")
Back to Journals » International Journal of Nanomedicine » Volume 12
Investigation of the effects of carbon-based nanomaterials on A53T alpha-synuclein aggregation using a whole-cell recombinant biosensor
Authors Mohammadi S, Nikkhah M , Hosseinkhani S
Received 24 June 2017
Accepted for publication 29 September 2017
Published 14 December 2017 Volume 2017:12 Pages 8831—8840
DOI https://doi.org/10.2147/IJN.S144764
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Soheila Mohammadi,1 Maryam Nikkhah,1 Saman Hosseinkhani2
1Department of Nanobiotechnology, 2Department of Biochemistry, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, Iran
Abstract: The aggregation of alpha-synuclein (αS), natively unstructured presynaptic protein, is a crucial factor leading to the pathogenesis of Parkinson’s disease (PD) and other related disorders. Recent studies have shown prefibrillar and oligomeric intermediates of αS as toxic to the cells. Herein, split-luciferase complementation assay is used to design a “signal-on” biosensor to monitor oligomerization of A53T αS inside the cells. Then, the effect of carbon-based nanomaterials, such as graphene quantum dots (GQDs) and graphene oxide quantum dots (GOQDs), on A53T αS oligomerization in vitro and in living cells is investigated. In this work, for the first time, it was found that GQDs at a concentration of 0.5 µg/mL can promote A53T αS aggregation by shortening the nucleation process, which is the key rate-determining step of fibrillation, thereby making a signal-on biosensor. While these nanomaterials may cross the blood–brain barrier because of their small sizes, the interaction between αS and GQDs may contribute to PD etiology.
Keywords: αS, aggregation, split luciferase, luminescent biosensor, GQDs
Introduction
Parkinson’s disease (PD) as the second most common progressive neurodegenerative disorder after Alzheimer’s disease affects 1% of the population over the age of 65 years.1,2 The classical symptoms of PD include resting tremor, muscular rigidity and bradykinesia1,3 as the consequence of progressive loss of dopaminergic neurons in the substantia nigra region of the brain.3,4 The association of mutations (A30P, E46K and A53T) and multiplication of the alpha-synuclein (αS) gene with early onset of familial forms of the disease implicate the role of αS in PD.5–8 It has been reported that aggregation of αS into oligomeric and fibrillar forms is linked to the pathogenesis of PD. Recent studies have implicated small soluble oligomeric and protofibrillar forms of αS as the most neurotoxic species.9–12
The influence of nanoparticles (NPs) on amyloid formation is of great interest due to their small sizes and high surface area-to-volume ratios. The small size of NPs allows them to access almost all parts of the human body; they even pass through the blood–brain barrier (BBB), which is the homeostatic defense mechanism of the brain. Various drug nano-carriers have been successfully synthesized to transport small molecules, especially hydrophilic ones, across the BBB.13–17 So, if NPs can inhibit the process of fibrillogenesis or disaggregate amyloid fibrils,18 they offer great potential to be used as drugs to control neurodegenerative amyloid disease. On the other hand, detailed information about the interactions between NPs and amyloid proteins could shed light on the mechanism of fibril formation at the molecular level, an area which still remains elusive. In addition, due to the increasing number of applications of NPs in the past decade, the toxicity of NPs has been of concern for the past years.19–21 If NPs facilitate the process of protein fibrillogenesis, they might be severely hazardous to humans and their safe use must be carefully ensured.
While several studies show that NPs with different structures interact with proteins and affect their aggregation,22–26 there are limited data on the interaction of αS with NPs. Some recent findings in this regard include the acceleration of αS fibrillation in the presence of αS-conjugated CdSe/ZnS quantum dots,27 the interactions of αS with negatively and positively charged gold NPs,28,29 the effect of negatively charged Au NPs with different sizes on the interaction of NPs,30 the effect of bare and surface-coated magnetic iron oxide (Fe3O4) NPs on αS aggregation31 and the influence of chemical composition and the amount and nature of the particle’s surface on the nucleation phase of αS aggregation.32
In this study, we adapted a protein-fragment complementation assay approach to observe the initial A53T αS–A53T αS interactions in living cells utilizing fragments of firefly luciferase that can reconstitute as active luciferase upon αS aggregation.33–35 Here, we generated and characterized a whole-cell bioluminescent sensor which reports A53T αS oligomerization in cell by a fast, sensitive and semiquantitative assay. We demonstrate that this cell-based biosensor in addition to tracking A53T mutant αS oligomerization in cells (Figure 1) can be exploited to assess the effects of NPs on αS oligomerization inside the cells.
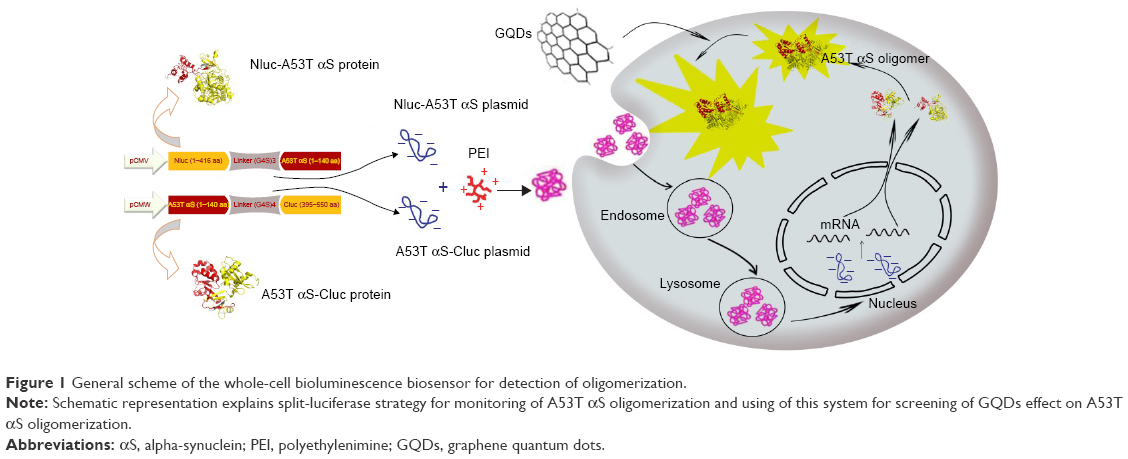 | Figure 1 General scheme of the whole-cell bioluminescence biosensor for detection of oligomerization. |
Materials and methods
Design of biosensor
