")
Back to Journals » OncoTargets and Therapy » Volume 9
Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-small-cell lung cancer with activating EGFR mutations
Authors Wang J, Wang B, Chu H, Yao Y
Received 14 February 2016
Accepted for publication 7 May 2016
Published 22 June 2016 Volume 2016:9 Pages 3711—3726
DOI https://doi.org/10.2147/OTT.S106399
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Min Li
Jun Wang, Baocheng Wang, Huili Chu, Yunfeng Yao
Department of Oncology, General Hospital, Jinan Command of the People’s Liberation Army, Jinan, People’s Republic of China
Abstract: Identifying activating EGFR mutations is a useful predictive strategy that helps select a population of advanced non-small-cell lung cancer (NSCLC) patients for treatment with EGFR tyrosine kinase inhibitors (TKIs). Patients with sensitizing EGFR mutations (predominantly an in-frame deletion in exon 19 and an L858R substitution) are highly responsive to first-generation EGFR TKIs, such as gefitinib and erlotinib, and show improved progression-free survival without serious side effects. However, all patients with activating EGFR mutations who are initially responsive to EGFR TKIs eventually develop acquired resistance after a median progression-free survival of 10–16 months, followed by disease progression. Moreover, ~20%–30% of NSCLC patients have no objective tumor regression on initial EGFR TKI treatment, although they harbor an activating EGFR mutation. These patients represent an NSCLC subgroup that is defined as having intrinsic or primary resistance to EGFR TKIs. Different mechanisms of acquired EGFR TKI resistance have been identified, and several novel compounds have been developed to reverse acquired resistance, but little is known about EGFR TKI intrinsic resistance. In this review, we summarize the latest findings involving mechanisms of intrinsic resistance to EGFR TKIs in advanced NSCLC with activating EGFR mutations and present possible therapeutic strategies to overcome this resistance.
Keywords: NSCLC, EGFR mutation, EGFR TKIs, intrinsic resistance, T790M
Introduction
Primary lung cancer is one of the most common malignancies and a major cause of cancer-related mortality worldwide, accounting for ~1.6 million deaths per year.1 Approximately 85% of all primary lung cancers are non-small-cell lung cancers (NSCLCs), and adenocarcinoma is the most common histologic subtype of NSCLC. A majority of NSCLC patients present with locally advanced or metastatic disease and cannot undergo surgical resection when they are initially diagnosed. The overall therapeutic outcome of NSCLC is far from satisfactory. The 5-year survival rate of metastatic NSCLC is <5%, with a median overall survival (OS) of <12 months. The benefits and efficacy of cytotoxic chemotherapy and radiation therapy are limited, and they cause relatively serious side effects, affecting the patients’ quality of life.2
In the past decade, significant improvements have been made due to the development of targeted therapies, such as EGFR tyrosine kinase inhibitors (TKIs), for advanced NSCLC. Several large Phase III clinical trials have demonstrated that patients with a sensitizing exon 19 deletion or an exon 21 substitution mutation are highly responsive to first-generation EGFR TKIs, such as gefitinib and erlotinib, compared to traditional platinum-based doublet chemotherapy, with a prolonged time to progression or improved progression-free survival (PFS) without serious drug-specific side effects (Table 1). However, all patients with activating mutations who are initially responsive to EGFR TKIs eventually develop acquired resistance after ~10–16 months of consistent clinical benefit, followed by disease progression. Moreover, ~20%–30% of NSCLC patients have no good initial clinical response to EGFR TKIs, although they harbor an activating EGFR mutation. These patients represent a subgroup that is intrinsically resistant to EGFR TKI treatment. Several potential mechanisms of acquired resistance to EGFR TKIs have been explored, and several novel strategies have been developed to target acquired resistance in many studies, but the mechanism of intrinsic resistance to EFGR TKIs is not clearly understood. Several reviews have been published addressing the clinical implications of EGFR mutations in lung cancer, as well as EGFR TKI resistance.3,4 This review focuses on the recently identified molecular mechanisms of intrinsic resistance to EGFR TKIs in advanced NSCLC, which will help improve patient stratification and develop new potential agents and therapeutic strategies to overcome this resistance.
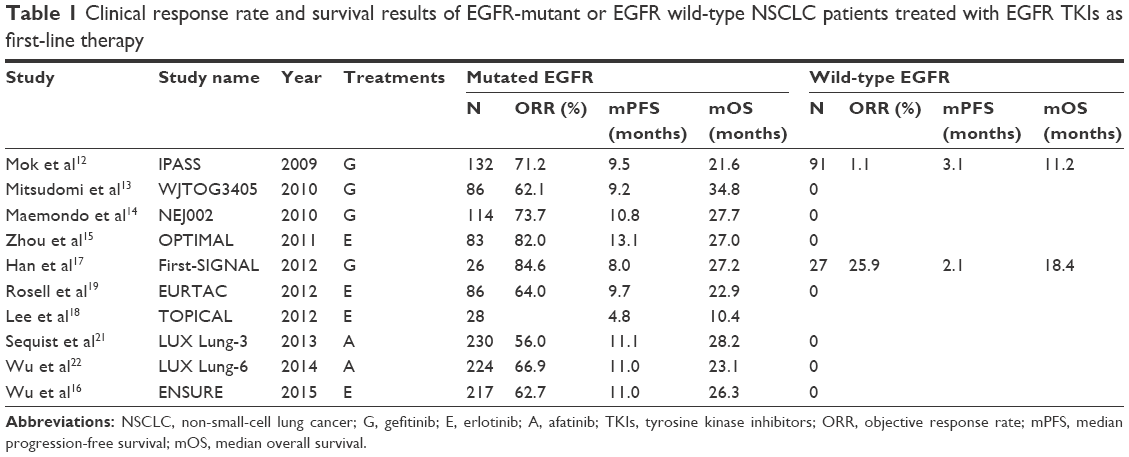 | Table 1 Clinical response rate and survival results of EGFR-mutant or EGFR wild-type NSCLC patients treated with EGFR TKIs as first-line therapy |
EGFR and activating EGFR mutations in NSCLC
EGFR is a member of the ErbB family, which also includes HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4). EGFR is auto-phosphorylated at tyrosine residues when it binds to its ligands, including EGF and transforming growth factor-α. As a potent oncogenic driver, EGFR activation further activates downstream signaling pathways, such as PI3K/Akt/mTOR and RAS/RAF/MAPK, which promote cell proliferation and survival and inhibit apoptosis.5 In 2004, somatic mutations in the tyrosine kinase domain of EGFR were characterized in NSCLC. The mutated EGFR is constitutively active in ~20% of NSCLC patients, with significantly increased proportions in adenocarcinoma, females, those of Asian ethnicity, and nonsmokers.6,7 EGFR expression and high EGFR gene copy number are also found in 10%–30% of NSCLC patients. Earlier studies showed that constitutive activation of the EGFR signaling pathway was initiated by EGFR gene amplification. Subsequent studies found that EGFR activation was triggered by EGFR mutations but not by EGFR amplification, although both activating EGFR mutations and amplification could occur in cancer tissues.8 Patients with EGFR gene amplifications or mutations have a different prognosis compared to those without these genetic alterations. Common activating mutations include an in-frame deletion in exon 19 and an L858R substitution in exon 21, which account for >90% of all EGFR mutations in NSCLC. Generally, activating mutations in the EGFR gene are mutually exclusive from other gene mutations and result in constitutive activation of EGFR and downstream signaling pathways independent of ligand binding and EGFR expression, eventually promoting the growth and spread of the cancer.9
Treatment of NSCLC with EGFR TKIs
Several small-molecule TKIs targeting EGFR, such as reversible competitive inhibitors of ATP, including erlotinib and gefitinib, and irreversible inhibitors, including afatinib, dacomitinib, and neratinib, have been developed to block EGFR-mediated downstream signaling activation. In lung cancer cells with EGFR mutations, gefitinib and erlotinib selectively bind to the tyrosine kinase region of the intracellular domain of EGFR and significantly attenuate the autophosphorylation of EGFR, reduce the subsequent activation of the PI3K/Akt/mTOR and RAS/RAF/MAPK pathways, and inhibit cell proliferation and promote apoptosis.10 Irreversible inhibitors also show potent activity against proliferation in cells with activating EGFR mutations and gatekeeper T790M mutations by binding covalently to EGFR.11
Several EGFR TKIs have been explored and evaluated as potent agents for the treatment of NSCLC with activating EGFR mutations in large randomized Phase III clinical trials (Table 1). Clinical use of EGFR TKIs has promoted a change in the approach to patients with advanced diseases, particularly lung adenocarcinomas. The IRESSA Pan-Asia Study (IPASS) was the first randomized clinical trial comparing gefitinib (n=609) to chemotherapy (paclitaxel plus carboplatin, n=608) as a first-line therapy for patients with advanced NSCLC, although only East Asian patients who were light or nonsmokers were enrolled. The objective response rate (ORR) in the overall population was significantly higher in patients treated with gefitinib monotherapy than those treated with chemotherapy (43.0% versus 32.2%, respectively). In the gefitinib-treated arm, the ORR was 71.2% in patients with EGFR mutations versus 1.1% in patients without EGFR mutations. Subgroup analysis in 261 patients with EGFR mutations showed that gefitinib resulted in a longer PFS than chemotherapy (9.5 versus 6.3 months, respectively).12 Several large Phase III trials further confirmed the superiority and efficacy of EGFR TKIs over standard chemotherapy, including the WJTOG3405, NEJ002, OPTIMAL, and ENSURE studies in Asian patients and the EURTAC study in European patients.13–19 These studies consistently found that NSCLC patients with EGFR mutations had a high ORR ranging from 58% to 83%, with a median PFS of 10–16 months, when they received reversible EGFR TKIs. However, EGFR TKIs were not more effective than chemotherapy as a second-line therapy for previously treated NSCLC patients who had wild-type EGFR.20 Although there was no benefit in terms of OS due to the influence of subsequent treatments in trials using first-generation EGFR TKIs, an irreversible EGFR inhibitor, afatinib, significantly prolonged OS of the subgroup of patients with exon 19 deletion mutations in the whole population from the LUX-Lung 3 and/or LUX-Lung 6 studies.21–23 However, no significant difference was found in the OS of patients with exon 21 mutations. Thus, the researchers concluded that the tumors with EGFR exon 19 deletions and L858R mutations should be considered two distinct populations that should be studied separately and will require different treatment strategies in the future. The clinical difference in OS observed with afatinib compared to traditional chemotherapy in patients with the exon 19 deletion could be attributed to a difference in mechanism between afatinib and the first-generation EGFR TKIs. Moreover, the rechallenge with EGFR TKIs could also affect survival.
