")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 4
Intravenous immunoglobulin for the treatment of Alzheimer's disease: current evidence and considerations
Authors Schindowski C, Zimmermann J, Schindowski K
Received 1 May 2014
Accepted for publication 28 May 2014
Published 5 September 2014 Volume 2014:4 Pages 121—130
DOI https://doi.org/10.2147/DNND.S51786
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Christina Schindowski,1,* Jürgen Zimmermann,2,* Katharina Schindowski3
1Vivantes Klinikum am Urban Hospital, Department of Psychiatry, Psychotherapy, and Psychosomatic Medicine, Berlin, Germany; 2Thermofisher Scientific, Langenselbold, Germany; 3Institute of Applied Biotechnology, Faculty for Biotechnology, Biberach University of Applied Sciences, Biberach/Riss, Germany
*These authors contributed equally to this work
Abstract: Alzheimer's disease (AD) is a devastating neurodegenerative form of dementia with increasing incidence rates in most countries. AD is characterized by amyloid plaques and neurofibrillary tangles in the brains of AD individuals accompanied by global neuronal loss. The peptide amyloid-β (Aβ) aggregates to amyloid plaques in AD brains. As a result, many therapeutic approaches target Aβ. Human plasma and the plasma product intravenous immunoglobulin (IVIG) contain naturally-occurring anti-Aβ antibodies (Nabs-Aβ) that appear to reduce risks of developing AD. IVIG sequesters Aβ and thus interferes with AD progression. This study reviews the role of different Aβ species, Nabs-Aβ, preclinical data, and clinical studies of IVIG as potential AD treatments. The focus of this study is the outcomes of a recent Gammaglobulin Alzheimer's Partnership Phase III trial that did not reach primary endpoints, as well as efforts to compare IVIG with current anti-Aβ monoclonals such as bapineuzumab, solanezumab, and BIIB037. Moreover, this study critically examines current market and ethical consequences of potential off-label uses of IVIG, limits in IVIG supply, and subsequent challenges.
Keywords: IVIG, amyloid-beta, Nabs-Aβ, Gammagard®, efficacy, target, market
Alzheimer’s disease dementia
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by cognitive decline accompanied by progressive loss of memory, orientation, and reasoning. AD is the most common form of dementia and accounts for 60%–70% of reported cases.1 Countries in demographic transition are predicted to experience a dramatic increase in AD in the next few decades. In 2010, the World Health Organization estimated the worldwide number of individuals with dementia to be 35.6 million; this is expected to double every 20 years.2 About 650,000 individuals are currently projected to develop AD in Germany. The USA has 5 million AD patients, and AD is now the sixth leading cause of death in that country. Within that patient population, 30% are considered to have mild, 40% moderate, and 30% severe AD.3 The estimated worldwide costs of dementia in 2010 were US$604 billion; on average, US$16,777 per patient, but with a projected US$33,000 cost per patient in higher income countries.4
The main histopathological hallmarks of AD are two types of misfolded proteins in the brain: intracellular hyper-phosphorylated tau in neurofibrillary tangles and extracellularly aggregated amyloid-β (Aβ) in senile plaques. Tau belongs to the microtubule-associated protein family predominantly found in neurons. In a low phosphorylated state, tau binds to microtubules and stabilizes their assembly. Properly assembled microtubules are essential for maintaining axonal transport processes. Hyper- and abnormal phosphorylation of tau result in impairment of axonal transport5,6 and neuronal death.7 The amyloid precursor protein (APP) is proteolytically processed to form Aβ, a peptide of predominantly 39–43 residues.8 The most common isoforms are Aβ40 and Aβ42, with Aβ42 being more hydrophobic and thereby more prone to aggregation.
Aβ42, total-tau, and phospho-tau in cerebrospinal fluid (CSF) serve as clinical biomarkers for AD and mild cognitive impairment (MCI),9 as evidenced by functional imaging such as MRI (magnetic resonance imaging) and PET (positron emission tomography).10 Biomarker strategies have had a significant impact on study outcomes but no biomarker has yet been approved as a successful predictor for AD.
Although AD was first described more than 100 years ago11 and is currently a subject of research in thousands of laboratories and renowned institutes worldwide, a clinically-proven disease-modifying therapy for AD remains elusive. Currently, the only method for combating AD is prevention of major risk factors such as vascular disease, type II diabetes, midlife hypertension, midlife obesity, smoking, and physical inactivity.4 A constant risk factor for AD is the Apolipoprotein E (ApoE) polymorphism.12 The major isoforms ApoE2, ApoE3, and ApoE4 carry different risks for developing AD: homozygous ApoE4 carriers pose the greatest risk for developing AD. ApoE3 is considered “normal” risk and ApoE2 is considered protective.
Aβ species: the dirty dozen?
