")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Intratracheal transplantation of endothelial progenitor cells attenuates smoking-induced COPD in mice
Authors Shi Z, Chen Y, Cao J, Zeng H, Yang Y, Chen P, Luo H, Peng H, Cai S, Guan C
Received 18 April 2016
Accepted for publication 2 July 2016
Published 20 March 2017 Volume 2017:12 Pages 947—960
DOI https://doi.org/10.2147/COPD.S110781
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Zhihui Shi,1 Yan Chen,1 Jun Cao,2 Huihui Zeng,1 Yue Yang,1 Ping Chen,1 Hong Luo,1 Hong Peng,1 Shan Cai,1 Chaxiang Guan3
1Department of Internal Medicine, Division of Respiratory Disease, The Second Xiangya Hospital, Central-South University, 2Department of Internal Medicine, Division of Respiratory Disease, The People’s Hospital of Hunan Province, 3Department of Physiology, Xiangya Medical School, Central-South University, Changsha, Hunan, People’s Republic of China
Background: Endothelial progenitor cells (EPCs) might play a protective role in COPD. The aim of this study was to investigate whether intratracheal allogeneic transplantation of bone-marrow-derived EPCs would attenuate the development of smoking-induced COPD in mice.
Methods: Isolated mononuclear cells from the bone marrow of C57BL/6J mice were cultured in endothelial cell growth medium-2 for 10 days, yielding EPCs. A murine model of COPD was established by passive 90-day exposure of cigarette smoke. On day 30, EPCs or phosphate-buffered saline alone was administered into the trachea. On day 90, EPCs or 30 µL phosphate-buffered saline alone was administered into the trachea, and on day 120, inflammatory cells, antioxidant activity, apoptosis, matrix metalloproteinase (MMP)-2, and MMP-9 were measured.
Results: After EPC treatment, the lung function of the mice had improved compared with the untreated mice. Mean linear intercept and destructive index were reduced in the EPCs-treated group compared with the untreated group. In addition, the EPCs-treated mice exhibited less antioxidant activity in bronchoalveolar lavage fluid compared with the untreated mice. Moreover, decreased activities of MMP-2, MMP-9, and TUNEL-positive cells in lung tissues were detected in EPCs-treated mice.
Conclusion: Intratracheal transplantation of EPCs attenuated the development of pulmonary emphysema and lung function disorder probably by alleviating inflammatory infiltration, decelerating apoptosis, inhibiting proteolytic enzyme activity, and improving antioxidant activity.
Keywords: COPD, cigarette smoke, endothelial progenitor cells, transplantation, lung function, matrix metalloproteinase
Introduction
COPD is a major and increasing global health problem and currently the fifth leading cause of death worldwide, as the prevalence and burden of COPD are expected to increase in the coming decades.1,2 The pathology of COPD includes chronic obstructive bronchiolitis with fibrosis and obstruction and closure of small airways, as well as emphysema with enlargement of airspaces and destruction of lung parenchyma leading to loss of lung elasticity. In addition to the well-known mechanisms, including airway inflammation, extracellular matrix destruction, and oxidative stress,3 our earlier studies demonstrated that endothelial cell apoptosis and endothelial dysfunction could contribute to the pathogenesis of COPD.4–8
Endothelial progenitor cells (EPCs) can be isolated from bone marrow or peripheral blood and are capable of transforming into mature, functional endothelial cells.9 EPCs have been found to participate in human neovascularization and maintenance of vascular integrity.10–12 Recently, some reports have focused on defining the role of EPCs in restoring damaged vascular endothelium and carrying out human clinical trials to observe the effect of EPCs in the repair of vascular injury and/or ischemic tissues.13,14 In addition, it was demonstrated that EPCs were decreased in patients with COPD and correlated with hypoxemia, severity of airway obstruction, and peak oxygen consumption.15,16 Some studies also suggest a possible link between systemic inflammation and a decrease in circulating progenitors.15–17 Taken together, we hypothesized that EPCs play an important role in the pathogenesis of COPD, and the possible protective role of increasing EPCs in a smoking-induced COPD model can provide more insight into the treatment of COPD. The aim of this study was to investigate whether intratracheal administration of EPCs can attenuate the development of smoking-induced COPD in mice by inhibiting proteolytic enzyme activities and other molecular pathogenesis pathways.
Methods
Isolation, cultivation, and characterization of EPCs
EPCs were isolated from the bone marrow of C57BL/6J mice and then cultured according to established methods with minor modifications.18,19 Briefly, bone marrow mononuclear cells were isolated from the femurs and tibias of mice by density gradient centrifugation using lymphocyte separation medium (LTS1092; BD biosciences) according to the manufacturer’s protocol. After two washing steps, mononuclear cells were plated onto a culture bottle (25 mL) at a density of 3×106 cells/mL and maintained in endothelial basal medium-2 (Lonza) supplemented with endothelial cell growth medium-2 (EGM-2MV) single aliquots (Lonza) containing vascular endothelial growth factor (VEGF), fibroblast growth factor-2, epidermal growth factor, insulin-like growth factor, ascorbic acid, hydrocortisone, gentamicin, and fetal bovine serum. Cells were cultured at 37°C with 5% CO2 in a humidified atmosphere. After 4 days of culture, non-adherent cells were removed by washing with phosphate-buffered saline (PBS) and fresh medium was added. EPCs after 7 days of culture were used for further analysis.
Characterization of EPCs
The EPCs were characterized as described earlier.20 In a separate experiment, isolated mice mononuclear cells were cultured on four-well glass slides coated with human fibronectin. On day 10, the attached EPCs were incubated subsequently with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate-labeled acetylated low-density lipoprotein (DiI-acLDL) (7.5 μg/mL; Molecular Probes, Carlsbad, CA, USA) and fluorescein isothiocyanate-labeled Ulex europaeus agglutinin I (FITC-UEA-I) (10 μg/mL; Sigma-Aldrich Co., St Louis, MO, USA). The staining of DiI-acLDL and FITC-UEA-I in cultured EPCs was detected under fluorescence confocal microscopy at absorption wavelengths of 549 nm and 492 nm, respectively.
Experimental protocols
Six-week-old male C57BL/6J mice were purchased from the Animal Centre of the Second Xiangya Hospital, each weighing 18–20 g. In the experiments, the mice were randomly assigned into five groups (n=8 in each group): control group, early and late sham-interfered groups, and early and late EPCs-interfered groups. The last four groups were exposed to cigarette smoking daily for 90 days according to the method as described earlier.7 Mice in the early interfered group were instilled vehicle (PBS) or EPCs after 30 days of smoking exposure, while mice in the late interfered group were instilled vehicle or EPCs after 90 days of smoking exposure. Immediately before transplantation, a fluorescent cell tracker, CM-DiI (Molecular Probes), was used to label EPCs to track their homing in the pulmonary vasculature.20 In addition, on day 120 of this approach, all the mice were sacrificed. The animal experiments were performed at the Animal Centre of the Second Xiangya Hospital. All experimental protocols were approved by the Animal Care and Use Committee of the Second Xiangya Hospital, Central South University, and were conducted in accordance with the committee’s animal care and use guidelines.
Pulmonary function
Lung function was measured in mice in all groups, according to our established methods.7 Upon anesthesia with intraperitoneal administration of chloral hydrate (3 mL/kg body weight [BW]), a Y-type endotracheal cannula was connected to a flow transducer (HX200 type; Beijing Xinghangxingye Corporation, Beijing, People’s Republic of China) for the measurement of airway resistance (Raw) and dynamic compliance (Cdyn).
Sampling of blood, bronchoalveolar lavage fluid, and lung tissues
On day 120 of the experiment, the mice were anesthetized with pentobarbital (50 mg/kg) intraperitoneally. Blood samples were collected from the inferior vena cava via the opened chest, collected into a polypropylene tube containing clot activator, and centrifuged at 3,000× g for 15 minutes. The serum was separated and stored at −70°C until the samples were assayed.4,5
The left main bronchus was cross-clamped, and bronchoalveolar lavage (BAL) was performed as suggested earlier in the right lung.7 Aliquots (0.5 mL) of PBS were slowly infused in the lungs through tracheostomy at a constant pressure of 25 cm H2O, and the aliquots were then withdrawn gently. The lavage was repeated (3×) using the same syringe. The collected lavage fluid was centrifuged at 2,000× g for 10 minutes within several minutes after the initial operation and the cell-free supernatants were stored in microcentrifuge tubes at −70°C in a freezer for subsequent cytokine analysis.
