")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
Investigational Rho Kinase Inhibitors for the Treatment of Glaucoma
Authors Al-Humimat G, Marashdeh I , Daradkeh D, Kooner K
Received 15 October 2020
Accepted for publication 7 January 2021
Published 25 February 2021 Volume 2021:13 Pages 197—212
DOI https://doi.org/10.2147/JEP.S259297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Ghadeer Al-Humimat1 12 2, Ibtisam Marashdeh2 2, Duaa Daradkeh2 2, Karanjit Kooner1 13 3
1Department of Ophthalmology, University of Texas Southwestern Medical Center, Dallas, TX, USA; 2Department of Ophthalmology, King Hussein Medical Center, Amman, Jordan; 3Department of Ophthalmology, Veteran Affairs North Texas Health Care System Medical Center Dallas, Dallas, TX, USA
Correspondence: Karanjit Kooner
Department of Ophthalmology, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX, 75390, USA
Tel +1 214 648-4733
Fax +1 214 648-2270
Email [email protected]
Abstract: This review provides a comprehensive update on emerging ROCK inhibitors as an innovative treatment option for lowering intraocular pressure (IOP) in glaucoma and aims to describe the structure, mechanism of action, pharmaceutical characteristics, desirable ocular effects, including side effects for each agent. A literature review was conducted using PubMed, Scopus, clinicaltrials.gov, ARVO journals, Cochrane library and Selleckchem. Databases were searched using “investigational Rho kinase inhibitors,” and “glaucoma” as keywords. In addition to this building block strategy, successive fractions were employed to further refine the results. Of the several ROCK inhibitors discovered, only two drugs are currently approved for glaucoma treatment; Netarsudil in the USA and Ripasudil in Japan and China. We identified and reviewed 15 agents currently in laboratory or clinical trials. These agents lower IOP mainly by decreasing outflow resistance through pharmacologic relaxation of the trabecular meshwork (TM) cells and reducing episcleral venous pressure. They have an optimistic safety profile; however, conjunctival hyperemia, conjunctival hemorrhage, pain on instillation, and corneal verticillata are common. Other properties such as neuroprotection (enhancing optic nerve blood flow and promoting axonal regeneration), anti-fibrotic activity, and endothelial cell proliferation may improve the visual prognosis and surgical outcomes in glaucoma. In addition, these agents have the potential to work synergistically with other topical glaucoma medications.
Keywords: rho kinase inhibitors, ROCK, glaucoma, intraocular pressure, trabecular meshwork, Schlemm’s canal
Introduction
Glaucoma is the second leading cause of blindness affecting over 76 million patients worldwide, including over 3 million in the USA.1,2 In glaucoma, there is an obstruction to the outflow of aqueous humor (AH) resulting in elevated intraocular pressure (IOP).3 The drainage of AH is mainly through the conventional pathways [trabecular meshwork (TM) and episcleral veins] and the uveoscleral-uveovortex pathways (Figure 1).3 The dysfunction of the conventional pathway is poorly understood, but increased TM contractility, change in extracellular matrix (ECM) composition, decrease in pore density of the inner wall of Schlemm’s canal and disruption of local regulatory mediators may increase outflow resistance.3
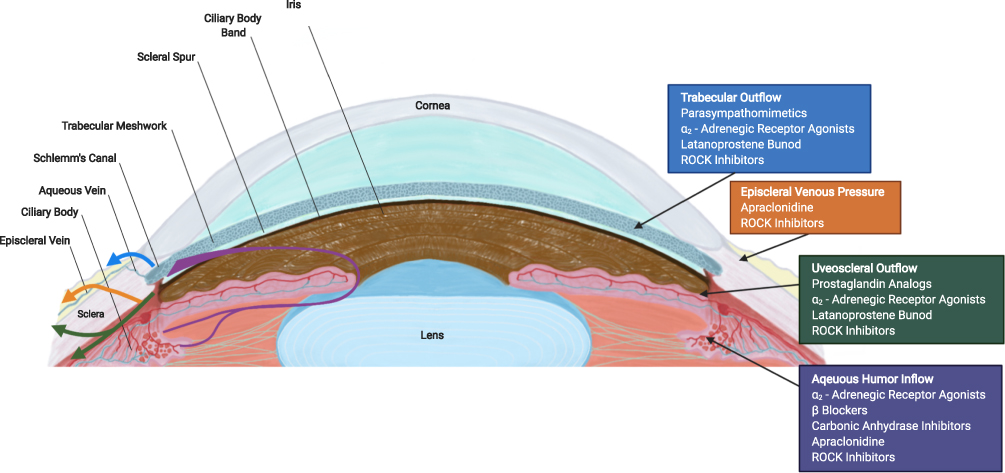 |
Figure 1 Cross section of an eye illustrating aqueous humor (AH) pathways (left) and site of action of antiglaucoma medications (right). AH formation occurs in the ciliary body and flows from the posterior chamber through the pupil to the anterior chamber angle. The drainage of AH is mainly facilitated by the conventional [trabecular meshwork (TM), Schlemm’s canal and episcleral veins] pathway and the non-conventional (uveoscleral-uveovortex) pathway. The current glaucoma hypotensive medications and their sites of action are shown on the right. Abbreviation: ROCK, rho kinase. |
Many studies have proven that reduction of IOP in glaucoma can slow damage to the optic nerve and preserve vision.4 The current glaucoma medications serve to reduce IOP by either decreasing production from the ciliary body or increasing AH drainage through the conventional or uveoscleral-uveovortex pathways (Figure 1). 4
The newest class of ocular hypotensive drugs, rho kinase (ROCK) inhibitors, serves to decrease IOP by inhibiting ROCK, a ubiquitous downstream effector protein that regulates the cell cytoskeleton.5 The role of ROCK in the control of multiple biological events has made it an attractive treatment modality for many eye diseases including glaucoma,5 corneal endothelial dysfunction,6 and diabetic retinopathy.7
Rho/ROCKs Structure and Mechanism of Action (Figures 2 and 3)
RhoA is a small guanosine triphosphatase (GTPase) that activates ROCK when bound to guanosine triphosphate (GTP).8 Two isoforms of ROCK have been identified: ROCK 1 and ROCK 2.9 In general, these kinases consist of an amino-terminal serine-threonine kinase domain that is followed by a coiled-coil-forming region, and other functional motifs at a carboxyl terminus (Figure 2). 8–10 In ROCKs, these motifs include a Rho-binding domain (RBD) and a pleckstrin homology (PH) domain, which has an internal cysteine-rich domain.8–10 The two ROCK isoforms share 65% overall identity in their amino acid sequences, with the highest similarity in their kinase domains (92% identity).11 The carboxyl terminal domain forms an autoinhibitory loop that folds back onto the kinase domain and inhibits the kinase activity of ROCK.12 GTP-bound RhoA binds to the RBD region in ROCK and renders the catalytic domain of ROCK to be accessible to its substrates, which results in the activation of ROCK.12 The guanine nucleotide dissociation inhibitors (GDI) keep the RhoA in the inactive form.13 The GTP-bound RhoA subsequently activates ROCK 1 and ROCK 2. There are other activators of ROCK including arachidonic acid that binds to the PH domain, caspase 3-dependent ROCK 1 cleavage of the inhibitory carboxyl terminus, and caspase 2 and granzyme B dependent ROCK 2 cleavage of the carboxyl terminus.10,14 In addition, the cycling between the two forms, GDP-bound RhoA and GTP-bound RhoA, is regulated by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs).13 The GEFs are themselves activated by receptor tyrosine kinases (RTKs), G protein-coupled receptors (GPCRs), cytokines, and integrins among many others (Figure 3). 8,14