Aβ exists not only as a monomer; there are more than a dozen different Aβ species. Despite much speculation, it remains unclear which Aβ species cause AD regardless of whether amyloid is responsible for AD at all. N-terminally truncated and pyro-Glu modified Aβ3-x (pE3-Aβ) account for >50% of Aβ accumulated in amyloid plaques.13,14 pE3-Aβ is more hydrophobic, which accelerates its neurotoxic aggregation.15
The self-association of Aβ peptides results in aggregates with varying morphologies. Monomeric Aβ peptides exist in rapid equilibrium with low molecular weight aggregates, further aggregating over different transient intermediates such as oligomers to mature insoluble Aβ-fibrils that finally accumulate as plaques.7,15 Many studies have described various types of higher molecular forms of synthetic Aβ but not much is known about which of these occur in vivo.16 Facing the meta-stability and ability for inter-conversion of different aggregation pathways, it is questionable whether to target a single “most-toxic” Aβ-species rather than depleting the whole spectrum of Aβ-aggregates.17
Active immunotherapy using aggregated Aβ1-42 was shown to be effective more than a decade ago in Aβ animal models by diminishing plaques and improving cognitive deficits.18 A subsequent clinical trial (AN-1792; NCT00021723) in which subjects with AD were vaccinated with Aβ1–42 was halted at Phase IIa after 6% of subjects developed meningoencephalitis.19 Of significance, active Aβ immunization in mice revealed similar data and led to encephalitis.20 A reduced cognitive decline in immunized patients who responded with a high titer and reduction in post-mortem amyloid load has been reported.21,22 However, amyloid load appears to not be correlated to cognition.23–25 Regardless, some studies have recommended continuation of anti-Aβ immunotherapy, but attention has been shifted to passive immunotherapy considered to be safer, controllable, and efficacious in AD animal models.26,27 Unfortunately, large Phase III studies of two anti-Aβ monoclonal antibodies (mAbs), bapineuzumab and solanezumab, recently failed to reach their primary endpoints.28,29
Natural anti-Aβ autoantibodies
Antibodies that react with self-molecules occur in healthy individuals and are named natural autoantibodies (Nabs). Nabs are mainly composed of immunoglobulin (Ig)M and display a moderate affinity for self-antigens and appear to be produced without any previous antigen exposure.30–32 Nabs probably act by eliminating circulating proteins before they can elicit a damaging response.33 However, the underlying mechanisms remain elusive.
Among these, Nabs against Aβ (Nabs-Aβ) have been found in the plasma of healthy individuals. Interestingly, Nabs-Aβ were found to be reduced in AD patients.34–36 Specifically, autoantibodies against Aβ42 were reduced but unfortunately without being a useful diagnostic marker for AD.37 Similarly to IgGs, levels of endogenous IgM autoantibodies against pE3-Aβ were significantly decreased in AD patients compared to healthy controls. A significant positive correlation between pE3-Aβ-IgM and cognitive decline has been reported by some MCI patients.38
In vitro, Nabs-Aβ were found to interfere with the oligomerization and fibrillization of Aβ, thereby blocking Aβ toxicity.39,40 Moreover, clearance of Aβ was successful but Nabs-Aβ did not clear senile plaques even though early fleecy-like plaques were reduced.41 Administration of Nabs-Aβ in transgenic mice has been shown to improve cognition.42 The N-terminal part of the fibrillar Aβ peptide in plaques was accessible at the plaque surface. Likewise, active immunization with aggregated Aβ42 generated predominantly antibodies that recognized the N-terminus of Aβ43, and so did most therapeutic anti-Aβ-mAbs. As revealed by epitope mapping, Nabs-Aβ detected mainly mid- and C-terminal epitopes of Aβ starting from amino acid 28. Nabs-Aβ appeared to recognize a common conformational epitope rather than a distinct peptide sequence, since most of them did not bind to native Aβ monomers.42 They seemed to preferentially capture apparent dimers and trimers and interfered with oligomerization.44
Intravenous immunoglobulin: an exciting and mysterious mixture
Intravenous immunoglobulin (IVIG) is a commercially available polyclonal Ig preparation offering important immunomodulatory and anti-inflammatory properties. Over the past 6 decades, polyvalent Igs have been approved and used as medications to treat rare immune-related and neurological conditions. Their effectiveness has been established in more than 80 diseases such as primary and secondary immunodeficiency and immune-mediated neurological diseases such as Guillan-Barré syndrome, chronic inflammatory demyelinating polyradiculoneuropathy, multifocal motor neuropathy, myasthenia gravis, autoimmune encephalitis, and stiff-person syndrome.
IVIG is commonly delivered intravenously, and approximately 16 different manufacturers have marketed IVIG products. IVIG is purified from human plasma pooled from more than 1,000 healthy donors by enzymatic, chemical, and chromatographic processes. A specific distribution of IgG subtypes and a functional Fc of native Igs are required for high-quality IVIG preparations. There is a specific safety concept that includes a selection of donors, gentle separation, and inactivation or removal of viral structures. That being said, IVIG is made from human plasma and carries a risk of transmitting infectious agents. Blood products have demonstrated an ability to induce severe hypersensitivity reactions, thrombotic events, hemolytic anemia, as well as renal dysfunction, acute renal failure, osmotic nephrosis, and death in predisposed patients. In addition, IVIG contains antibodies against blood group antigens that can act as hemolysins. Hyperproteinemia, increased serum viscosity, and hyponatremia, but also aseptic meningitis syndrome can occur in patients receiving IVIG.
Mechanisms of action of IVIG and how it produces its clinical effect in immune-mediated neurological disorders remains elusive. Direct actions on oligodendrocyte progenitors are presumed to be an effect of sustaining experimental remyelination.45,46 More relevant considerations include suppression of proinflammatory substrates and reduction of T-cell proliferation by IVIG.47 Furthermore, interference with the complement system has been thought to contribute to the cytopathic effects of IVIG.
IVIG for treatment of AD: preclinical evidence
Similarly to Nabs-Aβ that occur in healthy humans, Nabs-Aβ are also found in pooled preparations of IVIG derived from fractionated plasma of healthy donors.48 IVIG preparations contain IgGs that are cross-reactive against a conformational epitope on synthetic Aβ oligomers and fibrils as well as on amyloid in tissue sections.49 Binding in the nanomolar range has been reported from IgG1 subtypes Nabs-Aβ found in IVIG.
Attention has raised the observation that IVIG treatment due to other indications has been shown to reduce the risk of developing AD by >40%.50 As a result, IVIG treatment could be useful in the treatment of AD in humans as a type of passive anti-Aβ immunotherapy.51,52 Efflux of central Aβ into the periphery (peripheral sink) appears to be the main mechanism for Nabs-Aβ and IVIGs. However, a small amount of Nabs-Aβ from IVIG can cross the blood–brain barrier, as shown by using radiolabeled Nabs-Aβ.53 The few autoantibodies available in the brain have been suggested to be sufficient to induce uptake by opsonization in the brain (central sink).18,40,54,55 As a result, other mechanisms such as central degradation of Aβ oligomers in the presence of Nabs-Aβ by microglial cells could also have played an important role.
