")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Investigating the Role of Glutamate in Obsessive-Compulsive Disorder: Current Perspectives
Authors Karthik S , Sharma LP, Narayanaswamy JC
Received 6 February 2020
Accepted for publication 1 April 2020
Published 17 April 2020 Volume 2020:16 Pages 1003—1013
DOI https://doi.org/10.2147/NDT.S211703
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Sheshachala Karthik,* Lavanya P Sharma,* Janardhanan C Narayanaswamy
OCD Clinic, Department of Psychiatry, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore 560029, India
*These authors contributed equally to this work
Correspondence: Janardhanan C Narayanaswamy
OCD Clinic, Department of Psychiatry, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore 560029, India
Tel +91 8026995250
Email [email protected]
Abstract: Glutamate is a ubiquitous excitatory neurotransmitter, which is involved in normal physiology, a variety of central nervous system (CNS) functions, including excitotoxicity and neuronal migration. It is implicated in the pathogenesis of various neuropsychiatric disorders including epilepsy, Parkinson’s disease, Alzheimer’s dementia, schizophrenia and obsessive compulsive disorder (OCD). Over the years, a growing body of evidence has helped researchers understand the mechanisms underlying glutamatergic involvement in the pathogenesis of these disorders. In this review, we attempt to elucidate the role of glutamate in OCD, which is a chronic psychiatric condition with significant morbidity. This article provides current perspectives on the role played by glutamate in the pathogenesis, clinical symptoms and treatment response in OCD, a critical analysis of existing and emerging evidence, both clinical and preclinical, followed by a summary and future directions.
Keywords: glutamate, NMDA receptor, OCD, striatum, memantine, lamotrigine
Introduction
Obsessive compulsive disorder (OCD) is a chronic, often disabling disorder with onset generally in young adulthood. It is characterized by the presence of obsessions- repetitive and persistent thoughts, images, impulses or urges that are intrusive and unwanted, and are often associated with anxiety; and compulsions- repetitive behaviors or mental acts that the individual feels driven to perform stereotypically, in response to an obsession or to achieve a sense of ‘completeness’. It has been found to occur ubiquitously across sociodemographic and geographical boundaries with a lifetime prevalence of 2–3%. It contributes to significant impairment in functioning, both due to the primary illness (OCD), as well as by being highly comorbid with other psychiatric disorders. Despite the availability of treatment options (therapy and pharmacological), about half of all patients with OCD fail to respond adequately to treatment; about 30% are refractory to treatment.1
Given our current understanding of the pathophysiology of OCD, formed from indirect evidence of dysfunctional neurocircuitry and neurotransmitter systems, it is important to innovate therapeutic options for individuals who do not adequately benefit from SSRIs – implying that serotonergic dysfunction may not fully explain the illness. The cortico-striato-thalamo-cortical (CSTC) reverberating circuits, driven primarily by the excitatory neurotransmitter glutamate, were first described to be involved in OCD by Saxena and Rauch.2 Subsequent magnetic resonance spectroscopy (MRS), cerebrospinal fluid (CSF), and genetic studies, as well as animal models of OCD, have found evidence of glutamatergic dysfunction. There may, therefore, be a subset of individuals within the heterogeneous phenotype that is OCD, who would benefit from treatments aimed at modulating glutamatergic transmission. This review attempts to initially understand the neurobiological underpinnings of glutamate dysfunction in OCD, and then study the available evidence across genetic, imaging, pharmacological and animal studies.
Glutamate
Glutamate is the principal excitatory neurotransmitter in the adult brain. It is also a precursor for gamma-aminobutyric acid (GABA), the principal inhibitory neurotransmitter in the brain, as well as for the amino acid glutamine and the antioxidant molecule glutathione. Glutamatergic projection neurons are part of most cortical and subcortical systems. Glutamate receptors are of two types, ionotropic and metabotropic. Ionotropic glutamate receptors (AMPA, kainate, and NMDA), are cation channels that open when bound by glutamate. Metabotropic glutamate receptors are coupled to various intracellular signaling cascades that modulate neuronal function.3 Many postsynaptic glutamate receptors are embedded in a matrix of structural proteins known as the postsynaptic density (PSD), some of which have been studied in animal models of OCD.4
Glutamate can also diffuse out of the synaptic cleft. Whereas activation of postsynaptic NMDA receptors leads to synaptic transmission of information, synaptic plasticity, and trophic effects on neurons, activation of extrasynaptic NMDA receptors inhibits these processes and can lead to excitotoxicity – neuronal damage and death.5
Glutamate concentration is therefore tightly regulated through glutamate transporters on astrocytes and other glial cells, in addition to neurons. Excitatory amino acid transporters (EAAT) are responsible for the tight regulation of glutamate concentration and turnover, in addition to glutamate-cystine antiporter, which transports glutamate into glial cells in exchange for cystine. The polymorphisms of EAAT3/EAAC1 gene have been associated with OCD.
Glutamate plays an important role in various physiological processes including cognition, memory and learning.6 It is crucial for normal cortical neuronal maturation, by guiding cortical neuronal migration in the pyramidal cells of CNS and striatum, which is also abundant in glutamatergic neurons.7 Too much or too little glutamate (as evidenced by expression of NMDA receptors) is shown to affect cortical migration adversely. Glutamate influences neuronal migration mainly by acting on NMDA and AMPA receptors. Too few NMDA receptors reduce the speed of migration whereas supraphysiological levels are known to halt cortical neuronal migration.8 The striatum is the largest group of receptive neurons in the basal ganglia, receiving projections from the prefrontal cortex. Cell migration within the striatum is tightly controlled by glutamate and glutamatergic neurons are the most abundant type of neurons within it.7 Importantly, the striatum is a key brain region implicated in the pathophysiology of OCD. As a part of the CSTC circuit, it is responsible for planning cognitive and motor actions. Abnormalities of the CSTC have been widely recognized to be associated with the development of OCD. Given its crucial role in the striatum, glutamate is likely to play a key role in the pathogenesis of OCD.
