")
Back to Journals » Journal of Pain Research » Volume 9
Involvement of spinal glutamate transporter-1 in the development of mechanical allodynia and hyperalgesia associated with type 2 diabetes
Received 29 July 2016
Accepted for publication 27 October 2016
Published 28 November 2016 Volume 2016:9 Pages 1121—1129
DOI https://doi.org/10.2147/JPR.S118412
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Enrica Santarcangelo
Jinshan Shi,1,* Ke Jiang,2,* Zhaoduan Li,3
1Department of Anesthesiology, Guizhou Provincial People’s Hospital, 2Department of Anesthesiology, The Affiliated Hospital of Guizhou Medical University, Guiyang, 3Department of Anesthesiology, Tianjin Nankai Hospital, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Abstract: Little is known about the effects of the development of type 2 diabetes on glutamate homeostasis in the spinal cord. Therefore, we quantified the extracellular levels of glutamate in the spinal cord of Zucker diabetic fatty (ZDF) rats using in vivo microdialysis. In addition, protein levels of glutamate transporter-1 (GLT-1) in the spinal cord of ZDF rats were measured using Western blot. Finally, the effects of repeated intrathecal injections of ceftriaxone, which was previously shown to enhance GLT-1 expression, on the development of mechanical allodynia and hyperalgesia as well as on basal extracellular level of glutamate and the expression of GLT-1 in the spinal cord of ZDF rats were evaluated. It was found that ZDF rats developed mechanical hyperalgesia and allodynia, which were associated with increased basal extracellular levels of glutamate and attenuated levels of GLT-1 expression in the spinal cord, particularly in the dorsal horn. Furthermore, repeated intrathecal administrations of ceftriaxone dose-dependently prevented the development of mechanical hyperalgesia and allodynia in ZDF rats, which were correlated with enhanced GLT-1 expression without altering the basal glutamate levels in the spinal cord of ZDF rats. Overall, the results suggested that impaired glutamate reuptake in the spinal cord may contribute to the development of neuropathic pains in type 2 diabetes.
Keywords: diabetes, peripheral neuropathy, spinal cord, Zucker diabetic fatty rats, glutamate, glutamate transporter-1
Introduction
Peripheral neuropathy in diabetes is the most common complication of both type 1 and type 2 diabetes1,2 and can affect peripheral sensory neurons in a large percent of adult diabetic patients,2 who may experience excessive sensitivity to nociceptive stimuli or perceive normal stimuli as painful stimuli.3–5 This often results in severely reduced quality-of-life measures.6
It has been proposed that central sensitization can occur in the dorsal horn of the spinal cord, which may result in increased primary afferents, neuroplasticity in the spinal cord, and hyperactivity of dorsal horn neurons in the spinal cord.7–10 One of the critical mechanisms underlying the hyperactivity of dorsal horn neurons in the spinal cord may involve an increase in excitatory glutamatergic neurotransmission. For instance, studies have shown that administrations of N-methyl-d-aspartate (NMDA) induce a greater increase in the calcium influx in spinal lamina II neurons in nerve-ligated rats relative to control rats.11 In addition, NMDA receptor (NMDAR) antagonisms remarkably reduce evoked responses of dorsal horn neurons in spinal nerve-ligated rats.12 Thus, it is likely that enhanced excitatory glutamatergic neurotransmission may be critical in the perception of nociceptive signals in diabetic neuropathy.13 However, few studies have examined the effects of the development of diabetes on extracellular levels of glutamate in the spinal cord.
Most animal studies on diabetic neuropathy have been conducted using a diabetic rodent model by injections of streptozotocin (STZ), a pancreatic β-cell cytotoxin. This rodent model of diabetes resembles the development of type 1 diabetes. Using this model, previous studies have shown that type 1 diabetic neuropathy is associated with increased glutamate release from primary afferent terminals in the spinal cord.14,15 However, it remains unclear whether enhanced glutamatergic neurotransmission in the spinal cord is associated with diabetic neuropathy in type 2 diabetic animals. Furthermore, basal extracellular levels of glutamate are also likely a key component in regulating glutamate neurotransmission in the spinal cord. However, little is known about the effects of the development of type 2 diabetes on glutamate homeostasis. Given that type 2 diabetes accounts for most of all diagnosed cases of diabetes in human beings16,17 and obesity-associated type 2 diabetic animals exhibited diabetic neuropathy,18–20 it will be necessary to examine the role of basal extracellular levels of glutamate in the spinal cord in the development of neuropathy in type 2 diabetes.
The extracellular glutamate homeostasis is primarily regulated by the glutamate transporter system, which includes at least five types of glutamate transporters that have been identified as follows: EAAT1 (glutamate aspartate transporter, GLAST), EAAT2 (glutamate transporter-1, GLT-1), EAAT3, EAAT4, and EAAT5.21,22 Approximately 90% of glutamate reuptake is mediated by GLT-1 transporters, which play an important role in ending synaptic transmission of glutamate.21 Previous studies have shown that GLT-1 activation produces antinociceptive effects on visceral and nerve injury-induced neuropathic pain.23,24 Furthermore, systemic treatments of ceftriaxone, which is a beta-lactam antibiotic and has been shown to enhance GLT-1 expression,25 can alleviate mechanical allodynia and hyperalgesia in STZ-induced diabetic rats.26 While numerous studies have demonstrated that STZ-induced diabetic condition is associated with reduced expression and/or impaired activity of GLT-1 in various organs,21–23,27–29 little is known about the effects of the development of type 2 diabetes on the function/expression of GLT-1 in the spinal cord.