After BAL fluid collection, the left upper bronchus was cross-clamped and the left upper lung was excised and stored in liquid nitrogen for use. A part of this frozen lung tissue was sectioned and examined under fluorescence microscopy for the detection of DM-DiI-labeled cells. The left lower lung was filled with 10% formalin for half an hour, allowing for homogeneous expansion of lung parenchyma. Next, the lungs were fixed in 10% formalin for 24 hours. The paraffin-embedded sections from the same regions (upper and lower lobes) were used for histological examination.
Morphological assessment
The paraffin sections of the left lung were cut (thickness in 3.5 μm) and stained with hematoxylin and eosin (HE). Emphysematous changes were assessed by measurement of both the mean linear intercept (MLI) and the destructive index (DI). MLI is a measurement of mean inter-alveolar septal wall distance, which is widely used to examine alveolar space size. As described earlier,4,5 the MLI was measured by dividing the length of a line drawn across the lung section by a total number of intercepts counted within this line at ×100 magnification. A total of 36 lines per mouse lung were drawn and measured. The DI was calculated to quantify the parenchymal destruction by using a microscopic point count technique.4,5 The analysis was performed in duplicate with more than 3,000 alveoli randomly counted from 50 HE sections in each mouse at ×200 magnification. Destructive alveolus was defined if at least one of the following alveoli was observed: alveolar wall defects; intraluminal parenchymal rags in alveolar ducts; obvious abnormal morphology; and typical emphysematous changes. The DI is an index of parenchymal destruction that represents the percentage of destroyed space as a fraction of the total alveolar and duct space. The morphological assessment was repeated on coded samples.
Measurements of antioxidant activity in BAL fluid and serum
Antioxidant activity in BAL fluid was measured using commercially available T-AOC Kit (Nanjing Jiancheng Biotech Co., Ltd, Nanjing, People’s Republic of China) according to the manufacturer’s instructions.
Measurements of matrix metalloproteinase-2 and matrix metalloproteinase-9 in BAL fluid
Matrix metalloproteinase (MMP)-2, MMP-9, and tissue inhibitor metalloproteinase (TIMP)-1 concentrations in BAL fluid were measured in duplicate using commercially available enzyme-linked immunosorbent assay kits (Quantikine® Mice MMP-2 Kit and Quantikine® Mice MMP-9 Kit, respectively; R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer’s instructions.
Immunohistochemistry for MMP-2 and MMP-9 in lung tissue
The expression of MMP-2 and MMP-9 in “lung tissue” was determined using a rabbit polyclonal antibody against MMP-2 and MMP-9 (ab37150 and ab38898; Abcam Inc) by immunohistochemistry according to the manufacturer’s instructions.
Determination of MMP-2 and MMP-9 activity by gelatin zymography
MMP-2 and MMP-9 activities were determined by gelatin zymography.10,21 Proteins from each lung extract were electrophoresed on 10% sodium dodecyl sulfate–Tris–glycine gels containing 1 mg/mL gelatin (Thermo Fisher Scientific, Waltham, MA, USA). Sodium dodecyl sulfate was removed from gels by shaking in 2.5% Triton X-100 twice for 1 hour. The gels were then incubated overnight at 37°C in a substrate buffer (50 mM Tris–HCl [pH 7.5], 5 mM CaCl2, 0.5 μM ZnCl2). Gels were stained with Coomassie brilliant blue and destained with methanol. Proteolysis bands were visualized and quantified using computer software (Quantity One).
MMP-2 and MMP-9 mRNA expression in lung tissues
Gene expression in the lung tissue was examined by real-time polymerase chain reaction. The lungs were excised and homogenized using TRIzol (Thermo Fisher Scientific), and the total RNA was isolated according to the manufacturer’s protocol. RNA was reverse transcribed for 45 minutes at 42°C with oligo (dT) and Moloney murine leukemia virus reverse transcriptase (Thermo Fisher Scientific). The cDNA was amplified with various primers (Table 1) using Platinum Taq DNA polymerase (Thermo Fisher Scientific) according to the manufacturer’s protocol. The polymerase chain reaction products were electrophoresed in 1.2% agarose gels containing ethidium bromide and digitally visualized with an ultraviolet illuminator (Kodak). Band intensities were semiquantified using computer software (Quantity One).
 | Table 1 Sequence of PCR primers used to assess the expression of MMP-2 and MMP-9 |
TUNEL and quantitative analysis of apoptosis
The TUNEL assay was performed (In situ Apoptosis Detection Kit; Hoffman-La Roche Ltd., Basel, Switzerland) according to the manufacturer’s protocol. TUNEL-positive signals were visualized using a horseradish peroxidase-mediated diaminobenzidine reaction. Cell nuclei were counterstained with hematoxylin solution. We divided the whole slide into nine areas and selected five fields including each corner and the central area to measure the apoptotic index (AI). Each field was then photographed at a magnification of 400×. Fields containing nonparenchymal structures, such as large airways or vessels, were discarded. For each photomicrograph, a single observer who was blind to the related data manually counted the numbers of total alveolar septal cells and the number of alveolar septal cells tested positive for TUNEL. The percentage of positive alveolar septal cells against the total number of alveolar septal cells was computed for each image, and a mean value was obtained for each mouse. The results were expressed as AI.
Statistical analysis
A software package (SPSS 15.0; SPSS Inc., Chicago, IL, USA) was used to perform all statistical analyses. Continuous data were expressed as mean ± SD. Any differences among the five groups were evaluated by a one-way ANOVA. A P-value <0.05 was considered statistically significant.
Results
Characterization of phenotypes of EPCs and detection of transplanted EPCs in the lung tissue
Cultured EPCs showed the characteristics of forming capillary structure and “cobblestone” morphology (Figure 1). On day 10, EPCs exhibited phenotyping of endothelial cells, including incorporation of acetyl low-density lipoprotein.