Overall, patients receiving TKIs tolerated them well, and only displayed mild adverse events, including skin rash and diarrhea. TKIs improved the quality of life and delayed the deterioration of symptoms. There are more patients of Asian ethnicity who benefited from TKI treatment due to a high EGFR (nearly 50%) mutation frequency compared to those of Caucasian ethnicity (<20%).23 In the clinical management of advanced NSCLC, EGFR TKIs have been approved around the world as the standard first-line therapy for a subset of patients whose tumors harbor EGFR mutations. Because EGFR TKIs are costly and may have deleterious effects on individuals without activating mutations, EGFR mutation testing is recommended prior to the initiation of TKI treatment, which allows for selection of optimal advanced NSCLC.
Unfortunately, similar to standard platinum-based chemotherapy, all patients with mutant EGFR will ultimately develop resistance to EGFR TKI therapy after a period of promising clinical response.24 The efficacy of EGFR TKIs is largely limited by either acquired (secondary) or intrinsic (primary) resistance. The elucidation of the molecular mechanisms underlying acquired resistance to EGFR TKIs is an active area of clinical and translational research since Kobayashi et al first identified a secondary mutation associated with acquired resistance to gefitinib.25 These established mechanisms include development of a secondary EGFR T790M gatekeeper mutation, MET amplification, HER2 amplification, epithelial–mesenchymal transition (EMT), and transformation of NSCLC to small-cell histology.26,27 Moreover, ~20%–30% of patients with mutated EGFR have intrinsic resistance to EGFR TKI therapy, and some patients do not show an acceptable response to treatment. The mechanisms of intrinsic resistance to EGFR TKIs are currently not understood well.
Mechanisms of intrinsic resistance to EGFR TKIs in EGFR-mutated NSCLC
Similar to drug resistance associated with chemotherapy, resistance to EGFR TKIs includes intrinsic (primary) and acquired (secondary) resistance. Intrinsic resistance is generally defined as a de novo inactivation of EGFR TKIs, whereas acquired resistance is generally defined as relapse of the disease following a period of clinical response. The intrinsic resistance to EGFR TKIs in NSCLC patients can be divided into resistance in patients with mutant EGFR and resistance in patients with wild-type EGFR. Specifically, Jackman et al recently proposed detailed criteria for acquired resistance in patients with mutant EGFR. These include 1) patients harbor an activating EGFR mutation that is associated with clinical response to first-generation EGFR TKIs, 2) patients achieve a partial or complete response, or develop a stable disease in response to EGFR TKI monotherapy (>6 months), 3) disease progression occurs despite uninterrupted exposure to EGFR TKIs in accord with response evaluation criteria in solid tumors, and 4) patients receive additional systematic treatment after discontinuation of EGFR TKIs.24 However, clinical criteria for intrinsic resistance to EGFR TKIs have not been established. In all Phase III trials evaluating the efficacy of EGFR TKIs in previously untreated EGFR-mutant NSCLC, intrinsic resistance refers to development of progressive or stable disease as the best response to EGFR TKIs in ≤2 months. Other investigators defined intrinsic resistance as progressive or stable disease in <3 or 4 months without any evidence of an objective response when using EGFR TKIs.28–30 In contrast to acquired resistance to EGFR TKIs, intrinsic resistance is more complicated. Although the molecular basis of intrinsic resistance to EGFR TKIs remains to be clarified, genetic mutations and molecular alterations from several preclinical and retrospective studies present clues to direct further investigations.
Smoking status
It is well known that activating EGFR mutations are more frequently found in nonsmokers than smokers. Additionally, current smokers experience a lower clinical benefit from EGFR TKI therapy compared to never smokers as well as former smokers.31 A more recent meta-analysis showed that the PFS benefit for never smokers was better than that for ever smokers, although all patients had an activating EGFR mutation and received EGFR TKI treatment.32 Thus, smoking-related lung cancer is viewed as an independent disease that is different from nonsmoking-related lung cancer with respect to genetic profile, prognosis, and response to targeted therapy, as well as other treatment strategies. Cigarette smoke has been found to activate Src kinase and lead to aberrant activation of EGFR or cytochrome P450 1A1, which is involved in the metabolism of TKIs, or promote degradation of EGFR by inducing the release of reactive oxidative species.33–35 Nicotine exposure has been found to induce resistance to EGFR TKIs in EGFR-mutated NSCLC PC-9 cells via an EGFR signal.36 However, Lee et al identified eleven patients who showed primary resistance to EGFR TKIs (disease progression <3 months) among 197 consecutive EGFR-positive patients who received EGFR TKI therapy, in which EGFR genotype and smoking did not predict PFS for targeted treatments.28
Genetic alterations of EGFR
Some NSCLC patients exhibit intrinsic resistance to EGFR TKIs, although they have an activating EGFR mutation (Figure 1). It has been shown that insertion mutations in exon 20 of the EGFR gene are associated with oncogenic transformation of cells and confer primary resistance to EGFR TKIs, with the exception of the insertion A763_Y764insFQEA in the C-terminal helix of EGFR.37 This mutation represents ~5%–10% of all known EGFR gene mutations. In in vitro experiments, regular treatment with gefitinib or erlotinib was unable to inhibit EGFR signaling and induce cell apoptosis because this mutation decreased the affinity for EGFR TKIs, but the irreversible inhibitor CL-387,785 was efficacious.38 Further clinical investigations showed that the majority of patients with insertions in exon 20 of the EGFR gene did not demonstrate a clinical response when treated with TKIs, including reversible and irreversible inhibitors, although these patients had an OS similar to that of patients with EGFR exon 19 or 21 mutations.39
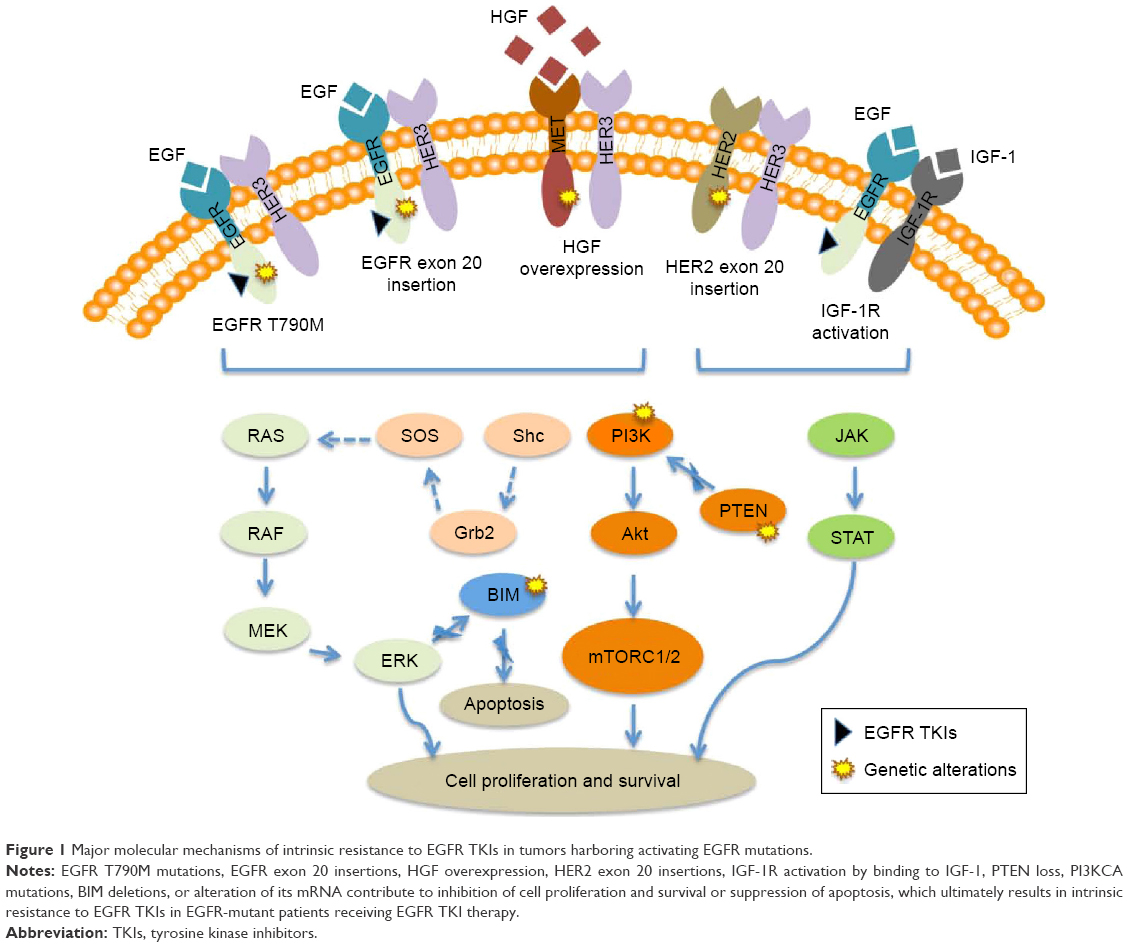 | Figure 1 Major molecular mechanisms of intrinsic resistance to EGFR TKIs in tumors harboring activating EGFR mutations. |
T790M is a frequently reported secondary mutation of the EGFR gene and is detected in >60% of patients with acquired resistance to EGFR TKI therapy.40 This point mutation causes a substitution of methionine for threonine at position 790 in exon 20 of the EGFR gene. The methionine side chain leads to steric hindrance that affects the ability of EGFR TKIs to bind to the ATP-kinase pocket. Interestingly, a recent report showed that the T790M resistance mutation may be associated with genetic susceptibility to lung cancer.41 The T790M mutation has been described as a minor clone in a small group of treatment-naïve NSCLCs harboring activating EGFR mutations, suggesting its association with intrinsic EGFR TKI resistance (Table 2).