In an AD mouse model, treatment with administration of IVIG (10%, 400 mg/kg) every 2 weeks for 3 months revealed a small but significant 15% decrease in tau hyper-phosphorylation of hippocampal neurons.56 Plasma levels of several modulators of neuronal plasticity, including synaptophysin, homer1, nestin, and neurofilament H increased by 40%–50% after IVIG treatment. Similarly, the IVIG treated group showed a mild beneficial effect on memory; an ~20% reduction of the soluble Aβ42/Aβ40 ratio and a 60% decrease of Aβ oligomers.57 In an Aβ mouse model, IVIG improved cognitive function, possibly through upregulation of the AMPA-CREB signaling pathway.58 In addition, human fetal neuronal cultures treated with 10 mg/mL and 20 mg/mL IVIG showed improved resistance against reactive oxygen species compared to vehicle- and placebo-treated cells.59 IVIG and pooled mouse IgG both reduced Aβ deposition and neuroinflammation to the same extent as anti-Aβ mAb but with slower kinetics.60 Importantly, Aβ concentration and structure appeared to have an impact on the blood–brain barrier’s transport of IVIG.61 As a result, levels of Aβ species could possibly serve as predictive biomarkers for outcomes of clinical IVIG studies.
Interestingly, antibodies that bind to Aβ typically also bind to the parent APP. This suggests that certain Nabs-Aβ can interfere with APP conformation and processing. Nabs-Aβ isolated from AD patients, but not from healthy controls promoted β-secretase activity in cultured cells.62 Moreover, some data indicate an autocatalytic function of Nabs-Aβ in hydrolyzing Aβ.63 Catalytic activity improved as a function of age, and subjects with AD produced catalytic antibodies at increased levels (>IgMs than IgGs). However, in the latter study, IVIG did not cleave Aβ, possibly since most IVIG preparations contained low quantities of IgMs.
IVIG for treatment of AD: clinical evidence
A 2004 pilot study examining the use of IVIG in AD enrolled five AD patients who received 400 mg/kg monthly of IVIG (Octagam, Octapharma, Lachen, Switzerland) for 6 months.36 Serum Aβ increased significantly subsequent to IVIG treatment, by more than 230%, while Aβ CSF levels decreased significantly by 30% compared to baseline. Cognition improved on the AD assessment scale-cognitive scale (ADAS-Cog) at 3.7 points but mini-mental state (MMSE) scores were not significantly altered following treatment (improved in four patients, stable in one patient).
In 2007, an interesting study investigated the effects of a washout period. Eight mild AD patients received 400 mg/kg–2,000 mg/kg IVIG (Gammagard®; Baxter, Deerfield, IL, USA) for 6 months in an open-label dose-ranging study, then discontinued the treatment as a washout and finally resumed treatment for an additional 9 months.64 IVIG was used as an add-on to approved standard AD therapies. The amount of Nabs-Aβ in patient serum increased dose-dependently and plasma Aβ levels increased transiently after each infusion. Interestingly, the half-life of Nabs-Aβ was reduced compared to non-Aβ binding IgGs. Levels of Aβ in CSF decreased significantly after 6 months of treatment, returned to baseline during the washout, and decreased again when IVIG was re-administered. This suggests that IVIG induced a peripheral sink-like efflux of Aβ from the brain to the periphery. Cognition measured by the 30 point MMSE scale increased by 2.5 points after 6 months, returned to baseline during washout, and remained stable during subsequent IVIG treatment. However, due to small sample size, the study was not powered to provide significant results and to discriminate from the placebo effect.
A subsequent small Phase II study (Gammagard; NCT00299988) with 24 patients showed a dose-dependent effect on brain atrophy.65 Enlargement of the cerebral lateral ventricles is known to occur as a consequence of brain atrophy in AD and has been correlated to cognitive decline and AD neuropathology.66 Among 14 IVIG-treated patients who underwent volumetric MRI at baseline and after 18 months, the yearly increase in lateral ventricle volume was found to be lower in patients treated with the highest dose of IVIG. The reduction in brain atrophy was significantly correlated with improvement in clinical outcomes at 18 months on the Clinical Global Impression of Change and the ADAS-Cog. Notably, patient baseline characteristics were not correlated with volumetric MRI outcomes.67 Again, Nabs-Aβ were apparently detected in CSF subsequent to treatment.68
These preliminary clinical results and preclinical data justified larger clinical trials with statistical power to validate the effects of IVIG in AD as follows: a Phase II trial by Octapharma (NCT00812565; Octapharma, Lachen, Switzerland) using Octagam 10%; a Phase II trial sponsored by Sutter Health (Sacramento, CA, USA) (NCT01300728; NewGam 10%); a Phase II trial (NCT01561053) by Grifols Biologicals Inc. (Los Angeles, CA, USA) evaluating the combination of Flebogamma® DIF 5% with human albumin (Albutein 20%) after plasmapheresis; and a Phase III trial (NCT00818662; Gammagard Baxter Gammaglobulin Alzheimer’s Partnership [GAP] study) sponsored by Baxter and the National Institutes of Health AD Cooperative Study.42
The Octagam Phase II study using 55 probable AD patients was a multicenter, placebo-controlled, randomized trial that was completed in 2011. The study tested doses of 1,000 mg/kg and 800 mg/kg IVIG every 2–4 weeks but did not meet its primary endpoints in improving or stabilizing cognition.69 As opposed to previous studies, no changes in plasma and CSF Aβ were detected. The only apparent benefit of the treatment was significant improvement in cerebral glucose metabolism.