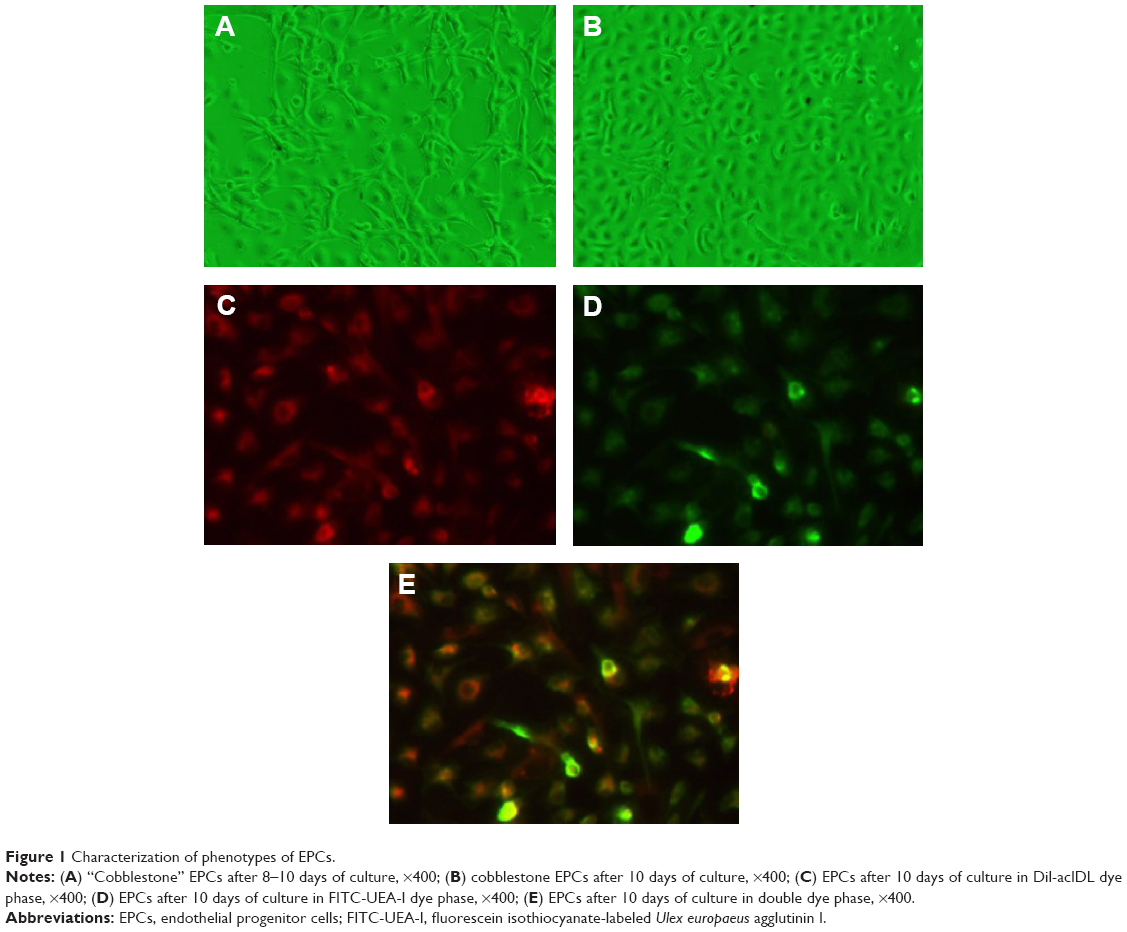 | Figure 1 Characterization of phenotypes of EPCs. |
Fluorescence-labeled EPCs were detected on the lumens of intralobular pulmonary vessel, indicating the homing of transplanted EPCs on the injured pulmonary endothelium. In addition, fluorescence-labeled EPCs were detected on the lumens of airway (Figure 2).
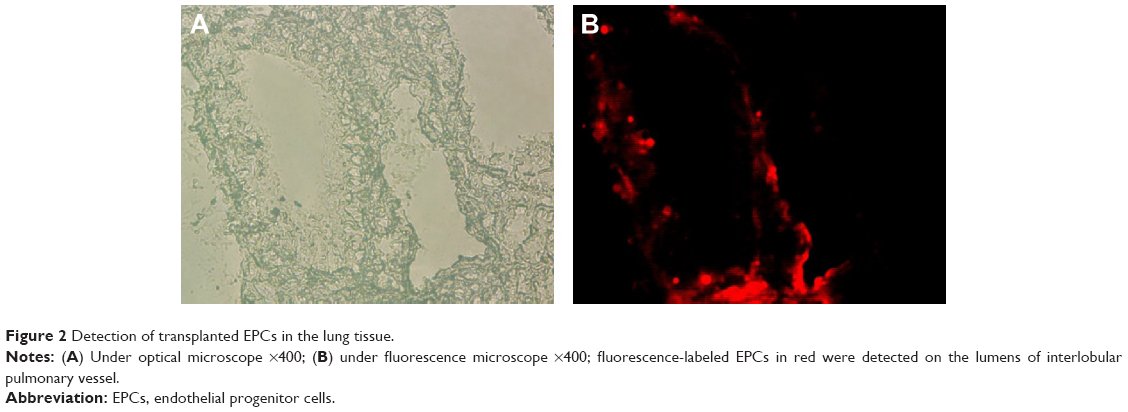 | Figure 2 Detection of transplanted EPCs in the lung tissue. |
Lung function
Compared with those in the control group, Raw was higher in the early and late sham-interfered groups and early and late EPCs-interfered groups (P<0.05; Figure 3). In comparison with the sham-interfered group, Raw was lower in the EPCs-interfered group in both the early and late groups (P<0.05). In addition, Raw was lower in the early EPCs-interfered group compared with those in the late EPCs-interfered group (P<0.05). Moreover, compared with the control group, the other four intervention groups exhibited lower Cdyn (P<0.05; Figure 3). However, the administration of early or late EPCs markedly increased the Cdyn compared with that in the early or late sham-interfered group (P<0.05). Cdyn was higher in the early EPCs-interfered group than that in the late EPCs-interfered group (P<0.05).
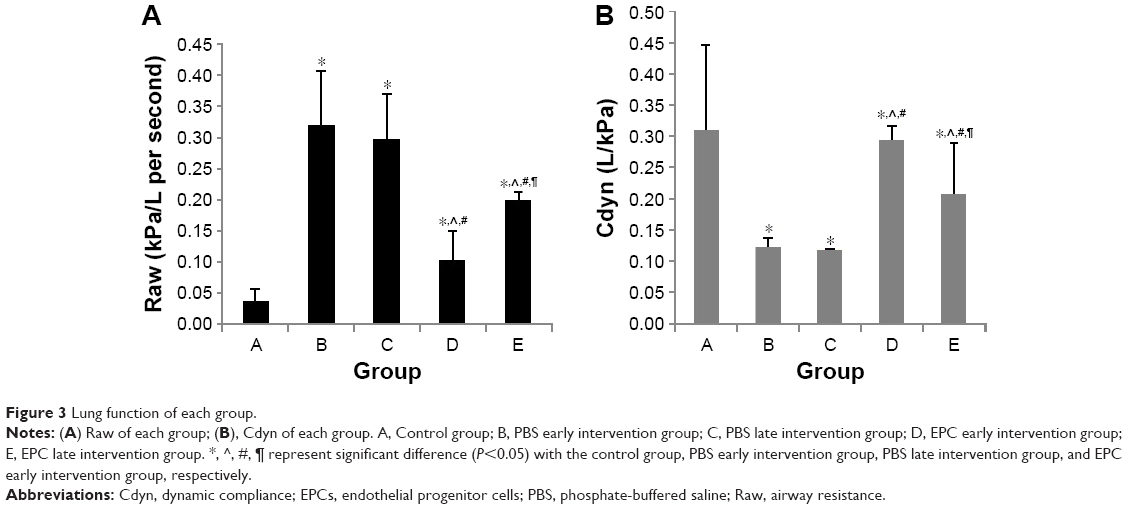 | Figure 3 Lung function of each group. |
Histological examinations
Compared with the normal alveolar architecture (control group, Figure 4A), HE staining of lung tissue sections from the early or late sham-interfered group (Figure 4B and C) and the early and late EPCs-interfered group (Figure 4D and E) showed histologically advanced emphysema. However, the administration of early or late EPCs markedly inhibited lung destruction induced by smoking exposure. Compared with the control group, the MLI and DI were significantly increased in the other four intervention groups (P<0.05). However, the lungs of mice treated by early or late EPCs showed less emphysematous changes, as the MLI and DI, than the sham-interfered mice (P<0.05). In addition, there were no significant differences of the MLI and DI between the early and late EPC treatment groups (P>0.05). Alternatively, compared with the control mice, the MLI and DI in the early or late EPC treatment group were still significantly augmented (P<0.05), indicating partly preventive effect of intratracheal administration of EPCs on emphysema (Figure 4F).
