upstroke and the current for conduction in such cells as muscles and neurons.41 The cardiac Na+ channel consists of a main pore-forming α-subunit and the auxiliary β-subunits. The α-subunit alone is sufficient to produce a functional channel. In mammals, the SCN1A–SCN11A genes encode a family of nine functionally expressed voltage-gated Na+ channels that are more than 50% homologous in their amino acid sequences. The Na+ channel isoform Nav1.5, encoded by the SCN5A gene, is the predominant α-subunit in the heart. Alteration in the Na+ channel, either in amino sequences or at the expression level, leads to arrhythmias.42,43
Abnormal splicing of SCN5A pre-mRNA has been linked with arrhythmia associated with heart failure. A decrease in both mRNA and protein levels of SCN5A has been shown in failing heart by Shang et al.11 In this condition, wild-type functional SCN5A is downregulated, and aberrantly spliced nonfunctional, truncated SCN5A variants, variant C and variant D, are increased concomitantly in both heart tissue and circulating white blood cells. Further work shows that hypoxia and angiotensin II are responsible for the abnormal splicing of SCN5A through upregulation of splicing factors RBM25 and Luc7A.9 SCN5A variants VC and VD result from aberrant pre-mRNA splicing of exon 27 to exon 28, and the resultant ion channel is truncated prematurely, is mostly trapped in the endoplasmic reticulum, and cannot conduct current. In addition, the truncated Na+ channel protein activates the unfold protein response, which further inhibits wild-type SCN5A protein synthesis.44 The expression levels of both myocardial SCN5A splicing variants VC and VD strongly correlated with those of circulating white blood cell samples, suggesting that circulating SCN5A variants might be used to predict cardiac events. Indeed, levels of circulating SCN5A variants are strongly associated with the risk for an appropriate implantable cardioverter-defibrillator (ICD) intervention. Therefore, circulating SCN5A variants might be useful biomarkers in the stratification of arrhythmic risk.8,45
Mutation-activated abnormal splicing of SCN5A is associated with Brugada syndrome (BrS). According to The Human Gene Mutation Database (HGMD® Professional, Fall Release 2014.3), many splicing-affecting mutations of ion channel genes are associated with arrhythmias. Most of the 30 splicing-affecting mutations in the SCN5A gene are associated with BrS, and five of the 30 mutations are associated with long QT syndrome (LQTS). Forty-one and 22 such mutations in KCNQ1 and KCNH2 genes, respectively, are related to LQTS as well. Here, we focus on examples of splicing-affecting mutations of ion channels wherein a mechanistic relationship to arrhythmia has been shown by in vivo or in vitro experiments. BrS is an inherited cardiac arrhythmia syndrome, characterized by the presence of ST segment elevation in leads V1 to V3 and sudden cardiac death in individuals with a structurally normal heart.46 Hong et al47 reported that an intronic insertion of TGGG at 5 base pairs (bp) downstream of the exon 27 of SCN5A in a family with BrS caused activation of an upstream 5′ splice site within exon 27 and led to exclusion of the last 96 bp sequences of exon 27 in the mature SCN5A mRNA. This mutation ultimately created an in-frame shortened, nonfunctional SCN5A protein. This shortened nonfunctional protein is thought to cause BrS, a condition of reduced Na+ current. A similar abnormal splicing pattern is also associated with another intronic insertion of GGGT at 6 bp from the end of exon 27.48 Mutation at the 5′ splice site of SCN5A exon 26 is also reported to result in activation of multiple cryptic 5′ splice sites, leading to BrS. A minigene with this mutation produced multiple mature transcripts with exclusion of the last 100 bp of exon 25, exclusion of the first four bp of exon 26, or intron 25 retention.49
Abnormal splicing of SCN5A without mutations has also been reported in patients with myotonic dystrophy type 1 (DM1) with BrS.50 DM1 is the most common inherited neuromuscular disease in adults and is caused by an expansion of a (CTG)n triplet repeat in the 3′ region of the gene encoding dystrophia myotonica protein kinase. The most frequent cardiac complications of DM1 are atrioventricular block, sinus node dysfunction, atrial fibrillation, and ventricular tachyarrhythmia. Up to one third of DM1 patients die suddenly, most often because of asystole after atrioventricular block or from ventricular fibrillation.51 Increased neonatal exon 6 splicing variant of SCN5A, called exon 6A, has been detected in ventricular myocardial specimens from BrS patients with DM1. Concomitantly, the adult splice variant of exon 6, called exon 6B, is downregulated. This aberrant splicing is not mutation-dependent and mostly likely represents alteration of trans-acting factors. Consistent with this idea, disturbance of the functional balance between splicing regulators, MBNL1 and CELF1, has been well documented in patients with DM1.52,53