The NewGam (NCT01300728) and the Flebogama Albutein (NCT00818662) studies are ongoing and data are expected at the end of 2014 and 2016, respectively.42 The NewGam study enrolled MCI instead of AD subjects and will hopefully result in new insights on the efficacy of earlier treatment. Meanwhile, to evaluate long-term effects, an open-label extension of the above-mentioned Phase II study (NCT00299988) was performed, in which 24 participants received 400 mg/kg IVIG (Gammagard Baxter) every 2 weeks for additional 30 months.70 Sixteen of the originally enrolled subjects received treatment until month 36; of these, five were given placebo and eleven were treated with various doses of IVIG during the first 6 months. The extension study showed a 3 year stabilization in AD symptoms. In addition, no decline in measures of cognition, memory, daily functioning, and mood were observed. Four subjects treated from the beginning with 400 mg/kg IVIG every 2 weeks for 36 months had the best outcomes with no signs of cognitive decline. The eleven participants who received IVIG for 36 months had favorable outcomes in terms of thinking abilities, behavior, and daily function. The five participants who were initially treated with placebo and then switched to IVIG declined while on placebo but experienced a less rapid decline during IVIG treatment. Although these results are considered significant within the AD community, it should be kept in mind that this study was not statistically powered.
There have been increased insights into the potential benefits of IVIG for AD subgroups; these could contribute to study results. The Phase III GAP study has been the largest placebo-controlled AD study.71 It was designed and powered to assess the safety and effectiveness of IVIG as a potential treatment for AD. The prospective, randomized, double-blind, placebo-controlled, multicenter trial included 390 patients with mild to moderate AD. Patients were randomized to receive 400 mg/kg IVIG, 200 mg/kg IVIG, or 0.25% human albumin placebo every 2 weeks for 18 months. Subjects between 50–89 years were required to maintain their treatment regimen of approved medications for AD symptom management. Randomization was balanced for baseline-modified MMSE test, examination scores, and ApoE genotype. Co-primary endpoints were ADAS-Cog versus (vs) placebo or change in functional ability according to the AD Cooperative Study Activities of Daily Living ADCS-ADL scale vs placebo. All patients underwent serial plasma collection and MRI imaging, and a subset of patients underwent collection of CSF and amyloid imaging using PET tracers. Follow-up occurred at 3 month intervals.
No new safety warnings associated with treatment were identified. The most common adverse reactions observed in at least 5% of patients during IVIG treatment were rash and decreased hemoglobin. There were no differences in rate of thromboembolic events in the treatment groups vs placebo. There were 17 serious adverse reactions considered to be treatment related (12 in the IVIG cohorts and five in the placebo cohort). In contrast to anti-Aβ-mAb trials, no apparent increase in risks for amyloid-related imaging abnormalities was observed.
Although IVIG showed acceptable safety and tolerability profiles, the GAP study did not meet its co-primary endpoints: after 18 months, there was no significant difference in rates of cognitive decline and functional ability (see Table 1). Nevertheless, biomarker analyses suggest that IVIG entered the brain while total IgG in CSF increased dose-dependently from baseline. In addition, Aβ was apparently mobilized from the brain as a dose-dependent increase in antibodies specific against oligomeric and fibrillar Aβ detected in the CSF of the subgroup that underwent CSF collection.72 In parallel, a dose-dependent reduction was observed in plasma levels of Aβ1–42 but not in Aβ1–40 in IVIG-treated patients compared to placebo. Finally, a reduction in brain fibrillar amyloid measured by PET using florbetapir showed central activity of IVIG treatment, particularly in ApoE4 carriers. This supports the hypothesis that IVIG can decrease amyloid burden in AD. However, this sub-study did not have a sufficient number of subjects to establish statistical significance. Finally, CSF levels of tau and phospho-tau were not influenced by the treatment. These signals, as observed on multiple biomarker measures, suggest the passage of antibodies across the blood–brain barrier and targeting of Aβ. Unfortunately, no data are available to explain how Aβ levels were measured since the method and masking by antibodies may have affected the quantification of biomarkers.73
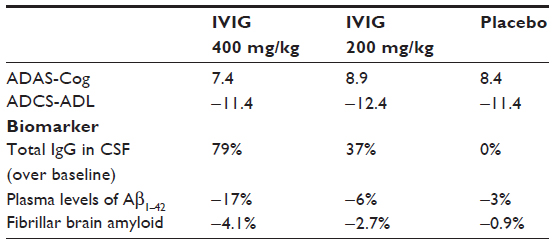 | Table 1 GAP study: co-primary endpoints and biomarker |
Since the third Phase III trial examining passive anti-Aβ immunotherapy after bapineuzumab and solanezumab failed, disappointment in the scientific community was significant. However, it should be kept in mind that until recently, it was not sufficiently shown that any preclinical effect of IVIG was due to anti-Aβ activity; hence, whether IVIG was comparable to anti-Aβ mAbs in any way. Similarly to the bapineuzumab and solanezumab trials, further analyses have been conducted with the hope of unraveling underlying mechanisms. Clearly, the GAP study was not designed to demonstrate statistical significance in these pre-planned subgroup analyses.72 Nevertheless, additional exploratory post hoc and pre-specified intention-to-treat subgroup analyses of the data showed some benefits. However, a type I error and other statistical effects could not be excluded and should be kept in mind when reviewing this study. With the knowledge acquired from the GAP study, an interpretation of the positive outcomes of the extension Gammagard Baxter Phase II study (NCT00299988) was possible. Interestingly, the 16 patients who benefited from IVIG fell into the subgroups “moderate disease” or “ApoE4 carrier”, which appeared to have a benefit in the GAP trial.74
After results of the GAP study were released, Baxter discontinued a second ongoing Phase III trial of IVIG in Europe and the US in patients with mild to moderate disease. Another Phase II trial of IVIG in patients with MCI using the same dose of IVIG but with a shorter treatment interval is ongoing.
There was an imbalance between the data available from Phase II studies compared to the Phase III study. That was mainly due to the fact that most Phase II studies were conducted by academic groups and were published. Data from the GAP study were published only in parts as press releases.