Therefore, this study was designed to examine the effects of the development of type 2 diabetes on the basal extracellular levels of glutamate and the expression of GLT-1 in the spinal cord. To this end, we used in vivo microdialysis on Zucker diabetic fatty (ZDF) rats to quantify the basal extracellular levels of glutamate in the spinal cord. In addition, we used Western blot to measure the protein levels of GLT-1 in the spinal cord of ZDF rats. Finally, we evaluated the effects of repeated intrathecal injections of ceftriaxone on the development of mechanical allodynia and hyperalgesia in ZDF rats, as well as the effects of ceftriaxone on basal extracellular levels of glutamate and the expression of GLT-1 in the spinal cord.
Materials and methods
Animals
Male ZDF (fa/fa) rats and control (lean; fa/+) rats at the age of 7 weeks were purchased from Charles River Laboratories (Beijing, China). Upon arrival, rats were acclimated in the Animal Center of Guizhou Provincial People’s Hospital for 1 week before subsequent experiments. All the rats were housed in separated cages in a room with a 12:12 light cycle at 21.0°C–23.0°C and 50%–60% humidity and were given food and water ad libitum. All animal experiments in the current study had been approved by the Institutional Animal Care and Use Committee of Guizhou Provincial People’s Hospital. The housing and treatment of the rats followed the guidelines of the “Guide for the Care and Use of Laboratory Rats” (Institute of Laboratory Animal Resources, Commission on Life Sciences, 2011). The experimental design is shown in Figure 1.
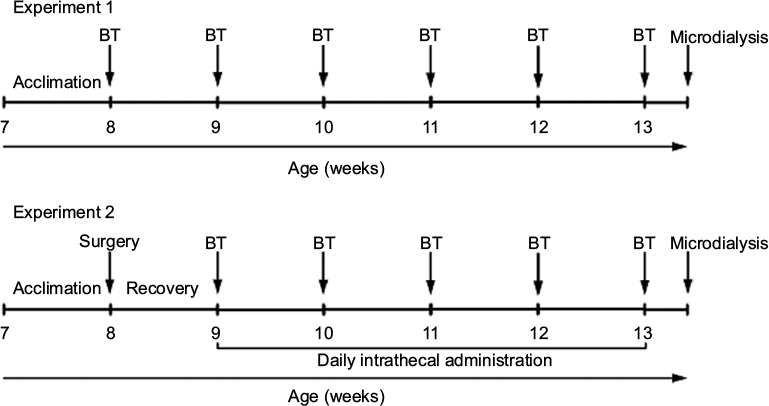 | Figure 1 Experimental design. Notes: In experiment 1, ZDF rats (n = 10) and lean rats (n = 10) were used. In experiment 2, ZDF rats (n = 8–10/dose; total n = 36) were used. BTs were conducted on Thursday and Friday of each week. One day after the last BT, rats underwent in vivo microdialysis experiment followed by sacrifice for spinal cord tissue collection for subsequent Western blot analysis. Abbreviations: BT, behavioral test; ZDF, Zucker diabetic fatty. |
Blood glucose and weight monitoring
Body weight was measured daily, and blood glucose measurements (glucose diagnostic reagents; Sigma-Aldrich, St. Louis, MO, USA) were recorded at the end of each week after the rats arrived in our animal facility. All the rats were fasted for 3 h before blood was collected from the tail. The onset of diabetic conditions was defined as blood glucose levels >13.3 mmol/L. Consistent with the literature, the animals in this study did not develop significant ketoacidosis or prostration during this time period.30,31
Intrathecal catheter implantation
To examine the effects of intrathecal administrations of ceftriaxone on mechanical allodynia and hyperalgesia, separate groups of ZDF rats received intrathecal catheter implantation 1 week after arrival (i.e., at the age of 8 weeks). To this end, rats were fully anesthetized using ketamine/xylazine (80–120 or 10–16 mg/kg, respectively; i.p.). To expose the L4–L5 vertebrae, we made a 1-cm midline incision on the dorsal surface and retracted the muscles. Sterile polyethylene (PE-10 catheter) tubing was then inserted into the subarachnoid space and was advanced 3.5 cm rostrally at the level of the enlarged spinal cord lumbar segments. The catheter was secured to the paraspinal muscle of the back and then tunneled subcutaneously to exit the dorsal neck region, where it was secured to the skin. After the surgery, rats were allowed to recover for 1 week prior to the rest of the experiment. To confirm the position of the PE-10 catheter, we conducted intrathecal injection of 2% lidocaine (15 μL) to observe whether there was paralysis of both hind limbs following injections. Intrathecal administrations of a total volume of 10 μL ceftriaxone (0, 5, 10, and 20 μg/μL; n = 8–10/group) were conducted daily for 4 consecutive weeks.
Behavioral analysis
All the behavioral tests were started 1 week after the rats arrived in our animal facility and were conducted on Thursday and Friday of each week until rats reached the age of 13 weeks. Before the first behavioral testing, rats were acclimated to the behavioral apparatus and equipment for a minimum of 2 days. On test days, rats were placed in the behavioral apparatus and allowed to acclimate to the environment for 30 min.