It is worth noting that SCN5A produces multiple mRNAs by means of alternative pre-mRNA splicing in normal conditions.54 Some particular isoforms have been linked to arrhythmias with the coexistence of other SCN5A mutations. For example, the 3′ splice site for exon 18 contains 2 consecutive CAG trinucleotides. The splicing machinery can select either the first or the second intronic “AG” acceptor site for coupling of exon 17, thereby including or skipping an additional CAG codon, respectively. While the CAG-exclusive isoform (Nav1.5) predominates in normal heart, the CAG-inclusive variant, called Nav1.5c, is also expressed at approximately 50% of the expression level of the CAG-exclusive variant. Although patch-clamp recordings indicated that the electrophysiological properties of the two variants are indistinguishable, increased persistent current has been noticed in the context of the CAG-exclusive variant Nav1.5 but not in the CAG-inclusive Nav1.5c background when these channels are expressed with three Nav1.5 mutations associated with sudden infant death syndrome (delAL586–587, R680H, and V1951L).55 Loss-of-function defects in the SCN5A missense mutation G1406R, a mutation reported to cause BrS, also demonstrate a more severe phenotype in the context of an Nav1.5c background compared with that of wild-type Nav1.5 background.56 These data suggest that not only selection of abnormal splice sites but also abnormal expression of normal splicing variants contributes to arrhythmogenesis.
Cardiac potassium channel pre-mRNA splicing and arrhythmias
In addition to alterations in the Na+ channel, mutation-activated abnormal splicing of the hyperpolarization activated cyclic nucleotide-gated potassium channel 4 (HCN4 channel or If channel) is also associated with BrS. HCN4 encodes a member of the hyperpolarization-activated cyclic nucleotide-gated potassium channels. The encoded protein shows slow kinetics of activation and inactivation and is necessary for the cardiac pacemaking process.57,58 Mutations in this gene have been linked to sick sinus syndrome 2, also known as atrial fibrillation with bradyarrhythmia or familial sinus bradycardia.59 Abnormal splicing of HCN4 was found in BrS patients without an SCN5A mutation. This abnormal splicing was activated by an insertion of GTGA at a splicing junction of exon 2 and intron 2.60 A minigene approach revealed that the additional four bases after the native splice donor site were incorporated into mRNA. The insertion caused a frame shift with an Ile-to-Val change at codon 404 and a subsequent 43 inappropriate amino acids. This truncated HCN4 protein is thought to be responsible for the idiopathic ventricular arrhythmia in patients.
Mutation-mediated abnormal splicing of another potassium channel gene, KCNQ1, is been associated with LQTS. LQTS is a life-threatening cardiac arrhythmia syndrome characterized by prolongation of the cardiac action potential, syncopal attacks, torsades de pointes arrhythmias, and sudden cardiac death. KCNQ1 encodes the α-subunit of the slow component of the delayed rectifier potassium K+ channel, Kv7.1, which generates the IKs current in the heart, and mutations in this channel have been identified as a cause for this disease.61 KCNQ1 is involved in approximately half of patients with LQTS type 1. Mutation of the last nucleotide of exon 7 (G1032A) has been reported to trigger abnormal exon skipping in patients with LQTS. Mature RNA products without exon 7, exon 8, or without both exon 7 and exon 8, have been detected in peripheral blood samples of the patients.62,63 In addition to this exonic mutation, another A-to-G intronic mutation at the third nucleotide of intron 7 (IVS7+3A>G) causes skipping of exon 7 or exon 8 splicing in patients with LQTS type 1.64 Imai et al reported that an intronic mutation at the first nucleotide of intron 9 (G-to-A) resulted in skipping exon 8 or skipping exon 8 and exon 9 in patients with LQTS.65 This is a rare case where a mutation in intron 9 causes exon 8 skipping rather than affecting the adjacent exons.
