")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Investigation and optimization of formulation parameters on preparation of targeted anti-CD205 tailored PLGA nanoparticles
Received 20 June 2015
Accepted for publication 28 August 2015
Published 10 December 2015 Volume 2015:10(1) Pages 7371—7384
DOI https://doi.org/10.2147/IJN.S90866
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Sheikh Tasnim Jahan, Azita Haddadi
Division of Pharmacy, College of Pharmacy and Nutrition, University of Saskatchewan, Saskatoon, SK, Canada
Abstract: The purpose of this study was to assess the effect of various formulation parameters on anti-CD205 antibody decorated poly(D, L-lactide co-glycolide) (PLGA) nanoparticles (NPs) in terms of their ability to target dendritic cells (DCs). In brief, emulsification solvent evaporation technique was adapted to design NP formulations using two different viscosity grades (low and high) of both ester and carboxylic acid terminated PLGA. Incorporation of ligand was achieved following physical adsorption or chemical conjugation processes. The physicochemical characterizations of formulations were executed to assess the effects of different solvents (chloroform and ethyl acetate), stabilizer percentage, polymer types, polymer viscosities, ligand-NP bonding types, cross-linkers, and cryoprotectants (sucrose and trehalose). Modification of any of these parameters shows significant improvement of physicochemical properties of NPs. Ethyl acetate was the solvent of choice for the formulations to ensure better emulsion formation. Infrared spectroscopy confirmed the presence of anti-CD205 antibody in the NP formulation. Finally, cytotoxicity assay confirmed the safety profile of the NPs for DCs. Thus, ligand modified structurally concealed PLGA NPs is a promising delivery tool for targeting DCs in vivo.
Keywords: nanoparticle, anti-CD205, PLGA, dendritic cells
Introduction
Dendritic cells (DCs) are known as the potent antigen presenting cells to induce adaptive immune responses. Manipulating DCs by targeted antigen delivery through various endocytic and secretory pathways is a consequence of delivering site-specific therapeutic delivery system. C-type lectin receptor CD205 (molecular weight of 205 kDa), exclusively expressed on DCs; is a widely studied DC target molecule for induction of immune response. Anti-CD205 monoclonal antibody (mAb) linked delivery system can efficiently deliver its cargo to the processing compartments of DCs in vivo.1 CD205 receptor possesses a fast internalization speed, where over 80% of surface CD205 are internalized within 90 minutes.2,3 The proportion of targeted molecules endocytosed by this receptor in both immature and mature DCs is exceptionally higher compared to other surface receptors. In addition to internalization, antigen presentation on major histocompatibility complex (MHC)-I and MHC-II, CD205 receptors elicited superior presentation compared to CD11c receptor. Thus, targeting this receptor would be promising in both steady-state and inflammatory conditions.2,4 Therefore, CD205 specific antibodies can induce efficient antigen processing and presentation, notably eliciting both T helper1 CD4+ T cell and CD8+ T cell responses. Engagement of anti-CD205 mAb to target CD205 receptors shows high consensus to deliver vaccine utilizing an appropriate delivery system.5
Over the past decade, nanoparticles (NPs) have gained increasing attention in the field of drug delivery. Particularly, polyester based NPs offer the advantage of effective delivery of drug to the target site, ensuring therapeutic benefit with minimum side effects. Industry has recently focused on the US Food and Drug Administration (FDA) approved poly(D, L-lactide co-glycolide) (PLGA) based NPs because of their biodegradability, biocompatibility, low toxicity, controlled release, and surface-modification properties.6,7 Hence, functionalization of PLGA NPs with ligands such as anti-CD205 antibody presents an opportunity for an innovative antibody-targeted vaccine delivery system. This coupling aims to provide increased payload of drug/antigen, thereby increasing response and reducing the number of doses required. The ligand itself might function in a non-activating manner, which is important for immunotherapeutic diseases.8 PLGA polymers are commercially available with different terminal groups, namely, free carboxylic acid (COOH) end groups (uncapped) or esterified terminal groups (capped). The end groups of PLGA can influence drug encapsulation efficiency, degradation, stability, and conjugation of ligands. For example, COOH terminated NPs can result in a slightly acidic environment, that may cause degradation of encapsulated antigen during formulation process or inside endosomal compartment.9
The present study focuses on the formulation optimization with anti-CD205 ligand using both capped and uncapped PLGA; each type offered with low and high viscosity grades (Figure 1).10 Discussions are based on the comparison and evaluation of how different process parameters affect these two subtypes of ester and COOH ended PLGA NPs for in vitro experiment setups. To serve this purpose, standardization of various parameters was executed to obtain NPs with suitable particle size, surface charge, polydispersity index (PDI), surface display, toxicity profile, and structural modification. Therefore, a structure-activity relationship is concluded after analyzing the results. As a consequence, the ultimate goal is to develop a delivery system with suitable formulation strategy that could simulate the in vitro responses in an animal model. Altogether, our results support the potential use of PLGA NPs as therapeutic delivery system to design a cancer vaccine.
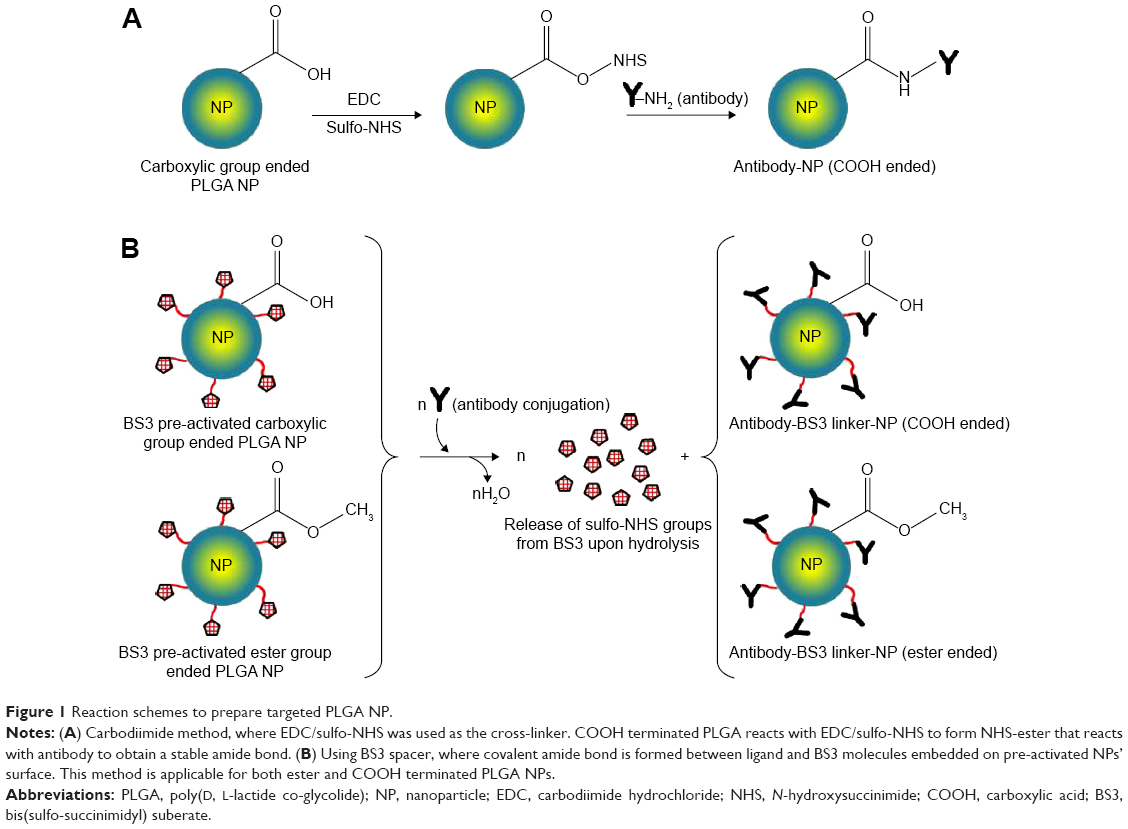 | Figure 1 Reaction schemes to prepare targeted PLGA NP. |
Materials and methods
Materials
Ester terminated PLGA (inherent viscosity 0.15–0.25 dL/g and 0.55–0.75 dL/g) and COOH terminated PLGA (inherent viscosity 0.18 dL/g and 0.55–0.75 dL/g) were purchased from LACTEL Absorbable Polymers, Birmingham, AL, USA. Polyvinyl alcohol (PVA), bis(sulfo-succinimidyl) suberate (BS3), alpha minimum essential medium, fetal bovine serum, 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay kit, and bicinchoninic acid (BCA) assay kit were purchased from Sigma-Aldrich Co., St Louis, MO, USA. Other reagents used were N-hydroxysuccinimide esters (sulfo-NHS) and 1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) from Thermo Fisher Scientific, Waltham, MA, USA. Biotin anti-mouse CD205 antibody was purchased from Biolegend (San Diego, CA, USA). JAWSII DC line was obtained from American Type Culture Collection (ATCC), Manassas, VA, USA. GM-CSF was purchased from Thermo Fisher Scientific. Solvents like chloroform and ethyl acetate were of analytical grade.
