")
Back to Journals » International Journal of Nanomedicine » Volume 14
Intestinal transport mechanism and in vivo anticancer efficacy of a solid oral formulation incorporating an ion-pairing complex of pemetrexed with deoxycholic acid derivative
Authors Pangeni R , Jha SK, Maharjan R, Choi JU , Chang KY , Choi YK, Byun Y, Park JW
Received 23 March 2019
Accepted for publication 27 July 2019
Published 8 August 2019 Volume 2019:14 Pages 6339—6356
DOI https://doi.org/10.2147/IJN.S209722
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Rudra Pangeni,1,* Saurav Kumar Jha,1,* Ruby Maharjan,2 Jeong Uk Choi,2 Kwan-Young Chang,3 Young Kweon Choi,3 Youngro Byun,4 Jin Woo Park1
1Department of Pharmacy, College of Pharmacy and Natural Medicine Research Institute, Mokpo National University, Jeonnam 58554, Republic of Korea; 2Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea; 3Global R&D Center, Icure BNP, Seoul 06170, Republic of Korea; 4Department of Molecular Medicine and Biopharmaceutical Science, Graduate School of Convergence Science and Technology, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea
*These authors contributed equally to this work
Objective: The rational combination of immunotherapy with standard chemotherapy shows synergistic clinical activities in cancer treatment. In the present study, an oral powder formulation of pemetrexed (PMX) was developed to enhance intestinal membrane permeability and investigate its application in metronomic chemotherapy in combination with immunotherapy.
Methods: PMX was ionically complexed with a bile acid derivative (Nα-deoxycholyl-l-lysyl-methylester; DCK) as a permeation enhancer and mixed with dispersing agents, such as poloxamer 188 (P188) and Labrasol, to form an amorphous oral powder formulation of PMX/DCK (PMX/DCK-OP).
Results: The apparent permeability (Papp) of PMX/DCK-OP across a Caco-2 cell monolayer was 2.46- and 8.26-fold greater than that of PMX/DCK and free PMX, respectively, which may have been due to the specific interaction of DCK with bile acid transporters, as well as the alteration of membrane fluidity due to Labrasol and P188. Furthermore, inhibition of bile acid transporters by actinomycin D in Caco-2 cell monolayers decreased the Papp of PMX/DCK-OP by 75.4%, suggesting a predominant role of bile acid transporters in the intestinal absorption of PMX/DCK-OP. In addition, caveola/lipid raft-dependent endocytosis, macropinocytosis, passive diffusion, and paracellular transport mechanisms significantly influenced the permeation of PMX/DCK-OP through the intestinal membrane. Therefore, the oral bioavailability of PMX/DCK-OP in rats was 19.8%±6.93%, which was 294% higher than that of oral PMX. Moreover, an in vivo anticancer efficacy study in B16F10 cell-bearing mice treated with a combination of oral PMX/DCK-OP and intraperitoneal anti-PD1 exhibited significant suppression of tumor growth, and the tumor volume was maximally inhibited by 2.03- and 3.16-fold compared to the oral PMX/DCK-OP and control groups, respectively.
Conclusion: These findings indicated the therapeutic potential of a combination of low-dose oral chemotherapy and immunotherapy for synergistic anticancer efficacy.
Keywords: pemetrexed, bile acid derivative, oral powder formulation, intestinal membrane permeability, oral delivery, oral metronomic chemotherapy
Introduction
Intravenous administration of the maximum tolerated dose of an antineoplastic chemotherapeutic agent has been widely accepted as a clinical strategy to obtain complete remission or cure of cancer. However, this strategy is associated with the development of chemoresistance and both long- and short-term toxicity, including myelosuppression, neutropenia, thrombocytopenia, and gastrointestinal dysfunction, along with the depletion of both innate and adaptive defense mechanisms. To address these issues related to conventional chemotherapy, a novel approach involving frequent low-dose oral administration, also known as metronomic chemotherapy, has been suggested, which maintains uniform serum levels of drugs, overcomes drug resistance by targeting the tumor vasculature, and avoids toxic effects.1 Moreover, metronomic chemotherapy inhibits tumor angiogenesis, promotes activation of innate and adaptive immunity, and induces tumor dormancy.2 However, metronomic scheduling for improved patient quality of life can be preferentially achieved by oral drug administration. In addition, oral administration is considered one of the safest and most convenient routes to maintaining prolonged exposure of cancerous cells to chemotherapeutic agents. However, oral delivery of these anticancer drugs is a major challenge due to their physicochemical properties, variable rate of degradation in gastrointestinal fluid, and high drug efflux through P-glycoprotein (P-gp).3,4 Furthermore, recent studies suggested the use of low-dose chemotherapeutics in combination with immunotherapy to overcome the immunosuppression induced by cancer, intensify host immunity to destroy tumor cells, produce highly active tumor-specific T cells, and synergistically optimize antitumor effects.5,6 Therefore, the rational combination of immunotherapy with oral low-dose chemotherapy is expected to control tumor growth and address the immunosuppressive tumor environment.