LQTS type 2 is caused by the loss of functional human ether-a-go-go-related gene (hERG, also called KCNH2).66,67 The potassium channel (Kv11.1) encoded by hERG is a tetrameric channel and a well characterized component of the rapidly activating delayed rectifier current in the heart. Besides missense and frame shift mutations of hERG, abnormal mRNA splicing of hERG has been increasingly associated with LQT2. In an LQT family, an intronic A-to-G substitution in hERG (IVS9-28A/G) at intron 9 disrupts the branch point of exon 10 and causes activation of a cryptic 3′ splice site within intron 9. Use of this cryptic splice site results in a 147 nucleotide intronic fragment of intron 9 being included in the mature mRNA.68 Abnormal splicing of hERG has also been found in LQT2 patients with a 5′ splice site mutation. A hERG G-to-A splice site mutation (2592+1G>A), which occurs at the first nucleotide of the 5′ splice site of intron 10, has been identified to cause LQTS.69 The minigene approach reported by Stump et al70 revealed that this mutation disrupts normal splicing and causes multiple splicing defects, including activation of cryptic splice sites within exon 10 and intron 10, as well as completely intron 10 retention. The resulting mature mRNA encoded a truncated protein with a 24 amino acid deletion at the C-terminus. Patch-clamp experiments reveal that the splice mutant does not generate hERG current, and immunostaining studies show that mutant channels do not traffic to the cell surface. This truncated protein results in significant dominant-negative suppression of hERG current via intracellular retention of the wild-type channels.
Another G-to-C 5′ splice site mutation (2398+1G>C) at the first nucleotide of intron 9 of hERG has been identified as the pathogenic mechanism of LQTS in three unrelated kindreds with LQTS.71 The effect of this mutation on mRNA splicing was verified by analysis of RNA isolated from lymphocytes of index patients and using minigenes expressed in HEK293 cells and neonatal rat ventricular myocytes. This mutation disrupted the normal splicing and activated a cryptic splice donor site within intron 9, leading to inclusion of 54 nucleotides of the intron 9 sequence in the hERG mRNA. This abnormal cryptic splicing results in an in-frame insertion of 18 amino acids in the middle of the cyclic nucleotide binding domain, causing the channel to lose the ability to generate hERG current. The abnormally spliced mRNA fails to reach the plasma membrane, and coexpression of the mutant and wild-type channels leads to a dominant negative suppression of wild-type channel function by intracellular retention of heteromeric channels.
Cardiac calcium channel pre-mRNA splicing and arrhythmias
Aberrant alternative splicing caused by mutations in genes related to the cardiac L-type calcium channel, coded by CACNA1C, has been reported to be causative of BrS. Fukuyama et al72 reported a G-to-A mutation at the 3′ splice site of exon 14 of CACNA1C in a BrS patient. Although this is a synonymous mutation, it results in loss of function of Cav1.2 by activation of abnormal pre-mRNA splicing. As a consequence, exons 14 and 15 are skipped from the mature mRNA, and exon 13 is spliced with exon 16. Subsequently, a premature stop codon is created within exon 16, and nonsense-mediated mRNA decay is activated to destroy this aberrant mRNA.
Dysregulation of mRNA turnover and arrhythmias
Ultimately, all mRNAs are degraded, and the abundance of steady-state mRNA is determined by the balance between transcription and mRNA decay. mRNA decay can influence timing, quantity, and perhaps even location of encoded proteins.73 In eukaryotes, the major pathway of mRNA decay begins with shortening of the poly(A) tail, followed by removal of the 5′ cap and degradation by exonucleases from both the 5′ and 3′ ends of the mRNA. This process is highly regulated by the interplay between cis-elements within the mRNA and the proteins that bind to them. The 3′ untranslated region is the most important mRNA structure for regulation of mRNA decay. In addition to RNA binding proteins, miRNAs can act as trans-acting factors to regulate mRNA decay.74 Dysregulation of RNA turnover has been shown to contribute to arrhythmogenesis.