The split firefly luciferase fragments according to the known split point of firefly luciferase36 were produced by polymerase chain reaction (PCR) amplification of pGL3-Control Vector (Promega) using forward (5′-TAACAGCTAGCACCATGGAAGACGCCAAAAAC-3′) and reverse (5′-CGGGGTACCTCCGCCTCCTCCAGATCCGCCTCCACCTGACCCGCCACCTCCACTATC-3′) primers which led to a fragment coding amino acids 1–416 (Nluc) of firefly luciferase and (G4S)3 linker. Forward (5′-TAACAGCTAGCACCATGGCTATGATTATGTCCGGTTATG-3′) and reverse (5′-TATGGTACCTCCGCCTCCACCTGATCCGCCTCCTCCAGATCCGCCTCCACCTGACCCGCCACCTCCACT
CAC-3′) primers were used to amplify the fragment which codes the amino acids 395–550 (Cluc) of firefly luciferase with (G4S)4. A flexible Gly-Ser linker was added to the reverse primers based on previous reports,35,37,38 and a Kozak consensus sequence was optimized with initiating ATG codon for proper initiation of translation in the forward primers of Nluc and Cluc fragments. The purified fragments were digested by NheI/KpnI and then cloned into the NheI/KpnI digested/dephosphorylated pcDNA3.1(+) vector (Invitrogen), harboring A53T αS gene between KpnI and XhoI sites with forward (5′-CGGGGTACCATGGATGTATTCATGAAAGGTCTTTCTAAGGC-3′) and reverse (5′-CCGCTCGAGTTAAGCTTCCGGTTCGTAGTCTTG-3′) primers. The constructs were transformed into Escherichia coli (DH5α).39
A53T αS-(G4S)4-Cluc was constructed by splicing-by-overlap-extension PCR using A53T αS (forward: 5′-CGGGGTACCACCATGGATGTATTCATGAAAGGTC-3′, reverse: 5′-TCCGCCTCCACCTGATCCTCCTCCTCCAGATCCTCCTCCACCTGACCCTCCACCTCCACTAGCTTCCGGTTCGT
AGTCTTG-3′) and Cluc (forward: 5′-AGTGGAGGTGGAGGGTCAGGTGGAGGAGGATCTGGAGGAGGAGGATCAGGTGGAGGCGGAATGATTATGT
CCGGTTATG-3′, reverse: 5′-CCGCTCGAGTTACACAGCGATCTTTCCGCCCTTCTTC-3′) primers, and the obtained fragment was subcloned in pcDNA3.1(+) vector with KpnI and XhoI restriction sites (Figure 2).
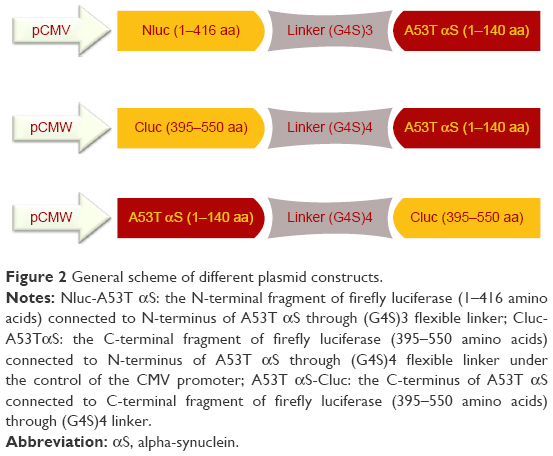 | Figure 2 General scheme of different plasmid constructs. |
Cell culture, transfection and bioluminescence assay of reconstituted reporter
Human embryonic kidney cells (HEK293T) (purchased from Iranian Biological Resource Center) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, high glucose; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Invitrogen) solution at 37°C in an incubator with 5% CO2.
For the transient transfection, HEK293T cells were seeded in a 48-well plate at 3.5×104 cells/well. The cells were cultured overnight to reach the confluency of 80% at the time of transfection, and were exposed to fresh DMEM without serum and antibiotics for 4 hours. Then, the plasmid mixtures were prepared at an N:P ratio of 12 with branched 25 kDa polyethylenimine (PEI) (Sigma-Aldrich, St Louis, MO, USA),40 and transfection mixture was applied dropwise to each well and incubated with 5% CO2 for 4 hours at 37°C. Finally, the transfection media were replaced with fresh DMEM supplemented with 10% FBS and antibiotics (complete growth medium) and incubated for another 48 hours. The psiCHECK plasmid was used to monitor firefly luciferase activity in transfected HEK293T cells (positive control). Finally, the transfected cells in each well were lysed with 30 μL of cell culture lysis buffer. The luciferase activity of lysed cells was monitored as a luminescence signal produced upon addition of 10 μL of luciferase assay complex (25 mM Tris, 10 mM MgSO4, 1 mM luciferin and 2 mM ATP) to 10 μL of cell lysate using a luminometer (Berthold Detection System, Pforzheim, Germany) as reported earlier.41 The results were indicated as relative light units per second. The data are reported as mean ± SD (n=3).
Cell toxicity assays of GQDs and GOQDs (cell viability study)
For cell toxicity assays, HEK293T cells were cultured in DMEM supplemented with 10% FBS in a humidified 5% CO2 atmosphere at 37°C. Then, the cells were transferred to sterile 96-well growth plates at a density of 10,000 cells/well, with three wells per treatment. The next day, the medium was changed, and the cells were treated with different amounts of graphene quantum dots (GQDs) and graphene oxide quantum dots (GOQDs) dispersed in phosphate-buffered saline (PBS, pH 7.4) to reach the final concentration of 0.25, 0.5, 0.75 and 1 μg/mL and were incubated for 24 hours. Doxorubicin which is a potent cytotoxic compound was used as positive control. The viability of the cells was measured by MTT assay, and the color intensity of formazan solution was measured by ELISA Microplate Reader (μQuant; BioTek Instruments, Winooski, VT, USA) at 570 nm. The untreated cells were assumed to show 100% viability.
Bioluminescence assay of reconstituted reporter in the presence of GQDs and GOQDs
A total of 3.5×104 cells/well was plated in 48-well plates. As mentioned earlier, the overnight culture of the cells was exposed to fresh DMEM without serum and antibiotics for 4 hours. Then, the complex of Nluc-A53T αS and A53T αS-Cluc plasmids with PEI was prepared at an N:P ratio of 12, and transfection mixture was applied to each well and incubated with 5% CO2 at 37°C for 4 hours. The medium was replaced with fresh DMEM and incubated for another 20 hours. The transfected cells were then treated with different concentrations of GQDs and GOQDs, curcumin (25 μM) and Fe3O4 NPs (50 μg/mL). Luciferase activity was determined 24 hours after addition of the nanomaterials as mentioned earlier.