Pemetrexed (N-[4-[2-(2-amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d] pyrimidin-5-yl)-ethyl]-benzoyl]-
To overcome these obstacles, various approaches, including coadministration of therapeutic agents with functional excipients (P-gp inhibitors) or use of permeation enhancers, have been developed.10–12 In addition, polymeric or lipid-based drug carriers, such as polymeric nanoparticles, polymeric micelles, microemulsions, self-emulsifying drug delivery systems, lipid-drug conjugates, and nanocrystals have been used to improve the oral bioavailability of drugs.13,14
There is increasing interest in the design of potential prodrug targets for carrier-mediated uptake via bile acid transporters to improve intestinal drug absorption.15,16 The prodrug approach includes chemical modification of drugs to improve their charge and lipophilicity, to in turn promote intestinal permeation. Prodrugs targeting endogenous transporters present on enterocytes can facilitate site-specific transport and represent a novel strategy for intestinal delivery of drugs with very low oral bioavailability.15 Various approaches to exploit the prodrug concept have already been applied to increase the central nervous system (CNS) delivery of several drugs. Bonina et al reported greater reserpine-induced hypolocomotion effects in rats when composites were linked via the C-3 glucose position.17 Bilsky et al demonstrated increased uptake of opioid peptide agonist through the blood-brain barrier (BBB) after conjugation with
In our previous studies, we prepared an ionic complex of hydrophilic drugs with a bile acid derivative (Nα-deoxycholyl-
The main objective of the present study was to design an oral powder formulation of PMX for application in metronomic chemotherapy, as well as for combination use with immunotherapy. PMX was ionically complexed with DCK in combination with a dispersing agent, such as P188 or Labrasol, forming an oral powder formulation of PMX/DCK (PMX/DCK-OP). This can be effectively absorbed across the intestinal membrane due to increased lipophilicity of PMX achieved by ion-pairing complex with DCK and interaction with bile acid transporters. In addition, stable formation of micelles in the aqueous solution, dispersant activity of P188, opening of tight junctions (TJs), and surfactant activity of Labrasol can also enhance the intestinal membrane permeability of PMX. Therefore, to confirm the increase in oral absorption of PMX after preparation of an oral powder formulation, we determined the artificial membrane and Caco-2 cell monolayer permeability of PMX/DCK and PMX/DCK-OP. The intestinal transport pathways of PMX/DCK-OP through a Caco-2 cell monolayer were then assessed using specific pharmacological inhibitors. Finally, the oral bioavailability of PMX/DCK-OP in rats, and its antitumor efficacy in combination with intraperitoneal anti-PD1 antibody in tumor-bearing mice, was evaluated.
Materials and methods
Materials
The PMX disodium hemipentahydrate was obtained from Shipla Medicare (Karnataka, India). Caprylocaproyl macrogol-8-glyceride (Labrasol) was provided by Gattefossé (Saint Priest, France). Polyoxyethylene (160) polyoxypropylene (30) glycol (P188) was purchased from BASF (Ludwigshafen, Germany). Chlorpromazine, methyl-β-cyclodextrin (MβCD), brefeldin A, genistein, actinomycin D (Act D), cyclosporine A, ethylene glycol-bis-(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), DOCA, and 4-[N-(2,4-diamino-6-pterinidinylmethyl)-N-methylamino]benzoic acid hemihydrochloride hydrate (DAMPA) were obtained from Sigma-Aldrich Inc. (St. Louis, MO). Anti-mouse PD-1 antibody (anti-PD1) was purchased from Bio X Cell (West Lebanon, NH). Solvents for high-performance liquid chromatography (HPLC) were obtained from Merck Millipore (Billerica, MA) and Thermo Fisher Scientific Inc. (Waltham, MA).
Animals
Sprague-Dawley rats (males, 6–7 weeks old, 200–250 g) and BALB/c mice (females, 6–7 weeks old, 20–25 g) were provided by Orient Bio (Gwangju, Republic of Korea). The animals were housed under environmentally controlled conditions of temperature (23 °C±2 °C), relative humidity (55%±10%), and light (12/12 hr light/dark cycle). The animals were fed a standard laboratory diet (Nestlé Purina, St. Louis, MO) and ion-sterilized tap water. Ethical approval for this study was obtained from the Institutional Animal Care and Use Committee (IACUC) of Mokpo National University (Jeonnam, Republic of Korea). All animal experiments were performed in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals and the guidelines of the IACUC.