miRNA-mediated mRNA alterations and arrhythmias
Mature miRNAs are a class of naturally occurring, small noncoding RNA molecules, approximately 21–25 nucleotides in length, which function in transcriptional and post-transcriptional regulation of gene expression.75 In addition to repression of translation,76 miRNAs are also involve in regulation of mRNA stability and degradation.77,78 miRNA-1 expression levels are significantly reduced in patients with atrial fibrillation.79 Decreased miRNA-1 correlates with an increase in both the mRNA and the protein of the Kir2.1 potassium channel, encoded by KCNJ2. Ex vivo rapid pacing of human atrial slices upregulates Kir2.1 while downregulating miR-1.79 These results suggest that one of the mechanisms of the involvement of miRNA-1 in atrial fibrillation is by increasing Kir2.1 mRNA stability. miR-133 and miR-30 have been associated with atrial fibrillation through downregulation of the mRNA for connective tissue growth factor (CTGF),80 which is a key molecule in the process of fibrosis. Fibrosis is thought to be a major risk factor for atrial fibrillation.81 Both miR-133 and miR-30 are downregulated while CTGF mRNA and protein levels are increased in atrial fibrillation. Overexpression of miR-133 and miR-30 decreases both CTGF mRNA abundance and protein expression in cultured cardiomyocyte and fibroblast cells. Knockdown of miR-133 or miR-30 significantly increases CTGF expression at both the mRNA and protein levels. Taken together, these data suggest that dysregulated mRNA turnover by cardiac miRNAs may contribute to arrhythmias.
mRNA process regulators and aberrant products as potential biomarkers for arrhythmias
As discussed previously, abnormal mRNA processing can be associated with cardiac arrhythmias. This provides the possibility to predict the risk of arrhythmia by assessment of aberrant products and regulators of the mRNA process. Some mRNA process regulators and aberrant products can not only be detected in heart but also in blood. Therefore, blood tests for the aberrant products and abnormal expression of mRNA process regulators may be valuable for stratification of arrhythmia risk. Gao et al45 have reported that expression levels of circulating abnormal SCN5A splicing variants VC and VD correlate strongly with the variant levels measured in myocardial tissue in patients with heart failure. Patients who have received appropriate intervention with an ICD have higher expression levels of both white blood cell-derived SCN5A variants compared with patients with heart failure who had not suffered a sudden death event. Further receiver operating characteristics analysis reveals that circulating SCN5A variant levels are excellent predictors of an appropriate ICD intervention within 1 year. Collectively, these variants may be used to predict arrhythmia with high sensitivity and specificity. The authors conclude that determination of white blood cell-derived SCN5A variant levels might help personalize ICD implantation by better defining a patient’s arrhythmic risk.
miRNA has been increasingly suggested as a biomarker for diseases.82 Circulating miRNAs are not only present in white blood cells but also present in all other compartments of the blood, including plasma, platelets, and erythrocytes. Heart tissue miRNAs can be released into the blood actively or passively by several mechanisms, such as in microvesicles, in exosomes, in apoptotic bodies, and in protein complexes.83,84 Therefore, miRNAs need not be produced by blood cells but can originate from the heart. They remain extremely stable because of their transport in association with microvesicles and exosomes or in tight association with RNA-binding proteins that protect them from ribonuclease degradation. Because of their stability in the circulation, miRNAs are currently being explored for their potential as biomarkers in a wide range of cardiovascular diseases.
There are preliminary results identifying individual miRNAs or miRNA signatures for diagnosis of cardiovascular diseases.85,86 It has been reported that miR-29 might be a potential biomarker in atrial fibrillation.87 Both miR29b plasma and atria levels are decreased in patients with chronic atrial fibrillation, and miR-29 expression is decreased by 87% within 24 hours in a dog model of atrial fibrillation induced by ventricular rapid pacing.
Conclusion
mRNA processing defects can clearly cause arrhythmia. Defects can be seen in such conditions as BrS, LQTS, heart failure, and atrial fibrillation. Noted defects include abnormal pre-mRNA splicing and altered mRNA turnover caused either by mutation of cis-elements within the RNA or alteration in the expression of trans-acting factors. Assessment of variant mRNA, trans-acting factors, or abnormally expressed mRNA process regulators is a logical approach to enhance stratification of arrhythmia risk. This same approach may be useful in directing current antiarrhythmic therapies, and novel therapeutics based on correcting mRNA defects could prove a novel approach to treating arrhythmias.