Expression and purification of A53T αS
For protein expression, recombinant pNIC28-Bsa4 vector containing A53T gene of αS was transferred to Escherichia coli C41 (DE3); starter culture was prepared by incubating a single colony of the bacteria in Luria–Bertani medium containing 50 μg/mL kanamycin. After reaching an optical density of 0.4 at 600 nm, 0.1 mM IPTG was added, and the culture was incubated for another 5 hours at 37°C.42 The induced cells were harvested by centrifugation at 5,000× g for 15 min. The cell pellet was resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazol, pH 8) and 1 mM phenylmethane sulfonyl fluoride, and then the cells were lysed by sonication on ice. The cell lysate was centrifuged at 16,000× g for 30 minutes at 4°C. The supernatant was applied to nickel-nitrilotriacetic acid sepharose column (Qiagen), and purification was done according to the manufacturer’s recommendations (Qiagen). The purified proteins were analyzed by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Characterization of GQDs
GQDs and GOQDs that have the epoxy and hydroxyl groups on both sides of single-layer GOQDs were synthesized through hydrothermal method.43 Fe3O4 NPs were synthesized by the coprecipitation method.44
UV/visible spectroscopy of the GQDS was performed with Alpha-1860S/1900S spectrophotometer (Laxco, Bothell, WA, USA). Fluorescence emission measurements were done using a Cytation3 Cell Imaging Multi-Mode Reader (BioTek Instruments). The zeta potential measurements were performed at room temperature by laser Doppler velocimetry using Malvern Nano ZS90 instrument and DTS software (Malvern Instruments, Malvern, UK).
The morphology of GQDs was analyzed using atomic force microscopy (AFM). For imaging, 100 μL of GQDs solution (2.5 μg/mL) was placed on a freshly cleaved mica, followed by drying at 70°C for 1 hour. The scan was done by contact mode using a reflex gold-coated rotated tip at 0.2 N/m with an AFM instrument (FemtoScan, Moscow, Russia). All AFM images were processed using Gwyddion analysis software.
ThT fluorescence spectroscopy for evaluation of effect of A53T αS aggregation by GQDs and GOQDs
Purified A53T αS was dialyzed against sterile PBS (pH 7.4) and was immediately incubated at a final concentration of 50 μM containing 0.02% NaN3 as an antiseptic agent, in the absence or presence of different concentrations of nanomaterials, at 37°C in a Turbo Thermo Shaker (TMS-200; Allsheng, Hangzhou, People’s Republic of China) with shaking at 850 rpm.
Fibril formation was monitored using thioflavin T (ThT) (Sigma-Aldrich), the fluorescence of which was dependent on the formation of amyloid fibrils. Fluorescence was measured in clear black 96-well plate reader using a Cytation3 Cell Imaging Multi-Mode Reader (BioTek Instruments) with excitation wavelength at 440 nm and emission at 485 nm. The protein concentration was 75 μM, and the ThT concentration was 25 μM. At different incubation times, aliquots of the A53T αS solution were taken for fluorescence measurements. Each sample was assayed in triplicates, and each experiment was repeated at least three times. The statistical analysis of the data was done by one-way ANOVA post hoc test.
Results
Detection of A53T αS dimer/oligomers using split-luciferase complementation assay