Mechanical hyperalgesia was quantified using an Ugo Basile analgesiometer (model 37215; probe tip diameter, 1 mm). Briefly, an increasing pressure was applied to the dorsal surface of the right hind paw. The nociceptive threshold was defined as the force, expressed as grams, at which the animals attempted to withdraw its paw. A maximal cutoff of 750 g was used to prevent tissue damage.
On the next day, mechanical allodynia was assessed using an Ugo Basile Dynamic Plantar Esthesiometer (Stoelting, IL, USA). The rigid tip of the esthesiometer, with a diameter of 0.5 mm, was applied perpendicularly to the plantar surface of the hind paw with an increasing force (0–50 g in 20 s) to cause brisk withdrawal. The paw withdrawal latency, defined as the time from the onset of the tip to the withdrawal of the paw, was detected with the electronic esthesiometer. Accordingly, the paw withdrawal threshold was digitally recorded in grams.
In vivo microdialysis
One day after the last behavioral test, microdialysis in the spinal cord was performed to examine the basal levels of glutamate. Briefly, rats were anesthetized using 4% isoflurane and then maintained with 2.5% isoflurane during the experiment. A heating pad was used to maintain the rectal temperature at 36.5 ± 0.5°C. The L3–L6 level of spinal cord was exposed by a T13-L1 laminectomy. A microdialysis probe (optical density (OD) = 0.22 mm, inner diameter (ID) = 0.20 mm, length = 1 mm) was prepared in the laboratory and inserted from just lateral to the right dorsal root and perfused with dialysis buffer (KCl 2.7 mM, NaCl 140 mM, CaCl2 1.2 mM, MgCl2 1.2 mM, plus 0.2 mM phosphate-buffered saline to achieve a pH of 7.4) at 1.0 μL/min.
To determine the basal concentration of extracellular glutamate, glutamate was added to the dialysis buffer to generate a series of points that were interpolated to measure the concentration at which no-net-flux of glutamate occurred across the dialysis membrane. Dialysis buffer was running for 1 h prior to the collection of samples; after 0, 1.0, 2.5, 5.0, and 10.0 µM, glutamate was delivered through the probe. Four 30 min dialysis samples were obtained at each concentration of glutamate, and the last three samples were averaged for the determination of the net flux of glutamate. Samples were then kept at -20°C until they were measured by a high-performance liquid chromatography (HPLC) system.
High-performance liquid chromatography
The glutamate HPLC system consisted of a 25 cm (5 μm) octadecasilane column, a Shimadzu LC-10AD solvent delivery system (Shimadzu Corporation, Kyoto, Japan), a Shimadzu SIL-10AD autosampler and a fluorescence spectrophotometer (Shimadzu RF-10AXL). Samples underwent derivatization (20 μL sample + 20 μL fluoraldehyde) before being injected onto the column via the autosampler. The flow rate of the mobile phase (62 mM NaH2PO4, 0.5% v/v tetrahydrofuran, and 40% v/v methanol, pH 6.3 with 6 N NaOH) was 1.0 mL/min. The fluorescence spectrophotometer detected glutamate with an excitation wavelength of 260 nm and an emission wavelength of 455 nm. The external standard curve ranged from 0.5 to 25 pmol for quantification.
Western blot analysis
Immediately after the microdialysis experiment, rats were sacrificed by live decapitation, and L4–L5 spinal cord tissues from each rat were collected and stored at –80°C. The tissue was homogenized using a Dounce homogenizer in 1 mL of lysis buffer (Sigma-Aldrich), followed by boiling at 100°C for 10 min in a dry heat block. The BioRad DC ProteinTM Assay Kit was used to determine the protein concentration of the samples. Proteins were separated on 9% polyacrylamide gel at 120 V for 1 h and transferred onto a nitrocellulose membrane at 100 V for 1 h. The membrane was then blocked in 3% milk for 1 h and incubated with rabbit polyclonal GLT-1 antibody (1:5000; ab106289; Abcam, Shanghai, China) overnight at 4°C. The membrane was then incubated for 1 h with horseradish peroxidase (HRP)-conjugated donkey antirabbit polyclonal secondary antibody (1:10,000; GE Healthcare, Piscataway, NJ, USA). The film was then developed using an enhanced chemiluminescence (ECL) system (Pierce Biotech, Rockford, IL, USA). Stripping buffer (62.5 mM Tris–HCl [pH 6.7], 2% sodium dodecyl sulfate, 100 mM β-mercaptoethanol) was used to wash the membrane to allow reprobing with antibodies to actin (1:50,000; Santa Cruz Biotechnology, Shanghai, China), which served as a loading control. NIH Image J software was used to quantify the proteins by densitometry. Data were analyzed after GLT-1 levels were normalized to the levels of β-actin and then to lean control or vehicle control.
Immunohistochemistry
Additional groups of rats were killed for immunohistochemistry experiments at the age of 13 weeks. The lumbar spinal cord was removed and immediately placed in a solution containing 60% ethyl alcohol, 10% acetic acid, and 30% chloroform. Spinal cords were placed in 70% ethanol until they were embedded in paraffin after 24 h. Serial sections of spinal cord were cut and used for immunohistochemical analysis. Deparaffinized sections were treated with a solution containing 20 mM Tris, 0.65 mM (EDTA), and 0.0005% Tween 20 (pH = 9.0) and heated by microwave for 10 min. After cooling the slides to room temperature, they were washed in Tris-buffered saline and soaked in 3% hydrogen peroxide. Sections were incubated for 1 h with rabbit polyclonal GLT-1 antibody (1:5000) at 4°C and then for 1 h with biotin-coupled secondary antibody (1:300; Abcam). 3,3-Diaminobenzidine tetrachloride was used for detection (Abcam). Control staining was performed without the primary antibodies. Zeiss Axioimager M1 microscope (Zeiss, Shanghai, China) and software Image-Pro Plus Windows 6.2 (Media Cybernetics, Inc, Bethesda, MD, USA) were used to conduct densitometric analysis on digital images. Densitometric analysis was performed by normalizing to the intensity of the background values of the unlabeled areas (i.e., the OD measured in the white matter interposed between the dorsal horns).