CSTC and Its Significance
The CSTC circuit is composed of numerous parallel open loops that link the fronto-cortical and subcortical areas (Figure 1). It is believed that different sets of fronto-cortical-subcortical loops participate in specialized motor and cognitive functions as determined by the specific cortical area involved.9 Cortical neurons send projections into the striatum which form glutamatergic synapses onto (GABA-ergic) medium spiny neurons, which link to the globus pallidus pars internalis (GPi) and substantia nigra pars reticulata (SNr). This occurs via two pathways- one mediated by dopamine receptor type 1 (D1R), called the “direct pathway” (striato-nigral), and the other by dopamine receptor type 2 (D2R) – the indirect pathway (striato-pallidal). Direct pathway axons terminate in GPi/SNr, whereas the indirect pathway involves a more complex relay via the globus pallidus pars externa and subthalamic nucleus (STN). These structures provide inhibitory tone to the thalamus, mediated by a balance of “accelerator- brake” activity through the direct and indirect pathways.10 An imbalance in this system with increased activity in the direct pathway, would disinhibit the thalamus and promote repetitive behavioral sequences.11 The most accepted model of pathogenesis of OCD is Baxter’s model2 which proposes dynamically balanced direct and indirect pathways within the CSTC which are tightly regulated by glutamate and gamma amino butyric acid (GABA). Glutamatergic signaling regulates neurotransmission between orbitofrontal cortex (OFC), anterior cingulate cortex (ACC) and striatal structures (caudate, putamen and others). Rosenberg et al proposed that glutamatergic hyperactivity associated with over-activity of direct pathway may underlie development of OCD.12 Indirect evidence for this comes from knockout mice and animal studies, some of which are described subsequently.
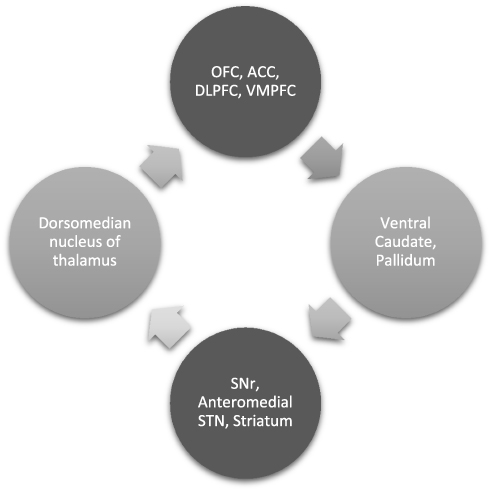 |
Figure 1 CSTC and its significance: OCD is said to be mediated by dysfunctional parallel, partly segregated ‘loops’ of cortico-striato-thalamo-cortical circuitry that are involved in specific motor and cognitive functions. A detailed description is in the paper. Abbreviations: OFC, orbito frontal cortex; ACC, anterior cingulate cortex; DLPFC, dorsolateral prefrontal cortex; VMPFC, ventromedial prefrontal cortex; SNr, substantia nigra pars reticulata; STN, subthalamic nucleus. |
Morphological and functional imaging studies have identified abnormalities in CSTC circuitry in patients with OCD.13,14 Studies have used MRS to investigate levels of glutamate and related molecules in this circuitry and have produced some evidence of glutamate dysregulation in patients with OCD.
Post synaptic glutamate receptors, which exist within the matrix of structural proteins known as the post synaptic density (PSD), play a key role in pathogenesis of OCD. Evidence from animal and genetic studies point towards this possibility, even though the exact nature of involvement is unclear.
Evidence from Animal Studies
Designing an animal model that mimics the behavioral and neurobiological manifestations of OCD is challenging. The defining characteristic of OCD – intrusive irrational obsessions and the performance of compulsions to relieve anxiety – have not, thus far, been accurately modeled in animals. However, repetitive behaviors such as grooming have been claimed to be phenomenologically similar to compulsive behaviors seen in OCD.15 These are not specific to OCD. Therefore, it is important to ensure the validity of the model, in terms of phenomenological similarity of the compulsive behavior modeled, to OCD (face validity), demonstrating the involvement of neurological structures known to be involved in OCD in the animal models (construct validity), and response to pharmacological agents known to be of benefit in OCD (predictive validity).15,16 With respect to involvement of glutamatergic dysfunction, the following genetic and behavioral models have been described.
Genetic Models
Various types of transgenic or knockout mice have been described to show repetitive behaviors similar to those observed in OCD.17 The presence of these behaviors prompted the further study of their etiology and response to treatment. The models listed below provide putative evidence for a glutamate centered hypothesis of OCD.
Sapap3 Knockout Mice
The Sapap3 gene encodes a family of proteins that are PSD components. They interact with other proteins to form a key scaffolding complex at glutamatergic synapses.18 Sapap3 is expressed in the striatum. Sapap3 knock-out mice had defects in synaptic transmission and the functioning of NMDA and AMPA receptors. Welch et al4 found that at the age of 4–6 months Sapap3 knockout mice displayed excessive self-grooming without any cutaneous defects, as well as increased anxiety-like behaviors, which subsided with repeated injections of fluoxetine. It was also found that Sapap3 knockout mice that received an intra-striatal injection of lentiviruses expressing SAPAP3 showed less anxiety-like behaviors, suggesting that altered functioning of the glutamatergic striatal system contributed to repetitive anxiety-like behaviors.4 The human counterpart of the Sapap3 gene is DLGAP3 gene.12 The discs large associated protein (DLGAP) family is a group of scaffold proteins that form an integral part of the PSD. They are also involved in multiple neuronal processes, including synaptic scaling (modifying the strength of excitatory synapses). DLGAP 1–4 have been linked with a variety of psychiatric disorders, including trichotillomania and OCD.19
Slitrk5 Knockout Mice
Mice deficient in neuron-specific transmembrane protein, SLIT and NTRK-like protein-5 (Slitrk5), showed excessive self-grooming, increased marble burying and increased anxiety-like behaviors, which improved with repeated administration (for 21 days) of fluoxetine. Slitrk5 knockout mice showed increased expression of FosB in the OFC, and anatomical abnormalities in the striatum, including decreased volume, decreased dendritic complexity of striatal neurons and a reduced number of glutamate receptors.20
D1CT 7 Mice
Burton et al 21 generated transgenic mice that expressed an intracellular form of cholera toxin (CT), a neuro-potentiating enzyme that chronically activates stimulatory G-protein (Gs) signal transduction and cAMP synthesis, under the control of the D1 promoter. DICT-7 mutant mice express CT under the control of the D1 promoter in a subset of neurons within the amygdala and in cortical areas that project to the OFC and striatum,17 which leads to hyperactivity of cortical afferents projecting to the striatum (the direct pathway). These mice were found to display OCD-like behaviors, including repeating normal behaviors, nonaggressive biting during grooming, repetitive leaping, anxiety, and tic-like movements.3 The transgene expressed in these mice enhances glutamatergic excitability. These mice also had a reduced seizure threshold, suggestive of a shifted balance between excitation and inhibition in the cortex.22 DICT 7 model provided indirect evidence for the role of glutamate dysfunction in OCD. This also leads to the cortical-limbic glutamatergic hyperactivity model for OCD and tics.23