Preparation of NPs by emulsification solvent evaporation method
Method 1
PLGA NPs were prepared by the double emulsification solvent evaporation technique as previously reported.11 Briefly, PLGA was dissolved in chloroform (25% weight/volume [w/v]). Aqueous phase comprised of 5% w/v PVA was added to the oil phase in a drop wise manner and sonicated by probe-sonicator. The resulting primary emulsion was transferred into water to form the secondary emulsion and stirred to evaporate the chloroform. NPs were then collected by centrifugation, washed with distilled water to remove the residual PVA, and resuspended in distilled water. Cryoprotectant was used in the formulations to minimize freeze-drying stress after washing.12
Method 2
The NPs were prepared following single oil in water emulsification solvent evaporation technique.13 In brief, PLGA was dissolved in ethyl acetate (6.5% w/v) and transferred into 2.2% of PVA to form the emulsion. The procedure was then followed as mentioned in Method 1 with some minor modifications. To prepare pre-activated NPs for the covalent attachment of ligand, presence of BS3 in the aqueous phase is mandatory. The prepared NPs were lyophilized with cryoprotectants to avoid aggregation. Parameters mentioned in Table 1 were considered in the preparation and evaluated for both methods 1 and 2. Discussion will be based on the effect of these parameters on the physicochemical properties of NPs.
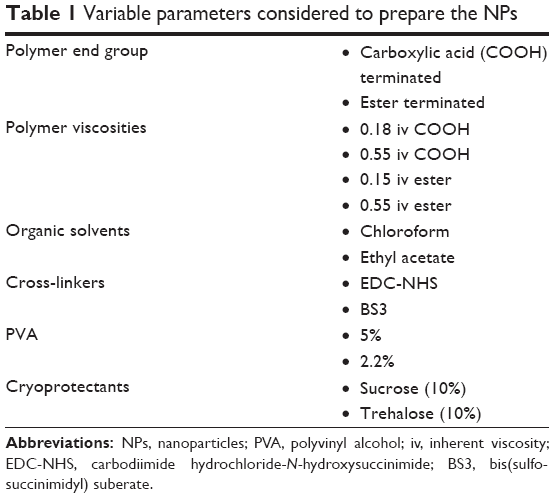 | Table 1 Variable parameters considered to prepare the NPs |
Antibody coupling to the particle surface
Antibody coupling was carried out through two approaches, physical adsorption and covalent conjugation.
Physical adsorption of antibody to NPs’ surface
Biotinylated anti-CD205 mAb was added at a certain concentration to previously lyophilized PLGA NPs in phosphate buffered saline (PBS) and stirred for 4 hours in ice. After that, excess PBS was centrifuged and washed out. The NPs were freeze-dried upon re-suspension followed by storage at −20°C. At each washing step, the obtained supernatants were stored to determine the amount of un-conjugated antibody by BCA assay. This is an indirect method to measure the actual amount of antibody attached to the NPs. The control groups for BCA assay were the supernatants obtained from antibody-free NPs of each formulation through the similar conditions maintained in antibody-NP conjugation process.14
Covalent attachment of antibody with NPs
Two different approaches were followed for covalent attachment where, one was using carbodiimide chemistry with EDC and sulfo-NHS (for method 1); and the other one with the use of BS3 (for method 2). For the first method, EDC/sulfo-NHS solution was added to antibody solution. The resulting suspension was stirred for 4 hours in ice, after which, centrifugation was done twice to remove excess reagents and soluble isourea by-product. Therefore, the amide bonds were formed between the primary amine groups of antibodies with the free carboxylic end groups of PLGA NPs.14 For the second method, BS3 pre-activated freeze-dried NPs were resuspended in PBS for the covalent attachment of antibody.13 Upon addition of antibody, the activated NPs underwent the process of conjugation followed by washing twice with PBS of pH 7.2. The covalent amide linkage was formed by replacing lysine groups of the antibody with free carboxylic group of BS3 upon releasing sulfo-NHS groups. The obtained NPs were further freeze-dried followed by storage at −20°C. The reaction schemes for both approaches are shown in Figure 1.
Determination of particle size, zeta potential (ZP), and PDI
Dynamic light scattering technique was used to measure the particle size, ZP, and PDI using Malvern ZetaSizer, Nano ZS (Malvern Instruments, Malvern, UK).15 ZP was measured on the basis of electrophoretic mobility in an electric field.16 The measurements were performed for both unmodified and modified NPs before and after freeze-drying. Recovery percentage or yield of the preparation technique was calculated from the amount of NPs obtained divided by the total amount of initial PLGA polymer used to prepare NPs (the amount of PVA is negligible).
Morphology by scanning electron microscopy (SEM)
Morphology of the NP surface was analyzed by SEM. In brief, PLGA NPs were dispersed in distilled water (0.2% w/v). Appropriate portion of nano-suspension was placed on the carbon tape of the metal stub and allowed to air-dry. The samples were then placed in a sputter coater (S150B; BOC Edwards, Sussex, UK) for 1 minute to produce a gold coating with ~20 nm thickness; and viewed under a scanning electron microscope at voltage of 20–25 kV (Carl Zeiss Evo 60; Carl Zeiss Meditec AG, Jena, Germany).17
Structural characterization by Fourier transform infrared spectroscopy (FTIR)
FTIR analysis of NPs was recorded on a Bruker IFS 66v/S infrared spectrophotometer (Bruker Optics Inc, Billerica, MA, USA) in the mid-infrared range at the Canadian Light Source (CLS), University of Saskatchewan, Saskatoon, SK, Canada. All samples were mixed with spectroscopic grade potassium bromide and mulled to prepare pellets. The spectra were taken for potassium bromide pellets in the range of 4,000–400 cm−1 in absorbance mode.13,18 Data analysis was performed with Bruker Opus software. Baseline correction was performed on all raw absorbance spectra.