In split-luciferase complementation assay, the luciferase reporter has to be rationally dissected into two fragments.34,36,37 The amino (1–416 amino acids) and carboxy (395–550 amino acids) terminal fragments of firefly luciferase were separately fused to the N-terminus of A53T αS. For enhancing the folding and the flexibility of luciferase fragments,36 a flexible linker composed of (G4S)n amino acids was inserted at the junction between the luciferase fragments and A53T αS ((G4S)3 linker for Nluc and (G4S)4 linker for Cluc fragment) (Figure 2). We presumed that by oligomerization of A53T αS, the N- and C-terminal fragments of firefly luciferase would interact and the functional structure of the enzyme would be reconstituted leading to exhibition of luminescence.
To evaluate the oligomerization of A53T αS within the living cell, transfection of the cells by Nluc-A53T αS and Cluc-A53T αS plasmids was done. The luciferase activity assay was performed after 48 hours of transfection (Figure 3).
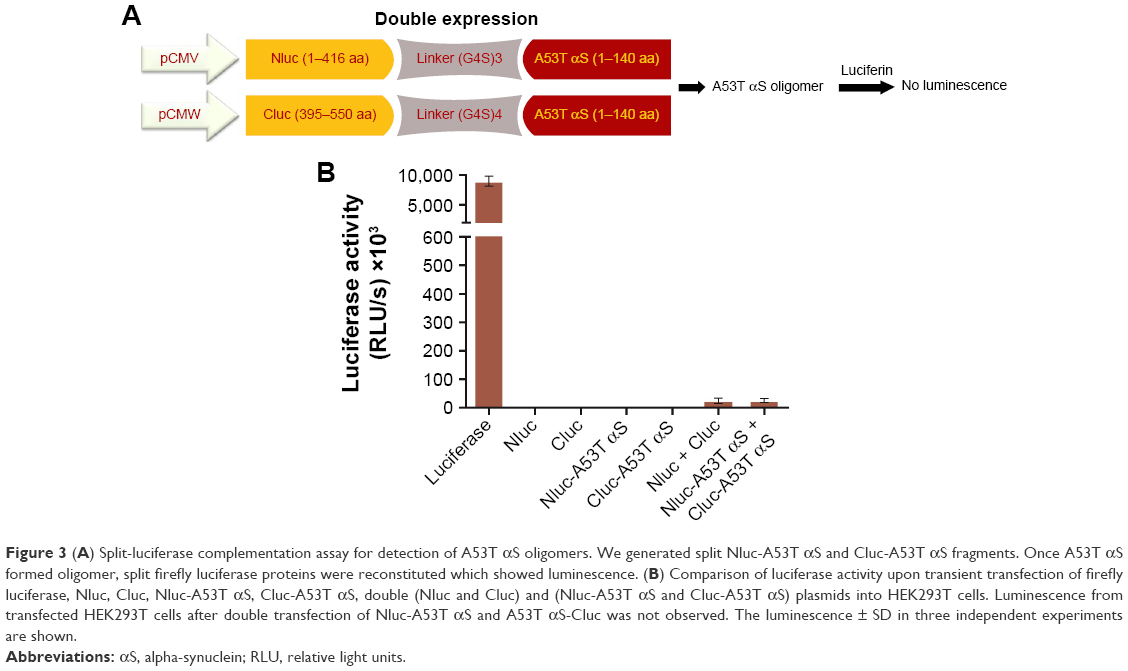 | Figure 3 (A) Split-luciferase complementation assay for detection of A53T αS oligomers. We generated split Nluc-A53T αS and Cluc-A53T αS fragments. Once A53T αS formed oligomer, split firefly luciferase proteins were reconstituted which showed luminescence. (B) Comparison of luciferase activity upon transient transfection of firefly luciferase, Nluc, Cluc, Nluc-A53T αS, Cluc-A53T αS, double (Nluc and Cluc) and (Nluc-A53T αS and Cluc-A53T αS) plasmids into HEK293T cells. Luminescence from transfected HEK293T cells after double transfection of Nluc-A53T αS and A53T αS-Cluc was not observed. The luminescence ± SD in three independent experiments are shown. |
Transient co-transfection of Nluc-A53T αS and A53T αS-Cluc plasmids led to more than 15-fold increase in luciferase activity compared to background which is the light observed by co-transfections of Nluc- and Cluc-containing plasmids. This observation is indicative of oligomerization of A53T αS (Figure 4) in the cells. It should be noted that fragment complementation and rise of bioluminescence signal occurred in cells expressing Nluc-A53T αS and A53T αS-Cluc, while in cells expressing Nluc-A53T αS and Cluc-A53T αS the light emission did not occur, supporting an antiparallel interaction between two A53T αS molecules. This finding is correlated with the results of bimolecular fluorescence complementation of wild-type (WT) αS oligomerization in living cells.45
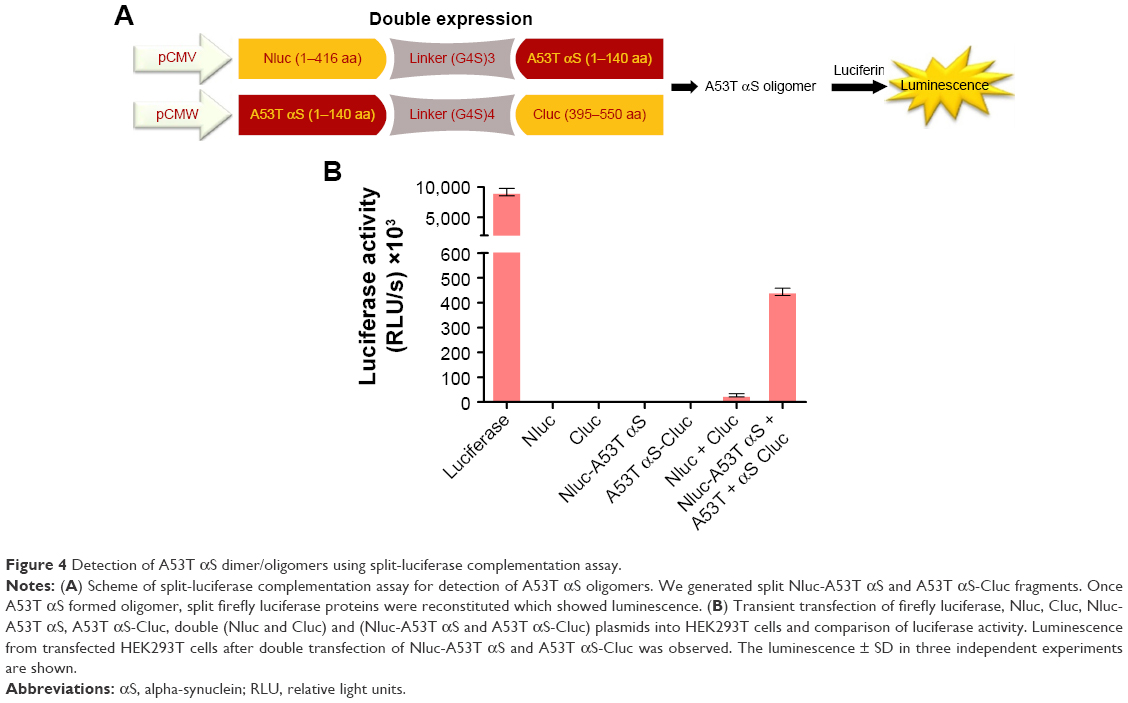 | Figure 4 Detection of A53T αS dimer/oligomers using split-luciferase complementation assay. |
Screening assay for GQDs and GOQDs
Cell toxicity assays of GQDs and GOQDs
The toxicity of nanomaterials on HEK293T cells was evaluated as described. The results showed (Figure 5) no significant decrease in cell viability upon treatment of the cells with nanomaterials at concentrations of 0.25, 0.5, 0.75 and 1 μg/mL.
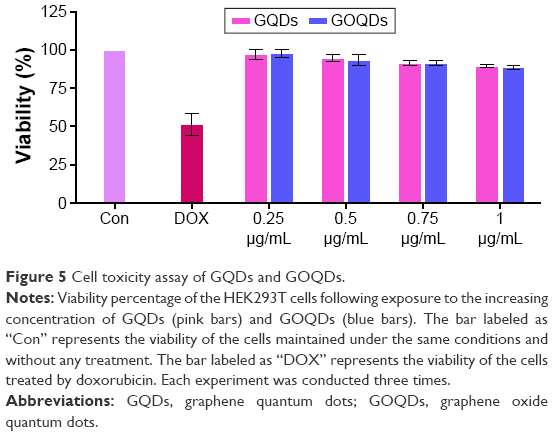 | Figure 5 Cell toxicity assay of GQDs and GOQDs. |
GQDs and GOQDs promote A53T αS oligomerization in cells
To evaluate the effect of GQDs and GOQDs on A53T αS oligomerization in cells, the HEK293T cells were transiently transfected with Nluc-A53T αS and A53T αS-Cluc plasmids as mentioned earlier. Cells were then treated with 0.5 μg/mL of GQDs and GOQDs in triplicate, 20 hours after transfection. The luciferase activity was measured 48 hours after transfection by addition of 10 μL of luciferase assay complex to 10 μL of cell lysate using a plate luminometer. It was demonstrated that the applied nanomaterials strongly increased split-luciferase activity which implicated augmentation of A53T αS oligomerization inside the cells (Figure 6). The lowest concentration of GQDs that could significantly increase split-luciferase activity was 0.25 μg/mL.