Preparation and characterization of PMX/DCK-OP
We synthesized DCK as an oral permeation enhancer, as described previously.16 Briefly, H-Lys(Boc)-OMe⋅HCl (20 g) dissolved in N-methyl morpholine (7.4 mL) and ethyl chloroformate (6.4 mL) were mixed with DOCA (26 g) in tetrahydrofuran (800 mL) with continuous stirring for 30 mins, followed by 2 hr reflux. The precipitates obtained after stirring of the reaction mixture overnight at room temperature were collected by filtration and the residual solvents in the precipitates were removed by evaporation. Purified Lys(Boc)DOCA was obtained by column chromatography (chloroform/methanol) of the dried precipitates, which was further dissolved in a mixture of acetyl chloride and methanol in an ice bath. After complete removal of solvent, the residue was dissolved in water and washed three times with chloroform. Then, the aqueous layer was collected and freeze-dried to obtain DCK powder. Next, ion-pairing complex of PMX with DCK (PMX/DCK) was prepared by dissolving 50 mg of PMX in 10 mL of deionized water. Then, 10 mL of DCK solution in deionized water (6.68 mg/mL) was prepared and added dropwise into the PMX solution at a molar ratio of 1:1, with vortex mixing after each addition. The solution mixture of PMX and DCK was freeze-dried at –70 °C to obtain PMX/DCK complex powder.
To enhance the intestinal membrane permeability of PMX, PMX/DCK-OP was prepared using the solid dispersion technique. Briefly, PMX (50 mg) was dissolved in P188 solution prepared by dissolving 500 mg of P188 in 10 mL of deionized water containing 375 µL of Labrasol. DCK solution was prepared in another tube by dissolving 66.8 mg of DCK in 8 mL of deionized water. At a molar ratio of 1:1, DCK solution was added dropwise into the PMX solution with regular vortex mixing. In addition, 80 µL of NaOH (1 M) was added with thorough mixing to maintain the PMX/DCK mixture at pH 7. The mixture was then freeze-dried at –70 °C to completely remove the water and obtain PMX/DCK-OP in powder form. Separately, to explore the role of DCK in enhancing the intestinal membrane permeability of PMX, an oral powder formulation of PMX containing Labrasol and P188 (PMX-OP) was prepared. Briefly, 50 mg PMX was dissolved in 10 mL of P188 solution (50 mg/mL) containing 375 µL of Labrasol. The mixture was then well vortexed for 5 min and freeze-dried at –70 °C to completely remove water and obtain PMX-OP in powder form. Furthermore, to confirm micelle formation of PMX/DCK-OP in the aqueous phase, the powder formulation was dispersed in deionized water (1:20, w/v) and its droplet size, polydispersity index (PDI), and zeta potential were measured with a dynamic laser light scattering (DLS) analyzer (Zetasizer Nano ZS90; Malvern Instruments, Malvern UK) at 25 °C. The morphology and particle size of PMX/DCK-OP micelles were further determined using transmission electron microscopy (TEM). After negative staining of the PMX/DCK-OP dispersed in deionized water (1:100, w/v) with 2% (w/v) phosphotungstic acid solution, the sample solution was dropped on a copper grid and examined using high-resolution transmission electron microscopy (HR-TEM; JEM-200; JEOL, Tokyo, Japan).
In vitro artificial intestinal membrane permeability
The in vitro artificial membrane permeability of free PMX, PMX/DCK, PMX-OP, and PMX/DCK-OP was determined using the parallel artificial membrane permeability assay. Sample solution (200 µL) at a concentration equivalent to 200 µg/mL of PMX was added to each well of the donor plates, which were prepared by dilution of free PMX, PMX/DCK, PMX-OP, and PMX/DCK-OP with phosphate-buffered saline (PBS, pH 6.8). We added 300 µL of PBS (pH 6.8) to each well of the receptor plates. After 5 hr incubation at room temperature, the plates were disassembled and samples were withdrawn from both the donor and acceptor plates. The concentration of PMX that permeated through the artificial membrane was measured by HPLC at 254 nm with a C18 column (4.6×250 mm, 5 µm, 100 Å) at 25 °C. The mobile phase consisted of water (pH 3.5 adjusted with phosphoric acid)-acetonitrile (80:20, v/v) and the flow rate was optimized to 1 mL/min.