Determination of amount of antibody attached to the NPs
A BCA protein assay was performed to measure the amount of antibody attached per mg of NPs. The antibody was analyzed both directly (on the NP surface) and indirectly (in the supernatant). Briefly, a sample of modified NP was taken and resuspended in water to assess the antibody attached to the surface. Unmodified NPs were used as controls. In indirect method, the collected supernatants during washing steps were stored for BCA assay to quantify the amount of unbound antibody. Ninety-six well plate was used and the instructions from the BCA assay kit were followed for the quantification. The standard curve was generated by plotting the absorbance versus various concentrations of the standard solution, bovine serum albumin. The standard curve was found to be linear over the range of 0–32 μg/mL. The absorbance was measured at a wavelength of 562 nm in a microplate reader.19
DC (JAWS II) culture
The DC line obtained from American Type Culture Collection was initiated as recommended. The cell culture was maintained at 37°C and 5% CO2 in complete media for few days to reach the optimum confluency. After that, the cells were stored in cryo-vials containing freezing solution (dimethyl sulfoxide). The complete media is as recommended by ATCC.
In vitro cytotoxicity assay
MTT assay was performed to investigate the cytotoxicity of NPs on DC lines using corresponding untreated cells as control. On day 1, the cells were seeded in 96-well plate at a density of 10,000 cells per mL. After overnight incubation, cells were exposed to various formulation treatments. On day 3, 10 μL of MTT solution was added to each well and further incubated for 4 hours. Finally, 100 μL of MTT solvent was added to dissolve the formazan crystals. The absorption intensity of the purple–blue color formed was measured at a wavelength of 570 nm by microplate reader.15 Cell viability was calculated using the following equation:20
|
|

Statistics
All data are presented as mean ± standard deviation. The significance of the differences between groups was analyzed by unpaired Student’s t-test or one-way analysis of variance followed by the Tukey’s post hoc test for multiple comparisons. P-value of <0.05 was considered as statistically significant. All the statistical analyses were performed with GraphPad Prism 5.03 software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Effect of polymers’ end groups and viscosities
The influence of polymer composition (functional end group and inherent viscosity) on physicochemical properties of NPs was investigated. Particle size is an important parameter that can affect the biopharmaceutical and biodistribution properties of formulations. The smaller particle size will lead to a higher total surface area relating to faster release of its payload.21 PDI is an index for size distribution where an enhanced PDI value indicates that the particles do not have a uniform distribution.22
In method 1, the average particle size and PDI for plain NPs prepared with COOH and ester end groups were found to range from 202 to 237 nm and 231 to 281 nm, respectively as shown in Figure 2A. Polymer viscosity (0.15 and 0.55 inherent viscosity) was indicated as a limiting factor for particle size in both COOH and ester terminated NPs. With an increase in polymer viscosity the particle size also increased as shown in Figure 2A and B.23 After statistical evaluation, there was no significant effect observed for the variable end groups of PLGA on plain NPs’ particle size, PDI, and ZP (Figure 2C) values (Table 2 and Figure 3). However, the effect of variable end groups and viscosities of the polymer on particle size was found significant for ligand modified NPs (Table 2) although these two variables had no effect on their PDI values.
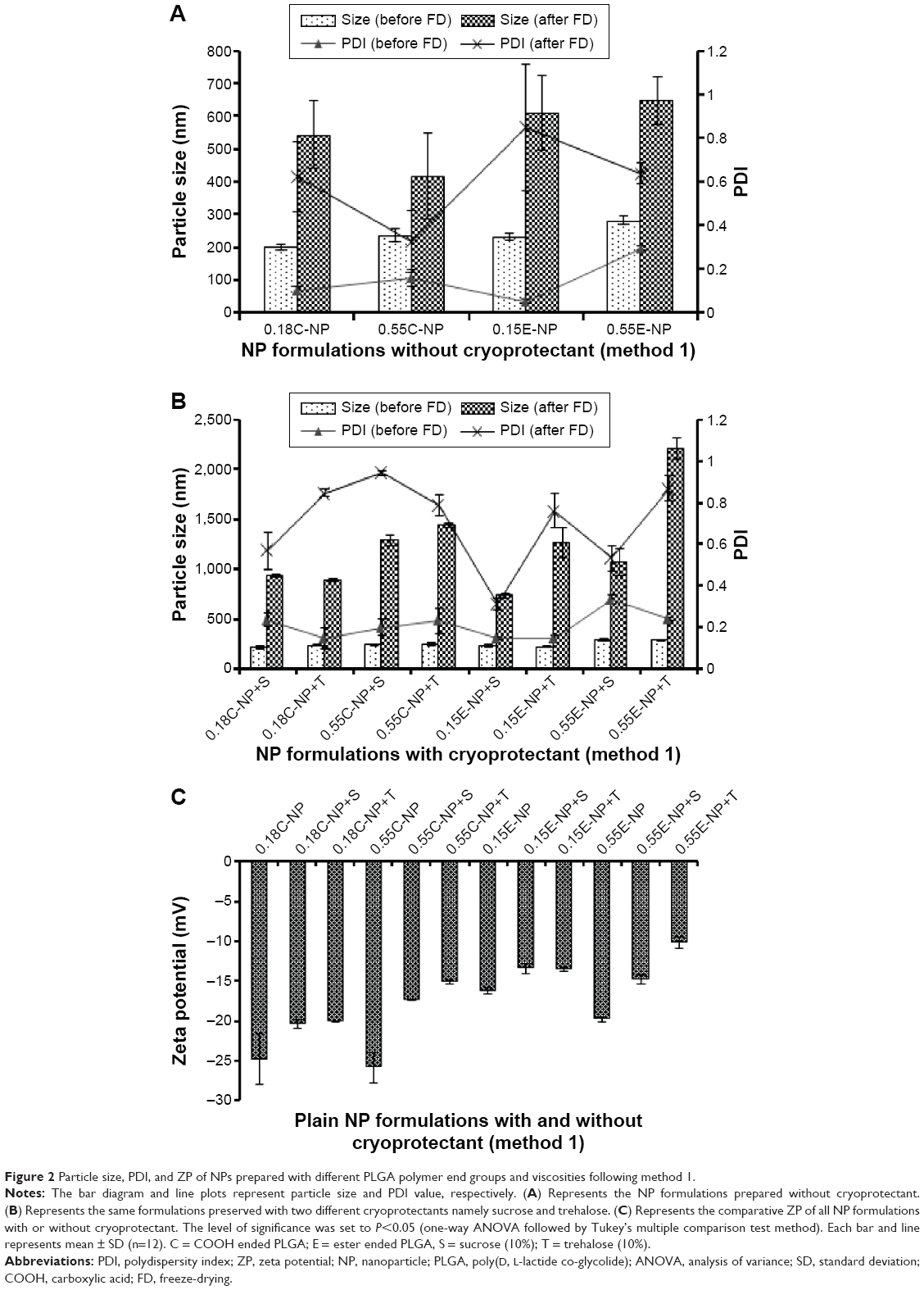 | Figure 2 Particle size, PDI, and ZP of NPs prepared with different PLGA polymer end groups and viscosities following method 1. |
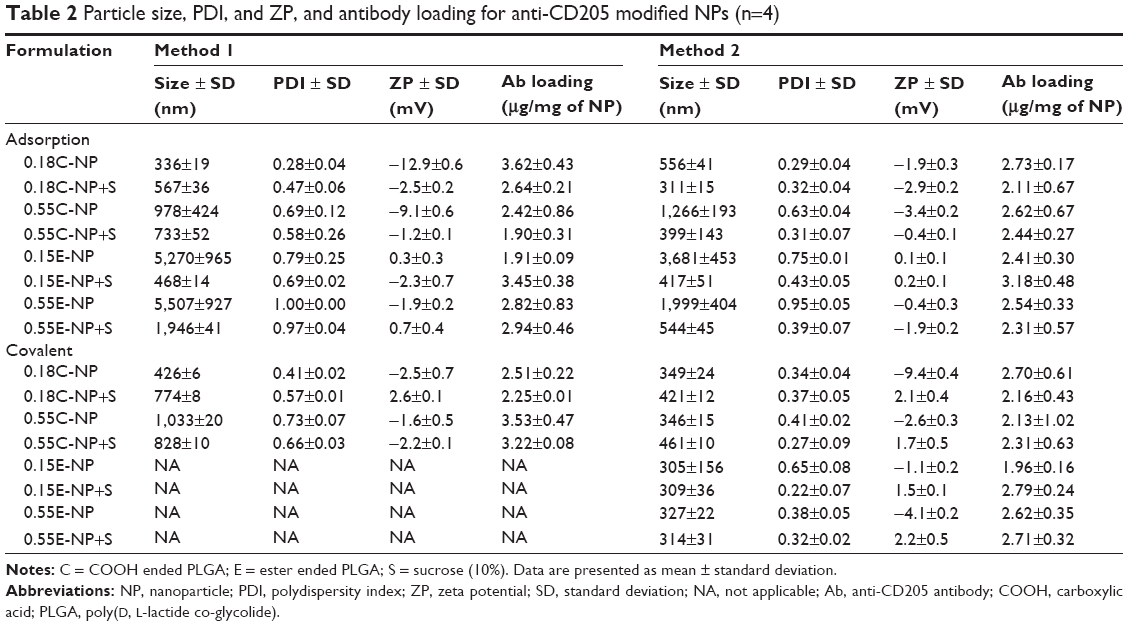 | Table 2 Particle size, PDI, and ZP, and antibody loading for anti-CD205 modified NPs (n=4) |
 | Figure 3 Particle size, PDI, and ZP of NPs prepared with different PLGA polymer end groups and viscosities following method 2. |