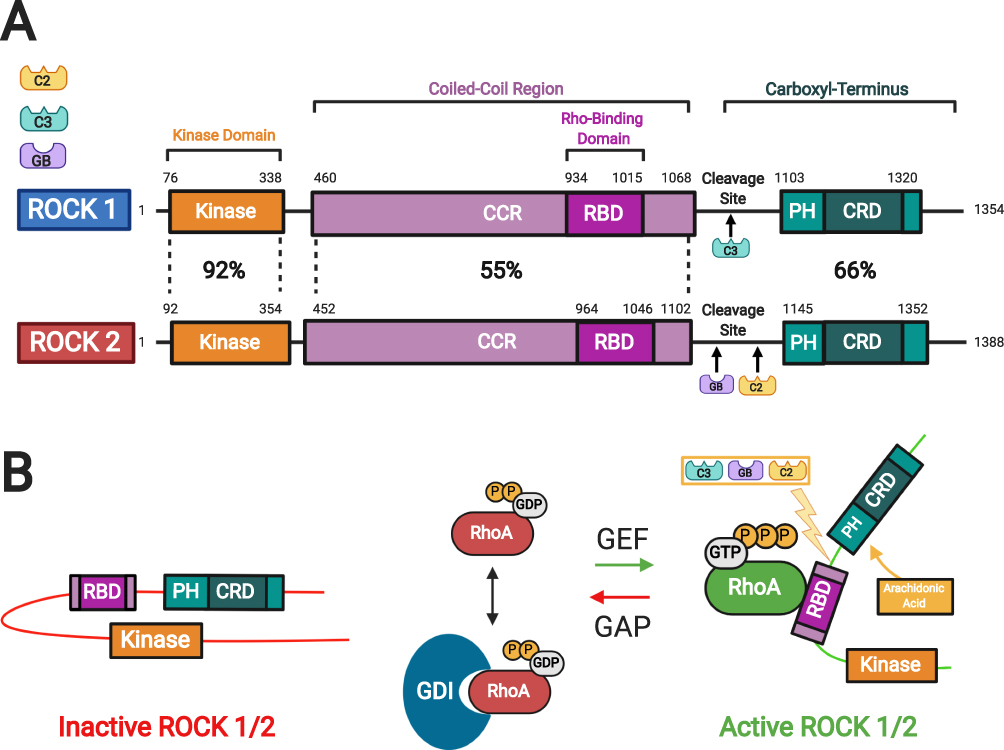 |
Figure 2 Rho kinase (ROCK) structure and mechanisms of activation. (A) Two isoforms of ROCKs have been identified: ROCK 1 and ROCK 2. They consist of a kinase domain, a coiled-coil region (CCR) containing the rho-binding domain (RBD), and the carboxyl terminus. The carboxyl terminus has a pleckstrin-homology (PH) domain with an internal cysteine-rich domain (CRD). The amino acid sequences of the two ROCK isoforms show the highest similarity at the kinase domain (92%). (B) In the inactive ROCK, both PH domain and RBD domain can bind independently to the kinase region forming an auto inhibitory loop. The GDP-bound RhoA is kept inactive by sequestration with guanine nucleotide dissociation inhibitors (GDI). The guanine nucleotide exchange factor (GEF) converts the inactive GDP-bound RhoA to active GTP-bound RhoA. In contrast, GTPase activating protein (GAP) converts the active RhoA to its inactive form. Binding of the GTP-bound RhoA to RBD results in an open conformation of the kinase and frees its catalytic activity. Similarly, ROCK can be activated by arachidonic acid, which binds to its PH domain. ROCK 1 can be activated by caspase-3-mediated cleavage near the carboxyl-terminus while ROCK 2 is activated by caspase-2 and granzyme B-mediated cleavage. Adapted with permission from Wirth A. Rho kinase and hypertension. Biochim Biophys Acta.2010;1802(12):1276–1284. Copyright © 2010 Elsevier B.V. All rights reserved.10 Abbreviations: C2, Caspase 2; C3, Caspase 3; GB, granzyme B; ROCK, rho kinase; CCR, coiled-coil region; RBD, rho-binding domain; PH, pleckstrin-homology; CRD, cysteine-rich domain; GDP, guanosine diphosphate; GTP, guanosine triphosphate; P, phosphate; GDI, guanine nucleotide dissociation inhibitor. |
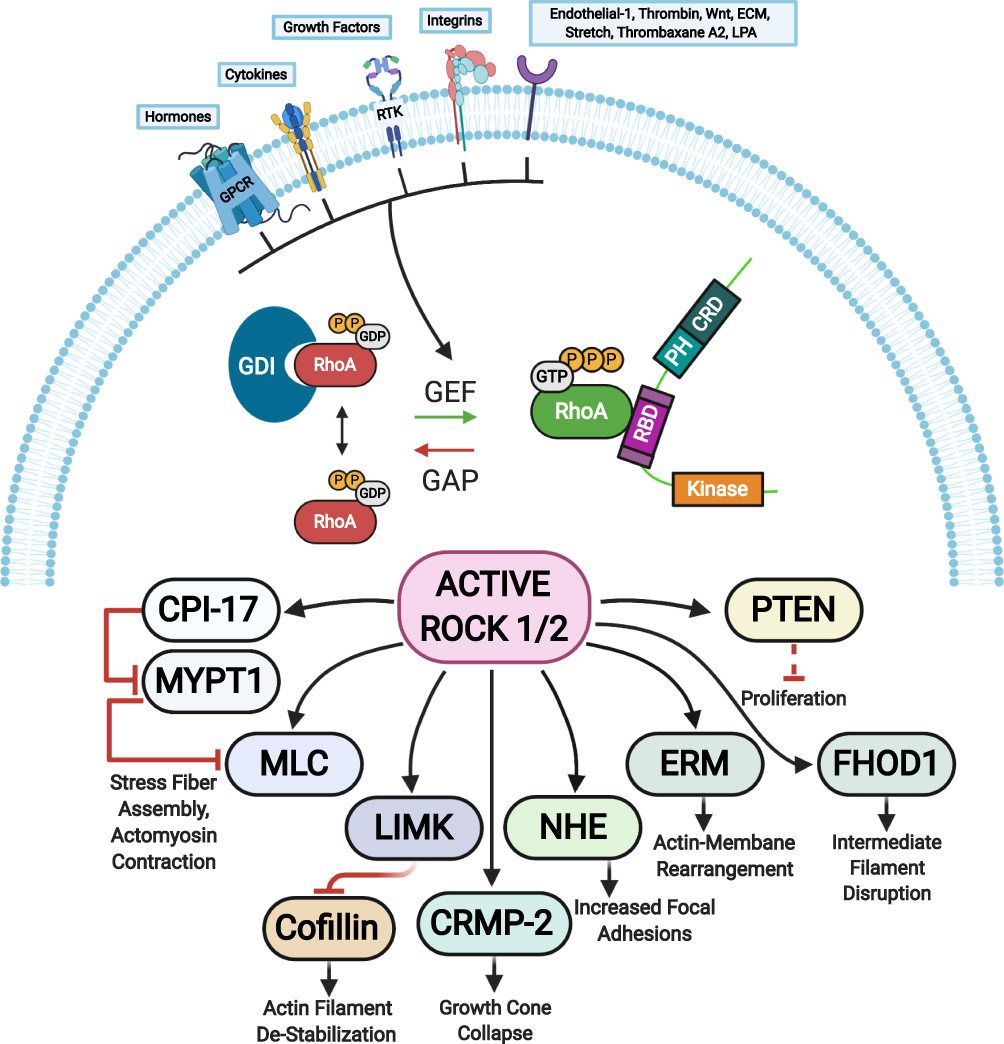 |
Figure 3 ROCK targets. Rho proteins can be activated by guanine nucleotide exchange factors (GEFs). GEFs are themselves activated and regulated via engagement of various receptors at the plasma membrane. The active GTP-bound Rho subsequently activates ROCK 1 and ROCK 2 that phosphorylate various substrates resulting in diverse cellular responses. The barred-line notation indicates inhibition and the arrows show the cascade of the molecular response. Adapted from Hartmann S, Ridley AJ, Lutz S. The function of rho-associated kinasesROCK1 and ROCK2 in the pathogenesis of cardiovascular disease.Front Pharmacol. 2015;6(276):1–16. Copyright © 2015 Hartmann, Ridley and Lutz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) https://creativecommons.org/licenses/by/4.0/.14 Abbreviations: CPI-17, kinase C-potentiated phosphatase inhibitor of 17 kDa; CRMP-2, collapsin response mediator protein 2; ECM, extracellular matrix; ERM, ezrin-radixin-moesin; FHOD1, formin homology 2 domain-containing 1; GAP, GTPase activating protein; GPCRs, G protein-coupled receptors; LIMK, LIM-kinase; LPA, lysophosphatidic acid; MLC, myosin II regulatory light chain; MYPT1, myosin phosphatase target subunit 1; NHE, Na+/H+ exchanger; PTEN, phosphatase and tensin homolog deleted on chromosome 10; RTK, receptor tyrosine kinases; GEF, guanine nucleotide exchange factor; GDI, guanine nucleotide dissociation inhibitor; P, phosphate; GTP, guanosine triphosphate; RhoA, Ras homolog family member A. |
The active ROCK phosphorylates a number of substrates including myosin light chain (MLC), myosin phosphatase substrate 1 (MLCP1), MLCP inhibitor phosphoprotein (CPI-17), formin homology domain protein 1 (FHOD 1), the thin filament regulator h-calponin, the ERM (ezrin, radixin, moesin) proteins, LIM (Lin11, Isl1 and Mec3) kinase, myosin phosphatase target subunit (MYPT-1), phosphatase and tensin homologue (PTEN) deleted on chromosome 10, Na/H-exchanger (NHE-1), and collapsin response mediator protein-2 (CRMP-2).8,14 These factors serve a number of cellular roles including cell adhesion, migration, proliferation, actomyosin contraction, transformation, neurite retraction, phagocytosis, and apoptosis.14
ROCK Inhibitors and Their Role in Glaucoma
The mechanism of ROCK inhibitors’ ability to decrease IOP is through the direct action of relaxation on the TM and Schlemm’s canal cells causing increased permeability, which reduces the resistance of AH outflow.8 ROCK inhibitors upregulate antioxidative molecules in TM cells, inhibit reactive oxygen species (ROS) production, and promote cell survival. Oxidative stress is a major physiological phenomenon, mediated through the production of ROS. ROS play an important role in cell homeostasis and pathogen response and are therefore essential in biological processes. However, increases in ROS are seen in various age-related diseases including glaucoma.15 Some ROCK inhibitors decrease AH production through inhibition of norepinephrine transporters (NETs) via vasodilatory effects on uveoscleral drainage and episcleral veins.16,17