The effective permeability (Pe) of each sample was calculated using the formula:
(1)
where Pe is the permeability (cm/s), S is the effective filter area (0.288 cm2), VD is the volume of the donor well (0.2 mL), VR is the volume of the receptor well (0.3 mL), t is the total time of incubation in seconds, CR(t) denotes the concentration of the drug in the receptor well at time t, and Cequilibrium represents [CD(t)×VD+CR(t)×VR]/(VD+VR), where CD(t) denotes the concentration of the drug in the donor well at time t.16
In vitro permeability across a Caco-2 monolayer
To determine the apparent permeability (Papp) of PMX, PMX/DCK, PMX-OP, and PMX/DCK-OP, Caco-2 cell monolayers were formed by seeding Caco-2 cells (ATCC® HTB-37™; American Type Culture Collection, Manassas, VA, USA), at a density of 1×105 cells/well, on polystyrene Transwell membranes in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin, followed by culture for 14 to 16 days. Media in the apical and basolateral compartments were replaced every other day until the transepithelial electrical resistance (TEER) reached >350 Ω·cm2. To investigate transcellular and ASBT-mediated transport, the culture medium in both the apical and basolateral compartments was removed and replaced with prewarmed HBSS. HBSS in the apical compartment was then replaced with 0.1 mL of PMX, PMX/DCK, PMX-OP, or PMX/DCK-OP diluted in HBSS (equivalent to 100 µg/mL PMX), and that in the basolateral compartment was replaced with 0.6 mL of fresh HBSS followed by incubation of the plates at 37 °C. Samples (0.1 mL) from each basolateral compartment were withdrawn at predetermined time points of 0, 0.5, 1, 2, 3, 4, and 5 hrs and replaced with the same volume of fresh HBSS. The collected samples were then filtered through polyvinylidene difluoride (PVDF) membrane filters (0.45 µm), and the concentrations of PMX in the samples were determined by HPLC with a UV detector at 254 nm, as described above.
The Papp for PMX in various formulations was calculated as follows:
(2)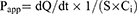
where dQ/dt represents the linear appearance rate of mass on the basolateral side (µg/s), S indicates the surface area of the monolayer (cm2), and Ci is the initial concentration of PMX on the apical side (µg/mL).26
Intestinal transport mechanism of PMX/DCK-OP
Next, we examined the role of ASBT in the permeation of PMX/DCK-OP, as well as the endocytosis pathways of PMX/DCK-OP micelles in the aqueous phase. Before performing the intestinal transport study, to determine the appropriate Act D concentration, the in vitro cytotoxicity assay results of Act D using Caco-2 cells were evaluated using the Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Rockville, MD, USA). Caco-2 cells were seeded at a density of 1×104 cells/well in 200 µL DMEM with 10% FBS in 96-well plates and cultured for 24 h. The cells were then treated with serially diluted sample solutions of 1, 2, 3.2, and 6 µM Act D in HBSS for 5 hrs. To evaluate the cytotoxic effect of Act D on the Caco-2 cells, a 10-µL WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt) solution was added to each well and incubated for 1 hr. The absorbance was then measured using a microplate reader (Multimode Plate Reader; PerkinElmer Inc., Waltham, MA, USA) at 450 nm. The results obtained for the treated cells are expressed as the percentage of viable cells compared to untreated cells. After determining the Act D concentration showing no toxic effect, Caco-2 cells were cultured on Transwell membranes and the medium in the apical and basolateral compartments was replaced every other day until the TEER value of the Caco-2 cell monolayer reached >350 Ω·cm2, to establish the in vitro transcellular model. To investigate the possible transcellular pathways of PMX/DCK-OP transport, 0.1 mL of HBSS containing inhibitors of the specific cellular uptake pathway (Table 1) and 0.6 mL of HBSS were then added to the apical and basolateral compartments of each well, respectively. After preincubation at 37 °C for 30 mins, the solution in the apical compartment was replaced with 0.1 mL of PMX/DCK-OP diluted in HBSS (equivalent to 100 µg/mL PMX) with corresponding inhibitors, and that in the basolateral compartment was replaced with 0.6 mL of fresh HBSS followed by incubation at 37 °C. Then, 200 µL of sample solution was withdrawn at 0, 0.5, 1, 2, 3, 4, and 5 hrs from the basolateral compartment of each well and replaced with the same volume of fresh HBSS.26,27
 |