In method 2, both particle size and PDI values were in a fairly desirable range for all NPs before freeze-drying (Figure 3A). A proportional relation between polymer viscosity and particle size was observed for plain (Figure 2A, B and Figure 3A, B) and modified (Table 2) NPs. In contrary, no significant correlation could be made between the variable polymer end groups and physicochemical properties.
Effect of ligand-NP bonding types
In method 1, low viscosity COOH terminated NP had a significantly higher (P<0.05) amount of antibody adsorbed compared to other groups and the particles were in an appropriate size range below 350 nm. The high viscosity COOH terminated NPs had relatively larger size with wide PDI values. In addition, ZP values showed a significant drop toward positive value upon attachment of ligand with NPs as shown in Table 2 (P<0.05). NPs obtained after covalent attachment of antibody, had the same particle size trend as observed in the adsorbed groups. While larger aggregates were formed with antibody-adsorbed NPs for ester terminated NPs, which was further minimized using cryoprotectant. The original particle size was above the nanometer range, indicating the presence of aggregates (PDI value >0.99).
In method 2, there was a significant decrease in particle size for the covalently modified NPs compared to antibody-adsorbed formulations. But no concrete correlation could be drawn to compare the antibody loading through adsorption and covalent attachment of ligand with NPs. In addition, the inclusion of antibody to NPs shifted the ZP toward positive values, which indirectly confirms the presence of antibody on the NP surface, as summarized in Table 2 and Figure 3C. This could be ascribed to the amphiphilic properties of antibody or shielding the negative charges on the surface by positively charged antibody.24,25 Table 2 represents the overall data for particle size, PDI, ZP, and antibody quantification for modified NPs. Furthermore, the SEM photographs confirmed that NPs form spherical shaped particles within the desired size range. SEM images of NPs were taken at a voltage of 20–25 kV at various magnifications as shown in Figure 4.
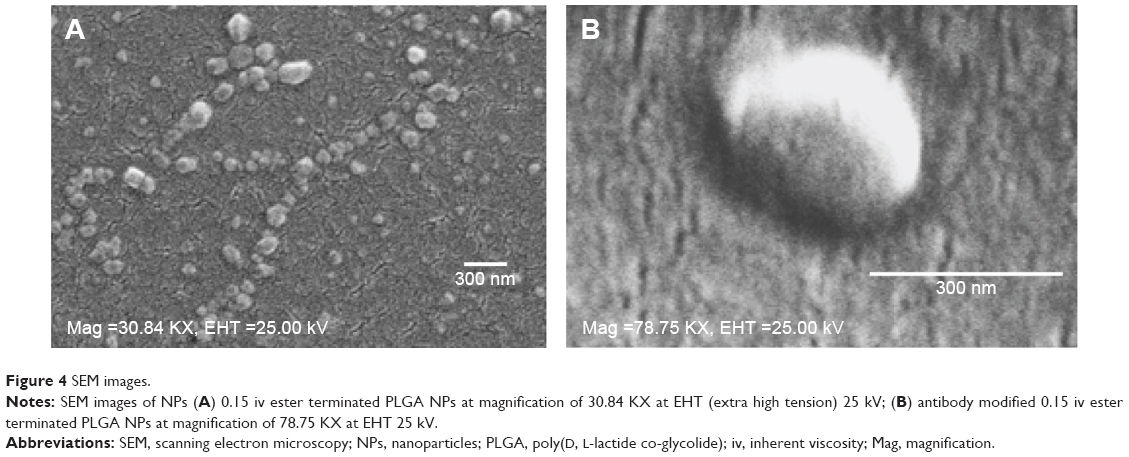 | Figure 4 SEM images. |
Effect of cryoprotectants
The NPs were in a well-dispersed suspension form before freeze-drying; however they formed aggregates upon reconstitution after freeze-drying, which could not be re-dispersed even after sonication. The reason behind this irreversible aggregation could be attributed to freeze-drying stress on the particles rendering a wide range of PDI value before ligand attachment.26 Use of cryoprotectants could overcome this stress to obtain aggregation-free fine suspension after ligand attachment. However, all the formulations had significant difference in particle size before and after freeze-drying (P<0.05) irrespective of the presence of cryoprotectant.
Notably, the use of trehalose (10%) could not fully minimize the aggregation produced between the prepared NPs following method 1 before ligand attachment compared to sucrose (10%). As a consequence, the rest of the formulations were continued with sucrose (10%). However, 10% sucrose was preferably chosen after comparative optimization based on the different percentages (1%, 5%, and 10%) of sucrose used as cryoprotectant (data not shown). Sucrose was found to be a better cryoprotectant to retain particle size when compared with trehalose used for plain NP formulations prepared following method 1. Thus, the effect of cryoprotectants was partially or not pronounced in formulations under method 1 as represented in Figure 2B. The ZP value for plain NPs without cryoprotectants was found to be more negative compared to formulations with sucrose. Even though the presence of sucrose should cause a higher negative charge on particles, the opposite was found.27
For method 2, there was significant change in ZP after addition of cryoprotectant (P<0.05). Also, the presence of cryoprotectant showed significant difference in particle size for antibody-adsorbed groups (P<0.05). However, the change in particle size was not significant when cryoprotectant was used in covalently modified formulations. Table 2 demonstrates the effect of sucrose after antibody attachment, where PDI was found to be below or equal to 0.43. Whereas formulations that were freeze-dried without cryoprotectant showed higher PDIs (highest PDI =0.95). There was significant reduction in particle size for all modified NPs after use of cryoprotectant, except the covalently modified formulations of method 2.