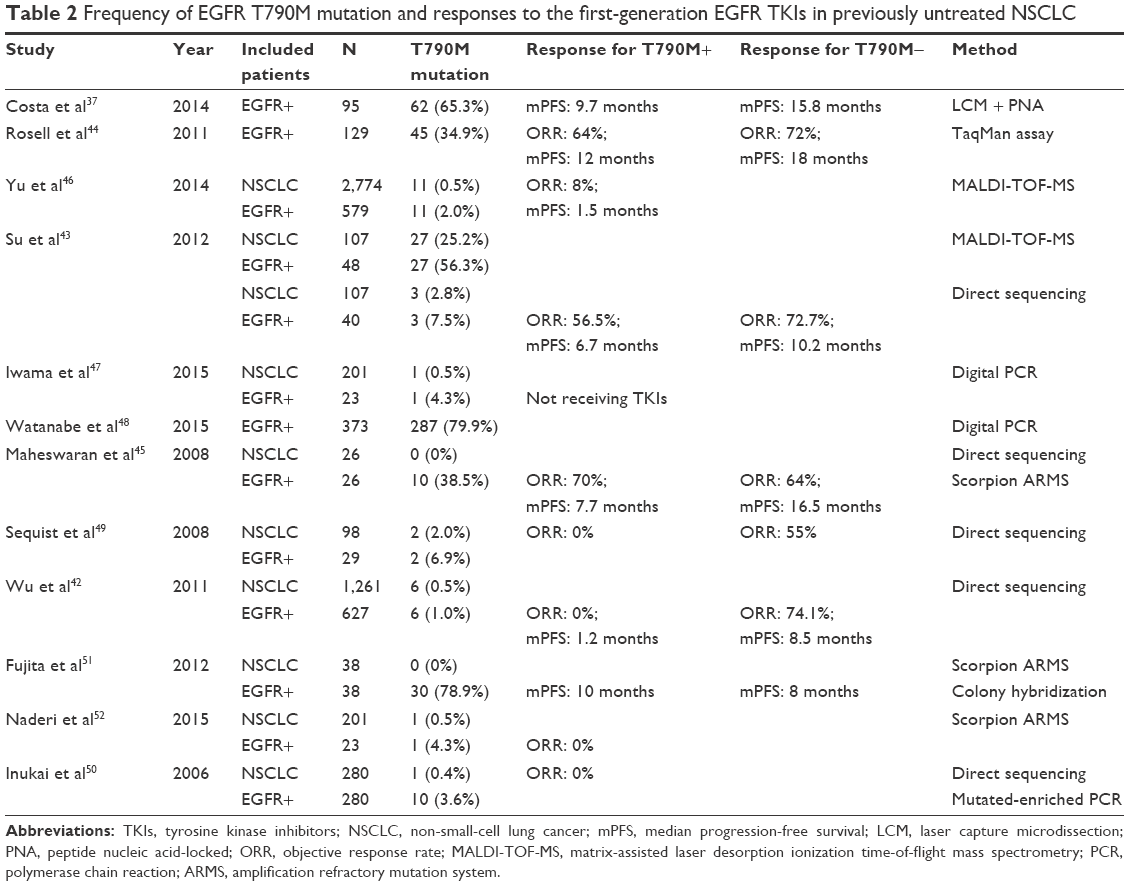 | Table 2 Frequency of EGFR T790M mutation and responses to the first-generation EGFR TKIs in previously untreated NSCLC |
Generally speaking, the T790M mutation leads to poor response to EGFR TKIs. Patients who harbor both resistant T790M and an activating EGFR mutation have a reduced ORR and a shorter PFS compared with those with an activating EGFR mutation but without T790M.37,42–45 However, there are no differences between the two groups regarding OS. Furthermore, patients with wild-type EGFR still have a significantly longer PFS.43,46 Thus, the EGFR TKI benefit for patients with a baseline EGFR T790M detected by standard molecular analysis is limited, and initial treatment with EGFR TKIs may not be a first-line option for these patients. However, it is currently unclear whether T790M-positive patients should be excluded from EGFR TKI therapy.
The frequency of the T790M mutation in pretreated tumor samples with activating EGFR mutations has been reported and ranges from 1% to 65%, which is dependent on the techniques used to analyze this mutation.37,42–45,47–52 For example, the T790M mutation was found in 25.2% of pretreated NSCLC tumors using a highly sensitive method, matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS), and only 2.8% by direct sequencing.43 A study reported that 79.9% of TKI-naïve tumor specimens with EGFR mutations were T790M-positive by digital polymerase chain reaction (PCR).48 The T790M mutation was detected by nanofluidic digital PCR in all EGFR-mutated NSCLC samples regardless of EGFR TKI treatment, and the ratio of the number of T790M alleles to that of activating mutation alleles (T/A) is associated with resistance to EGFR TKIs, indicating that highly sensitive and quantitative detection of T790M is also important for evaluation of the contribution of T790M to EGFR TKI resistance.47
Genetic alterations in bypass signaling
Genetic alterations in genes other than EGFR and its associated ErbB family members can occur with activating EGFR mutations and may be responsible for the decreased sensitivity of NSCLC to EGFR TKI treatment (Figure 1). These alterations include EGFR signal-related and non-EGFR signal-related mutations. Several studies have shown that genetic alterations are associated with acquired EGFR TKI resistance in TKI-treated tumors or intrinsic resistance in TKI-naïve tumors.
For example, somatic exon 20 insertions have been found in the HER2 gene in NSCLC specimens. These mutations lead to constitutive phosphorylation and activation of HER2 as well as EGFR and confer resistance to gefitinib or erlotinib.53 Somatic PIK3CA mutations in the catalytic domain of PIK3CA have been identified in NSCLC tumors after TKI treatment, which is one mechanism related to acquired resistance to EGFR TKI.54 These mutations have also been detected in 1%–3% of tumor samples harboring activating EGFR mutations prior to TKI therapy. In in vitro experiments using EGFR-mutant cell lines, PIK3CA mutations contributed to consistent activation of PI3K and attenuated TKI-induced apoptosis.55 Interestingly, PIK3CA mutations have been found in NSCLC with EGFR/KRAS mutations, and patients with single PIK3CA mutations had shorter OS than those with PIK3CA-EGFR/KRAS co-mutations or wild-type PIK3CA or wild-type EGFR/KRAS.56
The PTEN controls the PI3K/Akt signal pathway and acts as a tumor suppressor gene. Mathematical models showed that several downstream factors, including pAkt, pSTAT3, and pERK, were largely inhibited in the presence of erlotinib, but pAkt levels did not decrease with the loss of PTEN, suggesting that cells that lost PTEN were resistant to erlotinib.57 Sos et al found that loss of PTEN expression contributed to primary resistance to EGFR TKIs. In this study, there was one patient with PTEN loss among 24 patients with mutated EGFR. Further in vitro study indicated that H1650, a lung adenocarcinoma cell line with an exon 20 deletion in the EGFR gene, also harbored a PTEN loss of function and exhibited innate resistance to erlotinib. The effect of PTEN loss on TKI efficacy was related to activation of Akt and EGFR in EGFR-mutant cells.58 In another study by Cetin et al, reduced PTEN protein expression levels were observed in 17 (34%) of the cases. The loss of PTEN expression could be an intrinsic mechanism of EGFR TKI resistance in advanced NSCLC.59 In addition to the described genetic mutations, other pathways and molecules also contribute to intrinsic EGFR TKI resistance. A recent investigation identified >700 mutation hotspots in 46 cancer-related genes in 29 patients with EGFR L858R NSCLC by next-generation sequencing. Interestingly, the MLH1 V384D polymorphism was enriched in patients who were intrinsically resistant to EGFR TKIs and had a short PFS of <3 months.30 Thus, both “driver” mutations and candidate germline genetic variations could be genetic predictors of intrinsic resistance in EGFR mutation-positive NSCLC. Genetic testing of the EGFR pathway and other specific molecules should be considered in the era of personalized therapy before application of EGFR TKIs.