Besides the effect of ROCK inhibitors on the TM, animal studies have shown other potential mechanisms beneficial for treating glaucoma:8 reduced post-operative scarring after glaucoma filtration surgery;18 improved blood flow to the optic nerve, presumably through the relaxation of vascular endothelial smooth muscle cells;19 neuroprotection and regeneration of retinal ganglion cells (RGCs);19 and increase in corneal endothelial cell density and restoration of the damaged endothelial cells (Figure 4). 8
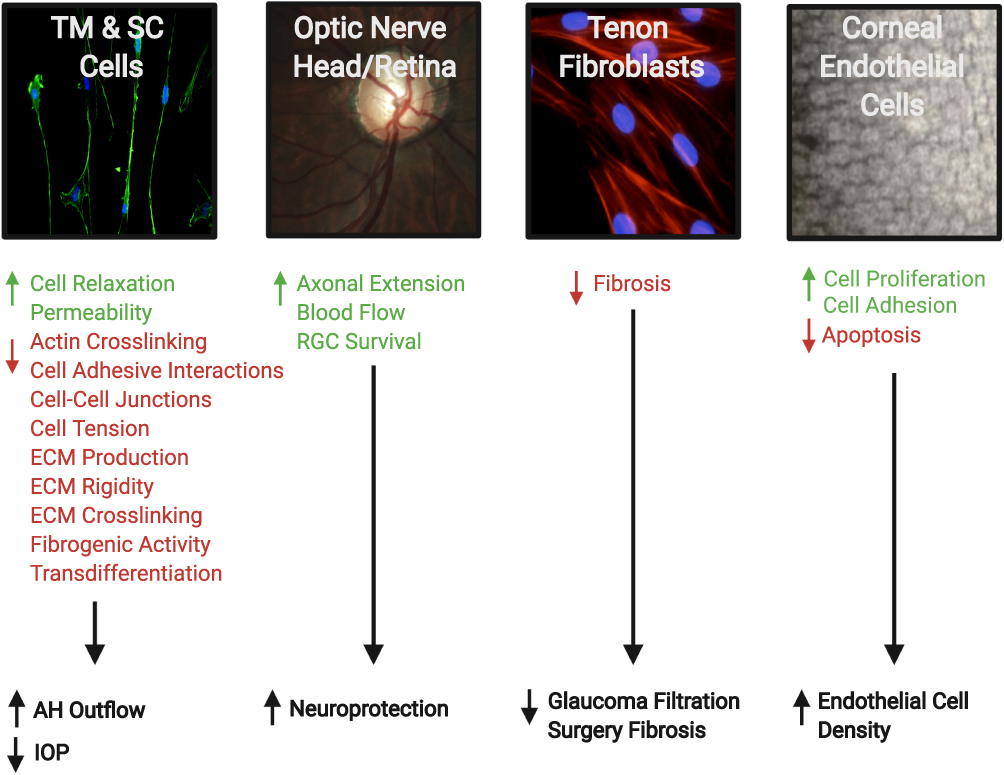 |
Figure 4 Therapeutic effects of Rho kinase (ROCK) inhibitors in treatment of glaucoma. They can increase aqueous humor (AH) outflow and decrease intraocular pressure (IOP) by targeting the cytoskeleton of the trabecular meshwork (TM) and Schlemm’s canal (SC) cells. ROCK inhibitors also have a neuroprotective effect by increasing blood flow to the retina and optic nerve, promoting axonal regeneration, and increasing retinal ganglion cell (RGC) survival. In addition, the inhibitory effect on Tenon fibroblasts decreases the fibrosis after filtration surgery. Finally, on the corneal endothelium they enhance cellular proliferation, promote adhesion, and suppress apoptosis. Accordingly, they provide a greater potential for the regeneration of damaged corneal endothelium and restoration of corneal transparency. The photos shown were taken at the Departments of Ophthalmology at UT Southwestern Medical Center Dallas, Texas, USA and King Hussein Medical Center Amman, Jordan. Adapted with permission from Rao PV, Pattabiraman PP, Kopczynski C. Role of the rho GTPase/rho kinase signaling pathway in pathogenesis and treatment of glaucoma:bench to bedside research. Exp Eye Res. 2017;158:23–32. © 2016 Elsevier Ltd. All rights reserved.8 Abbreviation: ECM, extracellular matrix. |
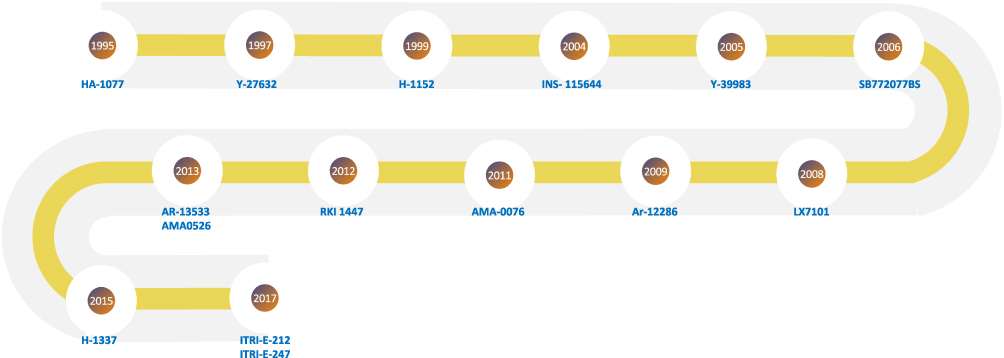 |
Figure 5 Timeline map showing the years the investigational agents were introduced (Table 1). |
Current ROCK Inhibitors
Netarsudil (AR-13,324): Netarsudil is a ROCK/NET inhibitor developed by Aerie-Pharmaceuticals Inc. (Durham, NC, USA) introduced for clinical use in the US in 2017.16 It is a novel ocular hypotensive agent used in patients with primary open-angle glaucoma (POAG) or ocular hypertension (OHT). It lowers IOP primarily by increasing outflow through TM in addition to decreasing both AH production and episcleral venous pressure.16,17 Metabolism of Netarsudil (AR-13,324) by esterases produces the main active metabolite AR-13,503.
The Rho kinase-elevated IOP treatment (ROCKET) trials investigated the efficacy and safety of Netarsudil (0.02%) and compared it with timolol maleate (0.5%) twice daily (BID).20 Efficacy data from the ROCKET series, including ROCKET-1, ROCKET-2, and ROCKET-4 (ROCKET-3 was solely a safety study), of Phase 3 clinical trials were pooled and analyzed. The pooled population included 839 patients treated with Netarsudil once daily (QD) and 839 treated with timolol BID. Analyses found that Netarsudil had IOP-lowering efficacy that was non-inferior to that of timolol and was generally well tolerated with respect to both systemic and ocular events. Mean treated IOP ranged from 16.4 to 18.1 mmHg among Netarsudil treated patients, and 16.8 to 17.6 mmHg among timolol-treated patients, with the mean IOP reductions from baseline up to 4.8 mmHg and 5.0 mmHg, respectively. The most frequent adverse events among Netarsudil QD treated patients were conjunctival hyperemia in nearly 50% of subjects, cornea verticillata (20.9%), and subconjunctival hemorrhages (17.2%).
PG324: In 2019, Aerie Pharmaceuticals also introduced PG324 (Rocklatan ®), an FDA approved fixed-dose combination of Netarsudil (0.02%) and latanoprost (0.005%).21 Two Phase 3 studies (MERCURY 1 and MERCURY 2) showed that this combination significantly lowered IOP and was superior to both Netarsudil and latanoprost alone.21 The most common adverse event was conjunctival hyperemia (63%), followed by instillation site pain (23%), conjunctival hemorrhage (13%), and cornea verticillata (17.6%).21
Ripasudil (K-115): Ripasudil hydrochloride hydrate, (0.4% or Ripasudil) a fluorinated analog of Fasudil, is a small molecule ROCK inhibitor developed and introduced by Kowa Company, Ltd (Naka-ku, Nagoya, Japan) for treatment of glaucoma and OHT in 2014.22 Phase 1 and Phase 2 clinical trials as well as a 24-hour time-course study established Ripasudil 0.4% BID as a clinically useful concentration and dosing frequency for the treatment of glaucoma and OHT.
The 0.4% solution lowered IOP on average by 2–4.4 mmHg 2-hour post-instillation in patients with glaucoma or OHT and continued to deliver statistically significant pressure reduction for at least 7 hours. The efficacy of Ripasudil as monotherapy and as an adjunctive medication to prostaglandin analogs, β blockers, and/or carbonic anhydrase inhibitors has been confirmed in clinical trials.
A non-comparative, year-long, open-label study reported IOP reduction of 2.6–3.7 mmHg from baseline with POAG or OHT patients after monotherapy.23 Side effects of Ripasudil included dose-dependent conjunctival hyperemia and non-dose dependent conjunctival hemorrhage. Nonocular side effects were rare and generally not severe: constipation (0.6%), headache (0.1%), dizziness (0.1%), nausea (0.1%), and others.
Additional ophthalmic uses include the ability to promote corneal endothelial cell proliferation in cultured human cells, prevent excessive scarring after glaucoma filtration surgery by attenuating the activation of conjunctival fibroblasts, and the potential to reduce foveal thickness in patients with diabetic retinopathy.24 A Phase 2 study is underway for the treatment of diabetic retinopathy. Ripasudil has also been shown to have neuroprotective activity in rodent optic nerve crush injury models.