Effect of cross-linkers
EDC/sulfo-NHS cross-linker was considered to conjugate antibody ligand with prepared NPs following method 1.28,29 The presence of cross-linkers could be attributed to larger particles after antibody attachment. In method 2, the BS3 (spacer) pre-activated NP formulations showed fairly considerable physicochemical properties after antibody modification. In both methods, successful attachment of antibody was obtained which was confirmed by BCA assay (Table 2).
Confirmation of structural modification by FTIR
The peak around 1,750 cm−1 is a marked peak that elicits the presence of a carbonyl bond (C=O stretching vibration), which is characteristic of PLGA.30 An amide stretching is present between 3,310 and 3,250 cm−1 in anti-CD205 antibody modified PLGA NP spectra, which corresponds to C=O stretching bond. Theoretically, amide-I vibrations result from C=O stretching vibration near 1,610 cm−1 as observed in the spectra for antibody modified NP.31 Amide-I bond is the most sensitive to prove the structural change in any compound containing proteins. Some contribution in the spectrum from the C=O groups in both BS3 free and BS3 containing NP formulations could also be observed.32,33 To confirm the presence of antibody on the NPs, the spectra for only high viscosity COOH ended PLGA NPs and their modification are represented here (Figure 5). Spectra for other polymer types and their subtypes are not shown here.
 | Figure 5 Infrared spectrum of NPs. |
Comparison of safety profiles for method 1 and 2
All NP formulations prepared by method 1 (both ester and COOH terminated NPs) retained DC viability (≥80%). The findings indicate that both plain and modified NPs were not toxic to the DCs confirming the safety profile of the NPs as shown in Figures 6 and 7. Similarly, method 2 based NP formulations demonstrate a viability of >90%.11 There was no significant difference in viability among different groups (plain and modified) for both methods. However, 0.15E-NP+S showed significant difference (P<0.05) compared to other formulations like 0.18C-NP+BS3, 0.55C-NP+BS3, 0.15E-NP+BS3, 0.15E-NP+BS3+S, and 0.55E-NP+BS3+S prepared following method 2.
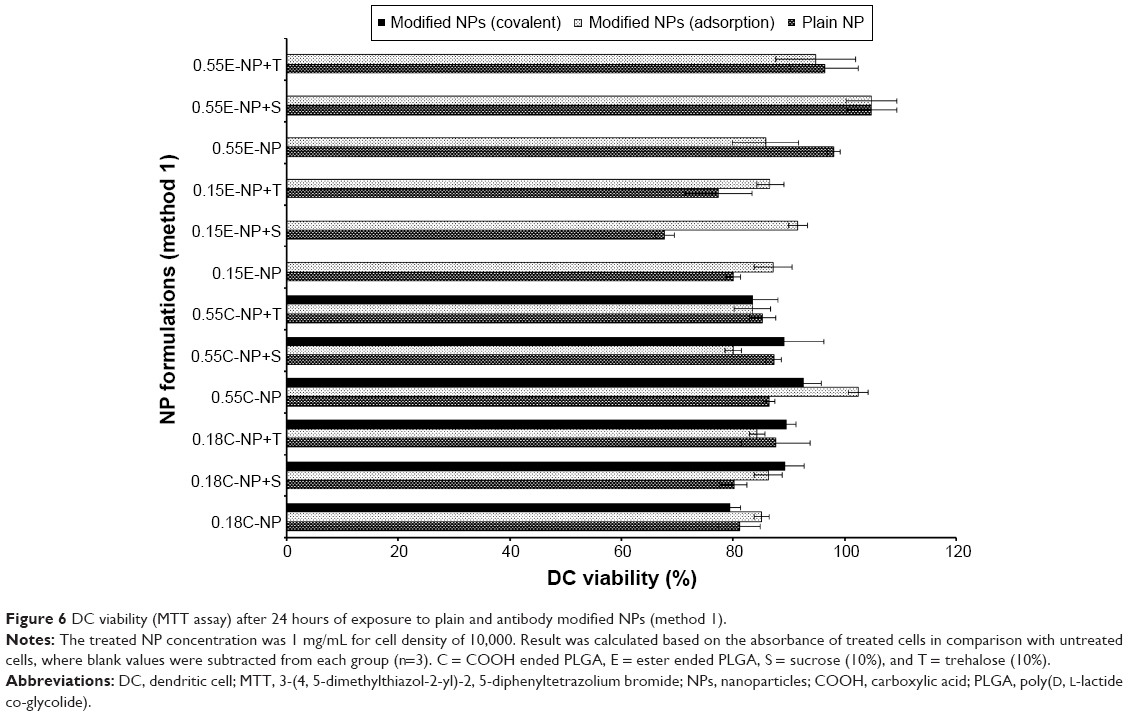 | Figure 6 DC viability (MTT assay) after 24 hours of exposure to plain and antibody modified NPs (method 1). |
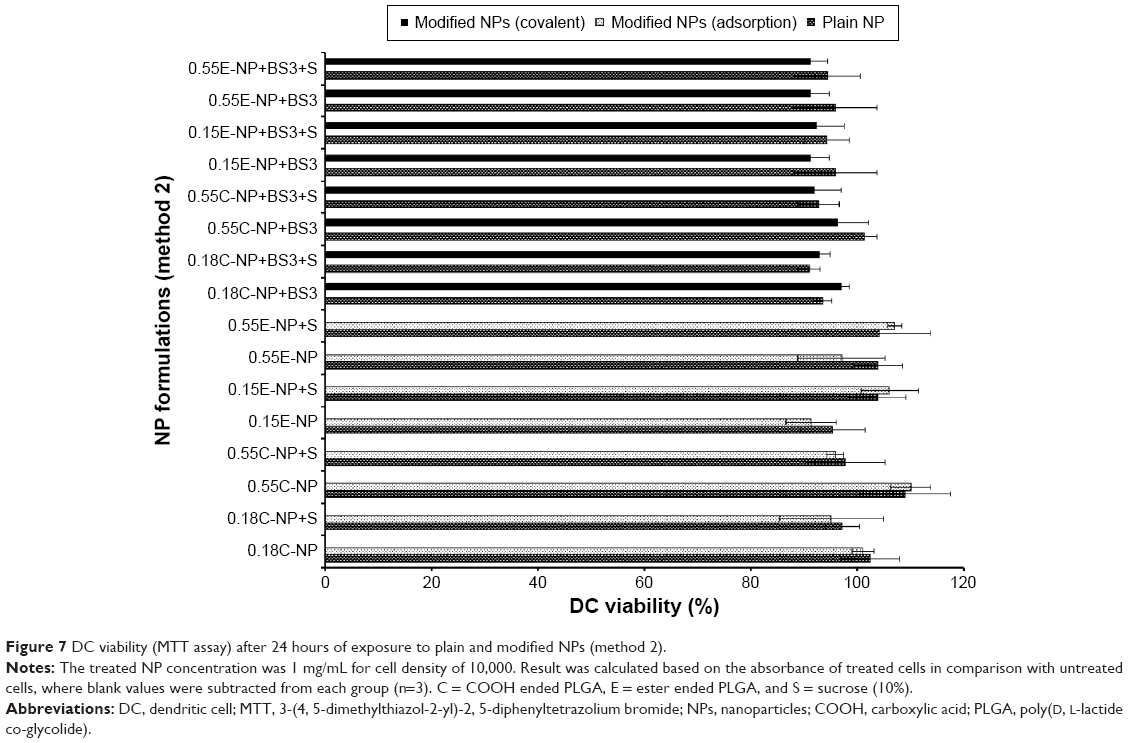 | Figure 7 DC viability (MTT assay) after 24 hours of exposure to plain and modified NPs (method 2). |
Discussion
The choice of a suitable NP formulation technique is dependent on the desired physicochemical properties. NP size is an important determinant of the formulation efficacy in vivo. Very large particles (around 1 μm) can cause irritant effects after injection.34 On the other hand, particles smaller than 5 nm are likely to be cleared by non-phagocytic cells.35 Alternatively, particles larger than 100 nm have the chance to be taken up by phagocytic cells (for example, DCs).34,36 The formulations prepared by the methods mentioned here, aim to target DC receptors in vivo. Thus the particle size range (309±36 to 544±45 nm) obtained by method 2 could be suitable for DC uptake via subcutaneous delivery.37,38 However, toxicity issue is a concern for emulsification solvent evaporation technique. Chlorinated solvents like chloroform and dichloromethane have issues regarding environmental challenges and human use. Considering these issues, ethyl acetate offers a better solubility and safety profile compared to chloroform. As a polar solvent ethyl acetate with a solubility of 8.1 volume/volume in water is comparatively safer.39 It has lower interfacial tension (1.7 dyne/cm) compared to chloroform (32.8 dyne/cm), which forms more stable emulsions and smaller NPs in PVA solutions.40 A considerable decrease in particle size was observed when ethyl acetate was used as solvent in method 2. In addition, all the formulations had improved PDI indicating mono-disperse NP suspension. Furthermore, method 2 showed a batch-to-batch reproducibility for all the formulations of interest that will be required for in vivo experiments. The represented SEM images confirmed the formation of spherical NPs within the desired size range (Figure 4). The images confirm the homogenous nature of particles with a uniform and aggregation-free distribution.41
There are different emulsion stabilizers available such as PVA, carbopol and polaxamer. Among these, PVA is found to provide homogenous particles with uniform size distribution.42 It acts on the boundary of aqueous and organic phase to modify the particle size as well as surface charge.43 PVA is able to maintain the interfacial tension at oil-water interface to obtain small sized particles.44 In addition, it prevents the aggregation and preserves the hydrophilicity of particles when used above a concentration of 2%.45 A high concentration of PVA is expected to produce particles with narrow granulometric distribution. Failure to maintain that would lead to the aggregated particles.46 Between method 1 (5% PVA) and method 2 (2.2% PVA), the difference in particle size could be attributed to not only the difference of PVA concentration but also the effect of solvents. Decrease in PVA concentration from method 1 to method 2 could lead to larger particles. But, PVA concentration of 5% did not play a better role to reduce particle size due to PVA’s optimum packing.47 In addition, PVA also renders slightly negative charges on the particles.48