Table 1 Inhibitors with different functions used in the transport study and their concentrations |
In addition, to evaluate the effects of TJ opening on the transport of PMX/DCK-OP, the extracellular Ca+2 chelating agent, EGTA, was applied to reversibly open the intracellular TJs in the Caco-2 cell monolayer. Moreover, to examine the effects of EGTA in combination with all inhibitors except cyclosporine A on the transport of PMX/DCK-OP, a Caco-2 cell monolayer with TEER value >350 Ω·cm2 was treated with 0.1 mL of HBSS (Ca+2-free medium) containing 2.5 mM EGTA with or without all inhibitors except cyclosporine A. After filling the basolateral side with 0.6 mL of HBSS (Ca+2-free medium), the Transwell membranes were preincubated at 37 °C for 45 mins and the integrity of the cell monolayer was monitored by measuring the TEER value as ≤70 Ω·cm2, to confirm opening of the junctional complex. The medium in the apical compartment was then replaced with 0.1 mL HBSS (with 1.8 mM Ca+2) containing PMX/DCK-OP (equivalent to 100 µg/mL PMX), alone or with all inhibitors except cyclosporine A, and 0.6 mL of HBSS (with 1.8 mM Ca+2) was added to the basolateral side of each well, followed by incubation at 37 °C for a further 5 hrs. At predetermined time points, 0.2 mL of medium on the basolateral side was withdrawn and replaced with the same volume of fresh HBSS. In addition, the integrity of the monolayer was checked by measuring the TEER value, which returned to ≥300 Ω·cm2 at 2 and 5 hrs after drug loading, respectively, to identify restoration of the transmembrane resistance.28
All collected samples were then filtered through PVDF membrane filters (0.45 µm) and the concentration of PMX that permeated across the Caco-2 cell monolayer was determined by HPLC at 254 nm as described above. Finally, the cumulative permeated amount of PMX from the PMX/DCK-OP was plotted as a function of time and Papp was determined from the linear slope of the plot using Equation (2).
In vivo pharmacokinetic study in rats
To evaluate the beneficial effects of ion-pairing complex formation of PMX with DCK on intestinal absorption, 400 µL of PMX in water (25 mg/kg), PMX/DCK in water (25 mg/kg PMX), or PMX/DCK-OP (25 mg/kg PMX) was orally administered to rats. In addition, 150 µL of an aqueous solution of PMX (5 mg/kg) was injected via the tail vein to evaluate the oral bioavailability. Next, blood samples of 150 µL were collected from the retroorbital plexus at predetermined time intervals into heparinized tubes, mixed with 50 µL of 3.8% sodium citrate solution, and immediately centrifuged (2,500×g, 15 mins, 4 °C). The plasma samples were separated and kept frozen at −70 °C until analysis. Liquid chromatography/mass spectrometry (LC/MS) was performed to determine the concentration of PMX in the plasma, as described previously.29 Briefly, 300 µL of 2% NH4OH in deionized water and 100 µL of standard solution were added to the plasma samples that were defrosted and centrifuged at 2,500×g for 5 mins at 4 °C, followed by vortex mixing. Next, 10 µL of DAMPA (5 µg/mL) was added to each sample as an internal standard, vortex-mixed for 2 mins, and subjected to solid-phase extraction using a Plexa Bond Elut PAX cartridge (30 mg, 1 mL; Agilent Technologies, Santa Clara, CA) in accordance with the manufacturer’s instructions. The cartridges were conditioned with 500 µL of methanol followed by 500 µL of deionized water, and standards and samples were loaded into the cartridges. After the unadsorbed samples and impurities in the cartridges were washed with 500 µL each of deionized water and methanol, elution of the adsorbed samples was performed with 2×250 µL of 5% formic acid prepared in methanol. The eluate was dried using a centrifugal evaporator (Genevac Ltd., Ipswich, UK), which was then reconstituted with 100 µL of 5% formic acid in methanol. The concentration of PMX in the reconstituted samples was determined using an Agilent 6120 Quadruple LC/MS system with a Phenomenex Luna C18 column (100×2 mm, 3 µm) as the stationary phase and acetonitrile:0.34% formic acid (15:85, v/v) as the mobile phase, at a flow rate of 0.2 mL/min. PMX and internal standard were ionized using an electrospray ionization (ESI) source in positive ion mode under the following source conditions: capillary voltage of 3.5 kV; drying gas flow rate of 3.1 L/min; and drying gas temperature of 300 °C. Quantitative analysis of protonated molecular ions was performed at ([M + H]+=428) and ([M + H]+=326.1) for PMX and DAMPA, respectively.