Statistical analysis
Data were expressed as mean ± standard error of the mean. Data were analyzed using unpaired t-test or one-way or mixed-factorial analyses of variance (ANOVA), where appropriate. Significant ANOVA main and interaction effects were further investigated using post hoc tests with Bonferroni correction, when appropriate. Alpha was set at 0.05.
Results
Monitoring body weights and blood glucose levels
We found that body weights of ZDF rats were generally increased faster than lean rats from the time when the rats arrived in our facility (i.e., at the age of 7 weeks) to the end of experiment (i.e., at the age of 13 weeks; all ANOVA main effects, F(1–6, 18–108) = 27.16–43.02, p = 0.0001; Table 1). While the blood glucose levels were similar at the first 2 weeks between lean rats and ZDF rats, blood glucose levels of ZDF rats raised faster and ZDF rats developed hyperglycemic in fasting state (13.8 ± 2.7 mmol/L) at the age of 10 weeks (ANOVA main and interaction effects, F(1–6, 18–108) = 28.89–56.03, p = 0.0001; Table 1).
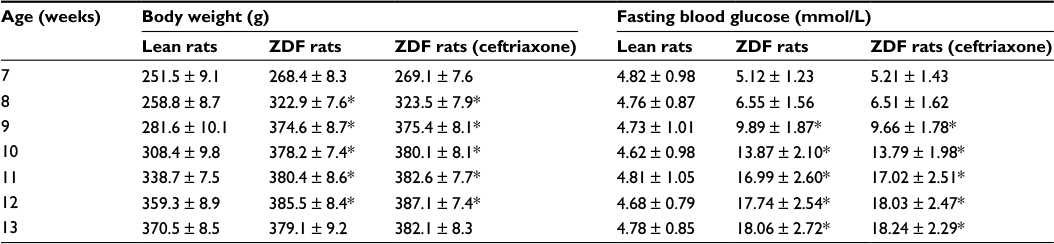 | Table 1 Monitoring body weight and blood glucose levels of ZDF rats Notes: Upon arrival, ZDF and lean rats were monitored for body weight daily and blood glucose every week. Rats were fasted for 3 h prior to the collection of blood from the tail. The onset of diabetic conditions was defined as blood glucose levels >13.3 mmol/L. *The significant difference relative to lean rats (ANOVA main and interaction effects, p < 0.05). Abbreviations: ANOVA, analyses of variance; ZDF, Zucker diabetic fatty. |
ZDF rats exhibited mechanical hyperalgesia and allodynia
We found that ZDF rats developed peripheral neuropathy. Specifically, ZDF rats exhibited mechanical hyperalgesia at the age of 12 weeks, as compared with lean rats (all ANOVA main and interaction effects, F(1–6, 18–108) = 26.79–34.51, p = 0.001–0.02; Figure 2A). Furthermore, ZDF rats also exhibited mechanical allodynia at the age of 12 weeks, as compared with lean rats (all ANOVA main and interaction effects, F(1–6, 18–108) = 21.39–31.27, p = 0.001–0.02; Figure 2B).
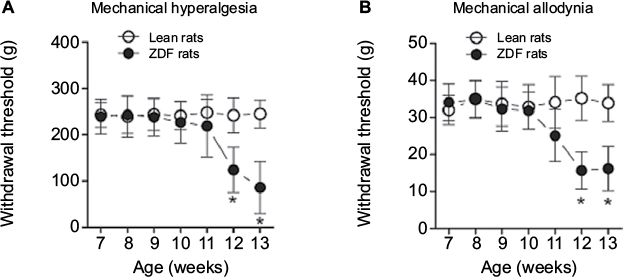 | Figure 2 The development of mechanical hyperalgesia and allodynia in rats (n = 10/rat type). Notes: All behavioral tests were conducted on Thursday and Friday of each week from week 7 to week 13 for 7 consecutive weeks. (A) Mechanical hyperalgesia was quantified using an Ugo Basile analgesiometer. (B) Mechanical allodynia was assessed using an Ugo Basile Dynamic Plantar Esthesiometer. *The significant difference relative to lean rats (ANOVA single week main effect, p < 0.05). Abbreviations: ANOVA, analyses of variance; ZDF, Zucker diabetic fatty. |
ZDF rats exhibited increased basal extracellular levels of glutamate and attenuated levels of GLT-1 expression in the spinal cord
We measured the basal glutamate levels in the spinal cord after 13 weeks and collected the spinal cord tissue for subsequent Western blot analysis. We found that the basal extracellular levels of glutamate in the spinal cord of ZDF rats were increased as compared with lean rats in a similar age (i.e., at the age of 13 weeks; t-test, t(18) = 9.04, p = 0.008; Figure 3A). Furthermore, GLT-1 expression was decreased in the spinal cord of ZDF rats, as compared with lean rats in a similar age (i.e., at the age of 13 weeks; t-test, t(18) = 13.08, p = 0.002; Figure 3B). Finally, immunohistochemistry analysis confirmed that GLT-1 expression was decreased in the dorsal horn of the spinal cord of ZDF rats, as compared with lean rats in a similar age (i.e., at the age of 13 weeks; t-test, t(18) = 11.77, p = 0.02; Figure 3C).