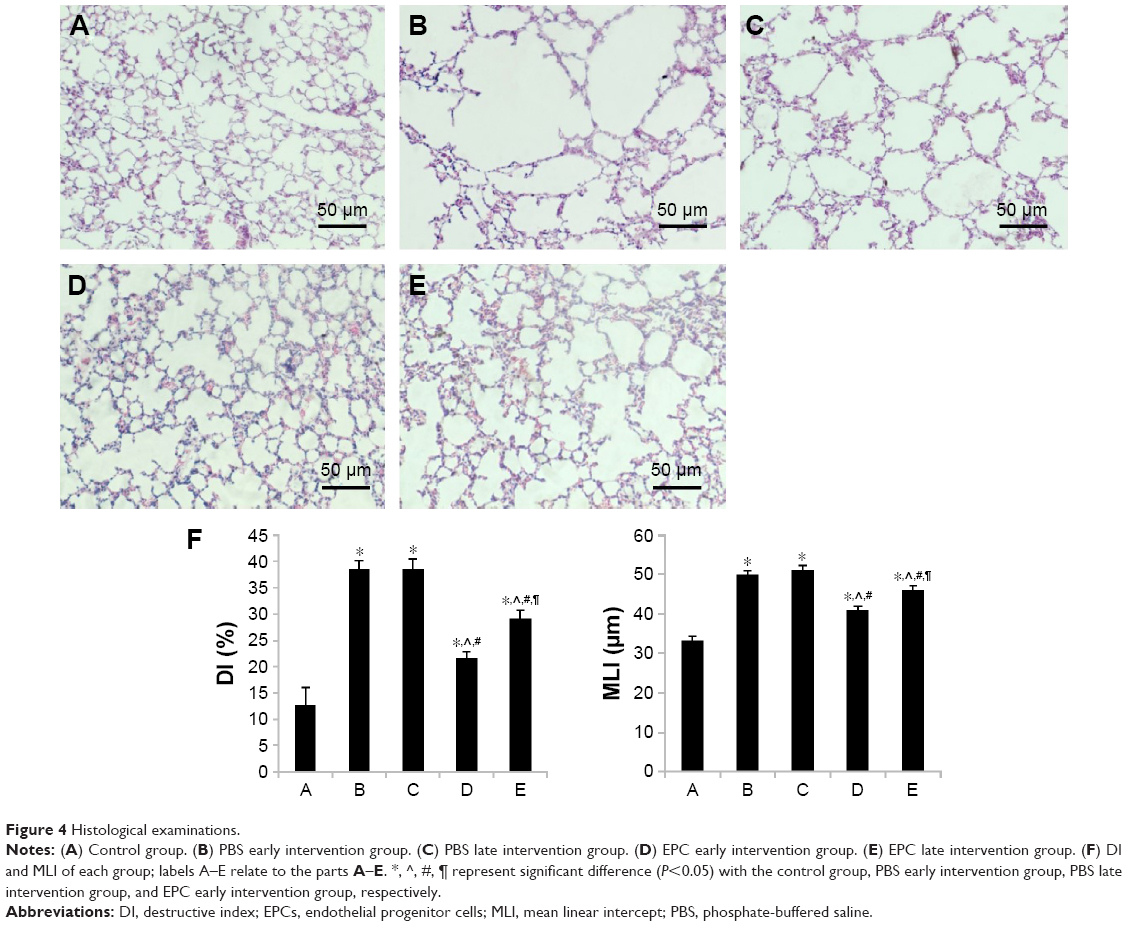 | Figure 4 Histological examinations. |
Effect of intratracheal administration of EPCs on antioxidant activity in the BAL fluid and serum of mice with COPD induced by smoking
We examined whether the protective effect of intratracheal administration of EPC of mice with COPD was associated with a reduction in biological antioxidant activity (Figure 5). The BAL fluid and serum antioxidant activity was significantly decreased in the early and late sham-interfered mice with COPD (early: 2.10±0.04 U/mL, 11.35±0.35 U/mL; late: 2.12±0.05 U/mL and 11.47±0.39 U/mL, respectively), compared with the control mice (3.27±0.10 U/mL and 18.49±0.44 U/mL; P<0.05). Early or late EPC treatment significantly prevented the reduction of antioxidant activity in the early and late sham-interfered mice with COPD (early: 2.74±0.08 U/mL and 16.03±0.40 U/mL; late: 2.40±0.04 U/mL and 14.83±0.17 U/mL; P<0.05). Moreover, the antioxidant activity was higher in the early EPC-interfered group compared with that in the late EPC-interfered group (P<0.05).
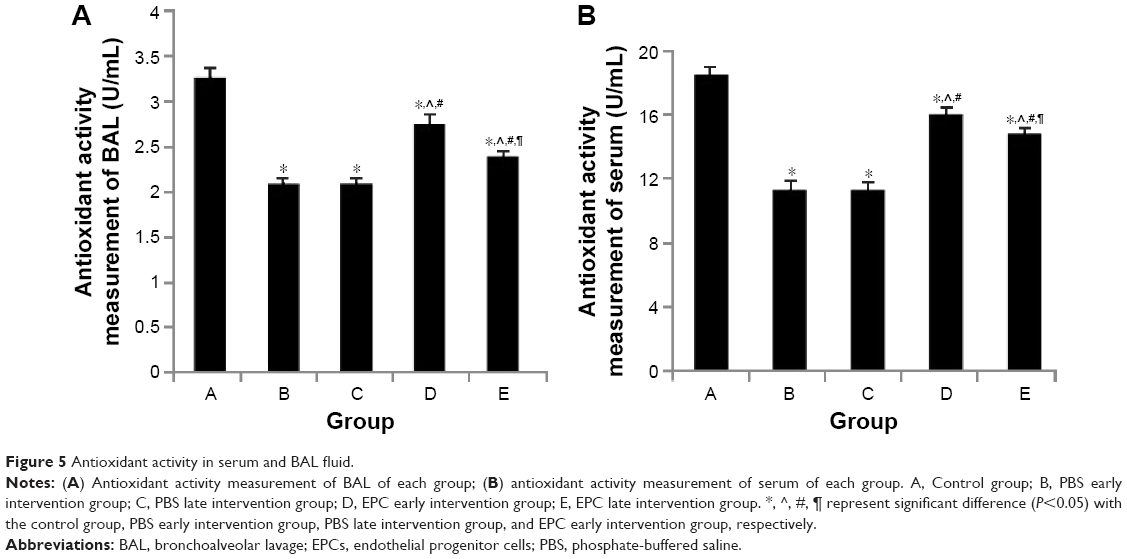 | Figure 5 Antioxidant activity in serum and BAL fluid. |
Effect of intratracheal administration of EPCs on MMP-2 and MMP-9 concentrations in the BAL fluid of mice with COPD induced by smoking
As shown in Figure 6, both sham- and EPCs-interfered mice with COPD showed increased MMP-2 and MMP-9 levels in the BAL fluid compared with those of the control mice (P<0.05). MMP-2 and MMP-9 levels were reduced in the two EPC treatment groups compared with those of sham-interfered mice (P<0.05). In addition, the MMP-2 and MMP-9 levels were lower in the early EPCs-interfered group compared with those in the late EPC-interfered group (P<0.05). In contrast, the level of TIMP-1 in the BAL fluid of sham- and EPCs-interfered mice with COPD was lower compared with that of the control mice (P<0.05). TIMP-1 level was increased in the two EPC treatment groups compared with that of sham-interfered mice (P<0.05). In addition, the early EPC treatment significantly increased the level of TIMP-1 compared with the late EPC treatment (P<0.05).
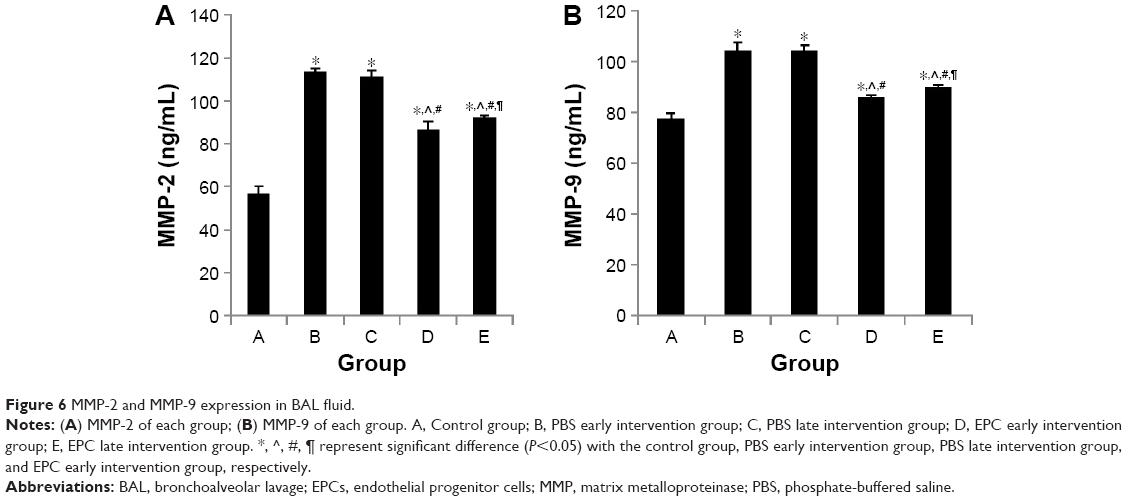 | Figure 6 MMP-2 and MMP-9 expression in BAL fluid. |
Effect of intratracheal administration of EPCs on MMP-2 and MMP-9 protein expression in the lung tissue of mice with COPD induced by smoking
MMP-2 and MMP-9 protein expression was analyzed by immunohistochemistry. The protein levels of MMP-2 and MMP-9 were increased in the four interfered mice groups with COPD compared with those of control mice (Figures 7 and 8). In addition, MMP-2 and MMP-9 expression was markedly reduced in the early and late EPC treatment groups compared with that in sham-interfered mice with COPD. In addition, the early EPC treatment significantly decreased the MMP-2 and MMP-9 protein expression compared with the late EPC treatment (P<0.05).