Microenvironment
The tumor microenvironment and stroma are critical for cancer progression or metastasis. Growing evidence has indicated the involvement of alteration of the tumor microenvironment in the development of resistance to EGFR TKIs. For example, cancer-associated fibroblasts (CAFs) in the tumor microenvironment contribute to intrinsic resistance to EGFR TKIs in NSCLC with mutated EGFR. In a mouse model, an EMT-derived subpopulation of CAFs expressed a marker of gefitinib resistance, epithelial membrane protein-1. Furthermore, EGFR TKI-resistant CAFs secreted paracrine factors that mitigated the EGFR TKI-mediated blockade of pEGFR and pMAPK.60 Lung adenocarcinoma cells with activating EGFR mutations (PC9 and HCC827) showed intrinsic resistance to gefitinib when they were cocultured with podoplanin-expressing CAFs by direct contact, whereas separate coculturing of lung cancer cells and CAFs did not induce resistance to gefitinib.61,62 The podoplanin-mediated pERK expression induced EMT and activation of the Hedgehog signaling pathway, which were associated with primary EGFR TKI resistance. Further clinicopathological investigations revealed that a significantly lower response was observed in EGFR-mutated patients with podoplanin-positive CAFs compared with those with podoplanin-negative CAFs following gefitinib therapy (53% versus 83%, respectively).62
Integrin and cadherin are responsible for cellular adhesion to the extracellular matrix, and play important roles in mediating cell–cell adhesion. Recently, EGFR-mutant NSCLC cells expressing increased levels of integrin β-1 were found to promote resistance to EGFR TKIs by activating the Akt signaling pathway. The expression levels of integrin β-1 in patients who were resistant to EGFR TKIs were significantly higher than those in patients who were sensitive to EGFR TKIs.63 Yamauchi et al found that N-cadherin expression was markedly upregulated in gefitinib-resistant PC9 cells with mutated EGFR as well as NSCLC cells with wild-type EGFR. Inhibition of N-cadherin expression induced caspase-dependent apoptosis in association with inactivation of the PI3K/Akt signaling pathway.64 Other cytokines and noncoding RNAs might play an important role in mediating primary resistance to EGFR TKIs. For example, microRNA (miRNA) expression signatures demonstrated that three miRNAs, miR-21, miR-27a, and miR-218, were related to primary resistance to EGFR TKIs in NSCLC patients who had EGFR exon 19 deletions.65 These results indicate that the clinical difference in outcome for patients with resistance to EGFR TKIs could be attributed to different miRNA expression patterns, which may distinguish between sensitive and resistant patients with activating EGFR mutations. However, these results are from a small sample of individuals.
Molecular alterations of related molecules
Molecular alterations of specific molecules might also play a role in primary resistance to EGFR TKIs in NSCLC patients with activating EGFR mutations, independent of the T790M mutation. MET protein expression and phosphorylation are commonly found in NSCLC tumors before EGFR TKI treatment. Furthermore, MET activation was significantly associated with poor response to subsequent EGFR TKI treatment, regardless of the EGFR status.66 As a ligand of the MET receptor, HGF can confer resistance on NSCLC cells harboring activating EGFR mutations by phosphorylation of MET and activation of the PI3K/Akt pathway.67 In a report involving 97 lung patients with mutant EGFR, high levels of HGF expression were found in 29% of tumors with intrinsic resistance compared to none with the EGFR T790M mutation and 4% with MET amplification. High HGF expression might be more common than other mutations in tumors with primary resistance and may promote intrinsic resistance to EGFR TKIs by activating the MET signaling pathway.68 Moreover, HGF was responsible for reducing susceptibility to irreversible EGFR TKIs in NSCLC with EGFR T790M mutations.69 Recent investigations by Park et al revealed that CRIPTO1 expression in NSCLC with mutated EGFR is likely a major mechanism that leads to intrinsic resistance to EGFR TKIs. All EGFR-mutated NSCLC tumors that were resistant to erlotinib expressed higher levels of CRIPTO1, whereas only 30% of EGFR-mutated NSCLC tumors that were sensitive to erlotinib showed CRIPTO1 expression. Further in vitro studies showed that CRIPTO1-induced erlotinib resistance was linked to activation of the SRC signaling pathway via downregulation of miR-205 expression.29
As a proapoptotic molecule of the Bcl-2 family, BIM is responsible for apoptosis triggered by a variety of molecules, including EGFR TKIs.70 In in vitro experiments, EGFR TKI-induced apoptosis in sensitive EGFR-mutated cells was associated with BIM expression, whereas cisplatin-induced apoptosis was not triggered by overexpression of BIM in different NSCLC cells regardless of the EGFR status.71 BIM deletions are more frequently found in East Asian than Caucasian patients, with a prevalence of 12.3%.72 Marker analyses from the EURTAC study showed that patients with erlotinib whose tumors expressed low or intermediate levels of BIM mRNA had a lower ORR (34.6% versus 87.5%, respectively) and unfavorable PFS (7.2 versus 12.9 months, respectively) and OS (22.1 versus 28.6 months, respectively) compared to those whose tumors expressed high levels of BIM mRNA.37 The molecular mechanisms underlying BIM regulation are still not fully understood. One possibility is that only specific active BIM isoforms affect the sensitivity of cancer cells to EGFR TKIs. A polymorphism may result in a BIM isoform that lacks the proapoptotic BH3 domain, promoting resistance to EGFR TKIs. Ng et al found that patients with this polymorphism showed a reduced median PFS compared to those without this polymorphism (6.6 versus 11.9 months, respectively).72 In a Chinese study involving 166 patients who received EGFR TKI therapy and had an activating mutation, the median PFS (4.7 versus 11.0 months; 25% versus 66%, respectively) and the ORR were lower in patients with BIM deletions than those without this variation.73 However, the effect of BIM deletion on the sensitivity to EGFR TKIs and prognosis of lung cancer patients remains controversial. Some investigators believe that this BIM deletion cannot account for the intrinsic resistance in EGFR-mutant NSCLC patients to erlotinib or gefitinib.28 A meta-analysis of six studies was conducted to evaluate the association between the BIM deletion and resistance to EGFR TKIs in NSCLC patients. The BIM deletion may represent a negative predictive biomarker for tumor response in NSCLC patients treated with EGFR TKIs, with an ORR of 0.39 (95% confidence interval [CI]: 0.23–0.67). This polymorphism was significantly correlated with poor PFS (hazard ratio [HR]: 1.37; 95% CI: 1.09–1.7) and OS (HR: 1.25; 95% CI: 1.08–1.45).74 Recently, Wu et al provided evidence suggesting that paxillin expression may confer primary resistance to EGFR TKIs in lung cancer cells with mutated EGFR in vitro and nude mouse models. Paxillin induced ERK activation and modulated the stability of BIM by phosphorylation at serine 69. Importantly, tumors expressing high levels of paxillin exhibited an unfavorable response to EGFR TKIs compared to those with low levels of paxillin (65% versus 26%). Furthermore, the OS and PFS were significantly shorter in patients with high levels of paxillin compared to those with low levels of paxillin.75 Thus, BIM deletion causes partial, but not absolute, intrinsic resistance to EGFR TKI treatment. Several molecular mechanisms could synergistically contribute to intrinsic resistance to EGFR TKIs, such as genetic alterations and posttranscriptional regulation of the BIM gene.