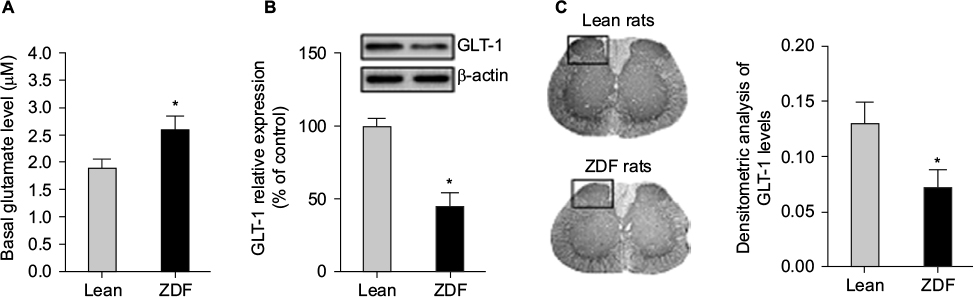 | Figure 3 The effects of the development of type 2 diabetes on glutamate homeostasis in the spinal cord of rats (n = 10/group). Notes: (A) One day after the last behavioral test, in vivo microdialysis with no-net-flux method was used to determine the basal levels of extracellular glutamate in the spinal cord. (B) The L4–L5 spinal cord tissue of ZDF and lean rats were collected immediately after microdialysis experiment. Protein levels of GLT-1 were measured using Western blot. GLT-1 expression levels were normalized to the levels of β-actin, then to lean rats control and are shown as the percentage of lean rat controls. (C) The lumbar spinal cord tissue of ZDF and lean rats were collected at the age of 13 weeks. Serial sections of spinal cord were cut and used for immunohistochemical analysis. Densitometric analysis was performed by normalizing to the intensity of the background values of the unlabeled areas (i.e., the OD measured in the white matter interposed between the dorsal horns). *The significant difference relative to lean rats (t-test, p < 0.05). Abbreviations: GLT-1, glutamate transporter-1; OD, optical density; ZDF, Zucker diabetic fatty. |
Repeated intrathecal administrations of ceftriaxone dose-dependently prevented the development of mechanical hyperalgesia and allodynia in ZDF rats
We started intrathecal administrations of ceftriaxone in ZDF rats at the age of 9 weeks. We found that intrathecal administrations of 5 μg ceftriaxone failed to prevent mechanical hyperalgesia and allodynia in ZDF rats, as compared with vehicle treatment (ANOVA main and interaction effects, F(3–12, 31–124) = 29.13–42.22, p = 0.01–0.001; Figure 4A and B). However, intrathecal administrations of 10 or 20 μg ceftriaxone prevented the development of mechanical hyperalgesia and allodynia in ZDF rats (p < 0.05; Figure 4A and B).
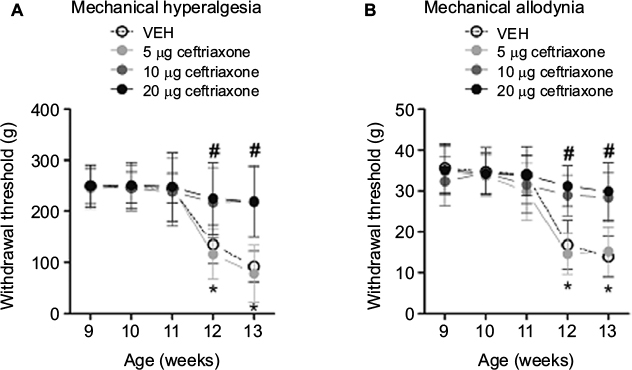 | Figure 4 Effects of repeated intrathecal administrations of ceftriaxone on mechanical hyperalgesia and allodynia in ZDF rats (n = 8–10/dose). Notes: Behavioral testing was conducted on Thursday and Friday of each week after the treatment started. (A) Mechanical hyperalgesia was quantified using an Ugo Basile analgesiometer. (B) Mechanical allodynia was assessed using an Ugo Basile Dynamic Plantar Esthesiometer. *The significant difference relative to week 9 (ANOVA single time main effect, p < 0.05). #The significant difference relative to vehicle (ANOVA single treatment main effect, p < 0.05). Abbreviations: ANOVA, analyses of variance; ZDF, Zucker diabetic fatty; VEH, vehicle. |
Repeated intrathecal administrations of ceftriaxone dose-dependently enhanced GLT-1 expression without altering the basal glutamate levels in the spinal cord of ZDF rats
We measured the basal glutamate levels in the spinal cord after 13 weeks and collected the spinal cord tissue for subsequent Western blot analysis from ceftriaxone-treated ZDF rats. We found that the basal extracellular levels of glutamate in the spinal cord of ZDF rats were not altered after ceftriaxone treatments, as compared with vehicle (F(3, 34) = 1.54, p = 0.17; Figure 5A). Furthermore, GLT-1 expression in the spinal cord of ZDF rats was increased after 10 or 20 μg, but not 5 μg, ceftriaxone treatments, as compared with vehicle (F(3, 34) = 9.72, p = 0.006; Figure 5B).