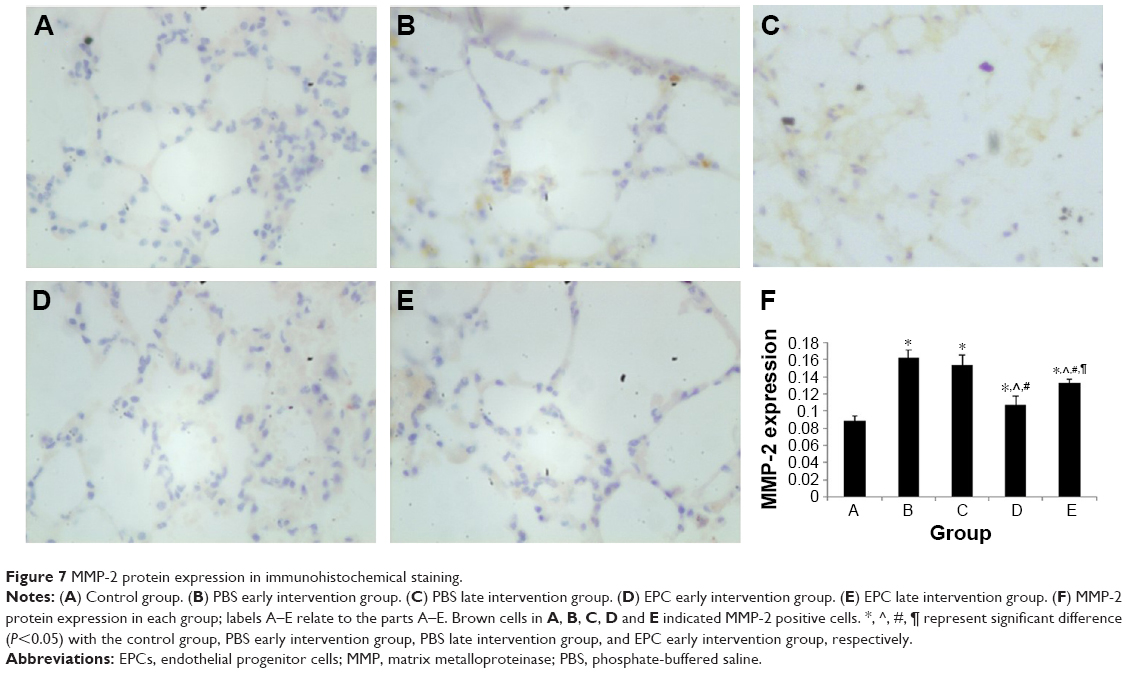 | Figure 7 MMP-2 protein expression in immunohistochemical staining. |
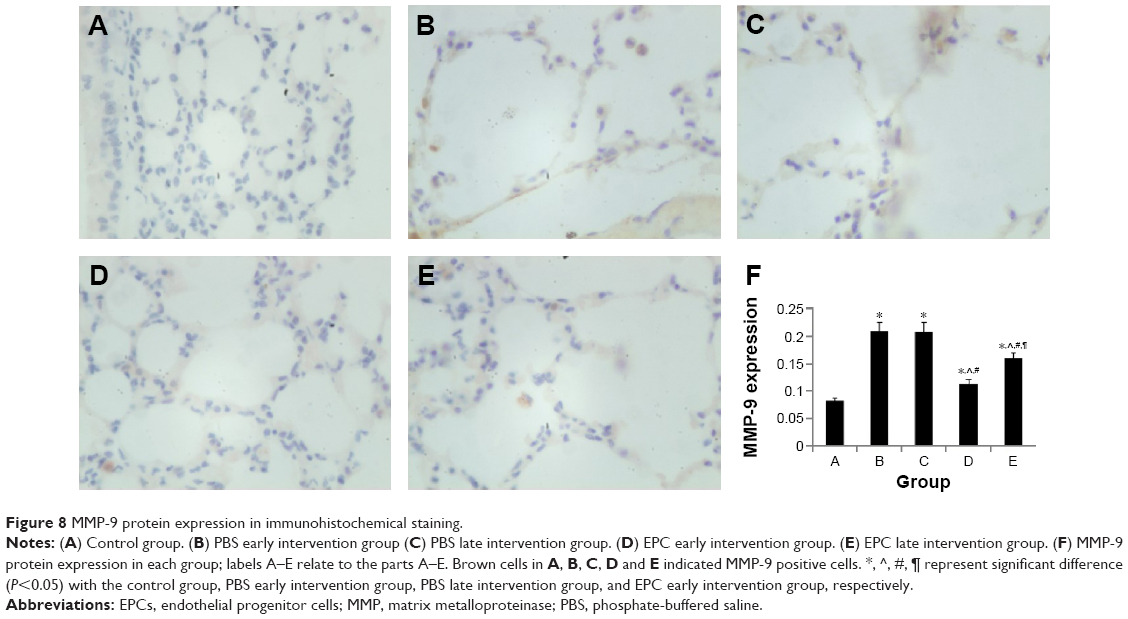 | Figure 8 MMP-9 protein expression in immunohistochemical staining. |
Effect of intratracheal administration of EPCs on MMP-2 and MMP-9 activity in the lung tissue of mice with COPD induced by smoking
As shown in Figure 9, the four interfered mice groups with COPD showed increased MMP-2 and MMP-9 activities in their lung tissue compared with the activity in the control mice (P<0.05). MMP-2 and MMP-9 activities were reduced in the two EPC treatment groups compared with those of sham-interfered mice (P<0.05). In addition, the early EPC treatment significantly decreased the MMP-2 and MMP-9 protein expression compared with the late EPC treatment (P<0.05).