Acknowledgments
This work was funded by National Institutes of Health grants RO1 HL104025 (SCD) and HL106592 (SCD), and a Veterans Affairs MERIT grant BX000859 (SCD).
Disclosure
SCD is the inventor in the following patent applications: SCN5A Splice Variants for Use in Methods Relating to Sudden Cardiac Death and Need for Implanted Cardiac Defibrillators, PCT/US2012/20564 and SCN5A Splicing Factors and Splice Variants for Use in Diagnostic and Prognostic Methods, 13/291,826. The authors declare no other conflicts of interest in this work.
References
Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125(4):620–637. | |
Amin AS, Tan HL, Wilde AA. Cardiac ion channels in health and disease. Heart Rhythm. 2010;7(1):117–126. | |
Clancy CE, Kass RS. Defective cardiac ion channels: from mutations to clinical syndromes. J Clin Invest. 2002;110(8):1075–1077. | |
Kirsch GE. Ion channel defects in cardiac arrhythmia. J Membr Biol. 1999;170(3):181–190. | |
Pfahnl AE, Viswanathan PC, Weiss R, et al. A sodium channel pore mutation causing Brugada syndrome. Heart Rhythm. 2007;4(1):46–53. | |
Remme CA. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J Physiol. 2013;591 Pt 17:4099–4116. | |
Voudris KV, Apostolakis S, Karyofillis P, et al. Genetic diversity of the KCNE1 gene and susceptibility to postoperative atrial fibrillation. Am Heart J. 2014;167(2):274–280. | |
Gao G, Dudley SC Jr. SCN5A splicing variants and the possibility of predicting heart failure-associated arrhythmia. Expert Rev Cardiovasc Ther. 2013;11(2):117–119. | |
Gao G, Xie A, Huang SC, et al. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure. Circulation. 2011;124(10):1124–1131. | |
Remme CA, Verkerk AO, Hoogaars WM, et al. The cardiac sodium channel displays differential distribution in the conduction system and transmural heterogeneity in the murine ventricular myocardium. Basic Res Cardiol. 2009;104(5):511–522. | |
Shang LL, Pfahnl AE, Sanyal S, et al. Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ Res. 2007;101(11):1146–1154. | |
Coronel R, Wilders R, Verkerk AO, Wiegerinck RF, Benoist D, Bernus O. Electrophysiological changes in heart failure and their implications for arrhythmogenesis. Biochim Biophys Acta. 2013;1832(12):2432–2441. | |
Duygu B, Poels EM, da Costa Martins PA. Genetics and epigenetics of arrhythmia and heart failure. Front Genet. 2013;4:219. | |
Farwell D, Gollob MH. Electrical heart disease: genetic and molecular basis of cardiac arrhythmias in normal structural hearts. Can J Cardiol. 2007;23 Suppl A:16A–22A. | |
Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112(16):2517–2529. | |
Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010;11(1):75–87. | |
Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8(7):533–543. | |
Hsin JP, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012;26(19):2119–2137. | |
Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15(2):108–121. | |
Melamed Z, Levy A, Ashwal-Fluss R, et al. Alternative splicing regulates biogenesis of miRNAs located across exon-intron junctions. Mol Cell. 2013;50(6):869–881. | |
Jiao X, Xiang S, Oh C, Martin CE, Tong L, Kiledjian M. Identification of a quality-control mechanism for mRNA 5’-end capping. Nature. 2010;467(7315):608–611. | |
Topisirovic I, Svitkin YV, Sonenberg N, Shatkin AJ. Cap and cap-binding proteins in the control of gene expression. Wiley Interdiscip Rev RNA. 2011;2(2):277–298. | |
Hoskins AA, Gelles J, Moore MJ. New insights into the spliceosome by single molecule fluorescence microscopy. Curr Opin Chem Biol. 2011;15(6):864–870. | |
Hegele A, Kamburov A, Grossmann A, et al. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell. 2012;45(4):567–580. | |
Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–718. | |
Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12(10):715–729. | |
Kelemen O, Convertini P, Zhang Z, et al. Function of alternative splicing. Gene. 2013;514(1):1–30. | |
Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6(5):386–398. | |
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–1415. | |