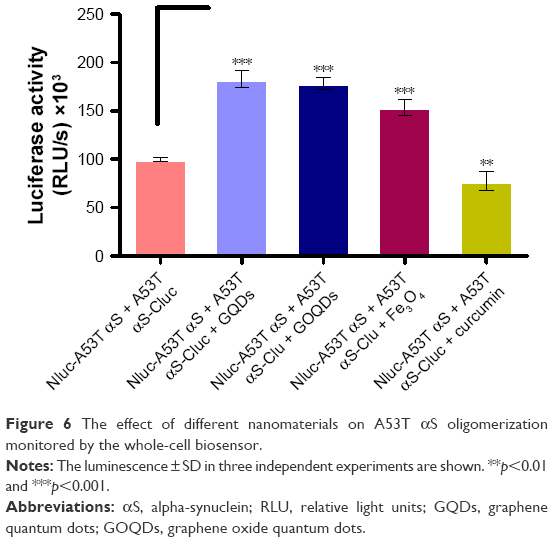 | Figure 6 The effect of different nanomaterials on A53T αS oligomerization monitored by the whole-cell biosensor. |
Moreover, in a proof-of-principle experiment, the effect of curcumin, a previously described known inhibitor of WT αS oligomerization, was checked.46–48 The results further confirmed the effectiveness of this rapid and sensitive cell model in monitoring A53T αS oligomerization affected by different treatments. Also, our cell-based reporter was used to assess the influence of Fe3O4 NPs on aggregation of αS. The increased luminescence compared to control attributed to enhancement of αS aggregation by magnetic NPs treatment (Figure 6). The inducing effect of bare Fe3O4 NPs has been reported previously for WT αS aggregation by spectroscopy.31
It should be noted that to verify the effect of GQDs on luciferase activity, HEK293T cells were transfected by psiCHECK plasmid, which contains full-length firefly luciferase. Moreover, the same cell line was co-transfected by luciferase fragments (Nluc and Cluc). No significant changes in luciferase activity were observed by treatment of the cells with GQDs at concentrations of 0.5 μg/mL (data not shown).
Characterization of GQDs
In order to assess the effect of GQDs on induction of luminescence in split-luciferase complementation assay, their structural properties were characterized. Particle size has been considered to play crucial role in controlling the level of cellular uptake. Several studies demonstrated that the NPs with a diameter about 200 nm are ideal for internalization through receptor-mediated endocytosis pathway.49,50
Zeta potential has been identified to play crucial roles in controlling the level of protein aggregation. Several reports suggest that negatively charged NPs exert significant effect on the kinetics of fibrillation of αS. For instance, various concentrations of negatively charged gold NPs of different sizes (10–22 nm) have been shown to strongly accelerate WT αS aggregation by increasing both the nucleation and growth rate of the aggregates.30 In addition, binding of WT αS to anionic lipid vesicles (20–100 nm) can enhance the rate of primary nucleation by three orders of magnitude.51
The surface charge studies revealed that the zeta potential of GQDs and Fe3O4 NPs was −57 and −28 mV, respectively. Moreover, Figure 7 shows the absorption and emission spectra of the blue luminescent GQDs. Typical AFM image of GQDs with an average diameter of ≈30 nm can be seen in Figure 7D.
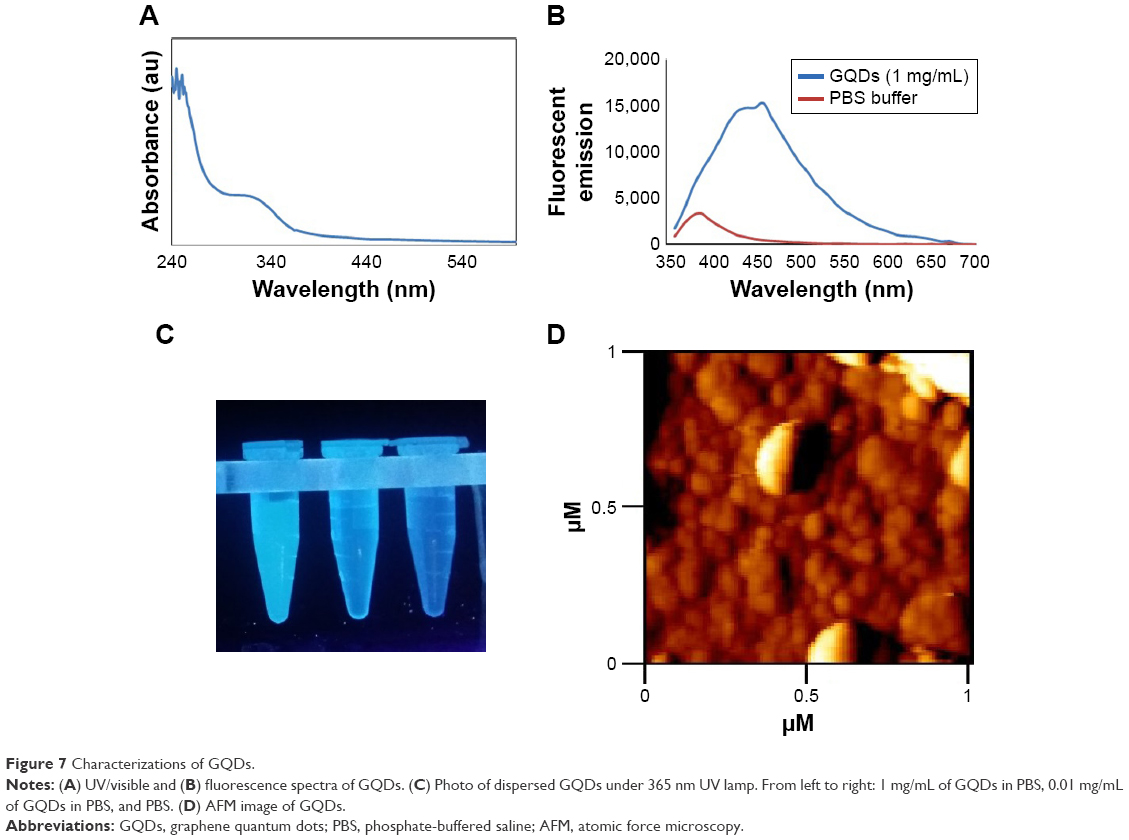 | Figure 7 Characterizations of GQDs. |
In vitro evaluation of A53T αS aggregation in the presence of GQDs and GOQDs
In order to discard any possible interfering effects of cellular components on the A53T αS aggregation, the in vitro effect of GQDs and GOQDs on aggregation of A53T αS was monitored (Figure 8). ThT is considered as a highly sensitive marker for the amyloid state of various aggregating proteins and peptides because of exhibiting a strong fluorescence upon binding to amyloid fibrils. The aggregation kinetics of 50 μM A53T αS protein in the presence of 50 μg/mL of GQDs and GOQDs (Figure 8B) and 50, 25, 5 and 0.5 μg/mL of GQDs (Figure 9) was studied by ThT fluorescence enhancement method. Our results showed GQDs and GOQDs promoted fibril formation. In this experiment, the effects of Fe3O4 NPs (50 μg/mL) and curcumin (25 μM) on protein aggregation were also studied (Figure 8B). In parallel with the results obtained by our cell-based biosensor (Figure 6), curcumin decreased A53T αS oligomerization significantly while considerable increase in aggregation of A53T αS was detected upon Fe3O4 NPs treatment (Figure 8). In the presence of GQDs at all concentrations mentioned, the lag times of A53T αS protein aggregation decreased. The acceleration of the fibrillation of A53T αS occurred in a dose-dependent manner (Figure 9). It can be suggested that the A53T αS monomers can efficiently adsorb on the large surface area of GQDs and significantly decrease the lag time and accelerate the fibrillation process.