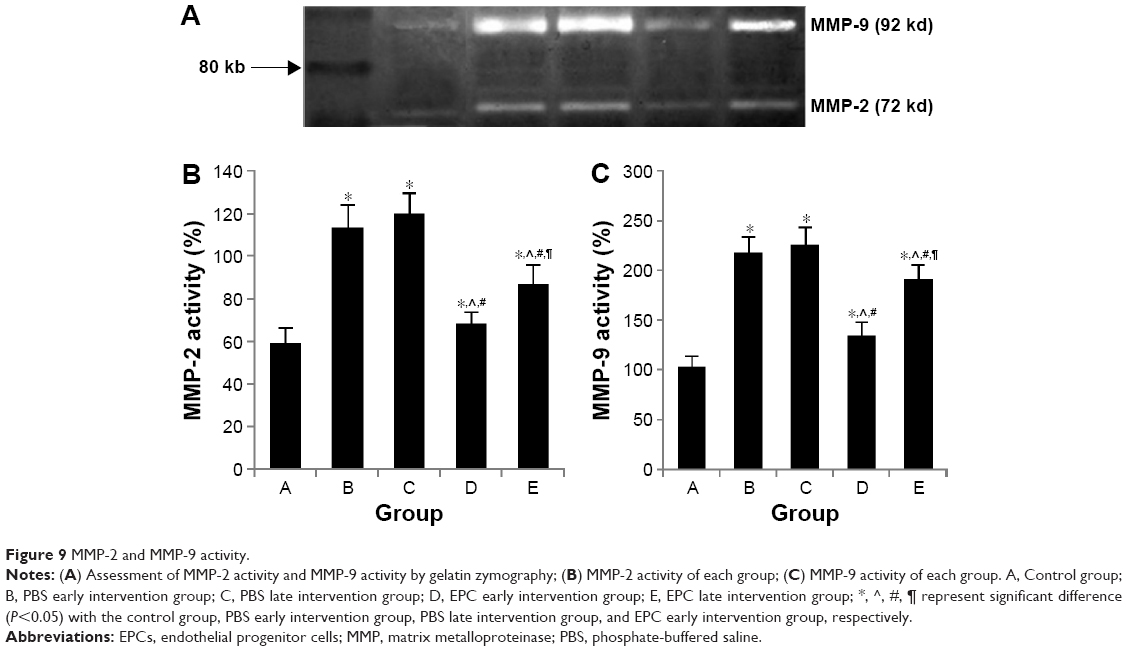 | Figure 9 MMP-2 and MMP-9 activity. |
Effect of intratracheal administration of EPCs on MMP-2 and MMP-9 mRNA expression in the lung tissue of mice with COPD induced by smoking
As shown in Figure 10, sham-interfered mice with COPD showed increased MMP-2 and MMP-9 mRNA expression in the lung tissue compared with that of the control mice (P<0.05). MMP-2 and MMP-9 mRNA expression was reduced in the two EPC treatment groups compared with that of sham-interfered mice (P<0.05). In addition, the MMP-2 and MMP-9 levels were lower in the early EPCs-interfered group than those in the late EPCs-interfered group (P<0.05).
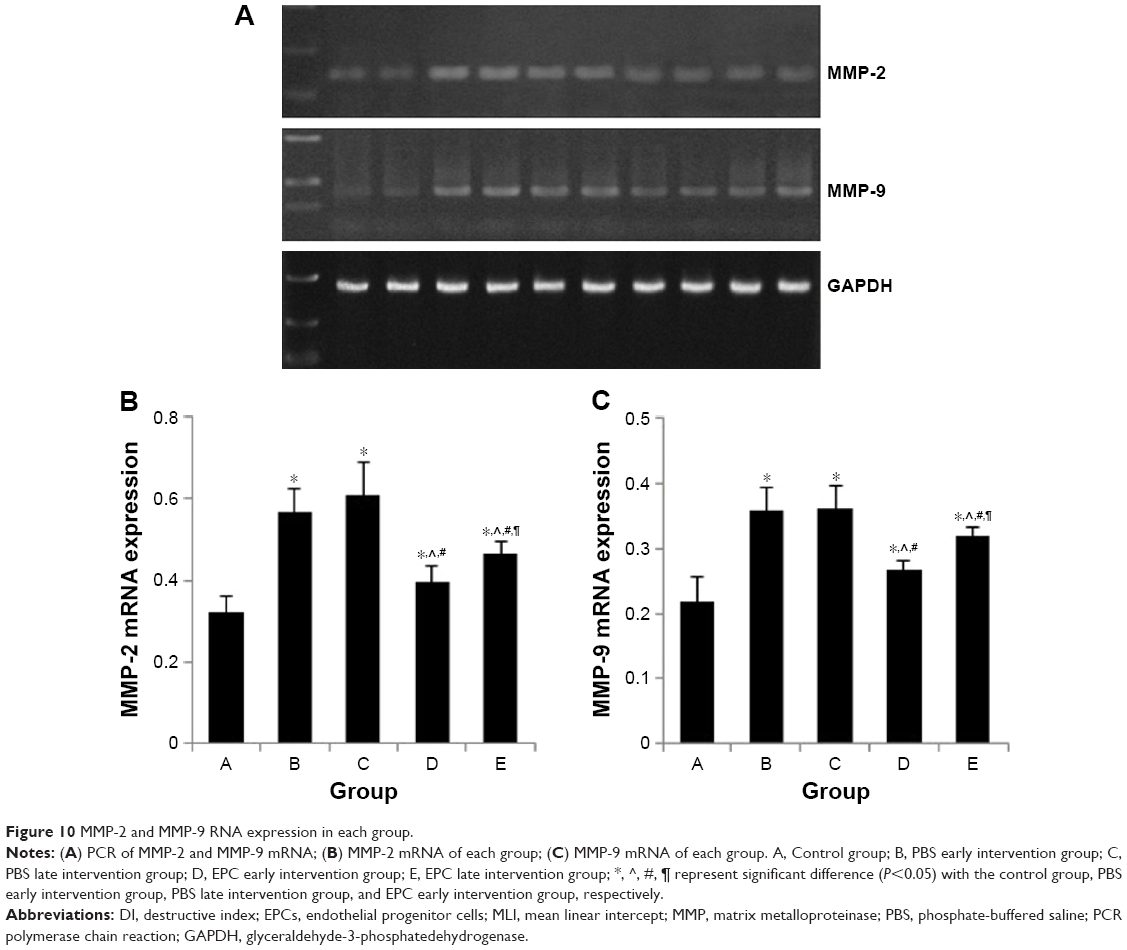 | Figure 10 MMP-2 and MMP-9 RNA expression in each group. |
Effect of intratracheal administration of EPCs on apoptosis in the lung tissue of mice with COPD induced by smoking
The TUNEL-positive (TUNEL+) cells were frequently localized in the alveolar septum and lung vascular endothelial cells in both the sham- and EPCs-interfered mice with COPD compared with those of the control mice (Figure 11). The (TUNEL+) cells were quantified as AI by which the amount of DNA-damaged cells was identified to evaluate the apoptosis. The protective effect of EPC treatment was related to a reduction in apoptosis as measured by AI. The DNA-damaged cells were very rare in the normal mice, while the TUNEL+ cells in the four interfered groups were much more frequent (P<0.05). The EPC treatment greatly reduced the number of TUNEL+ cells in the lungs of mice with COPD (P<0.05). Again, the early EPC treatment significantly reduced apoptosis of the alveolar septum and vascular endothelial cells compared with the late EPC treatment (P<0.05).