Other genes associated with glutamatergic system which have been implicated in OCD include SLC1A1(codes for EAAC1, EAAT3), GRIN2B (codes for the 2B subunit of NMDA receptors), GRIK2/3 (codes for Kainate receptor subunits), and DLGAP1(codes for members of PSD proteins). GRIN2B and SLC1A1 gene mutations have been associated with OCD in males.24 In addition, GRIN2B has been associated with symptom dimensions of contamination and cleaning.25
SLC1A1 Gene
SLC1A1 is a gene that encodes the glutamate transporter EAAC1, which is expressed in brain regions implicated in OCD. Although 9 different SLC1A1 polymorphisms have been studied and suggested to be involved in OCD, there are no confirmed susceptibility loci identified thus far.26 5. Evidence from GWAS studies:
However, two recently conducted GWAS by OCD collaborative genetic association studies and international OCD foundation genetic collaboration with large sample sizes have not identified any genes with genome-wide significance.27,28 Recently, using targeted sequencing and functional annotation techniques in a novel multispecies approach, Hyun et al identified four top genes – NRXN1, REEP3, CTTNBP2 and HTR2A. It was further noted that the NRXN1 gene, which codes for a synaptic adhesion protein neurexin1α, was associated with OCD at genome-wide significance.29
Behavioral Models
Signal Attenuation
Signal attenuation in rats is based on the premise that compulsive behaviors result from a deficit in feedback following performance of normal goal-directed responses. In this model, lever-pressing for food is followed by the presentation of a compound stimulus which serves as a feedback cue. This feedback is later attenuated by repeated presentations of the stimulus without food. Following this, lever pressing is assessed under conditions of extinction, where persistent lever pressing without attempting to collect a reward is said to be a compulsive behavior.30 In this model, the use of D-cycloserine (DCS), a partial agonist at the glycine co-agonist site on the NMDA receptor, decreased compulsive lever-pressing.15,31 DCS has also been found to help augment CBT in OCD.32
Marble Burying
Rodents use bedding material to bury objects. The finding that burying was reduced by serotonin reuptake inhibitors raised the possibility that this behavior may be related to OCD. Careful analysis of marble-burying behavior led to the conclusion that it does not model anxiety but may rather be related to compulsive behaviors, where a greater number of buried marbles represent a higher degree of compulsivity.15 Marble burying was attenuated by NMDA antagonists.33 Marble burying was also attenuated by presynaptic mGluR II/III receptor agonists; these agents are predicted to reduce glutamatergic outflow.3 These findings imply that modulation of glutamatergic transmission can affect repetitive behaviors.
Spontaneous Compulsive Behaviors
Naturally occurring OCD-like behaviors, including excessive grooming (“acral lick syndrome”), tail-chasing, and other behaviors, have been described in dogs and other species. The efficacy of SRIs in these conditions supports the similarity of these behaviors to OCD. Some evidence suggests that the glutamate modulator memantine may be of benefit in these OCD-like conditions in dogs.34 The occurrence of spontaneous OCD-like behaviors that responded to glutamatergic agents suggests an underlying glutamatergic dysfunction in these animals.
Evidence from Neurophysiological Studies
CSF Studies
Two studies have examined glutamate concentrations in CSF of unmedicated OCD patients. The first35 described measurements of glutamate in the CSF of 21 unmedicated adult patients and 18 controls and found statistically significantly higher glutamate levels in patients when compared with controls. Though lacking in diagnostic utility due to overlap between patients and controls, it provides strong support for a general dysregulation in glutamate homeostasis in these patients. A follow-up study replicated this finding.36 Both glutamate and glycine were found to be elevated in a group of 23 unmedicated adult OCD patients, compared to 23 controls. Although there is evidence of glutamatergic dysfunction in these studies, the inferences drawn may be limited. Hyperactivity of the ACC and OFC may be expected to increase glutamate in their projections and the striatum. However, glutamate reuptake is tightly regulated; this occurs via glutamate transporters, which efficiently remove glutamate from the perisynaptic and extrasynaptic spaces.3 As a result, increased activity in the ACC or OFC may not always reflect as tonic increments of glutamate in tissue or CSF. Generalized abnormalities in glutamate signaling or a disturbance in homeostasis may also affect its concentration in CSF.3
Imaging Studies
There is a growing body of evidence from neuroimaging studies, which implicate glutamatergic dysfunction in OCD. However, the evidence is divided regarding the exact nature of dysfunction. Structural MRI studies indicate volumetric differences in OFC, ACC and thalamus in patients with OCD as compared to healthy individuals, suggesting a structural basis for functional dysfunction of fronto-subcortical circuits in OCD.37 A wealth of neuroimaging evidence for glutamatergic dysfunction comes from proton magnetic resonance spectroscopy (1H-MRS) studies. Early studies by Rosenberg et al in the pediatric OCD population lead to a model of tonic phasic imbalance of glutamatergic transmission within CSTC which were thought to underlie OCD. Authors proposed that a tonic reduction of glutamatergic activity in ACC [as observed by decreased concentration of glutamine and glutamate (GLX) on MRS], resulted in phasic overactivity of glutamate in striatal regions and orbitofrontal cortex [as observed by increased GLX concentration].38 There are critical knowledge gaps in our understanding of nature of glutamatergic dysfunction as many neuroimaging studies conducted in adults with OCD who were medication free does not lend support to this hypothesis. Most studies conducted in adults with OCD have found no difference or minimal reduction in glutamate concentration in striatum and reduced glutamate levels in anterior cingulate cortex.39,41-45 Methodological issues exist for most studies. Medication status was a uniform confounding factor across most studies, with most patients being exposed to a stable dose of medications prior to the start of the study.39,44-46 This is known to influence the neuroanatomy and neurochemistry in different ways and may or may not be reflective