Investigational ROCK Inhibitors (Table 1)
HA-1077 [Fasudil (C14H17N3O2S)]
Since 1995, an oral form of HA-1077 has been available in Japan as Fasudil (Asahi Kasei Pharma Co., Tokyo, Japan) and has been used to treat a variety of vascular conditions including cerebral vasospasm, angina, hypertension, and coronary vasospasm.25
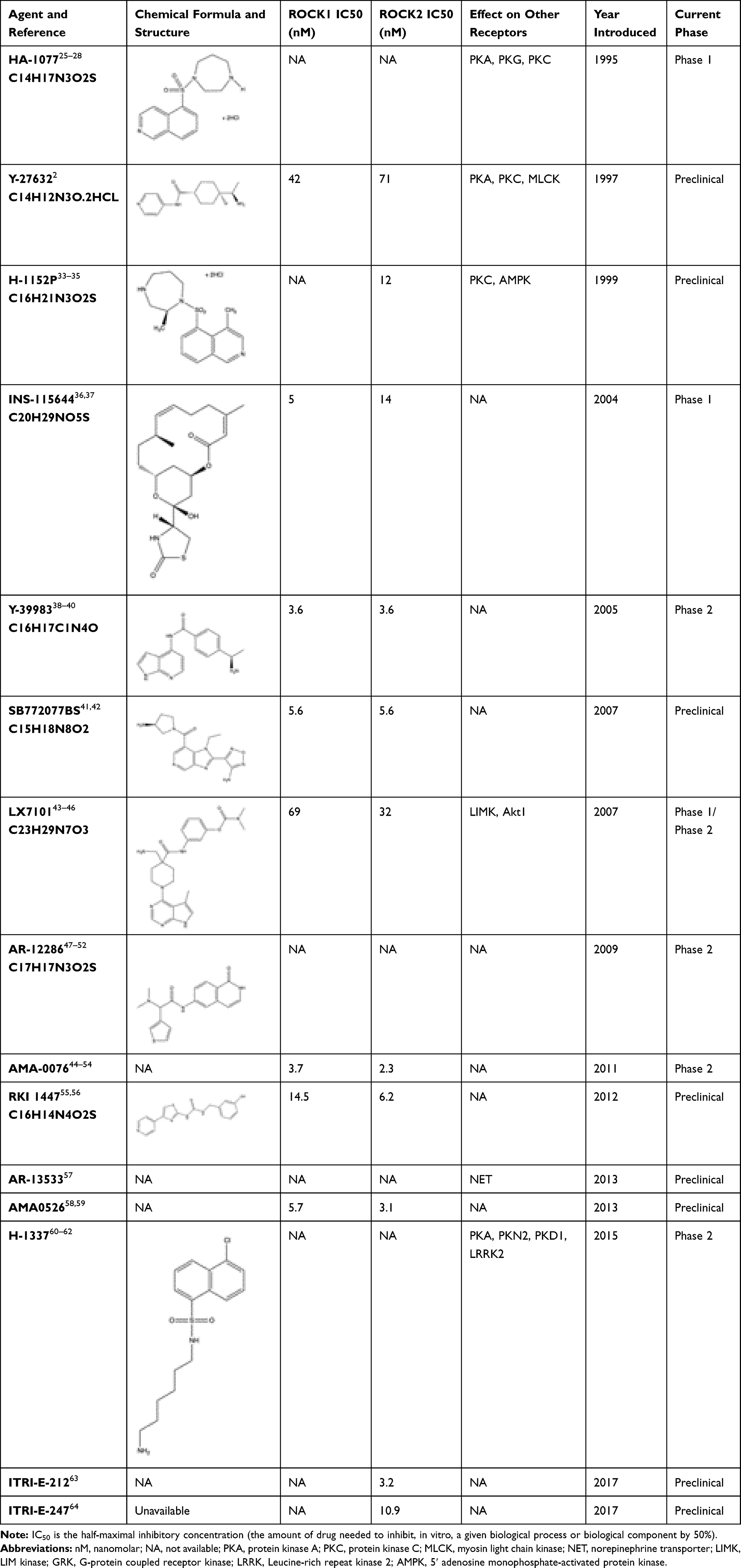 |
Table 1 Current Investigational Rho Kinase Inhibitors |
Fukunaga et al25 demonstrate the ocular hypotensive effects of Fasudil in hypertensive New Zealand White Rabbit models. After water loading to induce hypertension, 40 μL of 1 mM, 2 mM, and 3 mM HA-1077 was administered to the left eye of six rabbits in three different groups and 40 μL of phosphate-buffered saline was administered to all right eyes as control. Dose-dependent maximum IOP reductions were observed as 17.4%, 27.3%, and 46.6% for 1 mM, 2 mM, and 3 mM solutions, respectively, at 120 minutes after instillation. Of note, conjunctival injection in the 1 mM and 2 mM doses was not observed while a few 3 mM treated eyes were slightly injected.
Fasudil has also been shown to increase outflow facility (OF), induce retraction and rounding of TM cells, disrupt TM actin filament organization in rabbit eyes26 and reduce myofibroblast differentiation and proliferation of scleral fibroblasts induced by transforming growth factor-beta 1 (TGFβ1).27
In a small case series, Fasudil 0.5% was topically administered to three eyes and 1.2% to one eye with severe glaucoma with a mean morning IOP of 51–58 mmHg. The drug showed a dose-dependent response with IOP reductions of 21.2% for 1.2% Fasudil and 17.9% for 0.5% Fasudil. Adverse effects included conjunctival hyperemia with the 1.2% dose and instillation site irritation, but no apparent systemic side effects.28
Y‐27632 (C14H21N3O·2HCl)
Y‐27632 was introduced in 1997 and has been widely used in stem cell research.29 Administration of topical (100 and 1000 μM), intracameral (10, 100, and 1000 μM), and intravitreal (100 and 1000 μM) doses of this agent to rabbit eyes all resulted in decreased IOP, with maximal reduction observed between 1 and 3 hours for the 100 mM topical dose and 6 and 9 hours for the 1000 mM intracameral dose.30 Additionally, Y-27632 showed approximately two times higher OF compared with control eyes (0.24 ± 0.02 μL/min per mmHg).30
It has also been shown that in vitro administration of Y-27632 protects against post-surgical scarring by reduction of collagen gel contraction, unlike other anti-glaucoma drugs, eg timolol, latanoprost, which may induce fibrosis.31 This agent prevented fibrosis through inhibition of both the transdifferentiation of Tenon fibroblasts into activated myofibroblasts and TGF-β1 signaling through suppression of the mitogen‐activated protein kinase (MAPK) pathway.31
Additionally, Y-27632 selectively inhibits ROCK 1 (although it does inhibit other protein kinases at higher concentrations) and indirectly increases the activity of smooth muscle protein phosphatase 1 associated with myosin (SMPP-1M), leading to MLC dephosphorylation.32 It was found that 1, 10 and 100 μM administration of the agent dose-dependently increased OF in monkey eyes by 2 ± 19%, 55 ± 19%, and 206 ± 19%, respectively.32
H-1152 or H-1152P [Dimethylfasudil (C16H21N3O2S)]
In 1999, H-1152 was studied and developed by Ikenoya et al as a selective and potent ROCK inhibitor.33 This agent, derived from HA-1077 (Fasudil), exhibits superior ROCK selectivity and ROCK 2 specificity than its parent drug. H-1152 remains in the preclinical phase.
Pitha et al investigated the prevention of TGFβ1–induced transdifferentiation of cultured scleral fibroblasts to myofibroblasts.27 Using a 10 μM dose, they found that H-1152 in vitro reduced smooth muscle actin expression by 72% and collagen gel contraction by 33%.
Nishio et al studied the effects of H-1152 in normotensive and OHT rabbit eyes and found potent IOP lowering activity. The drug was topically administered at doses of 0.1, 1, 10, and 28 mM.34 In the normotensive eyes, the maximum IOP reduction observed was 3.6 ± 0.9 mmHg (26.9 ± 5.9%), 5.4 ± 0.7 mmHg (36.0 ± 4.0%), 6.8 ± 0.7 mmHg (48.4 ± 2.1%), and 7.2 ± 1.9 mmHg (46.1 ± 5.0%), respectively, with dose-dependent duration of IOP lowering. In the OHT model, basal IOP was elevated from 11.8 ± 1.5 mmHg to 26.3 ± 0.9 mmHg after 30 minutes of water loading, and maximum IOP reduction for 40 μL of 1% (28 mM) H-1152 was 10.6 ± 2.3 mmHg at 90 minutes after water loading. No serious side effects were seen in ocular tissues except for conjunctival congestion, which resolved within 3 hours.
The agent also proved to be species specific with IOP reduction efficacy stronger in rabbits (46.1 ± 5.0%) than rats (28.1%).34 Additionally, it increased OF in human-cultured trabecular meshwork cells (HTMCs) and enucleated porcine eyes.35
INS-115644 [Latrunculin-B (C20H29NO5S)]
Introduced in 2004, INS-115644 was produced by the Wisconsin Alumni Research Foundation and developed by Inspire Pharmaceuticals (Durham, NC, USA) in 2006, which was acquired by Merck & Co in 2011.
It is a unique marine toxin that increases OF by structural geometry changes of TM cells and the impairment of cellular contractility. Specifically, the agent disrupts microfilament organization and inhibits the assembly of actin microfilaments by 1:1 molecular binding of free actin monomers in the cytoplasm, resulting in cellular relaxation as well as loosened cell-cell junctions and weakened focal contacts between cells and the surrounding matrix.36
INS-115644 has also been reported to decrease TGFβ1-induced transformation of fibroblasts to myofibroblasts and modulate the expression of multiple ECM proteins in human TM cells. Thomasy et al hypothesized that this agent would decrease fibroblast to myofibroblast transformation particularly when cells were cultured on stiff substrates in vitro and would decrease corneal fibrosis post stromal injury in vivo.37 Using a dose and dose-frequency that reduced IOP in human glaucoma patients, they determined that topical 0.01% INS-115644 BID did not significantly impact corneal epithelial wound closure, stromal haze formation, or endothelial cell density in a rabbit phototherapeutic keratectomy model.37
In a Phase 1/Phase 2 study, INS-115644 was topically administered as 0.005%, 0.01%, 0.02%, and 0.05% to 14 subjects in each of four cohorts with early POAG or OHT to evaluate its safety and efficacy.36 After the fifth instillation of the drug, IOP decreased by 5.4 ± 2.4 mmHg (24%) from a baseline of 22.9 ± 2.4 mmHg, in the 0.02% cohort and 2.8 ± 2.7 mmHg (12%) from a baseline of 23.5 ± 3.1 mmHg in the 0.05% cohort at 4-hour post-instillation. Lower doses did not significantly decrease IOP. Adverse events included mild redness, irritation, conjunctival erythema/hyperemia, and a transient increase (≤2.5%) in corneal thickness.