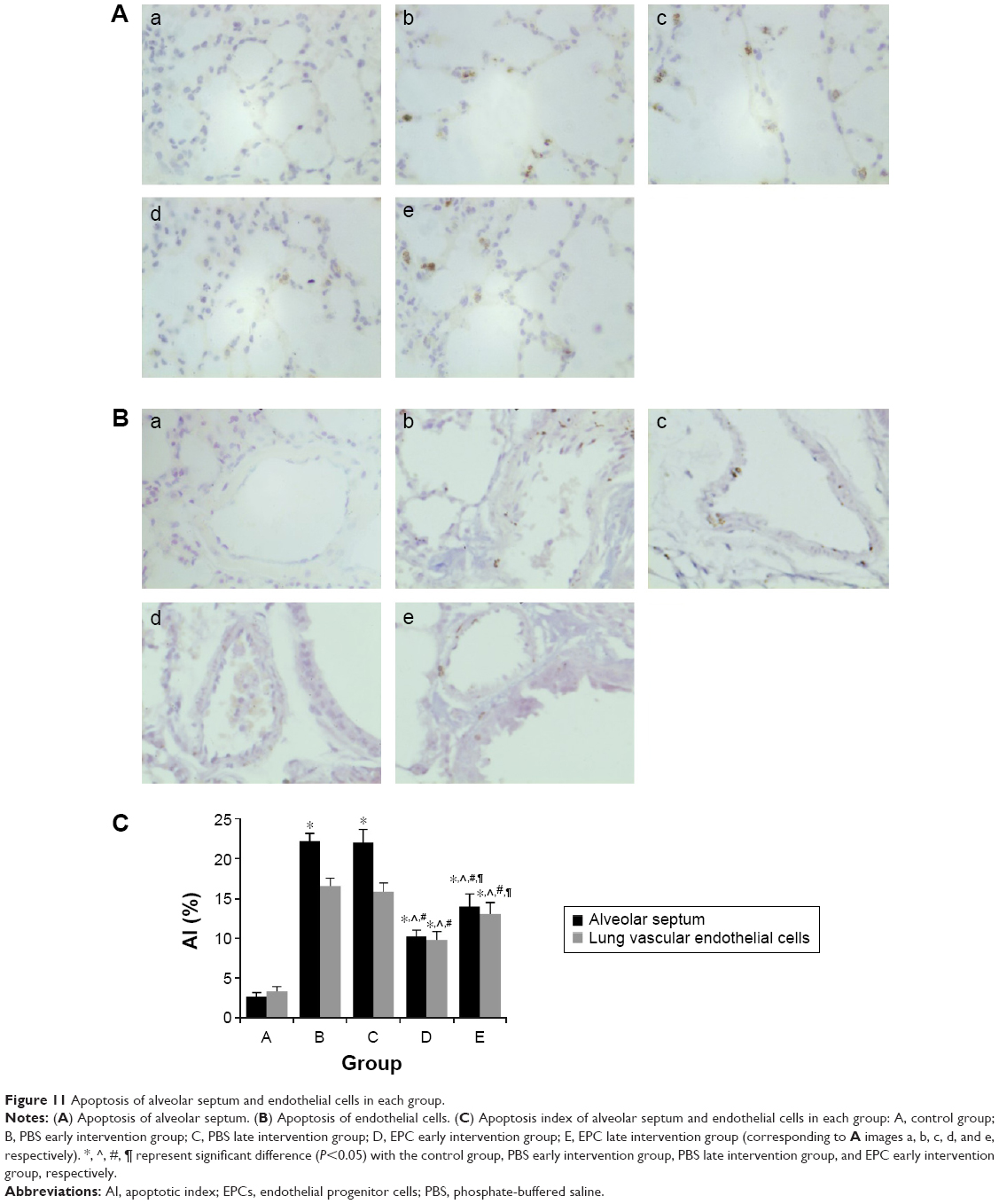 | Figure 11 Apoptosis of alveolar septum and endothelial cells in each group. |
Discussion
In the current study, a COPD model in mice was well established using cigarette smoking exposure for 90 days. The airspace enlargement, alveolar destruction, and altered lung function in our animal model of emphysema were evident through the increase in MLI, DI, and Raw and decrease in Cdyn, which is consistent with that of earlier studies.21,22 The most remarkable finding of this study is that intratracheal administration of EPCs partly prevented the development of airspace enlargement and improved the lung function including Raw and lung compliance in mice with COPD induced by smoking exposure. Activation of MMP-2 and MMP-9 in mice with smoking-induced COPD was inhibited by both the early and late EPC treatment. Moreover, we demonstrated that the EPC treatment decreased the lung alveolar septal cell and vascular endothelial cell apoptosis. Furthermore, the EPC treatment recovered antioxidant activity in BAL fluid and serum of mice with smoking-induced COPD.
EPCs, the so-called vascular blast cells or vascular endothelial stem cells, were characterized in the blood of adult mice by Asahara et al,10 as a population of CD34+ cells that could differentiate into endothelial cells in vitro. EPCs are mobilized from bone marrow to peripheral circulation by cytokines, growth factors, and ischemic conditions during endothelial injury and by pharmaceutics such as statins.23 Levels of EPCs may be a surrogate biologic marker for vascular endothelial function, and the absence of sufficient circulating progenitor cells contributes to the endothelial injury.24 During the past decade, evidence continues to accumulate on the relationship between EPCs and respiratory diseases and some studies had already focused on COPD. An earlier study25 has demonstrated that “on-and-off” cigarette smoking markedly influenced the number of circulating EPCs in apparently healthy chronic smokers. The number of circulating EPCs was reduced in chronic smokers, while smoking cessation led to a rapid restoration of EPC levels. Palange et al15 and Fadini et al16 had shown that circulating EPCs decreased in patients with COPD and could be correlated with disease severity. Moreover, Kim et al17 reported that EPCs may be associated with the development of emphysema, and circulating EPCs were an independent predictor of the presence of emphysema. Lam et al26 also revealed that autologous transplantation of EPCs preserves pulmonary endothelial function and maintains the integrity of pulmonary alveolar–capillary barrier by reducing infiltration of leukocytes and decreasing water content, hyaline membrane formation, and hemorrhage in the lung parenchyma of rabbits with acute lung injury and end-stage acute respiratory distress syndrome (ARDS). Granulocyte colony-stimulating factor and adrenomedullin treatment in mice with elastase-induced emphysema provided a significant reduction in emphysema by increasing circulating EPCs in peripheral blood and promoting lung regeneration.27,28 All of these studies strongly indicated that EPCs play an important role in the development of emphysema and COPD. However, whether the transplantation of EPCs improves smoking-induced COPD remains unknown. In the current study, transplanted EPCs were demonstrated to express red fluorescence and were distributed on the airway, lung vascular, and alveolar septum. Intratracheal administration of EPCs attenuated the emphysematous changes and improved lung function in mice with cigarette smoke-induced COPD.
It has been well known that MMPs, also called matrixins, belong to a family of zinc- and calcium-dependent enzymes, which are implicated in numerous physiological and pathological processes. The imbalance between proteases and the antiproteolytic defenses of the lung is believed to play an essential role in the development of pulmonary emphysema.3 Among the MMPs, MMP-2 and MMP-9, collectively known as the gelatinases, are particularly important in the pathogenesis of lung tissue remodeling and repair through degradation of collagen and different matrix proteins, including elastase.29 Our earlier study30 and data from animal models31 demonstrated that the secretion and/or activity of MMP-2 and MMP-9 increased in emphysema and COPD, suggesting that increased MMP-2 and MMP-9 production favors the development of emphysema. It was shown that MMP-9, induced in stem and progenitor cells, releases soluble Kit ligand, permitting the transfer of endothelial and hematopoietic stem cells from the quiescent to proliferative niche and the mobilization of EPCs was MMP-9 dependent.32 Moreover, recent studies found that MMP-2 is selectively required for the differentiation of EPCs33 and MMP-2 deficiency reduces the functional activities of EPCs, leading to impaired vasculogenesis.34 In this study, we demonstrated that the sham- and EPCs-interfered mice with COPD induced by cigarette smoking showed increased secretion of MMP-2 and MMP-9 in the BAL fluid and enhanced activation of MMP-2 and MMP-9 in lung tissue compared with those of the control mice. Furthermore, MMP-2 and MMP-9 levels were reduced in the two EPC treatment groups compared with those of sham-interfered mice. In addition, the MMP-2 and MMP-9 levels were lower in the early EPCs-interfered group compared with those in the late EPCs-interfered group. These data indicated that intratracheal administration of EPCs can partly protect against the development of smoking-induced emphysema and COPD by inhibiting proteolytic enzyme activation. In addition, a more effective trend was observed in the early EPC treatment compared with the late EPC treatment.
Recent evidence provided insight into the effects of EPC on endothelial cell survival. It was shown that the VEGF-A/B–vascular endothelial growth factor receptor 2 (VEGFR)-1/2–Erk1/2 signal pathway took major responsibility for the anti-apoptotic effects of EPC on pulmonary microvascular endothelial cells. In addition, transplanted EPC can inhibit cell apoptosis in the lung of pulmonary arterial hypertension by secreting growth factors in a paracrine manner and reducing caspase-3 expression.35 Yang et al36 found that EPC conditioned medium prevents oxidative stress-induced apoptosis by increasing the expression ratio of Bcl-2/Bax in mature endothelial cells. In agreement with these findings, our data demonstrated that AI of alveolar septum and lung vascular endothelial cells significantly decreased in the early or late EPC treatment group compared with those in the sham-interfered group with COPD, while it was still higher than that in the normal mice. The decreasing AI suggests that the induction of emphysema by alveolar wall and endothelial cell apoptosis may be partially reversible due to intratracheal administration of EPCs. In addition, the early EPC treatment significantly reduced apoptosis of the alveolar septum and vascular endothelial cells compared with the late EPC treatment. Recent data suggest that apoptosis is not an isolated event in the development of COPD and interacts with proteinase/anti-proteinase imbalance and oxidative stress, adding to the complexity of COPD.37 Consistently, we found an increasing biological antioxidant activity in both the serum and BAL fluid of mice with smoking-induced COPD after administration of early or late EPCs. In our case, we suspect that the recovered biological antioxidant activity after administration of EPCs is probably related to the inhibitory effect of bisphophonates (BPS) on apoptosis in this model.