Bielli P, Busa R, Di Stasi SM, et al. The transcription factor FBI-1 inhibits SAM68-mediated BCL-X alternative splicing and apoptosis. EMBO Rep. 2014;15(4):419–427. | |
Kornblihtt AR, de la Mata M, Fededa JP, Munoz MJ, Nogues G. Multiple links between transcription and splicing. RNA. 2004;10(10):1489–1498. | |
Munoz MJ, de la Mata M, Kornblihtt AR. The carboxy terminal domain of RNA polymerase II and alternative splicing. Trends Biochem Sci. 2010;35(9):497–504. | |
Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136(4):688–700. | |
Proudfoot N. New perspectives on connecting messenger RNA 3′ end formation to transcription. Curr Opin Cell Biol. 2004;16(3):272–278. | |
Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63(2):405–445. | |
Davidson L, West S. Splicing-coupled 3′ end formation requires a terminal splice acceptor site, but not intron excision. Nucleic Acids Res. 2013;41(14):7101–7114. | |
Mapendano CK, Lykke-Andersen S, Kjems J, Bertrand E, Jensen TH. Crosstalk between mRNA 3′ end processing and transcription initiation. Mol Cell. 2010;40(3):410–422. | |
Maquat LE, Tarn WY, Isken O. The pioneer round of translation: features and functions. Cell. 2010;142(3):368–374. | |
Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006;34(Database issue): D111–D114. | |
Beisang D, Rattenbacher B, Vlasova-St Louis IA, Bohjanen PR. Regulation of CUG-binding protein 1 (CUGBP1) binding to target transcripts upon T cell activation. J Biol Chem. 2012;287(2):950–960. | |
Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. | |
Rook MB, Evers MM, Vos MA, Bierhuizen MF. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res. 2012;93(1):12–23. | |
Zhang T, Yong SL, Tian XL, Wang QK. Cardiac-specific overexpression of SCN5A gene leads to shorter P wave duration and PR interval in transgenic mice. Biochem Biophys Res Commun. 2007;355(2):444–450. | |
Gao G, Xie A, Zhang J, et al. Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ Arrhythm Electrophysiol. 2013;6(5):1018–1024. | |
Gao G, Brahmanandam V, Raicu M, et al. Enhanced risk profiling of implanted defibrillator shocks with circulating SCN5A mRNA splicing variants: a pilot trial. J Am Coll Cardiol. 2014;63(21):2261–2269. | |
Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391–1396. | |
Hong K, Guerchicoff A, Pollevick GD, et al. Cryptic 5′ splice site activation in SCN5A associated with Brugada syndrome. J Mol Cell Cardiol. 2005;38(4):555–560. | |
Rossenbacker T, Schollen E, Kuiperi C, et al. Unconventional intronic splice site mutation in SCN5A associates with cardiac sodium channelopathy. J Med Genet. 2005;42(5):e29. | |
Shimada T, Ohkubo K, Abe K, Watanabe I, Makita N. A novel 5′ splice site mutation of SCN5A associated with Brugada syndrome resulting in multiple cryptic transcripts. Int J Cardiol. 2012;158(3):441–443. | |
Wahbi K, Algalarrondo V, Becane HM, et al. Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1. Arch Cardiovasc Dis. 2013;106(12):635–643. | |
Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358(25):2688–2697. | |
Buj-Bello A, Furling D, Tronchere H, et al. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet. 2002;11(19):2297–2307. | |
Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10(1):45–53. | |
Walzik S, Schroeter A, Benndorf K, Zimmer T. Alternative splicing of the cardiac sodium channel creates multiple variants of mutant T1620K channels. PLoS One. 2011;6(4):e19188. | |
Wang DW, Desai RR, Crotti L, et al. Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115(3):368–376. | |
Tan BH, Valdivia CR, Song C, Makielski JC. Partial expression defect for the SCN5A missense mutation G1406R depends on splice variant background Q1077 and rescue by mexiletine. Am J Physiol Heart Circ Physiol. 2006;291(4):H1822–H1828. | |
Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999;18(9):2323–2329. | |
Roden DM, Balser JR, George AL Jr, Anderson ME. Cardiac ion channels. Annu Rev Physiol. 2002;64:431–475. | |
Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med. 2006;354(2):151–157. | |