Off-label use of IVIG for AD: market and ethical considerations
Some AD patients are currently treated with IVIG as off-label use since no disease-modifying treatments are available for AD. This has been encouraged by the promising results of some Phase II studies and the fact that IVIG is well-known and already approved for other neurological conditions. So what does off-label use actually mean?
The European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) review manufacturer applications and data to grant approval of prescription drugs. When results do not support the use of a drug for unapproved indications or populations, the use can be marked as off-label. Physicians can prescribe such products but drug manufacturers may not promote their use. According to FDA guidelines, physicians can use a product that is not in the approved labeling; however, they have a responsibility to be well-informed about the product so as to base its use on solid scientific rationale and maintain records about the drug’s use and effects. Off-label use is common in older generic medications, particularly in oncology and pediatrics, where up to 50% of drug prescriptions are off-label. Up to 25% of all drugs are prescribed off-label and among psychiatric drugs, off-label use rises to 31%.75 Beyond that, there are numerous studies that support off-label use of non-approved drugs. Since the FDA provides a rigorous regulatory system for the protection of previous medical product consumers, companies are allowed to distribute peer-reviewed scientific articles and documents describing off-label use.76,77 There are various regulations for off-label use in different countries; in general, however, no laws prohibit the prescription of an approved drug for other reasons than the indications issued and approved. A medical consent discussion about the potential risks is compulsory.
Nevertheless, there remain legal disputes about coverage regarding costs of off-label drugs by different health care systems, such as the German Health Insurance. In Germany for example criteria were defined covering the use of off-label drugs. These criteria included both treatment of severe illness with no approved therapy available as well as a reasonable expectation of an assumed successful medical treatment based on current data. Beyond these facts, a growing off-label use of IVIG for AD could lead to significant costs to health care systems and engender an increasing demand for IVIG in additional payments. IVIG treatment in geriatric patients can quite frequently only be administered at reduced doses due to limited clearance as a result of renal insufficiency.78 It is not clear whether these patients can receive a sufficient dose of IVIG to be effective. Considering side effects and economic aspects, more multicenter studies with larger patient samples are necessary before such an expensive and burdensome treatment strategy can be widely recommended. Moreover, IVIG is in limited supply79 but is an important and lifesaving drug. The ethical considerations of off-label use for AD are therefore the other side of the story.
IVIG: a limited product
Considering off-label use and potential approval of IVIG for AD, how realistic is the scenario of using IVIG for a disease with a high incidence rate such as AD? The use of plasma products differs widely in different countries and has increased in the past years around the world. The demand for IVIG increased 14× from 1984 to 2013 from 7.41 to 103.4 metric tons.3 An increased need of 6% per year to 146.7 tons in 2018 was estimated in the case of non-approval of IVIG for AD, and 9%–12% per year to 186.5 metric tons in the case of approval. These numbers reflect the limit of global IVIG production. The demographic transition favors the limit in supply as ”healthy” donors grow older. The forecast of >40 metric tons was based on calculations assuming AD patients with a mean body weight of 70 kg receiving 400 mg to 500 mg IVIG/kg every 2 weeks, resulting in a total of 728 g to 840 g IVIG/year per AD patient. This implies that only 46,619 to 54,711 AD patients can be treated with 40 metric tons. Examining the actual number of AD patients and considering the benefits to the subgroups “moderate AD” and “ApoE4” only, it is clear that this would not cover the needs.
Is there a possibility of overcoming the supply problem? One option is to optimize the production of IVIG. Blood from healthy donors contains 7 g to 16 g IgG per liter. At present, 3.5 to 5.0 g of IVIG are yielded per liter of plasma by fractionation. Modified purification processes for IVIG have been examined for increasing yield and thus quantity;79 however, this would also significantly increase manufacturing costs.
Another option could be new sources of donors. In 2010, 32.2 million liters of plasma was collected globally for fractionation; >18 million were from the USA and ~3 million were from Germany. Currently, plasma fractionation is performed mainly in Europe, North America, and Australia, while only a handful of companies produce IVIG from donors in Asia and Africa. More donors in addition to high standards of safety and molecular diagnostics could increase the amount of available IVIG.
Is the present IVIG composition optimal as a potential future AD medication?
The concentration of Nabs-Aβ has been reported to be ~0.2% of all IgGs present in IVIG.52 Assuming that Nabs-Aβ are the active pharmaceutical ingredient in IVIG for AD, this would imply that 99.8% of IVIG is inactive and therefore useless. Moreover, different IVIG preparations have shown altered composition of antibodies and altered binding to Aβ42 monomers and soluble oligomers.80,81 This could imply that some preparations can reach clinical efficacy while others cannot. Clinical data and translational research are required to confirm the therapeutic action of anti-Aβ antibodies, regardless of whether they are endogenous/naturally occurring or exogenous such as bapineuzumab, solanezumab, and others.
Considering the appropriateness of anti-Aβ immunotherapies and that Aβ species and Aβ multimers provide many conformational epitopes, clinical data on IVIG potentially provides hints that a polyclonal anti-Aβ approach could be more effective than a monoclonal approach. It is evident to tackle as many forms of Aβ as possible.28 If this is true, an optimized polyclonal approach to AD would be the use of Nabs-Aβ-enriched IVIG or a combination of different anti-Aβ mAbs. While enrichment of Nabs-Aβ from body fluids has been recently patented, there are currently no other commercial sources other than IVIG.82
As such, the clinical outcomes of BIIB037-a mAb derived from an AD patient with an unusual stable clinical course are interesting since a single Nab-Aβ proceeded to clinical development.29 IVIG preparation from successfully-aged donors without any signs of AD could provide an optimal cocktail of highly-active Nabs-Aβ more suitable than those from young donors. However, the mechanisms underlying the effects of IVIG on cognitive outcomes remain unknown. IVIG could possibly act via an Aβ-independent mechanisms.83 AD has a strong neuroinflammatory component that is unquestionably not a cause of the disease but has significant impacts on progression. IVIG has been shown to improve efficacy, mainly by anti-inflammatory activity in neurological disorders apart from AD. For instance, sialylated IgG Fc fragments in IVIG increased secretion of soluble anti-inflammatory mediators but also had other anti-inflammatory effects.84,85 The anti-inflammatory activity of IVIG is probably important in AD. Current research does not understand the function of Nabs; however, it is known that IgM has mainly autoreactive functions. Moreover, in the central nervous system (CNS), antibodies bind surface antigens onto specific CNS cells, thereby activating intracellular repair-promoting signals.86 Most IVIG products do not contain >0.1% IgM. Considering that the mechanisms of action could be related to autoreactive IgMs regardless of the presence or absence of anti-Aβ IgM, this would mean that >99.9% of IVIG ingredients are inactive. In that case, an IgM-enriched preparation could provide better efficacy in the treatment of AD than conventional IVIG preparations.