of actual pathophysiology underlying OCD.47 Recent studies suggest that glutamatergic dysfunction as measured by standard MRS may not be specific to OCD alone.48 In a review comparing 59 1H-MRS studies across three related conditions, namely autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD) and OCD, it was found that there is a common hyperglutamatergic state in striatum in all three conditions.48 Findings were divided in terms of glutamate levels in ACC, with reduced levels being reported in adults with ASD and ADHD and increased levels being reported in pediatric and adolescents with ASD and ADHD. Currently, MRS studies cannot clearly differentiate between intracellular or extracellular glutamate. Nor can they differentiate between presynaptic and postsynaptic glutamate. Given this limitation, it may be difficult to confidently associate glutamate level differences to distinct pathogenetic process which may underlie OCD. Further, there may be multiple possible reasons for increased glutamate levels including glial cell dysfunction, altered transporter function in addition to pre-/post-synaptic glutamatergic dysfunction.49
Evidence from Pharmacological Studies
Modulation of glutamatergic neurotransmission is being evaluated as a treatment option in OCD. Glutamatergic medications which have been used to treat OCD mainly target NMDA receptors in different ways.3 The mechanisms of action range from receptor antagonism, reuptake inhibition, ion channel modulation and receptor co-agonism. So far, none have received FDA approval for use in OCD. However, these medications and the response observed across different studies provide critical insights in understanding the role of glutamate in treatment response in OCD. There is emerging evidence that serotonin is also involved in cortical glutamatergic regulation. Pre-synaptic 5HT1b receptors are involved in the gating of cortical and thalamic glutamate inputs onto principal neurons of basolateral amygdala (BLA), a key region implicated in the circuitry of emotion regulation, shown to be impaired in OCD.50 Some clinical evidence is derived from studies that have shown selective improvement in compulsive behaviours upon the addition of topiramate, N-acetyl cysteine (NAC),and lamotrigine.51–54 There appears to be a wider, more complex pharmacogenetic role for glutamate in treatment response in OCD.12 Antipsychotics like clozapine are shown to reduce expression of EAAC1 in ACC of rat,54 which is a protein, translated from mRNA of SLC1A1 gene. Further, variations in SLC1A1 gene, which regulates glutamate turnover, have been associated with induction of obsessive-compulsive symptoms during treatment with atypical antipsychotics.55 Pharmacogenetics of other glutamatergic genes mentioned above is sparsely studied, and the current level of understanding is insufficient to derive any plausible explanations at the molecular level for changes brought about by the pharmacologic agents.
Memantine
A non-competitive NMDA receptor antagonist, memantine acts on extrasynaptic NMDA receptors, blocking the ion channel pores, resulting in a reduced influx of calcium.56 Evidence suggests that it may decrease hyperactivity of the direct pathway of CSTC and modulate aberrant connectivity between OFC and ACC, hippocampus and amygdala.57 Open-label studies,58 single- and double-blind RCTs59,61,62 show a clear benefit of adding memantine to ongoing SSRI treatment in terms of reduction in YBOCS score. However, methodological issues exist with each of these trials, limiting the benefit that can be attributed to memantine addition.
Ketamine
The interest in the use of ketamine in OCD stems from several RCTs of its rapid antidepressant effect with a single intravenous infusion/intranasal spray.62,63 The rapid reduction in depressive symptom scores, even in those with refractory depression,64 with improvement that persisted at 1 week following infusion, led to its putative use in OCD to be investigated. Ketamine is a noncompetitive inhibitor of the NMDA glutamate receptor and can augment glutamate release once the NMDA receptor is already active. According to a study by Li et al, the rapid antidepressant actions of ketamine may be mediated by sustained elevation of synaptic proteins and spine number in the Prefrontal cortex, mediated by enhanced mTOR signaling, eventually resulting in enhanced serotonin levels – a process which is slower otherwise.65 Acute blockade of NMDA receptors leading to an enhanced activity of AMPA receptor is also thought to underlie the rapid response. The exact mechanism of action of ketamine in OCD is unclear. The literature on the use of ketamine in OCD is limited to a single RCT66 and open-label reports of use. With the exception of one study which recruited treatment refractory subjects,67 significant improvement in YBOCS/ VAS has been reported when used on a population of drug-naïve individuals with OCD. The RCT by Rodriguez et al66 and the open-label trial by Bloch et al67 reported transient effects of ketamine – lasting up until 1 week post a single infusion in the RCT, and up until 3 hours post-infusion in the open-label trial phase.
Glycine
Being an obligatory co-agonist at NMDA receptor, it modulates glutamatergic neurotransmission at NMDA receptors indirectly. Direct augmentation with glycine for OCD has shown disappointing results in the only RCT conducted by Greenberg’s group.68 Poor tolerability and high dropout rates were the main concerns noted by the group. However, recent studies involving sarcosine3 and rapastinel69 – a glycine reuptake inhibitor and glycine site co-agonist at NMDA receptor, respectively – have shown to be beneficial in a subset of medication-naïve OCD patients. At this point, glycine presents an opportunity to further elucidate mechanisms underlying the glutamatergic hypothesis of OCD. It requires more studies on its mechanism of action and clinical utility.
N-Acetyl Cysteine
N-acetyl cysteine is a molecule with antioxidant, hepatoprotective, mucolytic properties. It donates cysteine moiety for conversion into glutathione. Unconverted cysteine crosses the blood–brain barrier via sodium-dependent transport mechanisms and gets converted in CNS to cystine, which gets exchanged for glutamate by means of a cystine glutamate antiporter,70 leading to activation of mGLuR2/3 receptors. This in turn leads to inhibition of synaptic glutamate release and restoration of extracellular levels of glutamate.71 As can be seen, cysteine component of NAC plays an important role in the regulation of glutamate turnover and has been evaluated for its efficacy as an augmenting agent in OCD. The results have been mixed in terms of clinical improvement. Initial RCTs72,73 showed promising results which were not replicated in RCTs conducted subsequently.53,74 Therefore, despite favourable evidence, the data are inconsistent and needs further research.