Heterogeneity of mechanisms of resistance
Lung cancer is a disease with heterogeneous features, and a higher homogeneity has been found in EGFR mutation status. EGFR mutation heterogeneity in tumors could explain why a subset of patients had mixed response to EGFR TKIs. Thus, intratumor heterogeneity could be responsible for intrinsic resistance. Firstly, multiple intrinsic resistance mechanisms could co-occur in a single tumor sample. Bean et al found that MET amplification occurred with T790M mutations in EGFR-mutated tumors with acquired resistance to EGFR TKIs.76 Both EGFR T790M and MET amplification have been found in a minor population of NSCLC cells before exposure to EGFR TKI treatment.43,77 Secondly, mutation ambulance and category is different in different sites within same metastatic organ. As a sample, a large study of EGFR mutation heterogeneity between primary lung tumors and their metastases revealed that the overall discordance rate was 13.9% in 180 patients, but patients with multiple pulmonary nodules had the highest discordance rate of 24.4%.78 Although the T790M mutation is usually detected in tumors after exposure to EGFR TKIs, it also occurs prior to EGFR TKI treatment. In patients with acquired resistance, T790M mutation could be detected in one primary site, but it could disappear in another site and/or other resistance mutations could be found in another metastatic site including HER2 amplification and MET amplification.40 Thirdly, EGFR mutation status or mechanism of resistance could be different in different metastatic sites, despite the use of highly sensitive methods. Multiple biopsies might elucidate the relevance and degree of heterogeneity distribution of driver gene mutations regarding the metastatic locations and different parts of the same tumor at the time of initial diagnosis. However, single biopsy is sometimes not accessible. Liquid biopsy that measures the serum level of circulating tumor DNA, microdissection of small tumor foci, deep sequencing of small amounts of tumor tissues, and more sensitive detection techniques such as digital PCR and MALDI-TOF-MS would help answer the critical question of high intratumor heterogeneity in EGFR mutation and heterogeneity of clinical response to EGFR TKIs.
Overcoming intrinsic resistance to EGFR TKIs
Acquired resistance to EGFR TKIs has been widely investigated, and many treatment strategies have been explored. However, few studies have been specifically designed to delay or overcome intrinsic TKI resistance. In clinical practice, no standard approach exists to overcome this resistance. However, there are several overlapping mechanisms between intrinsic and acquired resistance, and strategies to overcome acquired resistance could also be applied to intrinsic resistance.
Second-generation EGFR TKIs
The irreversible EGFR TKIs, such as afatinib, neratinib, and dacomitinib, bind covalently to the Cys-797 residue of EGFR. These inhibitors suppress the activation of EGFR and PI3K/Akt and inhibit proliferation in cells with mutant EGFR, even those harboring a resistant T790M mutation. Nevertheless, the LUX-Lung 1 trial comparing afatinib and a placebo as a second-line or third-line therapy for patients who progressed following gefitinib or erlotinib therapy failed to demonstrate an improvement in OS for afatinib (10.8 versus 12 months). Patients treated with afatinib had a low ORR of 7% and a prolonged median PFS of only 2.2 months compared to those with the placebo.79 Clinical use of afatinib is presently limited because of dose-limiting toxicity related to concurrent inhibition of wild-type EGFR, such as diarrhea and rash. Currently, treatment with afatinib is not recommended for NSCLC patients with mutated EGFR when they are refractory to erlotinib or gefitinib. Therefore, second-generation EGFR TKI monotherapy does not appear to be an alternative strategy to overcome primary resistance.
Third-generation EGFR TKIs
The T790M mutation in exon 20 is the most common mechanism associated with acquired resistance to EGFR TKIs and accounts for ~50% of all acquired resistance in NSCLC patients who have lung cancer with an EGFR mutation. In addition, this mutation is most often present in pretreated patients with activating EGFR mutations. The third-generation EGFR TKIs, such as AZD9291, CO1686, and HM61713, have been developed to specifically target the T790M EGFR mutation (Table 3). These compounds were shown to be active in cell experiments and murine models of lung cancer driven by EGFR T790M. AZD9291 irreversibly and selectively inhibits T790M mutation-positive EGFR as well as sensitizing EGFR.80 Recently, Jänne et al reported the final results in a Phase I study, including a dose-escalation cohort of 31 patients and a dose-expansion cohort of 222 patients. The ORR among all patients was 51% (95% CI: 45%–58%). A total of 127 patients with centrally confirmed EGFR T790M mutations significantly responded to AZD9291, with an ORR of 61% (95% CI: 52%–70%) compared to 21% (95% CI: 12%–34%) in patients without the EGFR T790M mutation.81 The most frequently reported side effects were rash, pruritus, and diarrhea, without dose-limiting toxicity. In November 2015, a tablet formulation of AZD9291 (osimertinib) was granted accelerated approval by the US Food and Drug Administration as a second-line therapy for metastatic EGFR T790M mutation-positive NSCLC that progressed after erlotinib or gefitinib treatment.82 Several large prospective clinical trials evaluating the efficacy of AZD9291 in EGFR-mutated NSCLC are ongoing, including a Phase II, single-arm, open-label study (NCT02094261) in second-line T790M-positive patients and a Phase III study comparing AZD9291 to platinum/pemetrexed chemotherapy in second-line T790M-positive patients (NCT02151981). The results from the AURA2 Phase I/II study (NCT02094261) showed that pretreated patients with activating EGFR mutations and T790M mutations benefited from an 80 mg once-daily dose of AZD9291, with an ORR of 71% and an immature PFS of 8.6 months.83 An international Phase III study (NCT02296125) has been designed to assess the safety and efficacy of AZD9291 versus EGFR TKIs as a first-line treatment for patients with locally advanced or metastatic NSCLC with activating EGFR mutations.
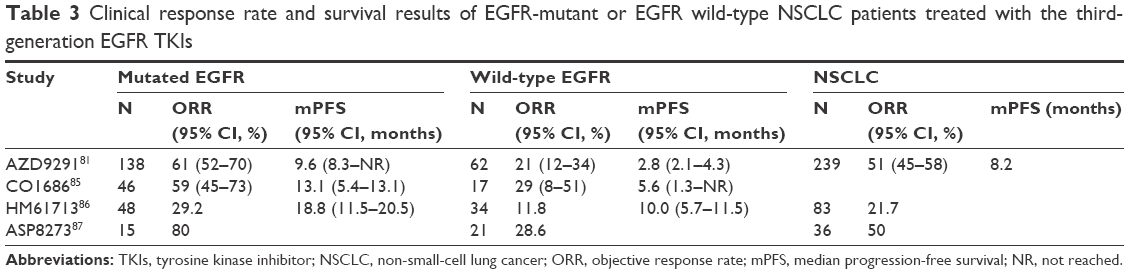 | Table 3 Clinical response rate and survival results of EGFR-mutant or EGFR wild-type NSCLC patients treated with the third-generation EGFR TKIs |
CO1686 (rociletinib) is another promising third-generation irreversible TKI that selectively inhibits mutant EGFR, in particular the T790M drug-resistant mutation, in NSCLC models.84 In an initial expansion of a Phase II/II study, 130 patients received the free-base form of rociletinib (150 mg once daily to 900 mg twice daily) or the hydrogen bromide salt form (500 mg twice daily to 1,000 mg twice daily). The ORR among the 46 patients with T790M mutations was 59% (95% CI: 45%–73%), and the rate among the 17 patients without T790M mutations was 29% (95% CI: 8%–51%).85 Subsequent interim Phase II data reported that the ORR among the 81 patients with T790M mutations was 51%. The T790M-positive subset with mature data showed an ORR of 60%. The frequent side effects included nausea, diarrhea, and grade 3 hyperglycemia. At the recommended Phase II dose (625 mg twice daily), the ORR was 60% with the duration of response ranging from 85 to 184+ days. In the T790M-negative group, the overall response rate was 28%. Two clinical trials, including a Phase II trial of rociletinib as a second-line therapy (NCT02147990) and a Phase III trial comparing rociletinib to erlotinib in a first-line setting (NCT02186301), are ongoing. Other compounds, including HM61713 and ASP8273, have also been shown to target T790M and common exon 19 deletions and exon 21 EGFR substitution mutations, with an ORR ranging from 29.2% to 77.8%. In particular, treatment with HM61713 delivered a comparable ORR of 29.6% in patients who progressed following treatment with first-generation TKIs within 4 weeks, which indicated that HM61713 was active in patients with intrinsic resistance to EGFR TKIs. In conclusion, the third-generation EGFR TKIs appear to be promising in patients who progressed on treatment with EGFR TKIs. Greater efficacy and benefits will be achieved in T790M mutation-positive patients than T790M mutation-negative patients. Ongoing studies may determine whether the third-generation EGFR TKIs are also active against advanced NSCLC with intrinsic resistance due to the presence of de novo T790M mutations as an initial treatment strategy.86,87
Chemotherapy plus EGFR TKIs
Platinum-based doublet chemotherapy is the current standard treatment for patients with advanced NSCLC who failed after receiving first-line first-generation EGFR TKIs if the patients are not eligible for clinical trial participation. Currently, standard chemotherapy should also be considered in patients who are primarily resistant to EGFR TKIs.
Several clinical studies have shown that concurrent administration of gefitinib or erlotinib with cytotoxic chemotherapy did not confer a survival advantage over chemotherapy alone in NSCLC patients with previously untreated advanced NSCLC.88–90 These early studies with a concurrent combination of chemotherapy and EGFR TKIs were designed before the discovery of the EGFR mutations and included unselected NSCLC patients. In contrast, sequential administration of EGFR TKIs after chemotherapy or intercalated chemotherapy and the EGFR TKI erlotinib might be more effective than chemotherapy plus placebo.91–93 A Phase III IMPRESS study showed no significant improvement in PFS with continued use of gefitinib plus pemetrexed/cisplatin doublet chemotherapy compared to chemotherapy alone in 265 EGFR mutation-positive patients who progressed on first-line treatment with EGFR TKIs.94 However, a recent Phase II study showed that 33 EGFR-mutant patients were treated with gefitinib (days 1–56 and restarted on day 134) and three cycles of cisplatin plus docetaxel, in which 12 patients achieved a 2-year PFS, with a rate of 40.2%.95 The results from this study suggested that the addition of standard chemotherapy might prevent the development of acquired resistance to EGFR TKIs only in specific patients with activating EGFR mutations. In conclusion, the validation of specific patients needs clarification when considering the application of chemotherapy plus EGFR TKIs for patients with acquired or intrinsic resistance in large clinical and translational studies.