However, it must be noted that the processes involved in the pathogenesis of COPD that we mentioned earlier are complicatedly connected, interacted, and networked in the development of COPD, and the cause–effect relationships are still unknown. In addition, when evaluating the therapeutic implications of intratracheal administration of EPCs, we were able to show only a protective and not a curative effect of EPC treatment because of the experimental murine model with smoking-induced COPD. Nevertheless, the therapeutic strategies aimed at protecting the lung from COPD may prove useful for slowing the presently unstoppable progression of COPD.
Conclusion
This study shows for the first time that intratracheal administration of EPCs partly protects against the development of cigarette smoking-induced COPD in mice. The mechanism of protective effect is possibly through attenuating the apoptosis of lung alveolar septum and vascular endothelial cells, inhibiting proteolytic enzymes, and recovering the antioxidant activity. Overall, these results suggest that transplantation of EPCs may therefore be a potentially effective therapeutic strategy to inhibit the progression of COPD, which is a highly irreversible disease.
Acknowledgment
This study was supported in part by the National Natural Science Foundation of China (81070039, 81170036, 81270100).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet. 2004;364(9434):613–620. | ||
Lopez AD, Mathers CD. Measuring the global burden of disease and epidemiological transitions: 2002–2030. Ann Trop Med Parasitol. 2006;100(5–6):481–499. | ||
Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. | ||
Chen Y, Hanaoka M, Chen P, Droma Y, Voelkel NF, Kubo K. Protective effect of beraprost sodium, a stable prostacyclin analog, in the development of cigarette smoke extract-induced emphysema. Am J Physiol Lung Cell Mol Physiol. 2009;296(4):L648–L656. | ||
Chen Y, Hanaoka M, Droma Y, Chen P, Voelkel NF, Kubo K. Endothelin-1 receptor antagonists prevent the development of pulmonary emphysema in rats. Eur Respir J. 2010;35(4):904–912. | ||
Zhang C, Cai S, Chen P, et al. Inhibition of tumor necrosis factor-alpha reduces alveolar septal cell apoptosis in passive smoking rats. Chin Med J (Engl). 2008;121(7):597–601. | ||
Cai S, Chen P, Zhang C, Chen JB, Wu J. Oral N-acetylcysteine attenuates pulmonary emphysema and alveolar septal cell apoptosis in smoking-induced COPD in rats. Respirology. 2009;14(3):354–359. | ||
Li J, Chen Y, Chen P, et al. AP-2α expression and cell apoptosis of the lung tissue of rats with COPD and ECV304 cells stimulated by cigarette smoke extract. Chin Sci Bull. 2011;56(15):1562–1568. | ||
Hristov M, Erl W, Weber PC. Endothelial progenitor cells: mobilization, differentiation, and homing. Arterioscler Thromb Vasc Biol. 2003;23(7):1185–1189. | ||
Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–967. | ||
Crosby JR, Kaminski WE, Schatteman G, et al. Endothelial cells of hematopoietic origin make a significant contribution to adult blood vessel formation. Circ Res. 2000;87(9):728–730. | ||
Cho HJ, Kim HS, Lee MM, et al. Mobilized endothelial progenitor cells by granulocyte-macrophage colony-stimulating factor accelerate reendothelialization and reduce vascular inflammation after intravascular radiation. Circulation. 2003;108(23):2918–2925. | ||
Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9(6):702–712. | ||
Ingram DA, Caplice NM, Yoder MC. Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood. 2005;106(5):1525–1531. | ||
Palange P, Testa U, Huertas A, et al. Circulating haemopoietic and endothelial progenitor cells are decreased in COPD. Eur Respir J. 2006;27(3):529–541. | ||
Fadini GP, Schiavon M, Cantini M, et al. Circulating progenitor cells are reduced in patients with severe lung disease. Stem Cells. 2006;24(7):1806–1813. | ||
Kim EK, Lee JH, Jeong HC, et al. Impaired colony-forming capacity of circulating endothelial progenitor cells in patients with emphysema. Tohoku J Exp Med. 2012;227(4):321–331. | ||
Marrotte EJ, Chen DD, Hakim JS, Chen AF. Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J Clin Invest. 2010;120(12):4207–4219. | ||
Zhao T, Li J, Chen AF. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am J Physiol Endocrinol Metab. 2010;299(1):E110–E116. | ||
He T, Smith LA, Harrington S, Nath KA, Caplice NM, Katusic ZS. Transplantation of circulating endothelial progenitor cells restores endothelial function of denuded rabbit carotid arteries. Stroke. 2004;35(10):2378–2384. | ||
Valenca SS, da Hora K, Castro P, Moraes VG, Carvalho L, Porto LC. Emphysema and metalloelastase expression in mouse lung induced by cigarette smoke. Toxicol Pathol. 2004;32(3):351–356. | ||
van der Strate BW, Postma DS, Brandsma CA, et al. Cigarette smoke-induced emphysema: a role for the B cell? Am J Respir Crit Care Med. 2006;173(7):751–758. | ||
Asahara T, Kawamoto A. Endothelial progenitor cells for postnatal vasculogenesis. Am J Physiol Cell Physiol. 2004;287(3):C572–C579. | ||
Shi Q, Rafii S, Wu MH, et al. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998;92(2):362–367. | ||
Kondo T, Hayashi M, Takeshita K, et al. Smoking cessation rapidly increases circulating progenitor cells in peripheral blood in chronic smokers. Arterioscler Thromb Vasc Biol. 2004;24(8):1442–1447. | ||
Lam CF, Liu YC, Hsu JK, et al. Autologous transplantation of endothelial progenitor cells attenuates acute lung injury in rabbits. Anesthesiology. 2008;108(3):392–401. | ||
Ishizawa K, Kubo H, Yamada M, et al. Bone marrow-derived cells contribute to lung regeneration after elastase-induced pulmonary emphysema. FEBS Lett. 2004;556(1–3):249–252. | ||
Murakami S, Nagaya N, Itoh T, et al. Adrenomedullin regenerates alveoli and vasculature in elastase-induced pulmonary emphysema in mice. Am J Respir Crit Care Med. 2005;172(5):581–589. | ||
Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol. 2003;28(1):12–24. | ||
Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology. 2008;13(7):1014–1021. | ||
Kirschvink N, Vincke G, Fievez L, et al. Repeated cadmium nebulizations induce pulmonary MMP-2 and MMP-9 production and emphysema in rats. Toxicology. 2005;211(1–2):36–48. | ||
Heissig B, Hattori K, Dias S, et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002;109(5):625–637. | ||
Wu Y, Dai J, Schmuckler NG, et al. Cleaved high molecular weight kininogen inhibits tube formation of endothelial progenitor cells via suppression of matrix metalloproteinase 2. J Thromb Haemost. 2010;8(1):185–193. | ||
Cheng XW, Kuzuya M, Nakamura K, et al. Mechanisms underlying the impairment of ischemia-induced neovascularization in matrix metalloproteinase 2-deficient mice. Circ Res. 2007;100(6):904–913. | ||
Xia L, Fu GS, Yang JX, Zhang FR, Wang XX. Endothelial progenitor cells may inhibit apoptosis of pulmonary microvascular endothelial cells: new insights into cell therapy for pulmonary arterial hypertension. Cytotherapy. 2009;11(4):492–502. | ||
Yang Z, von Ballmoos MW, Faessler D, et al. Paracrine factors secreted by endothelial progenitor cells prevent oxidative stress-induced apoptosis of mature endothelial cells. Atherosclerosis. 2010;211(1):103–109. | ||
Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7:53. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.