ZP value is an important parameter that reflects stability of a colloidal suspension.49 ZP values for COOH terminated NPs were found to be more negative (−25 to −24 mV) compared to ester terminated (−19 to −16 mV) NPs. This could be attributed to the presence of carboxyl group on the COOH terminated NPs. When the viscosity of the polymers increased, ZP value showed slightly more negative charge for uncapped NPs than the capped formulations.50 When the polymer of higher viscosity is used it increases the concentration of the media resulting in semi-folded particles.51 Increase in concentration of the organic phase also resulted in viscosity resistance against the net shear stress. Ultimately coalescence of particles occurs to provide increase in NP size.52,53 Particles with more positive (above +30 mV) or more negative (below −30 mV) ZP are considered colloidally stable but not pharmaceutically stable.16,54 This indicates that ZP beyond this range needs to be kept in solid and dry form rather than colloidal state. Our results show that ZP of antibody modified NPs was about neutral or slightly positive or negative. The prepared NPs were therefore lyophilized for pharmaceutical stability and should be reconstituted immediately before the administration.16,55
Freeze-drying is an essential part in retaining the stability of formulations after preparation. This process could be stressful leading to colloidal preparations having large aggregates, although there are several cryoprotectants available to increase the physical stability of NPs.56 Cryoprotectants could be water-soluble sugars such as glucose, trehalose, which are added to prevent NPs from aggregating during drying process. Hence, it is advantageous to prepare NPs with addition of cryoprotectants. There are two types of cryoprotectants: intracellular that prevents crystal formation and cell membrane rupture; and extracellular that minimizes hyperosmotic effect during freeze-drying process. Sucrose and trehalose are extracellular cryoprotectants that minimize freeze-drying stress of the formulations.57 For method 1, antibody modified NPs had higher particle size compared to the plain NPs which was predictable. The PDI value remained fairly high even after using cryoprotectants. No significant differences in the effect of sucrose and trehalose could be identified for these formulations. Addition of cryoprotectant might have formed hydrogen bond between their hydroxyl group and NP surface resulting in masking of the negative charges of the particles.
It has been reported that presence of stabilizers could also play a major role in NP aggregation in the drying process.58 The percentage of stabilizer is critical to maintain its influence on both particle size and ZP.59 In the NP formulations (following method 2), the PVA percentage was 2.2% below the optimum range (2.5%–5%) which necessities the use of cryoprotectant. A minimum of 5% cryoprotectant is necessary to ensure stability of formulations.60 In another study, aggregation-free PLGA NP suspension was obtained with 1%–2% PVA where 10% of different cryoprotectants (sucrose, trehalose, glucose) were used.61 Therefore, studies were continued with method 2 formulations using 10% sucrose as cryoprotectant due to its better cryopreservation.
The highest antibody incorporation was obtained with low viscosity ester terminated PLGA NPs (formulations with cryoprotectants for either conjugation method), whereas low viscosity COOH terminated PLGA NPs provided the lowest antibody incorporation (except antibody-adsorbed formulations of method 1). The low viscosity ester terminated PLGA had higher antibody attachment due to higher surface area (smaller size) of NPs. Besides, with the same amount of initial antibody, the COOH terminated PLGA had lower antibody incorporation which could be attributed to the rapid erosion of the polymer.62 For method 2 based formulations, covalently attached NPs were found to be smaller than physically adsorbed NPs although the antibody attachment between these groups was not significantly different. In addition, ZP did not show any rational correlation between antibody loading and conjugation method for both COOH and ester terminated NPs. It was observed that covalently attached low viscosity COOH and ester terminated polymers resulted in smaller particle size compared to high viscosity polymers, which is expected. Moreover, all the formulations had particle size within desired range for DC uptake.11,37 It was found that, the 4 hours stirring time during antibody-NP attachment should be enough to break down the aggregates and to form a fine suspension leaving the surface available for antibody attachment. This is observed from the smaller particle size with acceptable PDI obtained for the low viscosity ester terminated NPs after 4 hours incubation with continuous stirring. However, the particle size was above the target range with poor PDI values for the high viscosity ester terminated NPs prepared following method 1. On the contrary, ligand modified NP formulations prepared by method 2 showed equally acceptable range for PDI values with the highest of 0.43±0.05 and lowest of 0.22±0.07. The antibody attachment on NP surface was calculated to be in a desired range (2 μg/mg of NP). However, no significant correlation could be made between antibody-NP conjugation methods (adsorption and covalent attachment) and polymers’ viscosities. Considering the viability, it was slightly higher with high viscosity grade PLGA for most of the formulations; and conjugation method could not affect DC viability.