In addition, a combination of irreversible afatinib and the EGFR-specific antibody cetuximab overcame T790M-mediated resistance in preclinical models.96 In a Phase II study, 126 mutant EGFR-positive patients who progressed on erlotinib or gefitinib treatment were enrolled.97 The ORR was 29% in all patients, but the ORR was comparable in tumors with T790M mutations and tumors without T790M mutations (32% versus 25%). The median PFS and median duration of response to the combination of afatinib and cetuximab was 4.7 (95% CI: 4.3–6.4) and 5.7 months (range: 1.8–24.4 months), respectively. However, there is a lack of data regarding the efficacy of chemotherapy plus EGFR TKIs in advanced NSCLC with intrinsic resistance due to the presence of de novo T790M mutations.
Blockade of bypass signaling
MET amplification represents 5% of the mechanisms associated with acquired resistance to EGFR TKI treatment. Moreover, intrinsic resistance to EGFR TKIs can develop through upregulation of the MET ligand FGF or MET amplification in lung tumors previously untreated with gefitinib or erlotinib. In a single-arm Phase II trial investigating the safety and efficacy of the MET inhibitor INC280 plus gefitinib in EGFR-mutated and MET-positive NSCLC patients who progressed after prior EGFR TKI treatment, six unconfirmed patients (15%) had a partial response to the combined treatment out of 41 patients.98 A Phase III study compared the MET inhibitor tivantinib (ARQ197) plus erlotinib to erlotinib alone in previously treated patients with advanced NSCLC. Addition of the MET inhibitor to erlotinib did not increase the OS regardless of EGFR status.99 The dual MET-VEGF inhibitor cabozantinib plus erlotinib produced an unconfirmed partial response of 11% in EGFR-mutated NSCLC patients following progression on EGFR TKI therapy.100 In an in vivo model of intrinsic resistance to EGFR TKIs, a combination treatment targeting EGFR and MET simultaneously was highly active against tumors codriven by mutated EGFR and MET amplification.101
PIK3CA mutation has been reported as a mechanism associated with intrinsic and acquired resistance to EGFR TKIs. Targeting the PI3K pathway could be a novel strategy to overcome TKI resistance. A dual inhibitor of PI3K/mTOR, NVP-BEZ235, was found to effectively inhibit the growth of gefitinib-resistant NSCLC cells in vivo as well as in vitro.102 A Phase II study of the Akt inhibitor MK-2206 plus erlotinib showed a 9% ORR in advanced mutant EGFR-positive NSCLC previously treated with erlotinib, with a PFS of 4.4 months.103 Therefore, specific inhibitors targeting MET, PI3K, or other pathways may be promising treatments for NSCLC patients with mutations associated with intrinsic resistance to EGFR TKIs.
Other strategies against EGFR TKI resistance
Other approaches to overcome resistance include the use of antiangiogenic agents, immunotherapy, and other strategies. A recently published study of sorafenib in NSCLC patients who relapsed after EGFR TKI therapy showed that sorafenib failed to prolong survival with a disease control rate of 32.8%. The median PFS and OS were 3.7 (95% CI: 3.5–3.9 months) and 7.4 months (95% CI: 5.7–9.2 months), respectively.104 The disease control rate was not associated with BIM deletion and EGFR mutation status, and identification of biomarkers to determine the population who can benefit from sorafenib treatment is needed. Bevacizumab in combination with platinum has been approved for treating metastatic non-squamous NSCLC.105,106 The results from BeTa trial showed that combination of bevacizumab and erlotinib did not provide survival benefit to 636 previously treated unselected patients, regardless of the EGFR status.107 However, a small Phase II study in Japan investigated the efficacy of gefitinib plus bevacizumab as a first-line treatment in metastatic NSCLC with activating EGFR mutations. Although this clinical study did not meet the primary end point, the median PFS for overall patients and patients with exon 19 deletions were 14.4 and 18.0 months, respectively.108 Another trial (JO5567) in Japan was the first prospective randomized trial to compare the efficacy and safety of erlotinib plus bevacizumab to erlotinib alone in advanced EGFR-mutant non-squamous NSCLC in a first-line setting. The addition of bevacizumab to erlotinib improved PFS by ~6 months compared to erlotinib alone (16.0 versus 9.7 months).109 No new safety issues were identified, and the combination treatment did not cause a significant decrease of the quality of life. The BELIEF study recently presented at the 2015 European Cancer Congress demonstrated that addition of bevacizumab to erlotinib significantly improved the ORR (76.1%) and PFS (13.8 months; 95% CI: 10.3–21.3) independent of T790M status.110 In contrast, another retrospective study indicated that the T790M mutation might have an adverse effect on the combination of EGR TKIs and bevacizumab in patients with activating EGFR mutations. Both the ORR (0% versus 18%) and the PFS (3.3 versus 4.0 months) were significantly lower in T790M-positive patients compared with T790M-negative patients.111 Thus, the addition of bevacizumab to EGFR TKIs appears to be a favorable and well-tolerated strategy for EGFR mutation-positive individuals, even for those with intrinsic resistance to EGFR TKIs. Bevacizumab addition may enhance the antitumor activity of anti-EGFR therapy and/or partially reverse intrinsic resistance by increasing intratumoral concentration of EGFR TKIs.
The PD-1/PD-L1 checkpoint inhibitors, including nivolumab and pembrolizumab, showed an unprecedented survival benefit compared with chemotherapy and have been already approved for clinical use in advanced NSCLC.112–114 In squamous NSCLC, nivolumab was shown to be highly efficacious regardless of PD-L1 expression by immunohistochemistry. In the case of non-squamous NSCLC, nivolumab tended to be more effective in patients whose tumors expressed high levels of PD-L1 than those whose tumors expressed low levels of PD-L1.113 A small retrospective study demonstrated that the high initial expression of PD-L1 was correlated with the presence of activating EGFR mutations.114 Interestingly, the PD-L1 expression was significantly upregulated in cultured EGFR-mutant tumor cells and markedly increased in tumor cells in a subset of EGFR-mutant NSCLC patients after EGFR TKI treatment.115,116 A small study (NCT01454102) investigated the efficacy of the anti-PD1 antibody nivolumab plus erlotinib in EGFR mutation-positive NSCLC patients who progressed on TKI therapy. Of the 20 patients who progressed after receiving erlotinib, 15% achieved partial response.117 This subtype of EGFR mutation-positive NSCLC was highly eligible for PD-1/PD-L1 immunotherapy because the patients had high expression of PD-L1. Thus, PD-1/PD-L1 inhibitors appear to be an alternative strategy for EGFR-mutant patients who are intrinsically resistant to EGFR TKIs.118 Other targeted drugs also showed modest activity against NSCLC with mutant EGFR. In particular, the PPAR-gamma agonist rosiglitazone potentiated the antiproliferative effects of gefitinib by increasing PTEN expression.119 Treatment with vorinostat, a HDAC inhibitor, induced apoptosis by restoring the expression of BIM and overcoming gefitinib resistance in EGFR-mutated lung adenocarcinoma cells in vitro and in vivo due to the BIM polymorphism.120
Conclusion
In the past decade, a better understanding of the biological mechanisms of NSCLC had helped to identify critical biomarkers and oncogenic drivers. Additionally, individualized care has been explored to ensure that patients receive the correct clinical treatment. A growing number of therapeutic choices are available with excellent clinical responses. In particular, NSCLC patients benefit from first-generation EGFR TKIs if they have tumors with activating somatic EGFR mutations. Unfortunately, developing acquired resistance to EGFR TKIs ultimately limits the long-term effectiveness of such treatments, and a variety of mechanisms, including EGFR T790M secondary mutations, have been shown to contribute to this process. Many investigators have attempted to explore specific therapeutic strategies to overcome acquired resistance to EGFR TKIs. Additionally, intrinsic resistance remains another major challenge, and molecular mechanisms of intrinsic resistance have not been investigated widely. Although de novo EGFR T790M mutations, rare exon 20 insertions, or proapoptotic BIM gene deletions have been described in a small group of EGFR-mutant NSCLC patients, the majority of resistant cases cannot be explained by these mutations, and the mechanistic basis of intrinsic EGFR TKI resistance in patients supposed to be responsive remains largely unknown. Furthermore, lack of specific predictive makers associated with intrinsic resistance does not allow for effective selection of patients with initial resistance to EGFR TKIs prior to using erlotinib or gefitinib. Given that primary resistance is present in ~20%–30% of NSCLC patients with EGFR mutations, it is urgent to elucidate the mechanisms associated with innate resistance to EGFR TKIs and explore novel treatments to overcome this resistance. Future directions combating intrinsic resistance might focus on the following: 1) clinical standards, which should be established to identify patients who are initially resistant to EGFR TKIs, although they have sensitizing EGFR mutations; 2) cytotoxic chemotherapy, which remains the only standard therapy option for patients with intrinsic TKI resistance; 3) third-generation EGFR TKIs as they are a promising choice for a small group of untreated patients with T790M mutations; 4) coexistent genetic alteration, which might be responsible for de novo resistance to EGFR TKIs in a small subset of patients, and stratification analysis of global genetic mutations, which is needed before treatment choice; 5) novel specific and effective inhibitors of MET or other pathways or combination therapy with new targeted drugs to demonstrate improved efficacy in patients with intrinsic resistance due to activation of bypass signaling;28 and 6) further investigations of mechanisms associated with intrinsic resistance, including the microenvironment, epigenetic and genetic alterations, as well as expression and alterations of critical mediators considering concurrent resistance mechanisms and potential tumor heterogeneity in individual patients. New approaches, including initial global genetic investigations at the time of diagnosis, novel therapies targeting specific subgroups of lung cancers, and potential combined treatment strategies, are able to maximize the duration of clinical response to current targeted therapies and further improve long-term survival.