Conclusion
The outcomes of the GAP study and other Phase III AD trials highlight the importance of adapting clinical AD trials to the latest EMA and FDA recommendations. Many recent studies have been re-examined since they did not result in positive outcomes in clinical endpoints. As a result, clinical trials involving AD patients should be powered for subsequent sub-group analyses since AD patients seem to belong to a heterogeneous pool. AD research is beyond the point of trusting animal and cellular models since they do not often translate data to clinical applications. More retro-prospective and statistically-powered studies are required to better understand the mechanisms of degeneration in humans and how to interact with them. IVIG studies, regardless of whether they were successful or not, can provide important insights into the interactions between Aβ and/or tau and Nabs. It is possible that future studies will recognize that it was useless and too late to perform clinical trials with AD patients, and that only patients with MCI can benefit from any treatment.
Nevertheless, studies involving in-depth analyses could provide a significant step forward in understanding neurodegeneration. The costs of these studies are significant but not as significant as the costs of not treating AD. Additionally, approval of IVIG for treating AD is not an option for global therapy since this product is rather limited. It is important to identify the active pharmaceutical ingredients of IVIG and optimize its production.
Acknowledgment
The author thanks Chrystelle Mavoungou and Jens Moreth for helpful discussions and Iwona Walicka for proofreading.
Disclosure
Jürgen Zimmermann is an employee at Thermofisher Scientific Germany, Langenselbold, Germany. The authors report no other conflicts of interest in this work.
References
Alzheimer’s Disease International. World Alzheimer Report 2009. Prince M, Jackson J, editors. 2009. Available at: http://www.alz.co.uk/research/world-report. Accessed July 5th 2012. | |
Alzheimer’s Disease International. World Alzheimer Report 2010. Wimo A, Prince M, editors. 2010. Available at: http://www.alz.co.uk/research/world-report. Accessed July 5th 2012. | |
Robert P. Global plasma demand in 2015. Pharm Policy Law. 2009; 11(4):359–367. | |
World Health Organization and Alzheimer’s Disease International. Dementia: a public health priority; 2012. Available at: http://www.who.int/mental_health/publications/dementia_report_2012/en/index.html. Accessed July 5th 2012. | |
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. | |
Verdile G, Fuller S, Atwood CS, Laws SM, Gandy SE, Martins RN. The role of beta amyloid in Alzheimer’s disease: still a cause of everything or the only one who got caught? Pharmacol Res. 2004;50(4):397–409. | |
Bitan G, Vollers SS, Teplow DB. Elucidation of primary structure elements controlling early amyloid beta-protein oligomerization. J Biol Chem. 2003;278(37):34882–34889. | |
Katzman R. Alzheimer’s disease. N Engl J Med. 1986;314(15):964–973. | |
Spitzer P, Klafki HW, Blennow K, et al. cNEUPRO: Novel Biomarkers for Neurodegenerative Diseases. Int J Alzheimers Dis. 2010;2010.pii 548145. | |
Jack CR Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(5):1355–1365. | |
Alzheimer A. About a peculiar disease of the cerebral cortex. By Alois Alzheimer, 1907 (Translated by L. Jarvik and H. Greenson). Alzheimer Dis Assoc Disord. 1987;1(1):3–8. | |
Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342(8873):697–699. | |
Frost JL, Liu B, Kleinschmidt M, Schilling S, Demuth HU, Lemere CA. Passive immunization against pyroglutamate-3 amyloid-beta reduces plaque burden in Alzheimer-like transgenic mice: a pilot study. Neurodegener Dis. 2012;10(1–4):265–270. | |
Jawhar S, Wirths O, Bayer TA. Pyroglutamate amyloid-beta (Abeta): a hatchet man in Alzheimer disease. J Biol Chem. 2011;286(45):38825–38832. | |
Kuo YM, Emmerling MR, Vigo-Pelfrey C, et al. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271(8):4077–4081. | |
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. | |
Moreth J, Kroker KS, Schwanzar D, et al. Globular and protofibrillar Aβ aggregates impair neurotransmission by different mechanisms. Biochemistry. 2013;52(8):1466–1476. | |
Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. | |
Orgogozo J-M, Gilman S, Dartigues J-F, et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurol. 2003;61(1):46–54. | |
Furlan R, Brambilla E, Sanvito F, et al. Vaccination with amyloid-beta peptide induces autoimmune encephalomyelitis in C57/BL6 mice. Brain. 2003;126(Pt 2):285–291. | |
Vellas B, Black R, Thal LJ, et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009;6(2):144–151. | |
Boche D, Denham N, Holmes C, Nicoll JR. Neuropathology after active Aβ42 immunotherapy: implications for Alzheimer’s disease pathogenesis. Acta Neuropathol. 2010;120(3):369–384. | |
Förster S, Yousefi BH, Wester HJ, et al. Quantitative longitudinal interrelationships between brain metabolism and amyloid deposition during a 2-year follow-up in patients with early Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2012;39(12):1927–1936. | |
Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367. | |
Engler H, Forsberg A, Almkvist O, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006; 129(Pt 11):2856–2866. | |