Y-39983, SNJ-1656 or RKI983 (C16H16N4O·2HCl)
Y-39,983, SNJ-1656 or RKI983 were initially identified in 2005 by Mitsubishi Pharma Corporation (now Mitsubishi Tanabe Pharma, Chūō-ku, Japan) as a selective ROCK inhibitor with 30 times increased inhibitory potency than Y-27632.38
Shan et al cultured HTMCs from normal donors and incubated them with Y-39,983 (0.01–1 mM doses), and then observed cellular motility and morphological changes after 2 to 3 days of treatment.39 They found that the drug altered the intracellular architecture and cellular motility in a way that suppressed the migratory capacity of HTMCs. Additionally, 20 proteins were differentially regulated in the 1 mM treated cells, including downregulation of collagen-producing proteins (COL1A1) and proteins mediating cell-cell and cell-matrix interactions (TSP1) as well as upregulation of proteins facilitating glycosaminoglycan breakdown (GUSB).
Tokushige et al investigated the effects of Y-39,983 on blood flow in the optic nerve head in rabbits and axonal regeneration of RGCs in rats.19 The rabbits received a topical dose of 0.05% Y-39,983, and the maximum increase in blood flow (measured by the laser speckled method) was 122.84 ± 5.98% at 90-minute post-administration. This revealed that topical administration may reach the posterior retina at pharmacologic concentrations, possibly through the periocular and posterior scleral routes. The neurites of rats in vitro, after being treated with 10 μM Y-39,983, were found to be extended compared to those without treatment. Similarly, in vivo rat studies showed a dose-dependent increase in RGCs with regenerating axons; 99.3 ± 10.5 and 169.5 ± 43.3 cells/mm2 for 10 μM and 100 μM, respectively, a significant increase compared to control rats (43.3 ± 6.0 cells/mm2).
In 2007, a Phase 1 clinical trial was conducted to investigate the efficacy and safety of SNJ-1656 with single-instillation and prolonged-repeated instillation trials.38 For the single-instillation trial, 45 healthy volunteers were treated with SNJ-1656 in concentrations of 0.003%, 0.01%, 0.03%, 0.05%, and 0.1% in a stepwise fashion. Doses ranging from 0.003% to 0.1%, topical administration yielded a dose-dependent IOP reduction. Maximal IOP reduction was 3.00 ± 1.16 mmHg from baseline at 4-hour post-instillation with 0.1% dose, with IOP returning to near-baseline during the next 24 hours. In the repeated-instillation trial, 36 healthy volunteers received SNJ-1656 at 0.05% QD, 0.1% QD, 0.05% BID, or 0.1% BID. Daily administration decreased IOP levels post-instillation at 9 AM on each day, whereas such a pattern of changes was unclear with BID administration. Adverse effects for both trials included transient hyperemia of the bulbar and palpebral conjunctiva in the eyes for the 0.05% and 0.1% doses.
In 2015, a Phase 2 study was performed to evaluate the use of Y-39,983 in patients with POAG or OHT.40 Sixty-six subjects received placebo, 0.03%, 0.05% or 0.1% dose BID for 7 days. After 7 days, changes in IOP from pre-instillation baseline of morning eye drops were 2.2 ± 1.9, 3.8 ± 2.7, 4.3 ± 2.3 and 4.0 ± 2.5 mmHg, respectively. Seventy-five treatment-related adverse events in 41 subjects were reported (15 with 0.03%, 31 with 0.05%, 28 with 0.1%, one with placebo), with conjunctival hyperemia being the most frequently reported event and one subject developing hepatic dysfunction, which resolved after discontinuation of treatment.
SB772077B/SB77 (C₁₅H₂oCl₂N₈O₂)
SB772077B (SB77) was introduced in 2007 by GlaxoSmithKline (Brentford, England) as a novel and highly selective amino-furan ROCK inhibitor with an anti-inflammatory activity that is more selective for ROCK 1 than ROCK 2. The interaction of SB77 with other kinases has not been thoroughly studied and the drug remains in the preclinical stages of development.
In 2018, Ashwinbalaji et al analyzed the IOP lowering property of SB77 in a human anterior segment organ-culture.41 Anterior segments at baseline equilibrium were exchanged with 5 mL of SB77 at three separate doses (0.1 μM, 10 μM, and 50 μM) while the IOP was maintained at 8 mmHg. Spontaneous pressure was then re-established by continued perfusion at 3 μL/minute with the medium containing the test drug. Following a 24-hour treatment, SB77 induced dose-dependent effects and increased OF by 16%, 29%, and 39%, for the 0.1 μM, 10 μM, and 50 μM doses, respectively. Additionally, the group studied how SB77 treatment altered cell morphology to gain insight into the molecular mechanism responsible for its OF enhancing properties. They found that SB77 induced alterations in cell shape leading to stellate appearance and reduction in actin stress fiber formation, cytoskeletal and ECM proteins.
In 2020, the same group used a perfusion culture model to investigate the effect of cyclic biomechanical stress, in the presence or absence of SB77, induced by a positive displacement piston pump used to deliver IOP pulsations.42 They found that cyclic stress resulted in a decrease in aqueous OF by 19.79 ± 4.93% in perfused human eyes, and treatment with 50 µM SB77 significantly enhanced OF by 15%. Results were confirmed with the inactivation of ROCK signaling and decreased expression of ECM markers.
LX7101 (C23H29N7O3)
LX7101 is a dual LIM-kinase (LIMK) and ROCK inhibitor produced by Lexicon Pharmaceuticals (The Woodlands, TX, USA) in 2007, with LIMK inhibition hypothesized as the primary mechanism of action.43 LIMK are cytoskeletal regulators that phosphorylate and deactivate cofilin, a protein that depolymerizes actin. LIMK inhibitors are believed to lower IOP by inducing depolymerization of actin filaments in the TM, leading to relaxation of the tissue. LIMK is downstream from ROCK with effects more focused on actin polymerization, leading to the hypothesis that these drugs may lower IOP with fewer side effects than pure ROCK inhibitors. Potent inhibition of Akt1 was also identified with an IC50 of less than 1 nM.44
Harrison et al discovered a class of pyrrolopyrimidine LIMK inhibitors shown to reduce IOP by 3.6–3.7 mmHg with a 5 μg dose in dexamethasone-induced OHT mouse models.45 The compounds increased OF by 30% at 100 nM in porcine perfusion assays; however, it suffered from poor aqueous stability. The group revised the compound to a more stable form that achieved a mean IOP reduction of 4.2 mmHg following topical administration of 3 μL of 1 mg/mL hydroxypropyl methylcellulose (HPMC) based aqueous solution to OHT mouse models.43 The compound also achieved greater IOP reduction using 3 μL of 5 mg/mL HPMC-based aqueous solution when compared to timolol and latanoprost administered at clinically standard dosages to mouse models. This compound was chosen for use in Phase 1 clinical trials and its structure was disclosed in 2015.
The drug completed Phase 1/Phase 2 clinical trials from March 2012 to June 2012.46 Preliminary results of this double-masked, randomized, controlled study showed LX7101 was well tolerated amongst the 63 participants of the study at all doses evaluated (0.125% and 0.25%), with no serious side effects or withdrawals due to adverse events at 2 weeks. Mean IOP changes from baseline at day 14 for each LX7101 dose arm compared to vehicle at 8-hour post-dose were 3.18 mmHg and 2.32 mmHg for 0.125% and 0.25%, respectively, compared to the 0.40 mmHg vehicle. Reductions from baseline at day 14 in the diurnal mean IOP, representing mean changes across all daily time points, were 3.37 mmHg and 3.52 mmHg for 0.125% and 0.25%, respectively, compared to 2.17 mmHg for the vehicle.
AR-12286 [Verosudil (C₁₇H₁₇N₃O₂S)]
Discovered by Aerie pharmaceuticals in 2009, AR-12286, an aminoisoquinoline amide, is a selective ROCK inhibitor with dual ROCK 1/ROCK 2 activity. In 2009, Wang et al demonstrated the mechanism of IOP reduction by showing an increase in OF of 39% and reduction of IOP by 19% after topical administration of 0.6% AR-12286 in normotensive monkey eyes.47 Lin et al found AR-12286 lowered IOP by 5.6 ± 0.3 mmHg 4-hour post-dose after 3 days of topical administration of 0.5% solution in monkey eyes.48 Most recently, in 2020, AR-12286 was found to significantly decrease IOP in steroid-induced OHT in mouse models. Topical administration of 0.25% AR-12286 showed 2 mmHg reduction compared to discontinuing steroids alone and 4 mmHg reduction compared to continuous steroid treatment.49
AR-12286 has completed at least 12 clinical trials, investigating various aspects of the drug. In 2009, Williams et al investigated the efficacy of 3 dosages (0.05%, 0.1%, 0.25%) of AR-12286 compared with placebo in 89 participants with glaucoma or OHT.50 The drug was administered QD in the morning for a week, QD in the evening for a week, and BID for a final week. The study found dose-dependent significant mean IOP reductions at all three concentrations with maximum IOP reductions by 0.25% after BID dosing (25% reduction from baseline of 26–27 mmHg) with transient conjunctival hyperemia as the most common adverse effect. In 2010, Kopczynski et al investigated the safety, efficacy and systemic absorption of two formulations of 0.5% AR-12286 administered for 8 consecutive days in 18 healthy participants.51 The study demonstrated a significant average maximum IOP reduction of 42% (from baseline 16.5 mmHg) at 4-hour post-administration but was associated with frequent transient side effects including conjunctival hyperemia, ocular irritation, instillation site reaction, blurred vision, and headaches.