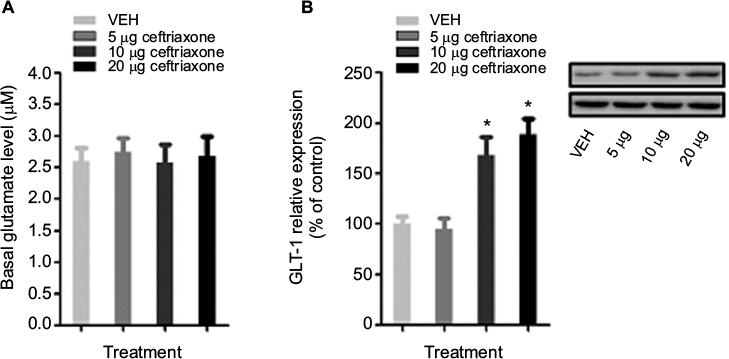 | Figure 5 Effects of repeated intrathecal administrations of ceftriaxone on glutamate homeostasis in the spinal cord of rats (n = 8–10/dose). Notes: (A) One day after the last behavioral test, in vivo microdialysis with no-net-flux method was used to determine the basal levels of extracellular glutamate in the spinal cord. (B) The L4–L5 spinal cord tissue of ZDF and lean rats were collected immediately after microdialysis experiment. Protein levels of GLT-1 were measured using Western blot. GLT-1 expression levels were normalized to the levels of β-actin, then to vehicle control and are shown as the percentage of vehicle controls. *The significant difference relative to lean rats (one-way ANOVA, Bonferroni post hoc test, p < 0.05). Abbreviations: ANOVA, analyses of variance; GLT-1, glutamate transporter-1; ZDF, Zucker diabetic fatty; VEH, vehicle. |
Discussion
This study was designed to evaluate the effects of the development of type 2 diabetes on glutamate homeostasis in the spinal cord. We found that ZDF rats developed mechanical hyperalgesia and allodynia over time, which were consistent with previous studies showing that ZDF rats develop peripheral neuropathy at a similar range of age.32,33 In addition, we found that ZDF rats exhibited enhanced basal extracellular levels of glutamate in the spinal cord, as compared with age-matched lean control rats. Furthermore, ZDF rats exhibited reduced expression levels of GLT-1 in the spinal cord, particularly in the dorsal horn, as compared with age-matched lean control rats. These results indicated that the development of type 2 diabetes may alter the extracellular glutamatergic tone and glutamate reuptake in the spinal cord dorsal horn. Finally, we showed that repeated intrathecal administrations of ceftriaxone dose-dependently prevented the development of mechanical hyperalgesia and allodynia in ZDF rats, as compared with vehicle control rats. This phenomenon is associated with the enhanced GLT-1 expression levels without altering the basal extracellular levels of glutamate in the spinal cord of ZDF rats. These results suggested that restoring impaired glutamate reuptake in the spinal cord may alleviate the neuropathic pain.
Our study suggests that the reduced GLT-1 protein in the spinal cord may be one of the underlying mechanisms that are responsible for the expression of mechanical hyperalgesia and allodynia in rats with type 2 diabetes. However, it should be noted that Western blot and immunohistochemistry analyses only measure protein expression levels of GLT-1, but not function/activity of GLT-1. Therefore, functional studies must be conducted to validate the reduced glutamate reuptake activity following the development of peripheral neuropathy in type 2 diabetes. While type 2 diabetes can induce the reduction of GLT-1 function in the spinal remains unclear, previous studies have shown that diabetes impairs the expression and/or function of various glutamate transporters, including GLT-1. For example, it has been shown that diabetes reduced the activity of GLAST in retinal Müller cells in Long-Evans rats.21 Furthermore, STZ-induced diabetes attenuate GLT-1 protein expression levels in various brain regions.22,23 Adding to this literature, our study indicated that impaired glutamate reuptake via GLT-1 may contribute to the development of peripheral neuropathy in type 2 diabetes.
While few studies have been conducted to investigate the molecular mechanisms that regulate the expression of GLT-1 during the development of type 2 diabetes, previous studies indicated that nuclear factor (NF)-κB may play a critical role in regulating GLT-1 expression. It has been demonstrated that p65 (a subunit of NF-κB) expression was upregulated in the dorsal root ganglions (DRGs) of rats with peripheral neuropathy.25,26 Moreover, several studies have shown that activation of NF-κB is able to increase GLT-1 expression in vitro and in vivo.27,28,34 Thus, future studies will be interesting to examine the effects of the development of type 2 diabetes on the expression and/or activation of NF-κB in astrocytes, where most of GLT-1 is located, in the spinal cord dorsal horn.
Our study also showed that rats with type 2 diabetes exhibited higher basal extracellular levels of glutamate in the spinal cord, as compared with nondiabetic lean rats. It seems that reduction of GLT-1 expression may impair the reuptake of glutamate, resulting in the increased basal extracellular levels of glutamate in the spinal cord. However, the basal extracellular tone of glutamate is also regulated by several other types of glutamate transporters. For example, the activity of GLAST in retinal Müller cells isolated from Long-Evans rats was reduced after STZ-induced diabetes.21,29 On the other hand, glutamate released from non-vesicular sources is regulated by the cystine/glutamate antiporter (xCT).35 While few studies have investigated the effects of the development of diabetes on the expression and/or function of cystine/glutamate antiporters, future studies may need to be undertaken to evaluate the effects of the development of diabetes on the expression and function of various types of glutamate transporters. These additional studies will help us understand the contributing components that are significant in altering the basal extracellular levels of glutamate in the spinal cord during the development of diabetic conditions.