The structural modification of BS3 activated formulations was well confirmed by infrared study. In the case of the anti-CD205 conjugated NPs spectrum, presence of N-H stretching of high intensity confirmed the conjugation. Second derivative of all spectra (Figure 5B) clearly shows the presence of an amide bond between BS3 and antibody. This characteristic peak stands out, reflecting the establishment of the antibody conjugation on the NPs.
The results from method 2 were more reproducible and consistent, whereas the formulations prepared by method 1 showed higher deviation from the average in the DC viability tests. Based on the confirmed safety profile of the NP formulations, we can further use them for DC uptake and targeting efficiency study.
Conclusion
In this research, the effects of various processing parameters were investigated. The formulation variables evaluated here could be manipulated to enhance the efficiency of the PLGA NPs. Based on the optimum potential parameters it is concluded that formulations prepared using ethyl acetate as solvent (method 2) are shown to be promising for NP preparation and will be further utilized in our in vitro experiments. Further in vitro investigations such as targeting efficiency, maturation of DCs, cytokine secretion profile, and activation of immune response leading to in vivo studies are being conducted in ongoing studies. Therefore, the structural characterizations reflected in those formulations will direct us to obtain optimum in vivo effects. This optimization would help other researchers to select the optimal parameters in their study. In conclusion, this systematic investigation could promote the development of PLGA NPs for further application to design a cancer vaccine.
Acknowledgments
This project was supported by funding from Natural Sciences and Engineering Research (NSERC) Discovery Grant and Saskatchewan Health Research Foundation (SHRF) New Investigator Establishment Grant. The authors thank Canadian Light Source (CLS), Saskatoon, Saskatchewan, for providing assistance with infrared spectroscopy and SEM.
Disclosure
The authors report no conflicts of interest in this work.
References
Bandyopadhyay A, Fine RL, Demento S, Bockenstedt LK, Fahmy TM. The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials. 2011;32(11):3094–3105. | ||
Reuter A, Panozza SE, Macri C, et al. Criteria for dendritic cell receptor selection for efficient antibody-targeted vaccination. J Immunol. 2015;194(6):2696–2705. | ||
Butler M, Morel AS, Jordan WJ, et al. Altered expression and endocytic function of CD205 in human dendritic cells, and detection of a CD205-DCL-1 fusion protein upon dendritic cell maturation. Immunology. 2007;120(3):362–371. | ||
Platzer B, Stout M, Fiebiger E. Antigen cross-presentation of immune complexes. Front Immunol. 2014;5:140. | ||
Barbuto S, Idoyaga J, Vila-Perello M, et al. Induction of innate and adaptive immunity by delivery of poly dA:dT to dendritic cells. Nat Chem Biol. 2013;9(4):250–256. | ||
Cooper DL, Harirforoosh S. Design and optimization of PLGA-based diclofenac loaded nanoparticles. PloS One. 2014;9(1):e87326. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Lewis JS, Zaveri TD, Crooks CP 2nd, Keselowsky BG. Microparticle surface modifications targeting dendritic cells for non-activating applications. Biomaterials. 2012;33(29):7221–7232. | ||
Haddadi A, Hamdy S, Ghotbi Z, Samuel J, Lavasanifar A. Immunoadjuvant activity of the nanoparticles’ surface modified with mannan. Nanotechnology. 2014;25(35):355101. | ||
Sah H, Thoma LA, Desu HR, Sah E, Wood GC. Concepts and practices used to develop functional PLGA-based nanoparticulate systems. Int J Nanomedicine. 2013;8:747–765. | ||
Ghotbi Z, Haddadi A, Hamdy S, Hung RW, Samuel J, Lavasanifar A. Active targeting of dendritic cells with mannan-decorated PLGA nanoparticles. J Drug Target. 2011;19(4):281–292. | ||
Keum CG, Noh YW, Baek JS, et al. Practical preparation procedures for docetaxel-loaded nanoparticles using polylactic acid-co-glycolic acid. Int J Nanomedicine. 2011;6:2225–2234. | ||
Thamake SI, Raut SL, Ranjan AP, Gryczynski Z, Vishwanatha JK. Surface functionalization of PLGA nanoparticles by non-covalent insertion of a homo-bifunctional spacer for active targeting in cancer therapy. Nanotechnology. 2011;22(3):035101. | ||
Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Control Release. 2007;120(1–2):18–26. | ||
Zou W, Liu C, Chen Z, Zhang N. Studies on bioadhesive PLGA nanoparticles: A promising gene delivery system for efficient gene therapy to lung cancer. Int J Pharm. 2009;370(1–2):187–195. | ||
Mukherjee B, Santra K, Pattnaik G, Ghosh S. Preparation, characterization and in-vitro evaluation of sustained release protein-loaded nanoparticles based on biodegradable polymers. Int J Nanomedicine. 2008;3(4):487–496. | ||
Al-Nemrawi NK, Dave RH. Formulation and characterization of acetaminophen nanoparticles in orally disintegrating films. Drug Deliv. 2015:1–10. | ||
Kaur R, Chitanda JM, Michel D, et al. Lysine-functionalized nanodiamonds: synthesis, physiochemical characterization, and nucleic acid binding studies. Int J Nanomedicine. 2012;7:3851–3866. | ||
Valencia PM, Hanewich-Hollatz MH, Gao W, et al. Effects of ligands with different water solubilities on self-assembly and properties of targeted nanoparticles. Biomaterials. 2011;32(26):6226–6233. | ||
Li F, Sun J, Zhu H, Wen X, Lin C, Shi D. Preparation and characterization novel polymer-coated magnetic nanoparticles as carriers for doxorubicin. Colloids Surf B Biointerfaces. 2011;88(1):58–62. | ||
Dillen K, Vandervoort J, Van den Mooter G, Verheyden L, Ludwig A. Factorial design, physicochemical characterisation and activity of ciprofloxacin-PLGA nanoparticles. Int J Pharm. 2004;275(1–2):171–187. | ||
Tsai YM, Chien CF, Lin LC, Tsai TH. Curcumin and its nano-formulation: the kinetics of tissue distribution and blood-brain barrier penetration. Int J Pharm. 2011;416(1):331–338. | ||
Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125(3):193–209. | ||
Copp JA, Fang RH, Luk BT, et al. Clearance of pathological antibodies using biomimetic nanoparticles. Proc Natl Acad Sci U S A. 2014;111(37):13481–13486. | ||
Sperling RA, Parak WJ. Surface modification, functionalization and bioconjugation of colloidal inorganic nanoparticles. Philos Trans A Math Phys Eng Sci. 2010;368(1915):1333–1383. | ||
Hermans K, Van den Plas D, Everaert A, Weyenberg W, Ludwig A. Full factorial design, physicochemical characterisation and biological assessment of cyclosporine A loaded cationic nanoparticles. Eur J Pharm Biopharm. 2012;82(1):27–35. | ||