In vivo tumor growth inhibition efficacy in mice
The antitumor effects of orally administered PMX/DCK-OP, alone or in combination with anti-PD1 administered intraperitoneally, were investigated in BALB/c mice subcutaneously inoculated with B16F10 cells at a density of 1×106 cells (in 100 µL of PBS, pH 7.4). Mice with an average tumor volume of 40–60 mm3 were randomly divided into five groups (10 mice per group): control (untreated), PMX-IV (twice-a-week intravenous administration of 20 mg/kg PMX), Anti-PD1 (once per 3 days intraperitoneal administration of 10 mg/kg anti-PD1), PMX/DCK-OP (once-a-day oral administration equivalent to 20 mg/kg PMX), and combination of PMX/DCK-OP and Anti-PD1 (once-a-day oral administration equivalent to 20 mg/kg PMX and once per 3 days intraperitoneal administration of 10 mg/kg anti-PD1). The body weight and tumor volumes of mice in all five groups were recorded every 2 days. Tumor volumes in each mouse were calculated based on the following formula: 0.52×w2×l, where w and l represent the width and length of the tumor, respectively. Two weeks after oral administration, mice were sacrificed and tumor masses were measured. Induction of apoptosis in the isolated tumor tissues was detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. Moreover, microvessel expression and cell proliferation were analyzed using epithelial cell marker CD31 and proliferating cell nuclear antigen (PCNA), respectively.
Pharmacokinetic and statistical analyses
Pharmacokinetic parameters were assessed using a non-compartmental method in WinNonlin® software (ver. 5.3; Pharsight Corporation, Mountain View, CA, USA). One-way analysis of variance (ANOVA) followed by Tukey’s multiple-comparison test was used to compare more than two mean values. All data were expressed as means ± standard deviation. In all analyses, a P-value less than 0.05 was considered statistically significant.
Results and discussion
Preparation and characterization of PMX/DCK-OP
To enhance the lipophilicity and intestinal membrane permeability of PMX, ion-pairing complex and oral powder formulations of PMX and DCK were prepared as shown in Figure 1A. The optimal PMX/DCK-OP diluted in deionized water showed a particle size and PDI of 134±0.098 nm and 0.114±0.005, respectively, with a zeta potential of 16.3±1.00 mV. The uniformly narrow particle size and high zeta potential supports the stable formation of micelles in the aqueous phase. In addition, TEM images of PMX/DCK-OP dispersed in water confirmed the formation of spherical and homogenous nano-sized micelles with diameters <100 nm, which was smaller than the size observed using a DLS analyzer (Figure 1B). The difference in particle size of the PMX-DCK-OP micelles may be caused by the difference in sample preparation between DLS and TEM measurements. Unlike DLS, a sample for TEM is prepared in a dried state and the loosely aligned micelles can shrink during drying. The presence of Labrasol and P188 as surfactant and dispersing agent, respectively, in the optimum powder formulation were expected to increase the long-term stability and oral bioavailability of PMX. Moreover, Labrasol showed a hydrophilic-lipophilic balance (HLB) value of 14, was less toxic with high tolerance, and enhanced the intestinal absorption of hydrophilic drugs.30,31 In addition, use of P188 in the formulation resulted in self-assembly into micelles in the presence of aqueous phase at concentrations above the critical micellar concentration (CMC). Structurally, P188, which is an amphiphilic triblock copolymer of poly(ethylene oxide) and poly(propylene oxide), consists of a hydrophobic central core and hydrophilic outer core that can accommodate lipophilic and water soluble drugs, respectively. Furthermore, P188 has been widely used as a dispersant and solubilizing agent, and is approved for oral drug delivery.32,33
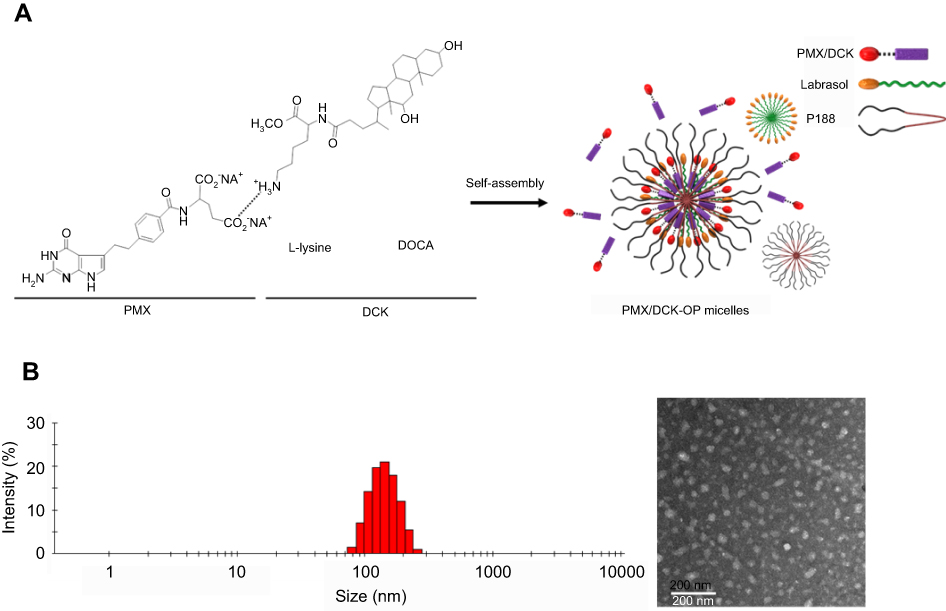 |