One of the important findings in the current study is that type-2-diabetes-induced peripheral neuropathy can be prevented by repeated intrathecal administrations of ceftriaxone. While our study was the first to report that intrathecal administrations of ceftriaxone can prevent the development of peripheral neuropathy in type 2 diabetes, a previous report has shown that systemic ceftriaxone treatment reduced both mechanical allodynia and hyperalgesia in STZ-induced diabetic rats.36 Thus, these results together indicated that ceftriaxone may be effective to alleviate diabetes-induced peripheral neuropathy in general.
It is not surprising that our study showed that ceftriaxone treatment prevented the loss of GLT-1 expression in the spinal cord. In fact, the effects of ceftriaxone treatments on GLT-1 expression were first described by Rothstein et al.37 After that, numerous studies have confirmed that ceftriaxone treatment can enhance the expression of GLT-1. Specifically, one study showed that ceftriaxone alone can induce the mRNA expression of GLT-1 in the frontal cortex and striatum, but not in the hippocampus.38 Another study reported that the protein levels of GLT-1 were increased in the hippocampus, frontal cortex, and striatum.39 While we cannot rule out that ceftriaxone treatment regimen, animal species, and experimental conditions may influence the effects of ceftriaxone on the dynamics of GLT-1 expression, our results suggested that mitigating the loss of GLT-1 expression in the spinal cord could prevent the development of peripheral neuropathy in diabetes. Surprisingly, ceftriaxone-induced expression of GLT-1 did not alter the basal extracellular levels of glutamate. These results further indicated that basal extracellular tone of glutamate in the spinal cord might not be critical for the expression of peripheral neuropathy in diabetes.
Conclusion
Our study showed that ZDF rats exhibited enhanced basal extracellular levels of glutamate and reduced expression levels of GLT-1 in the spinal cord. This altered glutamate homeostasis in the spinal cord is associated with the development of peripheral neuropathy in type 2 diabetes. Importantly, intrathecal delivery of ceftriaxone can prevent the development of peripheral neuropathy by increasing the GLT-1 expression in the spinal cord without altering the basal glutamate levels, suggesting glutamate reuptake in the spinal cord is critical for the expression of neuropathic pain. However, given the complexity of the system that are responsible for regulating glutamate homeostasis in the spinal cord, it will be important to dissect the putative role of components other than GLT-1 in the spinal cord in the modulation of peripheral neuropathy in diabetes in the future. Such a line of research would not only shed lights on the pathophysiological mechanisms of diabetes-induced peripheral neuropathy but also aid in the development of effective pharmacotherapies for diabetic patients with painful peripheral neuropathy.
Disclosure
The authors report no conflicts of interest in this work.
References
Griessenauer CJ, Raborn J, Foreman P, Shoja MM, Loukas M, Tubbs RS. Venous drainage of the spine and spinal cord: a comprehensive review of its history, embryology, anatomy, physiology, and pathology. Clin Anat. 2015;28(1):75–87. | ||
Sheerin F. Spinal cord injury: anatomy and physiology of the spinal cord. Emerg Nurse. 2004;12(8):30–36. | ||
Obrosova IG. Diabetes and the peripheral nerve. Biochim Biophys Acta. 2009;1792(10):931–940. | ||
Yasuda H, Terada M, Maeda K, et al. Diabetic neuropathy and nerve regeneration. Prog Neurobiol. 2003;69(4):229–285. | ||
Maritim AC, Sanders RA, Watkins JB 3rd. Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol. 2003;17(1):24–38. | ||
Schmader KE. Epidemiology and impact on quality of life of postherpetic neuralgia and painful diabetic neuropathy. Clin J Pain. 2002;18(6):350–354. | ||
Burchiel KJ, Russell LC, Lee RP, Sima AA. Spontaneous activity of primary afferent neurons in diabetic BB/Wistar rats. A possible mechanism of chronic diabetic neuropathic pain. Diabetes. 1985;34(11):1210–1213. | ||
Chen SR, Pan HL. Hypersensitivity of spinothalamic tract neurons associated with diabetic neuropathic pain in rats. J Neurophysiol. 2002;87(6):2726–2733. | ||
Chen X, Levine JD. Hyper-responsivity in a subset of C-fiber nociceptors in a model of painful diabetic neuropathy in the rat. Neuroscience. 2001;102(1):185–192. | ||
Khan GM, Chen SR, Pan HL. Role of primary afferent nerves in allodynia caused by diabetic neuropathy in rats. Neuroscience. 2002;114(2):291–299. | ||
Isaev D, Gerber G, Park SK, Chung JM, Randik M. Facilitation of NMDA-induced currents and Ca2+ transients in the rat substantia gelatinosa neurons after ligation of L5-L6 spinal nerves. Neuroreport. 2000;11(18):4055–4061. | ||