Acknowledgment
This study was supported by the National Nature Science Foundation of China (number: 81272619 and 81572875).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Klastersky J, Awada A. Milestones in the use of chemotherapy for the management of non-small cell lung cancer (NSCLC). Crit Rev Oncol Hematol. 2012;81(1):49–57. | ||
Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31(8):1070–1080. | ||
Cortot AB, Jänne PA. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur Respir Rev. 2014;23(133):356–366. | ||
Hynes NE, Lane HA. ERBB receptors and cancer. The complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. | ||
Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | ||
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. | ||
Okabe T, Okamoto I, Tamura K, et al. Differential constitutive activation of the epidermal growth factor receptor in non-small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res. 2007;67(5):2046–2053. | ||
Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118(2):257–262. | ||
Birnbaum A, Ready N. Gefitinib therapy for non-small cell lung cancer. Curr Treat Options Oncol. 2005;6(1):75–81. | ||
Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. | ||
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. | ||
Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. | ||
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. | ||
Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. | ||
Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015;26(9):1883–1889. | ||
Han JY, Park K, Kim SW, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol. 2012;30(10):1122–1128. | ||
Lee SM, Khan I, Upadhyay S, et al. First-line erlotinib in patients with advanced non-small-cell lung cancer unsuitable for chemotherapy (TOPICAL): a double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2012;13(11):1161–1170. | ||
Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. | ||
Garassino MC, Martelli O, Broggini M, et al. Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trial. Lancet Oncol. 2013;14(10):981–988. | ||
Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. | ||
Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–222. | ||
Yang JC, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16(2):141–151. | ||
Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28(2):357–360. | ||
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. | ||
Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–933. | ||
Uramoto H, Shimokawa H, Hanagiri T, Kuwano M, Ono M. Expression of selected gene for acquired drug resistance to EGFR-TKI in lung adenocarcinoma. Lung Cancer. 2011;73(3):361–365. | ||
Lee JK, Shin JY, Kim S, et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol. 2013;24(8):2080–2087. | ||
Park KS, Raffeld M, Moon YW, et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J Clin Invest. 2014;124(7):3003–3015. | ||
Chiu CH, Ho HL, Doong H, et al. MLH1 V384D polymorphism associates with poor response to EGFR tyrosine kinase inhibitors in patients with EGFR L858R-positive lung adenocarcinoma. Oncotarget. 2015;6(10):8407–8417. | ||
Gou LY, Niu FY, Wu YL, Zhong WZ. Differences in driver genes between smoking-related and non-smoking-related lung cancer in the Chinese population. Cancer. 2015;121 Suppl 17:3069–3079. | ||
Hasegawa Y, Ando M, Maemondo M, et al. The role of smoking status on the progression-free survival of non-small cell lung cancer patients harboring activating epidermal growth factor receptor (EGFR) mutations receiving first-line EGFR tyrosine kinase inhibitor versus platinum doublet chemotherapy: a meta-analysis of prospective randomized trials. Oncologist. 2015;20(3):307–315. | ||
Filosto S, Baston DS, Chung S, Becker CR, Goldkorn T. Src mediates cigarette smoke-induced resistance to tyrosine kinase inhibitors in NSCLC cells. Mol Cancer Ther. 2013;12(8):1579–1590. | ||
Filosto S, Becker CR, Goldkorn T. Cigarette smoke induces aberrant EGF receptor activation that mediates lung cancer development and resistance to tyrosine kinase inhibitors. Mol Cancer Ther. 2012;11(4):795–804. | ||
Alfieri RR, Galetti M, Tramonti S, et al. Metabolism of the EGFR tyrosin kinase inhibitor gefitinib by cytochrome P450 1A1 enzyme in EGFR-wild type non small cell lung cancer cell lines. Mol Cancer. 2011;10:143. | ||
Togashi Y, Hayashi H, Okamoto K, et al. Chronic nicotine exposure mediates resistance to EGFR-TKI in EGFR-mutated lung cancer via an EGFR signal. Lung Cancer. 2015;88(1):16–23. | ||
Costa C, Molina MA, Drozdowskyj A, et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin Cancer Res. 2014;20(7):2001–2010. | ||
Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2(11):e313. | ||
Naidoo J, Sima CS, Rodriguez K, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: clinical outcomes and response to erlotinib. Cancer. 2015;121(18):3212–3220. | ||
Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. | ||
Bell DW, Gore I, Okimoto RA, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. 2005;37(12):1315–1316. | ||
Wu JY, Yu CJ, Chang YC, Yang CH, Shih JY, Yang PC. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res. 2011;17(11):3812–3821. | ||
Su KY, Chen HY, Li KC, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012;30(4):433–440. | ||
Rosell R, Molina MA, Costa C, et al. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin Cancer Res. 2011;17(5):1160–1168. | ||
Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359(4):366–377. | ||
Yu HA, Arcila ME, Hellmann MD, Kris MG, Ladanyi M, Riely GJ. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann Oncol. 2014;25(2):423–428. | ||
Iwama E, Takayama K, Harada T, et al. Highly sensitive and quantitative evaluation of the EGFR T790M mutation by nanofluidic digital PCR. Oncotarget. 2015;6(24):20466–20473. | ||
Watanabe M, Kawaguchi T, Isa S, et al. Ultra-sensitive detection of the pretreatment EGFR T790M mutation in non-small cell lung cancer patients with an EGFR-activating mutation using droplet digital PCR. Clin Cancer Res. 2015;21(15):3552–3560. | ||
Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26(15):2442–2449. | ||
Inukai M, Toyooka S, Ito S, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66(16):7854–7858. | ||
Fujita Y, Suda K, Kimura H, et al. Highly sensitive detection of EGFR T790M mutation using colony hybridization predicts favorable prognosis of patients with lung cancer harboring activating EGFR mutation. J Thorac Oncol. 2012;7(11):1640–1644. | ||
Naderi S, Ghorra C, Haddad F, et al. EGFR mutation status in Middle Eastern patients with non-squamous non-small cell lung carcinoma: a single institution experience. Cancer Epidemiol. 2015;39(6):1099–1102. | ||
Wang SE, Narasanna A, Perez-Torres M, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10(1):25–38. | ||
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116(10):2695–2706. | ||
Wang L, Hu H, Pan Y, et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS One. 2014;9(2):e88291. | ||
Bidkhori G, Moeini A, Masoudi-Nejad A. Modeling of tumor progression in NSCLC and intrinsic resistance to TKI in loss of PTEN expression. PLoS One. 2012;7(10):e48004. | ||
Sos ML, Zander T, Thomas RK, Staratschek-Jox A, Claasen J, Wolf J. Expression of signaling mediators downstream of EGF-receptor predict sensitivity to small molecule inhibitors directed against the EGF-receptor pathway. J Thorac Oncol. 2008;3(2):170–173. | ||
Cetin Z, Ozbilim G, Erdogan A, Luleci G, Karauzum SB. Evaluation of PTEN and Mcl-1 expressions in NSCLC expressing wild-type or mutated EGFR. Med Oncol. 2010;27(3):853–860. | ||
Mink SR, Vashistha S, Zhang W, Hodge A, Agus DB, Jain A. Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol Cancer Res. 2010;8(6):809–820. | ||
Choe C, Shin YS, Kim C, et al. Crosstalk with cancer-associated fibroblasts induces resistance of non-small cell lung cancer cells to epidermal growth factor receptor tyrosine kinase inhibition. Onco Targets Ther. 2015;8:3665–3678. | ||
Yoshida T, Ishii G, Goto K, et al. Podoplanin-positive cancer-associated fibroblasts in the tumor microenvironment induce primary resistance to EGFR-TKIs in lung adenocarcinoma with EGFR mutation. Clin Cancer Res. 2015;21(3):642–651. | ||
Kanda R, Kawahara A, Watari K, et al. Erlotinib resistance in lung cancer cells mediated by integrin beta1/Src/Akt-driven bypass signaling. Cancer Res. 2013;73(20):6243–6253. | ||
Yamauchi M, Yoshino I, Yamaguchi R, et al. N-cadherin expression is a potential survival mechanism of gefitinib-resistant lung cancer cells. Am J Cancer Res. 2011;1(7):823–833. | ||
Wang S, Su X, Bai H, et al. Identification of plasma microRNA profiles for primary resistance to EGFR-TKIs in advanced non-small cell lung cancer (NSCLC) patients with EGFR activating mutation. J Hematol Oncol. 2015;8(1):127. | ||