Riluzole
Riluzole acts through various mechanisms to bring about reduced release and enhanced extracellular uptake of glutamate by glial cells.75 This may result in a reduction of CNS excitotoxicity theoretically, as glutamate is the principal mediator of the same. There is some evidence to suggest that autoimmune mechanisms involving glutamate may be operating in OCD76 as shown by the presence of elevated CSF levels of glycine and glutamine in patients of OCD who also had anti-basal ganglia (BG) antibodies.36 Other groups have also demonstrated the role of glutamate as a regulator of t-cell-mediated immunity, which is responsible for the development of compulsive behaviors in rats that have been infected by Borna disease virus (BDV) virus.12 However, it remains to be seen in humans, if a reduction in glutamate load, by riluzole or other molecules, actually results in a reversal of the immune mechanisms putated to be associated with the development of OCD. Clinically, riluzole was the first glutamatergic agent to be studied for its efficacy in the treatment of OCD. The results of riluzole augmentation have been mixed with modest benefits being reported in open-label trials,77,78 which have not been consistently replicated in RCTs.75,79
Topiramate
Topiramate is known to inhibit glutamate release and facilitate GABA release, via its actions on voltage-gated calcium channels. In addition, it is known to inhibit carbonic anhydrase enzyme. Topiramate has been examined as an augmenting agent in OCD. Studies indicate the anti-depressant like effects of topiramate may be mediated by inhibition of NMDA receptors.80 There is probably some evidence to support this mechanism, from clinical trials which have shown improvement in HAM-D scores, following a 12-week augmentation of topiramate in 41 treatment refractory OCD patients, along with a 32% reduction in YBOCS scores. There have been reports of modest benefit in the treatment of OCD from two of the three RCTs conducted so far. More importantly, the use of topiramate in this context sheds some light on the antidepressant role of NMDA receptor modulation, and thereby its effect on reducing OCD severity in patients with comorbid depression.
Lamotrigine
Lamotrigine has a mechanism of action similar to topiramate. It reduces glutamate release by inhibiting voltage-gated calcium channels. Lamotrigine augmentation for the treatment of OCD is supported by two double-blind placebo RCTs.81,82
D-Cycloserine (DCS)
DCS is a second line bacteriostatic anti-tubercular drug, which facilitates fear extinction by modulating glutamate signalling. The exact mechanisms remain unclear. There is evidence to suggest that it might help consolidate new memories. Given its transient nature and role in fear extinction, DCS has been evaluated for its use in facilitating fear extinction following CBT. However, studies so far83,84 have failed to show substantial benefit of DCS pre-administration in influencing the outcome.
Role of Glutamatergic Neurotransmission in the Pathogenesis and Treatment of OCD: A Summary
The existing evidence from animal studies, neurophysiological studies, genetic, neuroimaging and treatment studies, which support the role of glutamate in pathogenesis, clinical manifestations and treatment response of obsessive-compulsive disorder has been reviewed previously. Glutamate appears to play a key role in the development of normal CSTC circuits and maintaining a dynamic balance between direct and indirect pathways in health, along with GABA. Variations in genes involved in glutamate signaling and turnover may result in dysfunctional development of CSTC circuits which are thought to underlie the development of OCD. Glutamate has a role to play in immune modulation and is thought to underlie inflammatory processes which result in the development of OCD in some cases. Tight regulation of glutamate also appears to be essential for fear extinction and consolidation of new memories, which are impaired in OCD and result in compulsive behaviors in a bid to reduce fear or get rid of it. Glutamate may influence mood disturbances85 commonly seen comorbid with OCD, as some evidence suggests the involvement of NMDA receptors in mediating the anti-depressant like actions of topiramate in mice.80 An interesting observation that merits further investigation is that of reduction in compulsive behaviors following treatment with medications that enhance glutamate turnover/ reduce its release. Medications such as Lamotrigine, Topiramate, Riluzole, which mainly reduce glutamate transmission overall, have been observed to reduce compulsions specifically in a subset of patients even when global effects were not statistically significant.86 The scientific rationale for these observations, if any, require further research and understanding. Glutamate may also play a role in treatment resistance in OCD. A pharmacogenetic role for glutamate is present, as evidenced by the development of OC symptoms in people treated with atypical antipsychotics, who also expressed a genetic variant of SLC1A1 gene. Glutamate may have a role in altering the treatment response to OCD in connection with serotonin receptors, which are now shown to be involved in controlling cortical and thalamic glutamatergic input to basolateral amygdala.50
Overall, glutamatergic modulation might offer novel perspectives in understanding the pathophysiology of compulsivity. Such an enhanced perspective has the potential to widen the horizons of treatment options for the condition. Glutamatergic medications have been used in OCD with some success as augmentation strategies. More well-conducted in vivo and basic experimental studies and pharmacogenetic studies are essential to further elucidate the mechanisms of glutamatergic transmission with relevance to compulsive disease state. Furthermore, larger well-conducted systematic studies examining novel glutamatergic drugs appear to be the need of the hour.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Stein DJ, Costa DLC, Lochner C, et al. Obsessive–compulsive disorder. Nat Rev Dis Prim. 2019;5(1):1–21. doi:10.1038/s41572-019-0102-3
2. Saxena S, Rauch SL. Functional neuroimaging and the neuroanatomy of obsessive-compulsive disorder. Psychiatr Clin North Am. 2000;23(3):563–586. doi:10.1016/S0193-953X(05)70181-7
3. Pittenger C, Bloch MH, Williams K. Glutamate abnormalities in obsessive compulsive disorder: neurobiology, pathophysiology, and treatment. Pharmacol Ther. 2011;132(3):314–332. doi:10.1016/j.pharmthera.2011.09.006
4. Welch JM, Lu J, Rodriguiz RM, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448(7156):894–900. doi:10.1038/nature06104
5. Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11(10):682–696. doi:10.1038/nrn2911
6. Suzuki M, Nelson AD, Eickstaedt JB, Wallace K, Wright LS, Svendsen CN. Glutamate enhances proliferation and neurogenesis in human neural progenitor cell cultures derived from the fetal cortex. Eur J Neurosci. 2006;24(3):645–653. doi:10.1111/j.1460-9568.2006.04957.x
7. Hamasaki T, Goto S, Nishikawa S, Ushio Y. Neuronal cell migration for the developmental formation of the mammalian striatum. Brain Res Rev. 2003;41(1):1–12. doi:10.1016/S0165-0173(02)00216-3
8. Luhmann HJ, Fukuda A, Kilb W. Control of cortical neuronal migration by glutamate and GABA. Front Cell Neurosci. 2015;9(JAN). doi:10.3389/fncel.2015.00004
9. Tekin S, Cummings JL. Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002;53(2):647–654. doi:10.1016/S0022-3999(02)00428-2
10. Graybiel AM. The basal ganglia. Curr Biol. 2000;10:14. doi:10.1016/S0960-9822(00)00593-5
11. Ting JT, Feng G. Glutamatergic synaptic dysfunction and obsessive-compulsive disorder. Curr Chem Genomics. 2008;2:62–75. doi:10.2174/1875397300802010062
12. Wu K, Hanna GL, Rosenberg DR, Arnold PD. The role of glutamate signaling in the pathogenesis and treatment of obsessive-compulsive disorder. Pharmacol Biochem Behav. 2012;100(4):726–735. doi:10.1016/j.pbb.2011.10.007
13. Maia TV, Cooney RE, Peterson BS. The neural bases of obsessive - Compulsive disorder in children and adults. Dev Psychopathol. 2008;20(4):1251–1283. doi:10.1017/S0954579408000606
14. Rotge JY, Aouizerate B, Tignol J, Bioulac B, Burbaud P, Guehl D. The glutamate-based genetic immune hypothesis in obsessive-compulsive disorder. An integrative approach from genes to symptoms. Neuroscience. 2010;165(2):408–417. doi:10.1016/j.neuroscience.2009.10.043
15. Albelda N, Joel D. Review current animal models of obsessive compulsive disorder: an update. NSC. 2012;211:83–106. doi:10.1016/j.neuroscience.2011.08.070
16. Witkin JM. Animal models of obsessive-compulsive disorder. In: Current Protocols in Neuroscience. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2008. doi:10.1002/0471142301.ns0930s45.