In 2013, Skaat et al investigated AR-12286 in 10 patients with exfoliation syndrome and OHT or exfoliative glaucoma with participants randomized to 0.5% or 0.7% doses and IOP was measured at weeks 1, 4 and 12.52 The drug was then discontinued, IOP was measured at week 13 and the drug was resumed. At week 24, the drug was again discontinued with final IOP measurements at week 25. The study showed a statistically significant reduction in IOP of 23.6%, 30%, and 30.4% at 1 week, 4 weeks and 12 weeks, respectively (from a mean baseline IOP of 25 ± 2.4 mmHg); however, there was no significant decrease in IOP from baseline in the remaining 12 weeks. Several patients experienced conjunctival hyperemia leading to one participant dropping out. As of 2017, AR-12286 was abandoned by Aerie Pharmaceuticals, Inc. as Netarsudil was deemed to have a longer duration of action.
AMA0076 (PHP 201)
Introduced by Amakem Therapeutics (Hasselt, Limburg, Belgium) in 2009, AMA0076 is a soft ROCK inhibitor. This is a locally acting novel approach in which compounds undergo controlled metabolic inactivation outside of target tissues, which may minimize the risk of systemic and local side effects to increase the therapeutic window.53
AMA0076 is an inhibitor of ROCK 1 and ROCK 2 with similar potencies against each isoform, which is equipotent to Y-39,983.53 In vitro experiments in which HTMCs are cultured with AMA0076 showed reversible alterations in actin filament organization and focal adhesions, confirming the ability to modify TM cell morphology. Topical administration of 0.1%, 0.3% and 0.5% AMA0076 in vivo showed a dose-dependent response in New Zealand White (NZW) rabbits with maximum IOP reduction of 48%, 39% and 23%, respectively, when compared to baseline. Additionally, AMA0076 proved to be comparable in efficacy to latanoprost, when IOP was measured at night, with peak IOP lowering effects of 25% and 22%, respectively. However, unlike latanoprost, AMA0076 also demonstrated reductions in IOP during the day. In a hypertensive rabbit model, AMA0076 was compared with latanoprost and bimatoprost and was significantly more potent in preventing IOP elevation. This study found that the conjunctival hyperemic effects of AMA0076 were significantly lower than those treated with Y-39,983.
AMA0076 completed a first in human (FIH) study in 2013 followed by a Phase 1b study with optimized formulations.54 The FIH was a double-masked, randomized, placebo-controlled, dose-escalation study with topical administration of AMA0076 for 28 days in 82 POAG or OHT patients. The Phase 1b study was a double-masked, randomized, placebo-controlled, 3-period crossover study with 21 healthy participants. Both studies showed the drug was safe and well tolerated. The mean diurnal reduction in IOP was 3.7 mmHg and mild transient hyperemia was observed at a rate of 28.6%. Amakem announced their drug was the first to lower IOP without significant conjunctival hyperemia. A Phase 2 clinical trial was completed in 2014, but results have not yet been published.
RKI-1447 (C16H14N4O2S·2HCl)
Introduced in 2012, RKI-1447 is a potent and selective ROCK 1 and ROCK 2 inhibitor with a potentially increased safety profile due to lack of cross-reactivity with other kinases. It has been shown to display anti-invasive and anti-tumor activities in cancer cells.55 RKI-1447 likely lowers IOP and increases OF via disruption of actin stress fiber formation and increased TM phagocytosis, making it useful for the treatment of secondary glaucomas, which have an increased phagocytic load. Morphologic changes in TM cells are thought to be induced by other ROCK inhibitor activities, including cell rounding, contraction, and decreased adhesion. A study investigating the hypotensive effect of RKI-1447 in a porcine ex vivo pigmentary glaucoma model showed that treatment with this agent significantly reduced IOP from 20.14 ± 2.59 to 13.38 ± 0.91 mmHg (33.58%) and increased OF by 38.25%.56 This drug remains in preclinical development.
AR-13533
In 2013, Aerie pharmaceuticals introduced AR-13533, a second-generation ROCK inhibitor. It is a dual ROCK/NET inhibitor and is still in preclinical development. It does not require enzymatic conversion in the eye to achieve the maximal activity, and therefore may provide additional IOP-lowering effects beyond that obtained with Netarsudil. The company has not submitted an Investigational New Drug application for AR-13533 to the FDA and there have been no further updates.57
AMA0526
AMA0526, introduced by Amakem Therapeutics, is a locally active ROCK inhibitor in preclinical development with a focus on the prevention of excessive wound healing. In 2013, Sijnave et al investigated the effects of AMA0526 on corneal wound healing induced by alkali burns.58 Topical instillation of 0.1% AMA0526 showed reduced corneal opacity and neovascularization in mouse models. Results indicated AMA0526 is as efficient as dexamethasone and more efficient than bevacizumab in reducing excessive corneal wound healing.
In 2014, Velde et al studied the effects of AMA0526 on subconjunctival scarring after glaucoma filtration surgery.59 In vitro studies showed cells cultured with AMA0526 significantly reduced proliferation of human Tenon fibroblasts at 10 and 25 µM concentrations and inhibited fibroblast to myofibroblast differentiation induced by TGFβ1, further impeding the fibrotic process. Additionally, in vivo studies showed topical instillation of 0.3% AMA0526 significantly improved bleb area, lowered IOP, and prolonged bleb survival (35 vs 21 days) compared to control groups. Finally, AMA0526 also reduced inflammation, blood vessel density, and collagen deposition at the site of filtration surgery, all contributing to improved surgical outcomes.
H-1337
Introduced in 2015, H-1337 is a sulfonamide protein kinase inhibitor that has shown potent and long-lasting IOP-lowering activity. H-1337 is a multikinase inhibitor including inhibition of leucine-rich repeat kinase 2 (LRRK2).60 This agent is suggested to stimulate the drainage of AH from the main outflow tract via TM and Schlemm’s canal to lower IOP. The long-term persistence of this IOP-lowering effect has been demonstrated in vivo. Hidaka et al showed that H-1337 effectively inhibits LRRK2 (IC50: 0.045 μM) with maximum IOP reductions of 7.3 mmHg and 4.3 mmHg post-administration of 1% and 0.3% H-1337 in rabbits and monkeys, respectively. The long-lasting durations of IOP-lowering were over 14 hours and 24 hours in rabbits and monkeys, respectively, and neither systemic nor ocular toxicity was observed.
Kasai et al found that H-1337 inhibits PKN2, PKD1, and PKA (IC50: 0.47, 0.052, and 0.091 μM, respectively), which have been reported to regulate cytoskeletal reorganization.61 They emphasized that H-1337 induced morphological change of HTMC in a dose-dependent manner and that combination of H-1337 and other IOP lowering agents showed synergistic effects.
A Phase 1/Phase 2 randomized, double-masked, placebo-controlled, dose-ranging study of the safety and efficacy of H-1337 in subjects with POAG or OHT was carried out in 87 participants in 2018. Allysta Pharmaceuticals (San Francisco, California, USA) reported favorable results with formal publication not yet available.62
ITRI-E-212
Introduced in 2017, ITRI-E-212 is a highly selective ROCK 2 inhibitor and is an attractive amino-isoquinoline because of its adequate solubility and drug-loading percentage in eye drops. The in vivo IOP-lowering effect and tolerability of ITRI-E-212 were investigated in normotensive and OHT rabbits.63 A pharmacokinetics study was performed in vivo in the AH and hyperemia was assessed. The researchers found that post-administration of 1% ITRI-E-212 induced a maximum IOP reduction of 28.4% at 6 hours in an OHT model. In the normotensive model, a maximum IOP reduction of 25.3% was attained at 4 hours where the team observed transient, mild hyperemia. The mean IOP-lowering effect of ITRI-E-212 in normotensive and OHT models was 24.9% and 28.6%, respectively, with reductions sustained for at least 6 hours after each dose. The severity and duration of conjunctival hyperemia were minimal and virtually unobservable within 6 hours post-instillation. No significant difference was noted between the hyperemia scoring of controls and eyes treated with various concentrations of ITRI-E-212, and no severe adverse effects or profound conjunctival injections were observed. No information is currently available regarding clinical trials.
ITRI-E-247
Introduced in 2017, ITRI-E-247 is a second-generation ROCK inhibitor. Tu et al found that this agent inhibits MLC phosphorylation in a concentration-dependent manner and causes cell relaxation in vascular vasopressin receptors (A7r5 cells).64 They also studied the safety and efficacy of the drug in NZW rabbits using topical administration of 0.5% ITRI-E-247 and found that the agent significantly reduced IOP by 32% from baseline at 6-hour post-instillation. Additionally, no significant ocular toxicities, including conjunctival hyperemia, were observed in all groups throughout the study.
ITRI-E-247 is almost 100 times more selective towards ROCK than FGFRs (34% inhibition at 1000 nM). The agent achieved target IC50 following a single topical administration of 0.5% ITRI-E-247 with 4.5 ± 1.9 ng/mL and 3.2 ± 2.5 ng/mL for 4- and 6-hour post-instillation, respectively. No information is available regarding use in clinical trials.