Benedettini E, Sholl LM, Peyton M, et al. Met activation in non-small cell lung cancer is associated with de novo resistance to EGFR inhibitors and the development of brain metastasis. Am J Pathol. 2010;177(1):415–423. | ||
Gusenbauer S, Vlaicu P, Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene. 2013;32(33):3846–3856. | ||
Yano S, Yamada T, Takeuchi S, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6(12):2011–2017. | ||
Yamada T, Matsumoto K, Wang W, et al. Hepatocyte growth factor reduces susceptibility to an irreversible epidermal growth factor receptor inhibitor in EGFR-T790M mutant lung cancer. Clin Cancer Res. 2010;16(1):174–183. | ||
Gong Y, Somwar R, Politi K, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4(10):e294. | ||
Li Z, Zhou S, Zhang L, et al. BIM induction of apoptosis triggered by EGFR-sensitive and resistance cell lines of non-small-cell lung cancer. Med Oncol. 2011;28(2):572–577. | ||
Ng KP, Hillmer AM, Chuah CT, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18(4):521–528. | ||
Zhao M, Zhang Y, Cai W, et al. The Bim deletion polymorphism clinical profile and its relation with tyrosine kinase inhibitor resistance in Chinese patients with non-small cell lung cancer. Cancer. 2014;120(15):2299–2307. | ||
Ma JY, Yan HJ, Gu W. Association between BIM deletion polymorphism and clinical outcome of EGFR-mutated NSCLC patient with EGFR-TKI therapy: a meta-analysis. J Cancer Res Ther. 2015;11(2):397–402. | ||
Wu DW, Chen CY, Chu CL, Lee H. Paxillin confers resistance to tyrosine kinase inhibitors in EGFR-mutant lung cancers via modulating BIM and Mcl-1 protein stability. Oncogene. 2016;35(5):621–630. | ||
Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumours with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–20937. | ||
Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88. | ||
Chen ZY, Zhong WZ, Zhang XC, et al. EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist. 2012;17(7):978–985. | ||
Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. | ||
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. | ||
Jänne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. | ||
Greig SL. Osimertinib: first global approval. Drugs. 2016;76(2):263–273. | ||
Mitsudomi T, Tsai Ch, Shepeherd F, et al. AZD9291 in pre-treated T790M positive advanced NSCLC: AURA2 Phase II study. J Thorac Oncol. 2015;10(9 Suppl 2):MINI16.08. | ||
Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404–1415. | ||
Sequist LV, Soria JC, Goldman JW, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372(18):1700–1709. | ||
Kim D, Lee D, Kang J, et al. Clinical activity and safety of HM61713, an EGFR-mutant selective inhibitor, in advanced nonsmall cell lung cancer (NSCLC) patients (pts) with EGFR mutations who had received EGFR tyrosine kinase inhibitors (TKIs). J Clin Oncol. 2014;32(5 Suppl):abstr 8011. | ||
Marukami H. Antitumour activity of ASP8273, an irreversible mutant selective EGFR-TKI, in NSCLC patients with tumours harbouring EGFR activating mutations and T790M resistance mutation. Eur J Cancer. 2014;50(6 Suppl):abstr LBA9. | ||
Giaccone G, Herbst RS, Manegold C, et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial – INTACT 1. J Clin Oncol. 2004;22(5):777–784. | ||
Herbst RS, Giaccone G, Schiller JH, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial – INTACT 2. J Clin Oncol. 2004;22(5):785–794. | ||
Herbst RS, Prager D, Hermann R, et al. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol. 2005;23(25):5892–5899. | ||
Li T, Ling YH, Goldman ID, Perez-Soler R. Schedule-dependent cytotoxic synergism of pemetrexed and erlotinib in human non-small cell lung cancer cells. Clin Cancer Res. 2007;13(11):3413–3422. | ||
Cheng H, An SJ, Zhang XC, et al. In vitro sequence-dependent synergism between paclitaxel and gefitinib in human lung cancer cell lines. Cancer Chemother Pharmacol. 2011;67(3):637–646. | ||
Mok TS, Wu YL, Yu CJ, et al. Randomized, placebo-controlled, phase II study of sequential erlotinib and chemotherapy as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2009;27(30):5080–5087. | ||
Soria JC, Wu YL, Nakagawa K, et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol. 2015;16(8):990–998. | ||
Kanda S, Horinouchi H, Fujiwara Y, et al. Cytotoxic chemotherapy may overcome the development of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) therapy. Lung Cancer. 2015;89(3):287–293. | ||
Regales L, Gong Y, Shen R, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119(10):3000–3010. | ||
Janjigian YY, Smit EF, Groen HJ, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4(9):1036–1045. | ||
Wu Y, Yang J, Kim D, et al. Safety and efficacy of INC280 in combination with gefitinib (gef) in patients with EGFR-mutated (mut), MET-positive NSCLC: a single-arm phase lb/ll study. J Clin Oncol. 2014;32(5 Suppl):abstr 8017. | ||
Scagliotti G, von Pawel J, Novello S, et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol. 2015;33(24):2667–2674. | ||
Reckamp K, Frankel P, Mack P, et al. Phase II trial of XL184 (cabozantinib) plus erlotinib in patients (pts) with advanced EGFR-mutant non-small cell lung cancer (NSCLC) with progressive disease (PD) on epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) therapy: a California Cancer Consortium phase II trial (NCI 9303). J Clin Oncol. 2014;32(5 Suppl):abstr 8014. | ||
Xu L, Kikuchi E, Xu C, et al. Combined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res. 2012;72(13):3302–3311. | ||
Sun Z, Li Q, Zhang S, et al. NVP-BEZ235 overcomes gefitinib-acquired resistance by down-regulating PI3K/AKT/mTOR phosphorylation. Onco Targets Ther. 2015;8:269–277. | ||
Lara PN Jr, Longmate J, Mack PC, et al. Phase II study of the AKT inhibitor MK-2206 plus erlotinib in patients with advanced non-small cell lung cancer who previously progressed on erlotinib. Clin Cancer Res. 2015;21(19):4321–4326. | ||
Zhou Q, Zhou CC, Chen GY, et al. A multicenter phase II study of sorafenib monotherapy in clinically selected patients with advanced lung adenocarcinoma after failure of EGFR-TKI therapy (Chinese Thoracic Oncology Group, CTONG 0805). Lung Cancer. 2014;83(3):369–373. | ||
Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–2550. | ||
Reck M, von Pawel J, Zatloukal P, et al. Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: results from a randomised phase III trial (AVAiL). Ann Oncol. 2010;21(9):1804–1809. | ||
Herbst RS, Ansari R, Bustin F, et al. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trial. Lancet. 2011;377(9780):1846–1854. | ||
Ichihara E, Hotta K, Nogami N, et al. Phase II trial of gefitinib in combination with bevacizumab as first-line therapy for advanced non-small cell lung cancer with activating EGFR gene mutations: the Okayama Lung Cancer Study Group Trial 1001. J Thorac Oncol. 2015;10(3):486–491. | ||
Seto T, Kato T, Nishio M, et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014;15(11):1236–1244. | ||
Stahel R, Dafni U, Gautschi O, et al. A phase II trial of erlotinib (E) and bevacizumab (B) in patients with advanced non-small-cell lung cancer (NSCLC) with activating epidermal growth factor receptor (EGFR) mutations with and without T790M mutation. The Spanish Lung Cancer Group (SLCG) and the European Thoracic Oncology Platform (ETOP) BELIEF trial. In: Abstracts of the European Cancer Congress; September 25–29, 2015; Vienna, Austria. Abstract 3BA. | ||
Otsuka K, Hata A, Takeshita J, et al. EGFR-TKI rechallenge with bevacizumab in EGFR-mutant non-small cell lung cancer. Cancer Chemother Pharmacol. 2015;76(4):835–841. | ||
Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. | ||
Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. | ||
Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. | ||
Azuma K, Ota K, Kawahara A, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol. 2014;25(10):1935–1940. | ||
Han JJ, Kim DW, Koh J, et al. Change in PD-L1 expression after acquiring resistance to gefitinib in EGFR-mutant non-small-cell lung cancer. Clin Lung Cancer. Epub. 2015 Dec 1. | ||
Rizvi N, Chow L, Borghaei H, et al. Safety and response with nivolumab (anti-PD-1; BMS-936558, ONO-4538) plus erlotinib in patients (pts) with epidermal growth factor receptor mutant (EGFR MT) advanced NSCLC. J Clin Oncol. 2014;32(5 Suppl):abstr 8022. | ||
Chen N, Fang W, Zhan J, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10(6):910–923. | ||
Lee SY, Hur GY, Jung KH, et al. PPAR-gamma agonist increase gefitinib’s antitumor activity through PTEN expression. Lung Cancer. 2006;51(3):297–301. | ||
Nakagawa T, Takeuchi S, Yamada T, et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res. 2013;73(8):2428–2434. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.