17. Wang L, Simpson HB, Dulawa SC. Assessing the validity of current mouse genetic models of obsessive-compulsive disorder. Behav Pharmacol. 2009;20(2):119–133. doi:10.1097/FBP.0b013e32832a80ad
18. Bienvenu OJ, Wang Y, Shugart YY, et al. Sapap3 and pathological grooming in humans: results from the OCD collaborative genetics study. Am J Med Genet Part B Neuropsychiatr Genet. 2009;150(5):710–720. doi:10.1002/ajmg.b.30897
19. Rasmussen AH, Rasmussen HB, Silahtaroglu A. The DLGAP family: neuronal expression, function and role in brain disorders. Mol Brain. 2017;10(1):1–13. doi:10.1186/s13041-017-0324-9
20. Shmelkov SV, Hormigo A, Jing D, et al. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat Med. 2010;16(5):598–602. doi:10.1038/nm.2125
21. Campbell KM, de Lecea L, Severynse DM, et al. OCD-Like behaviors caused by a neuropotentiating transgene targeted to cortical and limbic D1+ neurons. J Neurosci. 1999;19(12):5044–5053. doi:10.1523/JNEUROSCI.19-12-05044.1999
22. Campbell KM, Veldman MB, McGrath MJ, Burton FH. TS+OCD-like neuropotentiated mice are supersensitive to seizure induction. Neuroreport. 2000;11(10):2335–2338. doi:10.1097/00001756-200007140-00053
23. Nordstrom EJ, Burton FH. A transgenic model of comorbid Tourette’s syndrome and obsessive-compulsive disorder circuitry. Mol Psychiatry. 2002;7(6):617–625. doi:10.1038/sj.mp.4001144
24. Stewart SE, Mayerfeld C, Arnold PD, et al. Meta-analysis of the glutamate transporter gene SLC1A1 in obsessive-compulsive disorder. Neuropsychopharmacology. 2010.
25. Alonso P, Gratacós M, Segalàs C, et al. Association between the NMDA glutamate receptor GRIN2B gene and obsessive-compulsive disorder. J Psychiatry Neurosci. 2012;37(4):273–281. doi:10.1503/jpn.110109
26. Stewart SE, Mayerfeld C, Arnold PD, et al. Meta-analysis of association between obsessive-compulsive disorder and the 3′ region of neuronal glutamate transporter gene SLC1A1. Am J Med Genet Part B Neuropsychiatr Genet. 2013;162(4):367–379. doi:10.1002/ajmg.b.32137
27. Mattheisen M, Samuels JF, Wang Y, et al. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry. 2015;20(3):337–344. doi:10.1038/mp.2014.43
28. Stewart SE, Yu D, Scharf JM, et al. Genome-wide association study of obsessive-compulsive disorder. Mol Psychiatry. 2013. doi:10.1038/mp.2012.85
29. Noh HJ, Tang R, Flannick J, et al. Integrating evolutionary and regulatory information with a multispecies approach implicates genes and pathways in obsessive-compulsive disorder. Nat Commun. 2017;8:1. doi:10.1038/s41467-017-00831-x
30. Joel D. The signal attenuation rat model of obsessive-compulsive disorder: a review. Psychopharmacology (Berl). 2006;186:487–503. doi:10.1007/s00213-006-0387-2
31. Albelda N, Bar-On N, Joel D. The role of NMDA receptors in the signal attenuation rat model of obsessive-compulsive disorder. Psychopharmacology (Berl). 2010;210(1):13–24. doi:10.1007/s00213-010-1808-9
32. Wilhelm S, Buhlmann U, Tolin DF, et al. Augmentation of behavior therapy with D-cycloserine for obsessive-compulsive disorder. Am J Psychiatry. 2008;165(3):335–341. doi:10.1176/appi.ajp.2007.07050776
33. Egashira N, Okuno R, Harada S, et al. Effects of glutamate-related drugs on marble-burying behavior in mice: implications for obsessive–compulsive disorder. Eur J Pharmacol. 2008;586(1–3):164–170. doi:10.1016/j.ejphar.2008.01.035
34. Schneider BM, Dodman NH, Maranda L. Use of memantine in treatment of canine compulsive disorders. J Vet Behav Clin Appl Res. 2009;4(3):118–126. doi:10.1016/j.jveb.2008.10.008
35. Chakrabarty K, Bhattacharyya S, Christopher R, Khanna S. Glutamatergic dysfunction in OCD. Neuropsychopharmacology. 2005;30(9):1735–1740. doi:10.1038/sj.npp.1300733
36. Bhattacharyya S, Khanna S, Chakrabarty K, Mahadevan A, Christopher R, Shankar SK. Anti-brain autoantibodies and altered excitatory neurotransmitters in obsessive-compulsive disorder. Neuropsychopharmacology. 2009;34(12):2489–2496. doi:10.1038/npp.2009.77
37. Rotge JY, Guehl D, Dilharreguy B, et al. Meta-analysis of brain volume changes in obsessive-compulsive disorder. Biol Psychiatry. 2009;65(1):75–83. doi:10.1016/j.biopsych.2008.06.019
38. Rosenberg DR, Mirza Y, Russell A, et al. Reduced anterior cingulate glutamatergic concentrations in childhood OCD and major depression versus healthy controls. J Am Acad Child Adolesc Psychiatry. 2004;43(9):1146–1153. doi:10.1097/01.chi.0000132812.44664.2d