Fonte P, Soares S, Costa A, et al. Effect of cryoprotectants on the porosity and stability of insulin-loaded PLGA nanoparticles after freeze-drying. Biomatter. 2012;2(4):329–339. | ||
Ikeda J, Sun YL, An KN, Amadio PC, Zhao C. Application of carbodiimide derivatized synovial fluid to enhance extrasynovial tendon gliding ability. J Hand Surg Am. 2011;36(3):456–463. | ||
Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60(15):1615–1626. | ||
Sampath M, Lakra R, Korrapati P, Sengottuvelan B. Curcumin loaded poly(lactic-co-glycolic) acid nanofiber for the treatment of carcinoma. Colloids Surf B Biointerfaces. 2014;117:128–134. | ||
Kountz SL. The effect of bioscience and technological momentum on the surgical treatment of chronic illness. Surgery. 1975;77(6):735–740. | ||
Barth A. Infrared spectroscopy of proteins. Biochim Biophys Acta. 2007;1767(9):1073–1101. | ||
Glassford SE, Byrne B, Kazarian SG. Recent applications of ATR FTIR spectroscopy and imaging to proteins. Biochim Biophys Acta. 2013;1834(12):2849–2858. | ||
Cohen-Sela E, Chorny M, Koroukhov N, Danenberg HD, Golomb G. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J Control Release. 2009;133(2):90–95. | ||
Choi HS, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165–1170. | ||
Kettiger H, Schipanski A, Wick P, Huwyler J. Engineered nanomaterial uptake and tissue distribution: from cell to organism. Int J Nanomedicine. 2013;8:3255–3269. | ||
Hamdy S, Haddadi A, Shayeganpour A, Samuel J, Lavasanifar A. Activation of antigen-specific T cell-responses by mannan-decorated PLGA nanoparticles. Pharm Res. 2011;28(9):2288–2301. | ||
Gutierro I, Hernandez RM, Igartua M, Gascon AR, Pedraz JL. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine. 2002;21(1–2):67–77. | ||
Soppimath KS, Aminabhavi TM. Ethyl acetate as a dispersing solvent in the production of poly(DL-lactide-co-glycolide) microspheres: effect of process parameters and polymer type. J Microencapsul. 2002;19(3):281–292. | ||
Sahana DK, Mittal G, Bhardwaj V, Kumar MN. PLGA nanoparticles for oral delivery of hydrophobic drugs: influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J Pharm Sci. 2008;97(4):1530–1542. | ||
Jose S, Sowmya S, Cinu TA, Aleykutty NA, Thomas S, Souto EB. Surface modified PLGA nanoparticles for brain targeting of Bacoside-A. Eur J Pharm Sci. 2014;63:29–35. | ||
Yoncheva K, Vandervoort J, Ludwig A. Influence of process parameters of high-pressure emulsification method on the properties of pilocarpine-loaded nanoparticles. J Microencapsul. 2003;20(4):449–458. | ||
Vandervoort J, Ludwig A. Biocompatible stabilizers in the preparation of PLGA nanoparticles: a factorial design study. Int J Pharm. 2002;238(1–2):77–92. | ||
Wang H, Jia Y, Hu W, Jiang H, Zhang J, Zhang L. Effect of preparation conditions on the size and encapsulation properties of mPEG-PLGA nanoparticles simultaneously loaded with vincristine sulfate and curcumin. Pharm Dev Technol. 2013;18(3):694–700. | ||
Mobarak DH, Salah S, Elkheshen SA. Formulation of ciprofloxacin hydrochloride loaded biodegradable nanoparticles: optimization of technique and process variables. Pharm Dev Technol. 2014;19(7):891–900. | ||
Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel). 2011;3(3):1377–1397. | ||
Sanad RA, Abdel Malak NS, El-Bayoomy TS, Badawi AA. Preparation and characterization of oxybenzone-loaded solid lipid nanoparticles (SLNs) with enhanced safety and sunscreening efficacy: SPF and UVA-PF. Drug Discov Ther. 2010;4(6):472–483. | ||
Vandervoort J, Yoncheva K, Ludwig A. Influence of the homogenisation procedure on the physicochemical properties of PLGA nanoparticles. Chem Pharm Bull (Tokyo). 2004;52(11):1273–1279. | ||
Nabi-Meibodi M, Vatanara A, Najafabadi AR, et al. The effective encapsulation of a hydrophobic lipid-insoluble drug in solid lipid nanoparticles using a modified double emulsion solvent evaporation method. Colloids Surf B Biointerfaces. 2013;112:408–414. | ||
Gomez-Gaete C, Bustos GL, Godoy RR, et al. Successful factorial design for the optimization of methylprednisolone encapsulation in biodegradable nanoparticles. Drug Dev Ind Pharm. 2013;39(2):310–320. | ||
Premaletha K, Licy CD, Jose S, Saraladevi A, Shirwaikar A, Shirwaikar A. Formulation, characterization and optimization of hepatitis B surface antigen (HBsAg)-loaded chitosan microspheres for oral delivery. Pharm Dev Technol. 2012;17(2):251–258. | ||
Awotwe-Otoo D, Zidan AS, Rahman Z, Habib MJ. Evaluation of anticancer drug-loaded nanoparticle characteristics by nondestructive methodologies. AAPS PharmSciTech. 2012;13(2):611–622. | ||
Li X, Xu Y, Chen G, Wei P, Ping Q. PLGA nanoparticles for the oral delivery of 5-Fluorouracil using high pressure homogenization-emulsification as the preparation method and in vitro/in vivo studies. Drug Dev Ind Pharm. 2008;34(1):107–115. | ||
Westesen K. Novel lipid-based colloidal dispersions as potential drug administration systems – expectations and reality. Colloid Polym Sci. 2000;278(7):608–618. | ||
Pinto Reis C, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomedicine. 2006;2(1):8–21. | ||
Rizkalla N, Range C, Lacasse FX, Hildgen P. Effect of various formulation parameters on the properties of polymeric nanoparticles prepared by multiple emulsion method. J Microencapsul. 2006;23(1):39–57. | ||
Janz Fde L, Debes Ade A, Cavaglieri Rde C, et al. Evaluation of distinct freezing methods and cryoprotectants for human amniotic fluid stem cells cryopreservation. J Biomed Biotechnol. 2012;2012:649353. | ||
Zhang X, Guan J, Ni R, Li LC, Mao S. Preparation and solidification of redispersible nanosuspensions. J Pharm Sci. 2014;103(7):2166–2176. | ||
Mura S, Hillaireau H, Nicolas J, et al. Influence of surface charge on the potential toxicity of PLGA nanoparticles towards Calu-3 cells. Int J Nanomedicine. 2011;6:2591–2605. | ||
Abdelwahed W, Degobert G, Stainmesse S, Fessi H. Freeze-drying of nanoparticles: formulation, process and storage considerations. Adv Drug Deliv Rev. 2006;58(15):1688–1713. | ||
Jeong YI, Shim YH, Kim C, Lim GT, Choi KC, Yoon C. Effect of cryoprotectants on the reconstitution of surfactant-free nanoparticles of poly(DL-lactide-co-glycolide). J Microencapsul. 2005;22(6):593–601. | ||
Lam XM, Duenas ET, Daugherty AL, Levin N, Cleland JL. Sustained release of recombinant human insulin-like growth factor-I for treatment of diabetes. J Control Release. 2000;67(2–3):281–292. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.