Ueda K, Hirano Y, Higashiuesato Y, et al. Role of HCN4 channel in preventing ventricular arrhythmia. J Hum Genet. 2009;54(2):115–121. | |
Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5(4):868–877. | |
Murray A, Donger C, Fenske C, et al. Splicing mutations in KCNQ1: a mutation hot spot at codon 344 that produces in frame transcripts. Circulation. 1999;100(10):1077–1084. | |
Tsuji K, Akao M, Ishii TM, et al. Mechanistic basis for the pathogenesis of long QT syndrome associated with a common splicing mutation in KCNQ1 gene. J Mol Cell Cardiol. 2007;42(3):662–669. | |
Tsuji-Wakisaka K, Akao M, Ishii TM, et al. Identification and functional characterization of KCNQ1 mutations around the exon 7-intron 7 junction affecting the splicing process. Biochim Biophys Acta. 2011;1812(11):1452–1459. | |
Imai M, Nakajima T, Kaneko Y, et al. A novel KCNQ1 splicing mutation in patients with forme fruste LQT1 aggravated by hypokalemia. J Cardiol. 2014;64(2):121–126. | |
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795–803. | |
Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104(4):569–580. | |
Crotti L, Lewandowska MA, Schwartz PJ, et al. A KCNH2 branch point mutation causing aberrant splicing contributes to an explanation of genotype-negative long QT syndrome. Heart Rhythm. 2009;6(2):212–218. | |
Berthet M, Denjoy I, Donger C, et al. C-terminal HERG mutations: the role of hypokalemia and a KCNQ1-associated mutation in cardiac event occurrence. Circulation. 1999;99(11):1464–1470. | |
Stump MR, Gong Q, Zhou Z. Multiple splicing defects caused by hERG splice site mutation 2592+1G.A associated with long QT syndrome. Am J Physiol Heart Circ Physiol. 2011;300(1):H312–H318. | |
Gong Q, Zhang L, Moss AJ, et al. A splice site mutation in hERG leads to cryptic splicing in human long QT syndrome. J Mol Cell Cardiol. 2008;44(3):502–509. | |
Fukuyama M, Ohno S, Wang Q, Shirayama T, Itoh H, Horie M. Nonsense-mediated mRNA decay due to a CACNA1C splicing mutation in a patient with Brugada syndrome. Heart Rhythm. 2014;11(4):629–634. | |
Farris S, Lewandowski G, Cox CD, Steward O. Selective localization of arc mRNA in dendrites involves activity- and translation-dependent mRNA degradation. J Neurosci. 2014;34(13):4481–4493. | |
Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Rev Genet. 2012;13(4):246–259. | |
Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8(2):93–103. | |
Pillai RS, Bhattacharyya SN, Artus CG, et al. Inhibition of translational initiation by Let-7 microRNA in human cells. Science. 2005;309(5740):1573–1576. | |
Bagga S, Bracht J, Hunter S, et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122(4):553–563. | |
Jing Q, Huang S, Guth S, et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120(5):623–634. | |
Girmatsion Z, Biliczki P, Bonauer A, et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm. 2009;6(12):1802–1809. | |
Duisters RF, Tijsen AJ, Schroen B, et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104(2):170–178. | |
Tanaka K, Zlochiver S, Vikstrom KL, et al. Spatial distribution of fibrosis governs fibrillation wave dynamics in the posterior left atrium during heart failure. Circ Res. 2007;101(8):839–847. | |
Wang GK, Zhu JQ, Zhang JT, et al. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J. 2010;31(6):659–666. | |
Fichtlscherer S, Zeiher AM, Dimmeler S. Circulating microRNAs: biomarkers or mediators of cardiovascular diseases? Arterioscler Thromb Vasc Biol. 2011;31(11):2383–2390. | |
Turchinovich A, Weiz L, Burwinkel B. Extracellular miRNAs: the mystery of their origin and function. Trends Biochem Sci. 2012;37(11):460–465. | |
Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. 2012;110(3):483–495. | |
Morley-Smith AC, Mills A, Jacobs S, et al. Circulating microRNAs for predicting and monitoring response to mechanical circulatory support from a left ventricular assist device. Eur J Heart Fail. 2014;16(8):871–879. | |
Dawson K, Wakili R, Ordog B, et al. MicroRNA29: a mechanistic contributor and potential biomarker in atrial fibrillation. Circulation 2013;127(14):1466–1475. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.