39. Starck G, Ljungberg M, Nilsson M, et al. A 1H magnetic resonance spectroscopy study in adults with obsessive compulsive disorder: relationship between metabolite concentrations and symptom severity. J Neural Transm. 2008;115(7):1051–1062. doi:10.1007/s00702-008-0045-4
40. Rosenberg DR, MacMaster FP, Keshavan MS, Fitzgerald KD, Stewart CM, Moore GJ. Decrease in caudate glutamatergic concentrations in pediatric obsessive-compulsive disorder patients taking paroxetine. J Am Acad Child Adolesc Psychiatry. 2000;39(9):1096–1103. doi:10.1097/00004583-200009000-00008
41. Strawn JR, Chu W-J, Whitsel RM, et al. A pilot study of anterior cingulate cortex neurochemistry in adolescents with generalized anxiety disorder. Neuropsychobiology. 2013;67(4):224–229. doi:10.1159/000347090
42. Yücel M, Wood SJ, Wellard RM, et al. Anterior cingulate glutamate–glutamine levels predict symptom severity in women with obsessive–compulsive disorder. Aust New Zeal J Psychiatry. 2008;42(6):467–477. doi:10.1080/00048670802050546
43. Yücel M, Harrison BJ, Wood SJ, et al. Functional and biochemical alterations of the medial frontal cortex in obsessive-compulsive disorder. Arch Gen Psychiatry. 2007;64(8):946. doi:10.1001/archpsyc.64.8.946
44. Lázaro L, Bargalló N, Andrés S, et al. Proton magnetic resonance spectroscopy in pediatric obsessive-compulsive disorder: longitudinal study before and after treatment. Psychiatry Res - Neuroimaging. 2012;201(1):17–24. doi:10.1016/j.pscychresns.2011.01.017
45. Bédard M-J, Chantal S. Brain magnetic resonance spectroscopy in obsessive-compulsive disorder: the importance of considering subclinical symptoms of anxiety and depression. Psychiatry Res. 2011;192(1):45–54. doi:10.1016/j.pscychresns.2010.10.008
46. Simpson HB, Shungu DC, Bender J, et al. Investigation of cortical glutamate–glutamine and γ-aminobutyric acid in obsessive–compulsive disorder by proton magnetic resonance spectroscopy. Neuropsychopharmacology. 2012;37(12):2684–2692. doi:10.1038/npp.2012.132
47. Benedetti F, Giacosa C, Radaelli D, et al. Widespread changes of white matter microstructure in obsessive-compulsive disorder: effect of drug status. Eur Neuropsychopharmacol. 2013;23(7):581–593. doi:10.1016/j.euroneuro.2012.07.002
48. Naaijen J, Lythgoe DJ, Amiri H, Buitelaar JK, Glennon JC. Fronto-striatal glutamatergic compounds in compulsive and impulsive syndromes: a review of magnetic resonance spectroscopy studies. Neurosci Biobehav Rev. 2015;52:74–88. doi:10.1016/j.neubiorev.2015.02.009
49. Pittenger C, Bloch M, Teitelbaum C, Coric V, Wegner R, Krystal JH. Glutamatergic dysfunction in obsessive-compulsive disorder and the potential clinical utility of glutamate-modulating agents. Prim Psychiatry. 2006;13(10):65–77.
50. Guo JD, O’Flaherty BM, Rainnie DG. Serotonin gating of cortical and thalamic glutamate inputs onto principal neurons of the basolateral amygdala. Neuropharmacology. 2017;126:224–232. doi:10.1016/j.neuropharm.2017.09.013
51. Mowla A, Khajeian AM, Sahraian A, Chohedri AH, Kashkoli F. Topiramate augmentation in resistant OCD: a double-blind placebo-controlled clinical trial. CNS Spectr. 2010;15(11):613–617. doi:10.1017/S1092852912000065
52. Afshar H, Akuchekian S, Mahaky B, Zarean E. Topiramate augmentation in refractory obsessive-compulsive disorder: a randomized, double-blind, placebo-controlled trial. J Res Med Sci. 2014;19(10):976–981.
53. Sarris J, Oliver G, Camfield DA, et al. N-Acetyl Cysteine (NAC) in the treatment of obsessive-compulsive disorder: a 16-week, double-blind, randomised, placebo-controlled study. CNS Drugs. 2015;29(9):801–809. doi:10.1007/s40263-015-0272-9
54. Kwon JS, Joo YH, Nam HJ, et al. Association of the glutamate transporter gene SLC1A1 with atypical antipsychotics–induced obsessive-compulsive symptoms. Arch Gen Psychiatry. 2009;66(11):1233. doi:10.1001/archgenpsychiatry.2009.155
55. Schirmbeck F, Nieratschker V, Frank J, et al Polymorphisms in the glutamate transporter gene SLC1A1 and obsessive-compulsive symptoms induced by second-generation antipsychotic agents. Psychiatr Genet. 2012.
56. Aboujaoude E, Barry JJ, Gamel N. Memantine augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. J Clin Psychopharmacol. 2009;29(1):51–55. doi:10.1097/JCP.0b013e318192e9a4
57. Vlček P, Polák J, Brunovský M, Horáček J. Role of glutamatergic system in obsessive-compulsive disorder with possible therapeutic implications. Pharmacopsychiatry. 2017.