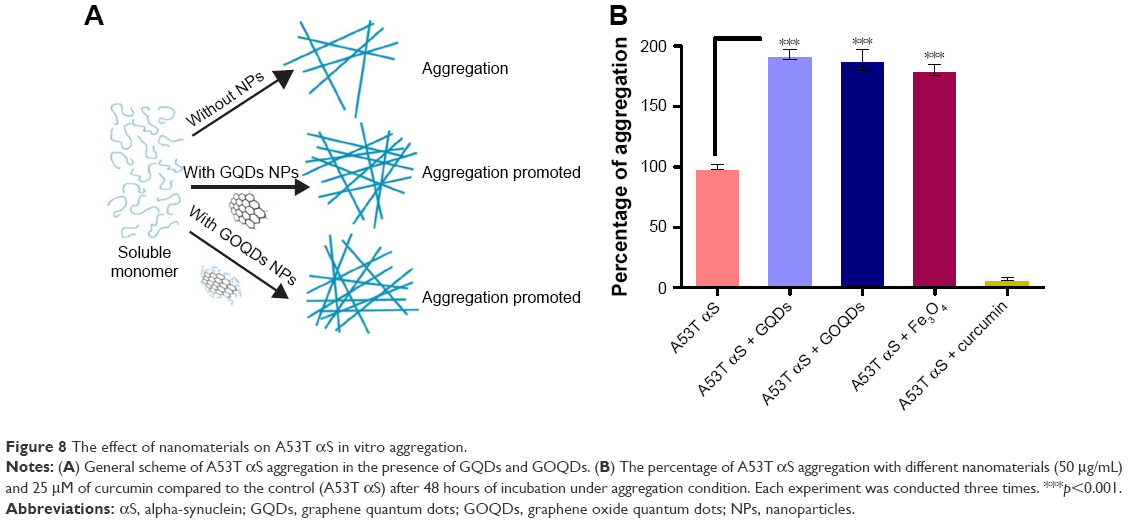 | Figure 8 The effect of nanomaterials on A53T αS in vitro aggregation. |
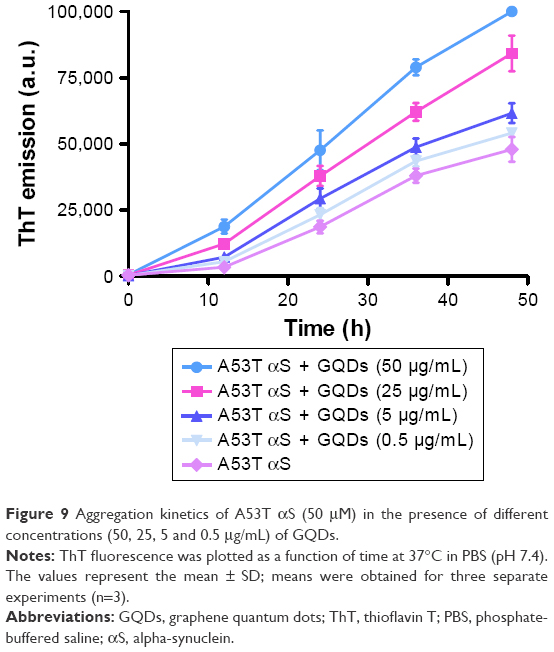 | Figure 9 Aggregation kinetics of A53T αS (50 μM) in the presence of different concentrations (50, 25, 5 and 0.5 μg/mL) of GQDs. |
Discussion
The interaction of nanomaterials with proteins may perturb both the protein structure and function. Therefore, it is not surprising that the interaction of nanomaterials and amyloidogenic proteins or peptides might inhibit or facilitate amyloid formation. The interaction between nanomaterials and intrinsically disordered protein αS, whose aggregation is crucial in the development of PD, is of great interest in the opinion of safety issues of nanomaterials or even of the drug development approaches. Although graphene and graphene oxide are being extensively studied in various biomedical applications,52 and GQDs have been considered one of the most promising nanomaterials for biological purposes and for not having the cytotoxic characteristic of other carbon-based nanomaterials,53 their effects on the aggregation of amyloidogenic proteins need to be further investigated. In this work, we explored the effects of GQDs on the aggregation of A53T αS which is a disease-relating mutant of αS. At first, a whole-cell recombinant bioluminescent sensor was developed based on the complementation of firefly luciferase fragments upon interaction of A53T αS proteins that fibrillate more profoundly than WT or other mutants of αS (Figure 1). It has been previously demonstrated that the firefly luciferase is an ideal reporter for in vitro and in vivo monitoring of αS aggregation by bioluminescence complementation approaches.54 It should be noted that the recovery of luciferase activity through intermolecular interaction of luciferase fragment was more effective in cells expressing Nluc-A53T αS and A53T αS-Cluc (Figure 4) than in cells expressing Nluc-A53T αS and Cluc-A53T αS (Figure 3). This observation supports an antiparallel interaction between two A53T αS molecules. It can be concluded that the positioning of A53T αS is critical in the success of designed biosensor and its aggregation-induced luminescence. As it has been shown, GQDs efficiently accelerate the aggregation of A53T αS in vitro (Figure 8) and also inside the cells (Figure 6).
To explore the in vitro effect of these nanomaterials on the kinetics of A53T αS aggregation, ThT assay was used to systemically study the kinetics of aggregation of A53T αS after mixing with the nanomaterials. Fibrillation of A53T mutant of αS is a two-step process: an initial lag period that reflects the thermodynamic barrier to the formation of nuclei or seeds, followed by the rapid fibril propagation. The introduction of 50 μg/mL of negatively charged GQDs and GOQDs to the A53T αS aggregation reaction imposed significant effects on the kinetics of fibrillation and decreased the lag time and enhanced A53T αS aggregation as monitored by ThT fluorescence measurements (Figures 8B and 9). This finding may suggest that GQDs are able to facilitate the nucleus formation. This effect seems to be dose dependent as checked using GQDs at 0.5, 5, 25 and 50 μg/mL (Figure 9). It can be suggested that strong interaction between A53T αS and GQDs probably alters the A53T αS secondary structure and accelerates the primary nucleation of A53T αS fibrillation. The accelerating effect of negatively charged GQDs and GOQDs is similar to that of other negatively charged nanomaterials on αS aggregation.30,51 In addition, by adsorption of proteins on the surface of GQDs, high local protein concentration favorable for amyloid fibril formation is provided55,56 which could result in the shortened lag time for A53T αS assembly into fibrils. The same explanation has been exploited to elucidate the inhibition mechanisms of fibril formation by some other nanomaterials. It has been suggested that binding between nanomaterials and amyloid proteins could block active sites for fibril formation and also lead to low protein concentration in solution, thus causing inhibition of fibril formation.57 More information is definitely needed to evaluate the exact mechanisms by which nanomaterials exert their effects on protein aggregation.