Adverse Effects of ROCK Inhibitors
Although ROCK inhibitors showed a promising safety profile, they have both local and systemic adverse events. ROCK inhibitors induce conjunctival hyperemia and subconjunctival hemorrhage due to their vasodilatory effect. The latter may increase the clearance of concomitantly administered topical drugs thereby reducing their intended ocular effects.65 Other local effects include: blepharitis, ocular irritation, increased lacrimation, and blurred vision. On the systemic level, they may cause blood pressure reduction and an associated increase in heart rate.66 Strategies allowing reduced systemic exposure as a soft drug approach have been applied to develop ROCK inhibitors for localized applications.67 In addition, systemic ROCK inhibition was found to induce a reversible reduction in lymphocyte counts in few individuals.68
Conclusion
ROCK inhibitors primarily lower IOP by decreasing resistance through the conventional pathway. Some investigational ROCK Inhibitors are exhibiting promising results in both animal and human studies. Y 39,983 has shown evidence of both increased blood flow to the optic nerve and an increase in RGCs including regeneration of their axons in rat models.19 In early human trials, AMA 0076 reduced IOP with minimal conjunctival hyperemia.54 In porcine ex vivo pigmentary models, RKI −1447 has shown a significant reduction in IOP.56 The drug also increases TM phagocytosis and may be useful in treating secondary glaucoma. AR-13533 does not require enzymatic conversion in the eye and therefore, may achieve an additional IOP lowering effect.57 In summary, because of the ubiquitous nature of ROCK, these investigational agents have the potential to not only reduce IOP but also provide neuroprotection of the optic nerve, anti-fibrotic activity after glaucoma surgery and restore corneal endothelial cells to improve visual prognosis and surgical outcomes of glaucoma.
Acknowledgments
Supported in part by an unrestricted grant from the Research to Prevent Blindness, New York, NY: Visual Sciences Core Grant EY020799. Figures 1–4 were created with BioRender (Toronto, ON, Canada). Chemical structures in Table 1 were created with ChemDraw (PerkinElmer, Waltham, Massachusettes, USA). Special Thanks to: Ashika Angirekula, Jacob Awkal, Hafsa Zuberi (University of Texas Southwestern Medical Center) and Emily Buchanan (University of Texas at Dallas) for their invaluable assistance in reviewing the background research including designing figures and tables.
Disclosure
The authors have no conflicts of interest in this work.
References
1. Gupta P, Zhao D, Guallar E, Ko F, Boland MV, Friedman DS. Prevalence of glaucoma in the United States: the 2005–2008 national health and nutrition examination survey. Invest Ophthalmol Vis Sci. 2016;57(6):2905–2913. doi:10.1167/iovs.15-18469
2. Tham YC, Hons B, Li X, et al. Global prevalence of glaucoma and projections of glaucoma burden through 2040. Ophthalmology. 2014;121(11):2081–2090. doi:10.1016/j.ophtha.2014.05.013
3. Goel M, Picciani RG, Lee RK. Aqueous humor dynamics: a review. Open J Ophthalmol. 2010;4:52–59. doi:10.2174/1874364101004010052
4. The advanced glaucoma intervention study (AGIS) group: 7. The relationship between control of intraocular pressure and visual field deterioration. The AGIS investigators. Am J Ophthalmol. 2000;130(2):429–440. doi:10.1016/S0002-9394(00)00538-9
5. Honjo M, Inatani M, Kido N, et al. A myosin light chain kinase inhibitor, ML-9, lowers the intraocular pressure in rabbit eyes. Exp Eye Res. 2002;75(2):135–142. doi:10.1006/exer.2002.2009
6. Okumura N, Okazaki Y, Inoue R, et al. Effect of the rho-associated kinase inhibitor eye drop (ripasudil) on corneal endothelial wound healing. Invest Ophthalmol Vis Sci. 2016;57(3):1284–1292. doi:10.1167/iovs.15-18586
7. Arita R, Hata Y, Ishibashi T. ROCK as a therapeutic target of diabetic retinopathy. J Ophthalmol. 2010;175–163. doi:10.1155/2010/175163
8. Rao PV, Pattabiraman PP, Kopczynski C. Role of the rho GTPase/rho kinase signaling pathway in pathogenesis and treatment of glaucoma: bench to bedside research. Exp Eye Res. 2017;158:23–32. doi:10.1016/j.exer.2016.08.023
9. Nakagawa O, Fujisawa K, Ishizaki T
10. Wirth A. Rho kinase and hypertension. Biochim Biophys Acta. 2010;1802(12):1276–1284. doi:10.1016/j.bbadis.2010.05.002
11. Riento K, Ridley AJ. ROCKS: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–456. doi:10.1038/nrm1128
12. Amano M, Chihara K, Nakamura N, Kaneko T, Matsuura Y, Kaibuchi K. The COOH terminus of rho-kinase negatively regulates rho-kinase activity. J Biol Chem. 1999;274(45):32418–32424. doi:10.1074/jbc.274.45.32418
13. Burridge K, Wennerberg K. Rho and rac take center stage. Cell. 2004;116(2):167–179. doi:10.1016/S0092-8674(04)00003-0
14. Hartmann S, Ridley AJ, Lutz S. The function of rho-associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front Pharmacol. 2015;6(276):1–16. doi:10.3389/fphar.2015.00276
15. Fujimoto T, Inoue T
16. Kazemi A, McLaren J, Kopczynski C, et al. The effects of netarsudil ophthalmic solution on aqueous humor dynamics in a randomized study in humans. J Ocul Pharmacol Ther. 2018;34(5):380–386. doi:10.1089/jop.2017.0138
17. Ren R, Li G, Le TD. Netarsudil increases outflow facility in human eyes through multiple mechanisms. IOVS. 2016;57(14):6197–6209.
18. Honjo M, Tanihara H, Kameda T, et al. Potential role of rho-associated protein kinase inhibitor Y-27632 in glaucoma filtration surgery. Invest Ophthalmol Vis Sci. 2007;48(12):5549–5557. doi:10.1167/iovs.07-0878
19. Tokushige H, Waki M, Takayama Y, Tanihara H. Effects of Y-39983, a selective rho-associated protein kinase inhibitor, on blood flow in optic nerve head in rabbits and axonal regeneration of retinal ganglion cells in rats. Curr Eye Res. 2011;36(10):964–970. doi:10.3109/02713683.2011.599106
20. Singh IP, Fechtner RD, Myers JS, et al. Pooled efficacy and safety profile of netarsudil ophthalmic solution 0.02% in patients with open-angle glaucoma or ocular hypertension. J Glaucoma. 2020;29(10):878–884. doi:10.1097/IJG.0000000000001634
21. Asrani S, Bacharach J, Holland E, et al. Fixed-dose combination of netarsudil and latanoprost in ocular hypertension and open-angle glaucoma: pooled efficacy/safety analysis of phase 3 MERCURY-1 and −2. Adv Ther. 2020;37:1620–1631. doi:10.1007/s12325-020-01277-2
22. Garnok-Johns KP. Ripasudil first global approval. Drugs. 2014;74(18):2211–2215. doi:10.1007/s40265-014-0333-2
23. Kaneko Y, Ohta M, Inoue T, et al. Effect of K-115 (ripasudil), a novel rock inhibitor, on trabecular meshwork and Schlemm’s canal and endothelial cells. Sci Rep. 2016;6:19640. doi:10.1038/srep19640
24. Yamamoto K, Maruyama K, Himori N, et al. The novel rho kinase (ROCK) inhibitor K-115: a new candidate drug for neuroprotective treatment in glaucoma. Invest Ophthalmol Vis Sci. 2014;55(11):7126–7136. doi:10.1167/iovs.13-13842
25. Fukunaga T, Ikesugi K, Nishio M, et al. The effect of the rho-associated protein kinase inhibitor, HA-1077, in the rabbit ocular hypertension model induced by water loading. Curr Eye Res. 2009;34(1):42–47. doi:10.1080/02713680802531353
26. Honjo M, Inatani M, Kido N, et al. Effects of protein kinase inhibitor, HA1077, on intraocular pressure and outflow facility in rabbit eyes. Arch Ophthalmol. 2001;119(8):1171–1178. doi:10.1001/archopht.119.8.1171
27. Pitha I, Oglesby E, Chow A, et al. Rho-kinase inhibition reduces myofibroblast differentiation and proliferation of scleral fibroblasts induced by transforming growth factor β and experimental glaucoma. Trans Vis Sci Tech. 2018;7(6):6. doi:10.1167/tvst.7.6.6
28. Pakravan M, Beni AN, Ghahari E, et al. The ocular hypotensive efficacy of topical fasudil, a rho-associated protein kinase inhibitor, in patients with end-stage glaucoma. Am J Ther. 2017;24(6):e676–e680. doi:10.1097/MJT.0000000000000362
29. Jung D, Kim W, Williams D. Reprogram or reboot: small molecule approaches for the production of induced pluripotent stem cells and direct cell reprogramming. ACS Chem Biol. 2014;9:80–95. doi:10.1021/cb400754f
30. Honjo M, Tanihara H, Inatani M, et al. Effects of rho-associated protein kinase inhibitor Y-27632 on intraocular pressure and outflow facility. Invest Ophthalmol Vis Sci. 2001;42:137–144.