58. Chaudhury S, Bakhla A, Verma V, Soren S, Sarkhel S. An open-label trial of memantine in treatment-resistant obsessive-compulsive disorder. Ind Psychiatry J. 2013;22(2):149. doi:10.4103/0972-6748.132930
59. Stewart SE, Jenike EA, Hezel DM, et al. A single-blinded case-control study of memantine in severe obsessive-compulsive disorder. J Clin Psychopharmacol. 2010;30(1):34–39. doi:10.1097/JCP.0b013e3181c856de
60. Ghaleiha A, Entezari N, Modabbernia A, et al. Memantine add-on in moderate to severe obsessive-compulsive disorder: randomized double-blind placebo-controlled study. J Psychiatr Res. 2013;47(2):175–180. doi:10.1016/j.jpsychires.2012.09.015
61. Haghighi M, Jahangard L, Mohammad-Beigi H, et al. In a double-blind, randomized and placebo-controlled trial, adjuvant memantine improved symptoms in inpatients suffering from refractory obsessive-compulsive disorders (OCD). Psychopharmacology (Berl). 2013;228(4):633–640. doi:10.1007/s00213-013-3067-z
62. Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–354. doi:10.1016/S0006-3223(99)00230-9
63. Aan Het Rot M, Zarate CA, Charney DS, Mathew SJ. Ketamine for depression: where do we go from here? Biol Psychiatry. 2012;72(7):537–547. doi:10.1016/j.biopsych.2012.05.003
64. Niciu MJ, Luckenbaugh DA, Ionescu DF, et al. Clinical predictors of ketamine response in treatment-resistant major depression. J Clin Psychiatry. 2014;75(05):e417–e423. doi:10.4088/JCP.13m08698
65. Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. doi:10.1126/science.1190287
66. Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38(12):2475–2483. doi:10.1038/npp.2013.150
67. Bloch MH, Wasylink S, Landeros-weisenberger A, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. BPS. 2012;72(11):964–970. doi:10.1016/j.biopsych.2012.05.028
68. Greenberg WM, Benedict MM, Doerfer J, et al. Adjunctive glycine in the treatment of obsessive-compulsive disorder in adults. J Psychiatr Res. 2009;43(6):664–670. doi:10.1016/j.jpsychires.2008.10.007
69. Köse S, Çetin M. Ketamine and rapastinel: NMDA receptor modulators in the rapid treatment of obsessive-compulsive disorder. Psychiatry Clin Psychopharmacol. 2017;27(3):213–214. doi:10.1080/24750573.2017.1357355
70. Zhou Y, Danbolt NC. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm. 2014;121(8):799–817. doi:10.1007/s00702-014-1180-8
71. Oliver G, Dean O, Camfield D, et al. N-acetyl cysteine in the treatment of obsessive compulsive and related disorders: a systematic review. Clin Psychopharmacol Neurosci. 2015;13(1):12–24. doi:10.9758/cpn.2015.13.1.12
72. Afshar H, Roohafza H, Mohammad-Beigi H, et al. N-acetylcysteine add-on treatment in refractory obsessive-compulsive disorder. J Clin Psychopharmacol. 2012;32(6):797–803. doi:10.1097/JCP.0b013e318272677d
73. Paydary K, Akamaloo A, Ahmadipour A, Pishgar F, Emamzadehfard S, Akhondzadeh S. N-acetylcysteine augmentation therapy for moderate-to-severe obsessive-compulsive disorder: randomized, double-blind, placebo-controlled trial. J Clin Pharm Ther. 2016;41(2):214–219. doi:10.1111/jcpt.12370
74. Costa DLC, Diniz JB, Requena G, et al. Randomized, double-blind, placebo-controlled trial of N-acetylcysteine augmentation for treatment-resistant obsessive-compulsive disorder. J Clin Psychiatry. 2017;78(7):e766–e773. doi:10.4088/JCP.16m11101
75. Pittenger C, Bloch MH, Wasylink S, et al. Riluzole augmentation in treatment-refractory obsessive-compulsive disorder: a pilot randomized placebo-controlled trial. J Clin Psychiatry. 2015;76(8):1075–1084. doi:10.4088/JCP.14m09123
76. Gerentes M, Pelissolo A, Rajagopal K, Tamouza R, Hamdani N. Obsessive-compulsive disorder: autoimmunity and neuroinflammation. Curr Psychiatry Rep. 2019;21:8. doi:10.1007/s11920-019-1062-8
77. Grant P, Lougee L, Hirschtritt M, Swedo SE. An open-label trial of riluzole, a glutamate antagonist, in children with treatment-resistant obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. 2007;17(6):761–767. doi:10.1089/cap.2007.0021
78. Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive–compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424–428. doi:10.1016/j.biopsych.2005.04.043
79. Emamzadehfard S, Kamaloo A, Paydary K, et al. Riluzole in augmentation of fluvoxamine for moderate to severe obsessive–compulsive disorder: randomized, double-blind, placebo-controlled study. Psychiatry Clin Neurosci. 2016;70(8):332–341. doi:10.1111/pcn.12394
80. Ostadhadi S, Khan MI, Norouzi-Javidan A, et al. Involvement of NMDA receptors and l-arginine/nitric oxide/cyclic guanosine monophosphate pathway in the antidepressant-like effects of topiramate in mice forced swimming test. Brain Res Bull. 2016;122:62–70. doi:10.1016/j.brainresbull.2016.03.004
81. Khalkhali M, Aram S, Zarrabi H, Kafie M, Heidarzadeh A. Lamotrigine augmentation versus placebo in serotonin reuptake inhibitors-resistant obsessive-compulsive disorder: a randomized controlled trial. Iran J Psychiatry. 2016;11(2):104–114.
82. Bruno A, Micò U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive–compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456–1462. doi:10.1177/0269881111431751
83. Storch EA, Wilhelm S, Sprich S, et al. Efficacy of augmentation of cognitive behavior therapy with weight-adjusted d-cycloserine vs placebo in pediatric obsessive-compulsive disorder: a randomized clinical trial. JAMA Psychiatry. 2016;73(8):779–788. doi:10.1001/jamapsychiatry.2016.1128
84. Kushner MG, Kim SW, Donahue C, et al. D-cycloserine augmented exposure therapy for obsessive-compulsive disorder. Biol Psychiatry. 2007;62(8):835–838. doi:10.1016/j.biopsych.2006.12.020
85. Rubio-Casillas A, Fernández-Guasti A. The dose makes the poison: from glutamate-mediated neurogenesis to neuronal atrophy and depression. Rev Neurosci. 2016;27(6):599622. doi:10.1515/revneuro-2015-0066
86. Sheshachala K, Narayanaswamy J. Glutamatergic augmentation strategies in obsessive-compulsive disorder. Indian J Psychiatry. 2019;61(7):S58–S65. doi:10.4103/psychiatry.IndianJPsychiatry_520_18
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.