Our results are contrary to those reported previously that negatively charged GQDs can inhibit Aβ peptide aggregation and rescue the cells from the Aβ-induced cytotoxicity, thereby showing that they play a therapeutic role.58 Moreover, another carbon-based nanomaterial, fullerene, was found to inhibit significantly the amyloid formation of Aβ by specifically binding to its central hydrophobic motif, KLVFF.59 As the physical properties of GQDs greatly depend on the condition and the methods of their synthesis, this controversy might be attributed to the differences between the proteins and also the physical properties of the GQDs used in the experiment. However, our findings clearly show that extreme care must be taken for the use of graphene in biomedical applications and drug development approaches.
Herein, we showed that the interaction of negatively charged GQDs leads to a significant alteration in A53T αS aggregation kinetics. The nucleation process, which is the rate-determining step in fibrillogenesis, is dependent on GQDs concentration.
Conclusion
By developing a cell-based biosensor, we have shown the effect of GQDs and GOQDs on aggregation of A53T αS inside the cells. GQDs were demonstrated to promote efficiently the aggregation of A53T αS as indicated by the whole-cell bioluminescent biosensor, promising its application as a tool for high-throughput screening of amyloid-targeting compounds. Moreover, in parallel with intracellular data, GQDs were shown to exhibit a promoting effect on the aggregation of A53T αS in vitro. According to the results obtained here, while GQDs possess special physicochemical properties which make them an excellent candidate for biomedical applications, their interactions with cell components may hinder their applications. Our findings suggest the need for further investigation on the capability of the GQDs in passing through the BBB and also on the effects of these nanomaterials on αS aggregation in animal models.
Acknowledgment
This work was supported by a grant from the research council of Tarbiat Modares University.
Disclosure
The authors report no conflicts of interest in this work.
References
Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P. α-Synuclein and Parkinson’s disease. FASEB J. 2004;18(6):617–626. | ||
Hoehn MM, Yahr MD. Parkinsonism: onset, progression, and mortality. Neurology. 1998;50(2):318–333. | ||
Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and α-synuclein. Nat Rev Neurosci. 2002;3(12):932–942. | ||
Gomez-Tortosa E, Newell K, Irizarry M, Albert M, Growdon J, Hyman B. Clinical and quantitative pathologic correlates of dementia with Lewy bodies. Neurology. 1999;53(6):1284–1291. | ||
Singleton A, Farrer M, Johnson J, et al. α-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. | ||
Li J, Uversky VN, Fink AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry. 2001;40(38):11604–11613. | ||
Conway KA, Lee S-J, Rochet J-C, Ding TT, Williamson RE, Lansbury PT. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97(2):571–576. | ||
Pandey N, Schmidt RE, Galvin JE. The alpha-synuclein mutation E46K promotes aggregation in cultured cells. Exp Neurol. 2006;197(2):515–520. | ||
Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418(6895):291. | ||
Emadi S, Barkhordarian H, Wang MS, Schulz P, Sierks MR. Isolation of a human single chain antibody fragment against oligomeric α-synuclein that inhibits aggregation and prevents α-synuclein-induced toxicity. J Mol Biol. 2007;368(4):1132–1144. | ||
El-Agnaf OM, Salem SA, Paleologou KE, et al. Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006;20(3):419–425. | ||
Emadi S, Kasturirangan S, Wang MS, Schulz P, Sierks MR. Detecting morphologically distinct oligomeric forms of α-synuclein. J Biol Chem. 2009;284(17):11048–11058. | ||
Kanazawa T, Akiyama F, Kakizaki S, Takashima Y, Seta Y. Delivery of siRNA to the brain using a combination of nose-to-brain delivery and cell-penetrating peptide-modified nano-micelles. Biomaterials. 2013;34(36):9220–9226. | ||
Patel PJ, Acharya NS, Acharya SR. Development and characterization of glutathione-conjugated albumin nanoparticles for improved brain delivery of hydrophilic fluorescent marker. Drug Deliv. 2013;20(3–4):143–155. | ||
Chopra D, Gulati M, Saluja V, Pathak P, Bansal P. Brain permeable nanoparticles. Recent Pat CNS Drug Discov. 2008;3(3):216–225. | ||
Wang X-Y, Lei R, Huang H-D, et al. The permeability and transport mechanism of graphene quantum dots (GQDs) across the biological barrier. Nanoscale. 2015;7(5):2034–2041. | ||
Kong SD, Lee J, Ramachandran S, et al. Magnetic targeting of nanoparticles across the intact blood–brain barrier. J Control Release. 2012;164(1):49–57. | ||
Triulzi RC, Dai Q, Zou J, et al. Photothermal ablation of amyloid aggregates by gold nanoparticles. Colloids Surf B Biointerfaces. 2008;63(2):200–208. | ||
Marcato PD, Durán N. New aspects of nanopharmaceutical delivery systems. J Nanosci Nanotechnol. 2008;8(5):2216–2229. | ||
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. IntJ Nanomed. 2008;3(2):133. | ||
Nie S, Xing Y, Kim GJ, Simons JW. Nanotechnology applications in cancer. Annu Rev Biomed Eng. 2007;9:257–288. | ||
O’Brien EP, Straub JE, Brooks BR, Thirumalai D. Influence of nanoparticle size and shape on oligomer formation of an amyloidogenic peptide. J Phys Chem Lett. 2011;2(10):1171–1177. | ||
Mirsadeghi S, Dinarvand R, Ghahremani MH, et al. Protein corona composition of gold nanoparticles/nanorods affects amyloid beta fibrillation process. Nanoscale. 2015;7(11):5004–5013. | ||
Huang R, Carney RP, Stellacci F, Lau BL. Protein–nanoparticle interactions: the effects of surface compositional and structural heterogeneity are scale dependent. Nanoscale. 2013;5(15):6928–6935. | ||
Cabaleiro-Lago C, Szczepankiewicz O, Linse S. The effect of nanoparticles on amyloid aggregation depends on the protein stability and intrinsic aggregation rate. Langmuir. 2012;28(3):1852–1857. | ||