Figure 1 (A) Schematic illustration of ion-pairing complex formation between PMX and DCK, and self-assembled micelle formation of PMX/DCK-OP in the aqueous phase. (B) Transmission electron micrograph of PMX/DCK-OP micelles. Scale bar represents 200 nm. Abbreviations: PMX, pemetrexed; DCK, Nα-deoxycholyl-L-lysyl-methylester; PMX/DCK, ion-pairing complex between PMX and DCK; PMX/DCK-OP, oral powder formulation of PMX/DCK. |
In vitro permeability study of PMX/DCK-OP
The artificial intestinal membrane permeability of PMX was significantly increased after ion-pairing complex formation with DCK and formulation of PMX/DCK-OP. The Pe of PMX after ionic complex formation with DCK was increased by 36.2- and 7.29-fold compared to free PMX and PMX-OP, respectively (Table 2). The enhanced permeability of PMX/DCK may be due to the increased lipophilicity of PMX after complex formation with the oral permeation enhancer, DCK.34,35 On the other hand, the permeability of PMX was improved 4.97-fold compared to that of free PMX after formulation with P188 and Labrasol. However, the Pe of PMX/DCK-OP was increased by 3.87-, 28.2-, and 140-fold compared to PMX/DCK, PMX-OP, and PMX, respectively. Therefore, the enhanced artificial membrane permeability of PMX/DCK-OP could be ascribed to the synergistic action of DCK, Labrasol, and P188 in the membrane permeation of PMX. Moreover, the presence of Labrasol and P188 in the oral powder formulation may have further increased the amphiphilicity of PMX, thus facilitating passive diffusion.
 |
Table 2 Effective and apparent permeability of PMX, PMX/DCK, and PMX/DCK-OP |
The in vitro permeability data of PMX, PMX/DCK, PMX-OP, and PMX/DCK-OP across Caco-2 cell monolayers are presented in Table 2. The Papp of PMX after ion-pairing complex formation with DCK was 3.35- and 1.73-fold higher than those of free PMX and PMX-OP, respectively. The improved permeability of PMX may have been due to the increased membrane flexibility and opening of transient TJs caused by binding of bile acids to the calcium channels, which enhanced paracellular drug transport.36 Moreover, DOCA induces phosphorylation of epidermal growth factor receptor, occludin dephosphorylation, and occludin redistribution, which modulates intestinal membrane permeability.37 In addition, transcellular transport of hydrophilic drugs could be enhanced by the incorporation of bile acids into the cell membrane; this forms reverse micelles and results in the creation of aqueous channels, thereby increasing membrane permeability.38 The permeability of PMX across a Caco-2 cell monolayer was only improved 1.94-fold by adding P188 and Labrasol, while the Papp of PMX/DCK-OP was further increased by 2.46-, 4.26-, and 8.26-fold compared to those of PMX/DCK, PMX-OP, and free PMX, respectively. This increase in the Papp of PMX/DCK-OP may have been due to the formation of PMX/DCK micelles in combination with Labrasol and P188, which could in turn increase drug absorption through endocytosis and micropinocytosis.39 In addition, nonionic surfactants, such as Labrasol and P188, can modify intestinal membrane fluidity, followed by alteration of the epithelial barrier function.40 Labrasol has been shown to act as a permeation enhancer by disrupting TJs, causing redistribution of zonula occludens-1 (ZO-1).41 Therefore, the synergistic activity of DCK, Labrasol, and P188 may have significantly increased the Papp of PMX/DCK-OP. However, further permeability studies using gastrointestinal tissues are required to determine the stability and association constant of PMX/DCK and its oral formulation in the gastric and intestinal fluids. In addition, future studies should investigate the effects of ion-pairing complex formation with DCK and formulation with Labrasol and P188 on the intestinal permeability of PMX, and the absorption-enhancing mechanism of PMX/DCK-OP.