Suzuki R, Matthews EA, Dickenson AH. Comparison of the effects of MK-801, ketamine and memantine on responses of spinal dorsal horn neurones in a rat model of mononeuropathy. Pain. 2001;91(1–2):101–109. | ||
Chen SR, Pan HL. Spinal GABAB receptors mediate antinociceptive actions of cholinergic agents in normal and diabetic rats. Brain Res. 2003;965(1–2):67–74. | ||
Li JQ, Chen SR, Chen H, Cai YQ, Pan HL. Regulation of increased glutamatergic input to spinal dorsal horn neurons by mGluR5 in diabetic neuropathic pain. J Neurochem. 2010;112(1):162–172. | ||
Wang XL, Zhang HM, Chen SR, Pan HL. Altered synaptic input and GABAB receptor function in spinal superficial dorsal horn neurons in rats with diabetic neuropathy. J Physiol. 2007;579(pt 3):849–861. | ||
Mohamadi A, Cooke DW. Type 2 diabetes mellitus in children and adolescents. Adolesc Med State Art Rev. 2010;21(1):103–119,x. | ||
Campbell RK, Neumiller JJ, White J, Sisson E, Kuhn C. Type 2 diabetes: epidemiology and treatment, pathophysiology, new therapeutics, and the evolving role of the pharmacist. J Am Pharm Assoc (2003). 2009;49(suppl 1):S2. | ||
Otto KJ, Wyse BD, Cabot PJ, Smith MT. Longitudinal study of painful diabetic neuropathy in the Zucker diabetic fatty rat model of type 2 diabetes: impaired basal G-protein activity appears to underpin marked morphine hyposensitivity at 6 months. Pain Med. 2011;12(3):437–450. | ||
Oltman CL, Davidson EP, Coppey LJ, Kleinschmidt TL, Lund DD, Adebara ET, Yorek MA. Vascular and neural dysfunction in Zucker diabetic fatty rats: a difficult condition to reverse. Diabetes Obes Metab. 2008;10(1):64–74. | ||
Romanovsky D, Walker JC, Dobretsov M. Pressure pain precedes development of type 2 disease in Zucker rat model of diabetes. Neurosci Lett. 2008;445(3):220–223. | ||
Li Q, Puro DG. Diabetes-induced dysfunction of the glutamate transporter in retinal Muller cells. Invest Ophthalmol Vis Sci. 2002;43(9):3109–3116. | ||
Nagayach A, Patro N, Patro I. Experimentally induced diabetes causes glial activation, glutamate toxicity and cellular damage leading to changes in motor function. Front Cell Neurosci. 2014;8:355. | ||
Coleman E, Judd R, Hoe L, Dennis J, Posner P. Effects of diabetes mellitus on astrocyte GFAP and glutamate transporters in the CNS. Glia. 2004;48(2):166–178. | ||
Zhang HH, Hu J, Zhou YL, et al. Promoted interaction of nuclear factor-kappaB with demethylated purinergic P2×3 receptor gene contributes to neuropathic pain in rats with diabetes. Diabetes. 2015;64(12):4272–4284. | ||
Zhang HH, Hu J, Zhou YL, et al. Promoted interaction of nuclear factor-kappaB with demethylated cystathionine-beta-synthetase gene contributes to gastric hypersensitivity in diabetic rats. J Neurosci. 2013;33(21):9028–9038. | ||
Shi L, Zhang HH, Xiao Y, Hu J, Xu GY. Electroacupuncture suppresses mechanical allodynia and nuclear factor kappa B signaling in streptozotocin-induced diabetic rats. CNS Neurosci Ther. 2013;19(2):83–90. | ||
Ghosh M, Yang Y, Rothstein JD, Robinson MB. Nuclear factor-kappaB contributes to neuron-dependent induction of glutamate transporter-1 expression in astrocytes. J Neurosci. 2011;31(25):9159–9169. | ||
Gupta RK, Prasad S. Differential regulation of GLT-1/EAAT2 gene expression by NF-kappaB and N-myc in male mouse brain during postnatal development. Neurochem Res. 2014;39(1):150–160. | ||
Ward MM, Jobling AI, Kalloniatis M, Fletcher EL. Glutamate uptake in retinal glial cells during diabetes. Diabetologia. 2005;48(2):351–360. | ||
Chen X, Levine JD. Altered temporal pattern of mechanically evoked C-fiber activity in a model of diabetic neuropathy in the rat. Neuroscience. 2003;121(4):1007–1015. | ||
Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol Chem. 2004;279(28):29341–29350. | ||
Yokoi N, Hoshino M, Hidaka S, et al. A novel rat model of type 2 diabetes: the Zucker fatty diabetes mellitus ZFDM rat. J Diabetes Res. 2013;2013:103731. | ||
Shiota M, Printz RL. Diabetes in Zucker diabetic fatty rat. Methods Mol Biol. 2012;933:103–123. | ||
Ji YF, Zhou L, Xie YJ, et al. Upregulation of glutamate transporter GLT-1 by mTOR-Akt-NF-small ka, CyrillicB cascade in astrocytic oxygen-glucose deprivation. Glia. 2013;61(12):1959–1975. | ||
Bridges RJ, Natale NR, Patel SA. System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165(1):20–34. | ||
Gunduz O, Oltulu C, Buldum D, Guven R, Ulugol A. Anti-allodynic and anti-hyperalgesic effects of ceftriaxone in streptozocin-induced diabetic rats. Neurosci Lett. 2011;491(1):23–25. | ||
Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73–77. | ||
Krzyzanowska W, Pomierny B, Budziszewska B, Filip M, Pera J. N-acetylcysteine and ceftriaxone as preconditioning strategies in focal brain ischemia: influence on glutamate transporters expression. Neurotox Res. 2016;29(4):539–550. | ||
Verma R, Mishra V, Sasmal D, Raghubir R. Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury. Eur J Pharmacol. 2010;638(1–3):65–71. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.