Marshall KE, Morris KL, Charlton D, et al. Hydrophobic, aromatic, and electrostatic interactions play a central role in amyloid fibril formation and stability. Biochemistry. 2011;50(12):2061–2071. | ||
Roberti MJ, Morgan M, Menéndez G, Pietrasanta LI, Jovin TM, Jares-Erijman EA. Quantum dots as ultrasensitive nanoactuators and sensors of amyloid aggregation in live cells. J Am Chem Soc. 2009;131(23):8102–8107. | ||
Yang JA, Lin W, Woods WS, George JM, Murphy CJ. α-Synuclein’s adsorption, conformation, and orientation on cationic gold nanoparticle surfaces seeds global conformation change. J Phys Chem B. 2014;118(13):3559–3571. | ||
Yang JA, Johnson BJ, Wu S, Woods WS, George JM, Murphy CJ. Study of wild-type α-synuclein binding and orientation on gold nanoparticles. Langmuir. 2013;29(14):4603–4615. | ||
Álvarez YD, Fauerbach JA, Pellegrotti JV, Jovin TM, Jares-Erijman EA, Stefani FD. Influence of gold nanoparticles on the kinetics of α-synuclein aggregation. Nano Lett. 2013;13(12):6156–6163. | ||
Joshi N, Basak S, Kundu S, De G, Mukhopadhyay A, Chattopadhyay K. Attenuation of the early events of α-synuclein aggregation: a fluorescence correlation spectroscopy and laser scanning microscopy study in the presence of surface-coated Fe3O4 nanoparticles. Langmuir. 2015;31(4):1469–1478. | ||
Zaman M, Ahmad E, Qadeer A, Rabbani G, Khan RH. Nanoparticles in relation to peptide and protein aggregation. Int J Nanomedicine. 2014;9:899–912. | ||
Hashimoto T, Adams KW, Fan Z, McLean PJ, Hyman BT. Characterization of oligomer formation of amyloid-β peptide using a split-luciferase complementation assay. J Biol Chem. 2011;286(31):27081–27091. | ||
Azad T, Tashakor A, Hosseinkhani S. Split-luciferase complementary assay: applications, recent developments, and future perspectives. Anal Bioanal Chem. 2014;406(23):5541–5560. | ||
Torkzadeh-Mahani M, Ataei F, Nikkhah M, Hosseinkhani S. Design and development of a whole-cell luminescent biosensor for detection of early-stage of apoptosis. Biosens Bioelectron. 2012;38(1):362–328. | ||
Ozawa T. Designing split reporter proteins for analytical tools. Anal Chim Acta. 2006;556(1):58–68. | ||
Takeuchi M, Nagaoka Y, Yamada T, Takakura H, Ozawa T. Ratiometric bioluminescence indicators for monitoring cyclic adenosine 3′,5′-monophosphate in live cells based on luciferase-fragment complementation. Anal Chem. 2010;82(22):9306–9313. | ||
Misawa N, Kafi A, Hattori M, Miura K, Masuda K, Ozawa T. Rapid and high-sensitivity cell-based assays of protein–protein interactions using split click beetle luciferase complementation: an approach to the study of G-protein-coupled receptors. Anal Chem. 2010;82(6):2552–2560. | ||
Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. New York: Cold Spring Harbor Laboratory Press; 1989. | ||
Choosakoonkriang S, Lobo BA, Koe GS, Koe JG, Middaugh CR. Biophysical characterization of PEI/DNA complexes. J Pharm Sci. 2003;92(8):1710–1722. | ||
Nazari M, Hosseinkhani S. Design of disulfide bridge as an alternative mechanism for color shift in firefly luciferase and development of secreted luciferase. Photochem Photobiol Sci. 2011;10(7):1203–1215. | ||
Caldinelli L, Albani D, Pollegioni L. One single method to produce native and Tat-fused recombinant human α-synuclein in Escherichia coli. BMC Biotechnol. 2013;13(1):1. | ||
Salehnia F, Faridbod F, Dezfuli AS, Ganjali MR, Norouzi P. Cerium (III) ion sensing based on graphene quantum dots fluorescent turn-off. J Fluoresc. 2017;27(1):331–338. | ||
Unsoy G, Yalcin S, Khodadust R, Gunduz G, Gunduz U. Synthesis optimization and characterization of chitosan-coated iron oxide nanoparticles produced for biomedical applications. J Nanopart Res. 2012;14(11):1–13. | ||
Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, et al. Formation of toxic oligomeric α-synuclein species in living cells. PLoS One. 2008;3(4):e1867. | ||
Wang MS, Boddapati S, Emadi S, Sierks MR. Curcumin reduces α-synuclein induced cytotoxicity in Parkinson’s disease cell model. BMC Neurosci. 2010;11(1):1. | ||
Ahmad B, Lapidus LJ. Curcumin prevents aggregation in α-synuclein by increasing reconfiguration rate. J Biol Chem. 2012;287(12):9193–9199. | ||
Erfani-Moghadam V, Nomani A, Zamani M, Yazdani Y, Najafi F, Sadeghizadeh M. A novel diblock of copolymer of (monomethoxy poly [ethylene glycol]-oleate) with a small hydrophobic fraction to make stable micelles/polymersomes for curcumin delivery to cancer cells. Int J Nanomedicine. 2014;9(1):5541–5554. | ||
Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin-and caveolae-mediated endocytosis. Biochem J. 2004;377(1):159–169. | ||
Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44. | ||
Galvagnion C, Buell AK, Meisl G, et al. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol. 2015;11(3):229–234. | ||
Akhavan O, Ghaderi E. Escherichia coli bacteria reduce graphene oxide to bactericidal graphene in a self-limiting manner. Carbon. 2012;50(5):1853–1860. | ||
Zhu S, Zhang J, Qiao C, et al. Strongly green-photoluminescent graphene quantum dots for bioimaging applications. Chem Commun (Camb). 2011;47(24):6858–6860. | ||
Aelvoet SA, Ibrahimi A, Macchi F, et al. Noninvasive bioluminescence imaging of α-synuclein oligomerization in mouse brain using split firefly luciferase reporters. J Neurosci. 2014;34(49):16518–16532. | ||
Linse S, Cabaleiro-Lago C, Xue WF, et al. Nucleation of protein fibrillation by nanoparticles. Proc Natl Acad Sci U S A. 2007;104(21):8691–8696. | ||
Colvin VL, Kulinowski KM. Nanoparticles as catalysts for protein fibrillation. Proc Natl Acad Sci U S A. 2007;104(21):8679–8680. | ||
Cabaleiro-Lago C, Quinlan-Pluck F, Lynch I, et al. Inhibition of amyloid β protein fibrillation by polymeric nanoparticles. J Am Chem Soc. 2008;130(46):15437–15443. | ||
Liu Y, Xu L-P, Dai W, Dong H, Wen Y, Zhang X. Graphene quantum dots for the inhibition of β amyloid aggregation. Nanoscale. 2015;7(45):19060–19065. | ||
Kim JE, Lee M. Fullerene inhibits β-amyloid peptide aggregation. Biochem Biophys Res Commun. 2003;303(2):576–579. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.