31. Ibrahim D, Ko J, Iwata W, Okumichi H, Kiuchi Y. An in vitro study of scarring formation mediated by human tenon fibroblasts: effect of Y-27632, a rho kinase inhibitor. Cell Biochem Funct. 2019;37:113–124. doi:10.1002/cbf.3382
32. Tian B, Kaufman P. Effects of the rho kinase inhibitor Y-27632 and the phosphatase inhibitor calyculin A on outflow facility in monkeys. Exp Eye Res. 2005;80(2):215–225. doi:10.1016/j.exer.2004.09.002
33. Ikenoya M, Hidaka H, Hosoya T, Suzuki M, Yamamoto N, Sasaki Y. Inhibition of rho-kinase-induced myristoylated alanine-rich C kinase substrate (MARCKS) phosphorylation in human neuronal cells by H-1152, a novel and specific rho-kinase inhibitor. J Neurochem. 2002;81(1):9–16. doi:10.1046/j.1471-4159.2002.00801.x
34. Nishio M, Fukunaga T, Sugimoto M, et al. The effect of the H-1152P, a potent rho-associated coiled coil-formed protein kinase inhibitor, in rabbit normal and ocular hypertensive eyes. Curr Eye Res. 2009;34(4):282–286. doi:10.1080/02713680902783763
35. Rao PV, Deng P, Sasaki Y, Epstein DL. Regulation of myosin light chain phosphorylation in the trabecular meshwork: role in aqueous humour outflow facility. Exp Eye Res. 2005;80(2):197–206. doi:10.1016/j.exer.2004.08.029
36. Rasmussen C, Kaufman P, Ritch R, et al. Latrunculin B reduces intraocular pressure in human ocular hypertension and primary open-angle glaucoma. Transl Vis Sci Tech. 2014;3(5):1–9. doi:10.1167/tvst.3.5.1
37. Thomasy SM, Raghunathan VK, Miyagi H, et al. Latrunculin B and substratum stiffness regulate corneal fibroblast to myofibroblast transformation. Exp Eye Res. 2018;170:101–107. doi:10.1016/j.exer.2018.02.003
38. Tanihara H, Inatani M, Honjo M, et al. Intraocular pressure-lowering effects and safety of topical administration of a selective ROCK inhibitor, SNJ-1656, in healthy volunteers. Arch Ophthalmol. 2008;126(3):309–315. doi:10.1001/archophthalmol.2007.76
39. Shan S, Lam T, Stamer W, et al. The effects of a rho-associated protein kinase (ROCK) inhibitor (Y39983) on human trabecular meshwork cells – a morphological and proteomic study. Invest Ophthalmol Vis Sci. 2019;60:5138.
40. Inoue T, Tanihara H, Tokushige H, Araie M. Efficacy and safety of SNJ-1656 in primary open-angle glaucoma or ocular hypertension. Acta Ophthalmol. 2015;93(5):e393–e395. doi:10.1111/aos.12641
41. Ashwinbalaji S, Senthilkumari S, Gowripriya C, et al. SB772077B, a new rho kinase inhibitor enhances aqueous humour outflow facility in human eyes. Sci Rep. 2018;8:15472.
42. Ashwinbalaji S, Haribalaganesh R, Krishnadas S, et al. SB772077B (SB77) alleviated the aqueous outflow resistance mediated by cyclic mechanical stress in perfused human cadaveric eyes. Sci Rep. 2020;10(1):10202. doi:10.1038/s41598-020-67087-2
43. Harrison B, Almstead Z, Burgoon H, et al. Discovery and development of LX7101, a dual LIM-kinase and ROCK inhibitor for the treatment of glaucoma. ACS Med Chem Lett. 2015;6:84–88. doi:10.1021/ml500367g
44. Boland S, Bourin A, Alen J, et al. Design, synthesis and biological characterization of selective LIMK inhibitors. Bioorg Med Chem Lett. 2015;25(18):4005–4010. doi:10.1016/j.bmcl.2015.07.009
45. Harrison BA, Whitlock NA, Voronkov MV, et al. Novel class of LIM-kinase 2 inhibitors for the treatment of ocular hypertension and associated glaucoma. J Med Chem. 2009;52(21):6515–6518. doi:10.1021/jm901226j
46. Lexicon pharmaceuticals reports preliminary results from two phase 1 studies. Lexicon pharmaceuticals, Inc. 2012. Available from: http://www.lexpharma.com/media-center/news/543-lexicon-pharmaceuticals-reports-preliminary-results-from-two-phase-1-studies.
47. Wang RF, Serle JB, Kopczynski C. Effect of 0.6% AR-12286 on aqueous humor dynamics in 6 normotensive monkey eyes. Invest Ophthalmol Vis Sci. 2009;50(13):1465.
48. Lin C, Sherman B, Moore L, et al. Discovery and preclinical development of netarsudil, a novel ocular hypotensive agent for the treatment of glaucoma. J Ocul Pharmacol Ther. 2018;34:40–51. doi:10.1089/jop.2017.0023
49. Ren R, Humphrey A, Kopczynski C, et al. Rho kinase inhibitor AR-12286 reverses steroid-induced changes in mouse eyes. Investig Ophthalmol Vis Sci. 2020;61:2720.
50. Williams RD, Novack GD, Haarlem TV, Kopczynski C. Ocular hypotensive effect of the Rho kinase inhibitor AR-12286 in patients with glaucoma and ocular hypertension. Am J Ophthalmol. 2011;152(5):5. doi:10.1016/j.ajo.2011.04.012
51. Kopczynski C, Novack GD, Swearingen D, Haarlem TV. Ocular hypotensive efficacy, safety and systemic absorption of AR-12286 ophthalmic solution in normal volunteers. Br J Ophthalmol. 2013;97(5):567–572. doi:10.1136/bjophthalmol-2012-302466
52. Skaat A, Jasien JV, Ritch R. Efficacy of topically administered rho-kinase inhibitor AR-12286 in patients with exfoliation syndrome and ocular hypertension or glaucoma. J Glaucoma. 2016;25(9):807–814. doi:10.1097/IJG.0000000000000508
53. Velde SV, Bergen TV, Sijnave D. AMA0076, a novel, locally acting rho kinase inhibitor, potently lowers intraocular pressure in New Zealand white rabbits with minimal hyperemia. Investig Ophthalmol Vis Sci. 2014;55:2.
54. John H. Initial evaluation of safety, tolerability and pharmacodynamics of the locally-acting ROCK inhibitor AMA0076.
55. Patel R, Forinash K, Pireddu R, et al. RKI-1447 is a potent inhibitor of the rho-associated ROCK kinases with anti-invasive and antitumor activities in breast cancer. Cancer Res. 2012;72(19):5025. doi:10.1158/0008-5472.CAN-12-0954
56. Dang Y, Wang C, Shah P, Waxman S, Loewen RT, Loewen NA. RKI-1447, a rho kinase inhibitor, causes ocular hypotension, actin stress fiber disruption, and increased phagocytosis. Graefes Arch Clin Exp Ophthalmol. 2019;257(1):101–109. doi:10.1007/s00417-018-4175-6
57. United States Securities and Exchange Commission Form 10-K. December 31, 2013. Available from: https://investors.aeriepharma.com/static-files/2cc9950a-b0ae-4777-ac94-c74636a16911.
58. Sijnave D, Hollanders K, Van Bergen T. A RHO kinase inhibitor, AMA0526 improves corneal wound healing after alkali burn injury. Acta Ophthalmol. 2013;91(252). doi:10.1111/j.1755-3768.2013.1633.x
59. Velde SV, Bergen TV, Vandewllae E. Rho kinase inhibitor AMA0526 improves surgical outcome in a rabbit model of glaucoma filtration surgery. Acta Ophthalmol. 2014;92:253.
60. Hidaka H. A novel isoquinoline sulfonamide protein kinase inhibitor (H-1337) produces long-lasting reduction of IOP.
61. Kasai A. Elucidation of molecular mechanism of H-1337, an antiglaucoma agent.
62. A rare growing bio-venture with multiple stable sources of revenue. April 22, 2020. Available from: http://pdf.irpocket.com/C4576/yCGV/Fnnr/DXyw.pdf.
63. Hsu C-R, Chen Y-H, Liu C-P, et al. A highly selective rho-kinase inhibitor (ITRI-E-212) potentially treats glaucoma upon topical administration with low incidence of ocular hyperemia. Invest Ophthalmol Vis Sci. 2019;60:624–633. doi:10.1167/iovs.18-25252
64. Tu C, Liu C-P, Chen CH, et al. ITRI-E-247, a novel selective rho-associated kinase inhibitor, characterized with the potent intraocular pressure lowering effect and less conjunctival hyperemia. Invest Ophthalmol Vis Sci. 2019;60:37–68.
65. Arnold JJ, Hansen MS, Gorman GS, et al. The effect of rho-associated kinase inhibition on the ocular penetration of timolol maleate. Invest Ophthalmol Vis Sci. 2013;54(2):1118–1126. doi:10.1167/iovs.12-10583
66. Lohn M, Plettenburg O, Kannt A, et al. End-organ protection in hypertension by the novel and selective rho-kinase inhibitor, SAR407899. World J Cardiol. 2015;7:31–42. doi:10.4330/wjc.v7.i1.31
67. Bolanda S, Deferta O, Alenet J, et al. 3-[2-(aminomethyl)-5-[(pyridin-4-yl)carbamoyl]phenyl] benzoates as soft ROCK inhibitors. Bioorg Med Chem Lett. 2013;23(23):6442–6446. doi:10.1016/j.bmcl.2013.09.040
68. Shaw D, Hollingworth G, Soldermann N, et al. Novel ROCK inhibitors for the treatment of pulmonary arterial hypertension. Bioorg Med Chem Lett. 2014;24:4812–4817. doi:10.1016/j.bmcl.2014.09.002
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.