Chauhan NB, Siegel GJ. Intracerebroventricular passive immunization with anti-Abeta antibody in Tg2576. J Neurosci Res. 2003;74(1):142–147. | |
Dodart JC, Bales KR, Gannon KS, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5(5):452–457. | |
Moreth J, Mavoungou C, Schindowski K. Passive anti-amyloid immunotherapy in Alzheimer’s disease: What are the most promising targets? Immun Ageing. 2013;10(1):18. | |
Mavoungou C, Schindowski K. Immunotherapy with anti-amyloid-beta antibodies in Alzheimer’s disease: A critical review on the molecules in the pipelines with regulatory considerations. In: Rahman A, editor. Front Clin Drug Res – Alzheimer Disord. 2013. | |
Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7(6):812–818. | |
Elkon K, Casali P. Nature and functions of autoantibodies. Nat Clin Pract Rheumatol. 2008;4(9):491–498. | |
Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. 2010;10(11):778–786. | |
Maetzler W, Berg D, Synofzik M, et al. Autoantibodies against amyloid and glial-derived antigens are increased in serum and cerebrospinal fluid of lewy body-associated dementias. J Alzheimers Dis. 2011;26(1):171–179. | |
Du Y, Dodel R, Hampel H, et al. Reduced levels of amyloid beta-peptide antibody in Alzheimer disease. Neurology. 2001;57(5):801–805. | |
Weksler ME, Relkin N, Turkenich R, LaRusse S, Zhou L, Szabo P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp Gerontol. 2002;37(7):943–948. | |
Dodel RC, Du Y, Depboylu C, et al. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75(10):1472–1474. | |
Brettschneider S, Morgenthaler NG, Teipel SJ, et al. Decreased serum amyloid β1-42 autoantibody levels in Alzheimer’s disease, determined by a newly developed immuno-precipitation assay with radiolabeled amyloid β1-42 peptide. Biol Psychiatry. 2005;57(7):813–816. | |
Marcello A, Wirths O, Schneider-Axmann T, Degerman-Gunnarsson M, Lannfelt L, Bayer TA. Reduced levels of IgM autoantibodies against N-truncated pyroglutamate Abeta in plasma of patients with Alzheimer’s disease. Neurobiol Aging. 2011;32(8):1379–1387. | |
Adekar SP, Klyubin I, Macy S, et al. Inherent anti-amyloidogenic activity of human immunoglobulin γ heavy chains. J Biol Chem. 2010;285(2):1066–1074. | |
Magga J, Puli L, Pihlaja R, et al. Human intravenous immunoglobulin provides protection against Aβ toxicity by multiple mechanisms in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2010;7:90. | |
Weksler ME. The immunotherapy of Alzheimer’s disease. Immun Ageing. 2004;1(1):2. | |
Dodel R, Balakrishnan K, Keyvani K, et al. Naturally occurring autoantibodies against beta-amyloid: investigating their role in transgenic animal and in vitro models of Alzheimer’s disease. J Neurosci. 2011;31(15):5847–5854. | |
McLaurin J, Cecal R, Kierstead ME, et al. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med. 2002;8(11):1263–1269. | |
Britschgi M, Olin CE, Johns HT, et al. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106(29):12145–12150. | |
Asakura K, Miller DJ, Murray K, Bansal R, Pfeiffer SE, Rodriguez M. Monoclonal autoantibody SCH94.03, which promotes central nervous system remyelination, recognizes an antigen on the surface of oligodendrocytes. J Neurosci Res. 1996;43(3):273–281. | |
Ciric B, Van Keulen V, Paz Soldan M, Rodriguez M, Pease LR. Antibody-mediated remyelination operates through mechanism independent of immunomodulation. J Neuroimmunol. 2004;146(1–2):153–161. | |
Aktas O, Waiczies S, Grieger U, Wendling U, Zschenderlein R, Zipp F. Polyspecific immunoglobulins (IVIg) suppress proliferation of human (auto)antigen-specific T cells without inducing apoptosis. J Neuroimmunol. 2001;114(1–2):160–167. | |
Dodel R, Hampel H, Depboylu C, et al. Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer’s disease. Ann Neurol. 2002;52(2):253–256. | |
Klaver AC, Patrias LM, Coffey MP, Finke JM, Loeffler DA. Measurement of anti-Aβ1-42 antibodies in intravenous immunoglobulin with indirect ELISA: The problem of nonspecific binding. J Neurosci Methods. 2010;187(2):263–269. | |
Fillit H, Hess G, Hill J, Bonnet P, Toso C. IV immunoglobulin is associated with a reduced risk of Alzheimer disease and related disorders. Neurology. 2009;73(3):180–185. | |
Fu HJ, Liu B, Frost JL, Lemere CA. Amyloid-beta immunotherapy for Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):197–206. | |
O’Nuallain B, Hrncic R, Wall JS, Weiss DT, Solomon A. Diagnostic and therapeutic potential of amyloid-reactive IgG antibodies contained in human sera. J Immunol. 2006;176(11):7071–7078. | |
Bacher M, Depboylu C, Du Y, et al. Peripheral and central biodistribution of (111)In-labeled anti-beta-amyloid autoantibodies in a transgenic mouse model of Alzheimer’s disease. Neurosci Lett. 2009;449(3):240–245. | |
Hock C, Konietzko U, Papassotiropoulos A, et al. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8(11):1270–1275. | |
Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. | |
Counts S, Perez S, Mufson E. IVIG reduces tau pathology and increases neuroplastic gene expression in the 3xTg mouse model of Alzheimer’s disease. Alzheimer’s Dement J Alzheimer’s Assoc. 2013;9(4):P356. | |
St-Amour I, Paré I, Tremblay C, Coulombe K, Bazin R, Calon F. IVIg protects the 3xTg-AD mouse model of Alzheimer’s disease from memory deficit and Abeta pathology. J Neuroinflammation. 2014;11(1):54. | |
Gong B, Pan Y, Zhao W, et al. IVIG immunotherapy protects against synaptic dysfunction in Alzheimer’s disease through complement anaphylatoxin C5a-mediated AMPA-CREB-C/EBP signaling pathway. Mol Immunol. 2013;56(4):619–629. | |
Lahiri D, Ray B. Effect of IVIg in preserving human primary neurons and protecting them against oxidative stress. Alzheimer’s Dement J Alzheimer’s Assoc. 2013;9(4):P800. | |
Sudduth TL, Greenstein A, Wilcock DM. Intracranial injection of Gammagard, a human IVIg, modulates the inflammatory response of the brain and lowers Aβ in APP/PS1 mice along a different time course than anti-Aβ antibodies. J Neurosci. 2013;33(23):9684–9692. | |
Wuest DM, Lee KH. Amyloid-β concentration and structure influences the transport and immunomodulatory effects of IVIG. J Neurochem. 2014;130(1):136–144. | |
Deng J, Hou H, Giunta B, et al. Autoreactive-Aβ antibodies promote APP beta-secretase processing. J Neurochem. 2012;120(5):732–740. | |
Paul S, Planque S, Nishiyama Y. Immunological origin and functional properties of catalytic autoantibodies to amyloid beta peptide. J Clin Immunol. 2010;30 Suppl 1:S43–S49. | |
Relkin NR, Szabo P, Adamiak B, et al. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30(11):1728–1736. | |
Evans J. IVIG reduced brain atrophy in Alzheimer’s. Clin Psychiatry News. 2010;38(6):12. | |
Evans J. IVIG reduced brain atrophy, improved cognition in AD. Fam Pract News. 2010;40(8):47. | |
Relkin N. Use of ventricular enlargement raqte in intavenous immunoglobin treatment of Alzeimer’s disease. 2011; available at http://www.faqs.org/patents/app/20110251479. Accessed July 20th 2014. | |
Wurster U, Haas J. Passage of intravenous immunoglobulin and interaction with the CNS. J Neurol Neurosurg Psychiatry. 1994;57(Suppl):21–25. | |
Dodel R, Rominger A, Bartenstein P, et al. Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol. 2013;12(3):233–243. | |
Alzheimer’s Association International Conference. Alzheimer’s Treatment Study Reports Three Years with No Decline in Memory and Function at AAIC 2012. Available at: http://www.prnewswire.com/news-releases/alzheimers-treatment-study-reports-three-years-with-no-decline-in-memory-and-function-at-aaic-2012-162727566.html. Accessed on July 20th 2012. | |
Baxter International Inc. Baxter Presents Additional Data from Phase III Study of Immunoglobulin for Alzheimer’s Disease at AAIC. [Press Release]. Deerfield [Jul 2013]. Available from: http://www.baxter.com/press_room/press_releases/2013/07_16_13_aaic_gap_data.html. Accessed September 1, 2013. | |
Baxter Internatioal Inc. Baxter announces top line results of phase iii study of immunoglobulin for Alzheimer’s disease. [Press Release]. Deerfield [May 2013]. Available at: http://www.baxter.com/downloads/press_room/press_releases/2013/05_07_12_gap_study.pdf. Accessed September 1, 2013. | |
Moreth J, Mavoungou C, Schindowski K. Is abeta a sufficient biomarker for monitoring anti-abeta clinical studies? A critical review. Front Aging Neurosci. 2013;5:25. | |
Medscape.com. GAP: IVIG negative in Alzheimer’s, but some hints of benefit. Conf News. 2013. Available at: http://www.medscape.com/viewarticle/807970. Accessed September 1, 2013. | |
Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021–1026. | |
Kesselheim AS, Mello MM, Avorn J. FDA regulation of off-label drug promotion under attack. JAMA. 2013;309(5):445–446. | |
O’Reilly J, Dalal A. Off-label or out of bounds? Prescriber and marketer liability for unapproved uses of FDA-approved drugs. Ann Health Law. 2003;12(2):295–324. | |
Siegel J. IVIG medication safety: a stepwise guide to product selection and use. Pharm Pr News. 2010:1–8. | |
Hofbauer L, Bruckschwaiger L, Butterweck HA, Teschner W. Affinity chromatography for purification of IgG from human plasma. In: Magdeldin S, editor. Affinity Chromatography; 2012. | |
Djoumerska I, Tchorbanov A, Pashov A, Vassilev T. The autoreactivity of therapeutic intravenous immunoglobulin (IVIG) preparations depends on the fractionation methods used. Scand J Immunol. 2005;61(4):357–363. | |
Klaver AC, Finke JM, Digambaranath J, Balasubramaniam M, Loeffler DA. Antibody concentrations to Aβ1-42 monomer and soluble oligomers in untreated and antibody-antigen-dissociated intravenous immunoglobulin preparations. Int Immunopharmacol. 2010;10(1):115–119. | |
Du Y, Dodel R. Human beta-amyloid antibody and use thereof for treatment of alzheimer’s disease. 2001; available at http://www.google.it/patents/EP1172378A1?cl=en. Accessed March 22nd 2014. | |
Loeffler DA. Intravenous immunoglobulin and Alzheimer’s disease: what now? J Neuroinflammation. 2013;10(1):70. | |
Anthony RM, Wermeling F, Karlsson MCI, Ravetch J V. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A. 2008;105(50):19571–19578. | |
Bruley-Rosset M, Mouthon L, Chanseaud Y, Dhainaut F, Lirochon J, Bourel D. Polyreactive autoantibodies purified from human intravenous immunoglobulins prevent the development of experimental autoimmune diseases. Lab Invest. 2003;83(7):1013–1023. | |
Wright BR, Warrington AE, Edberg DD, Rodriguez M. Cellular mechanisms of central nervous system repair by natural autoreactive monoclonal antibodies. Arch Neurol. 2009;66(12):1456–1459. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.