Intestinal transport mechanism of PMX/DCK-OP
To determine the transport pathway involved in the active transcellular transport of PMX/DCK-OP, Caco-2 monolayers were pretreated with various biochemical inhibitors of different pathways (Figure 2). In the aqueous phase, PMX/DCK-OP may exist as PMX/DCK, which can be further incorporated into micelles together with P188 and Labrasol, as confirmed in the preparation and characterization section. The main goal of ionic complex formation of PMX with DCK was to increase the lipophilic properties and oral bioavailability of PMX via the bile acid transporter-mediated approach. The transporter specificity, affinity, and capacity are the three major factors taken into consideration in prodrug design. Bile acids are mostly reclaimed in the distal ileum by ASBT.42 Moreover, ASBT exhibits high capacity and affinity for native bile acid, suggesting that ASBT may be a useful prodrug target.15 Coupling of PMX with DCK can increase the uptake of PMX through ASBT-mediated transport. To verify the involvement of ASBT in the uptake of PMX/DCK, we inhibited ASBT using Act D at a concentration that had no cytotoxic effect on Caco-2 cells; the cell viability was greater than 95% and the concentration ranged from 1 to 6 µM Act D. The cell viability at 3.2 µM Act D was 105±6.81% (Figure S1).43 As a result, the Papp of PMX/DCK-OP was markedly decreased, by 75.4%, compared to the control (without inhibitors) (Figure 2A). This observation was suggestive of active involvement of ASBT in the uptake of PMX/DCK-OP. After interacting with ASBT, PMX/DCK from PMX/DCK-OP may have followed a pathway similar to that of bile acid: entry into the enterocytes; binding with intestinal bile acid-binding protein (IBABP); shuttling across the enterocytes to the basolateral membrane; and effusing into the portal circulation via the recently identified heteromeric transporters, organic solute transporter alpha and beta (OSTα and OSTβ) (Figure 3).19,44
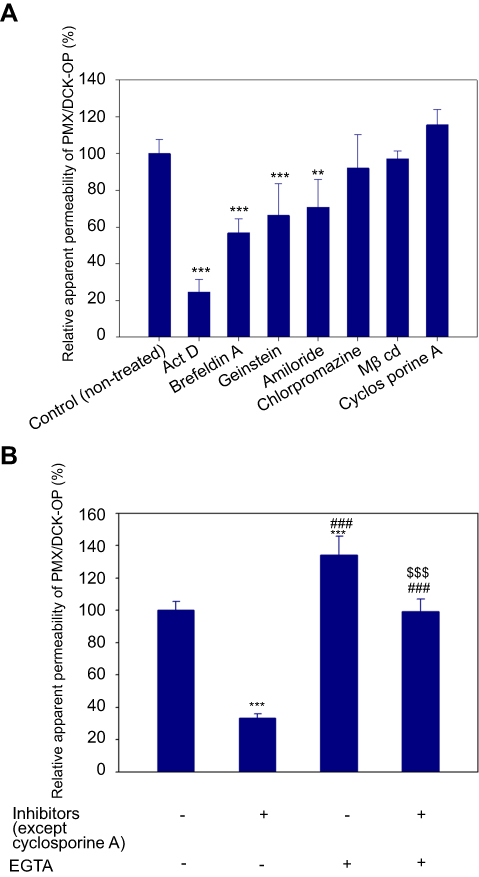 |
Figure 2 (A) Relative apparent permeability of PMX/DCK-OP across a Caco-2 cell monolayer after incubation with various transport inhibitors and (B) with or without EGTA in the presence or absence of all inhibitors except cyclosporine A. Notes: Each value represents the mean ± standard deviation (n=6). **P<0.01, ***P<0.001 compared to the Papp of the untreated control group. ###P<0.001 compared to the apparent permeability of PMX/DCK-OP after treatment with all inhibitors except cyclosporine A in the absence of EGTA. $$$P<0.001 compared to the Papp of PMX/DCK-OP after treatment with EGTA in the presence of all inhibitors except cyclosporine A.Abbreviations: PMX, pemetrexed; DCK, Nα-deoxycholyl-L-lysyl-methylester; PMX/DCK, ion-pairing complex between PMX and DCK; PMX/DCK-OP, oral powder formulation of PMX/DCK; Act D, actinomycin D; MβCD, methyl-β-cyclodextrin; ASBT; EGTA, ethylene glycol-bis-(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid; Papp, apparent permeability of PMX/DCK-OP across a Caco-2 cell monolayer. |
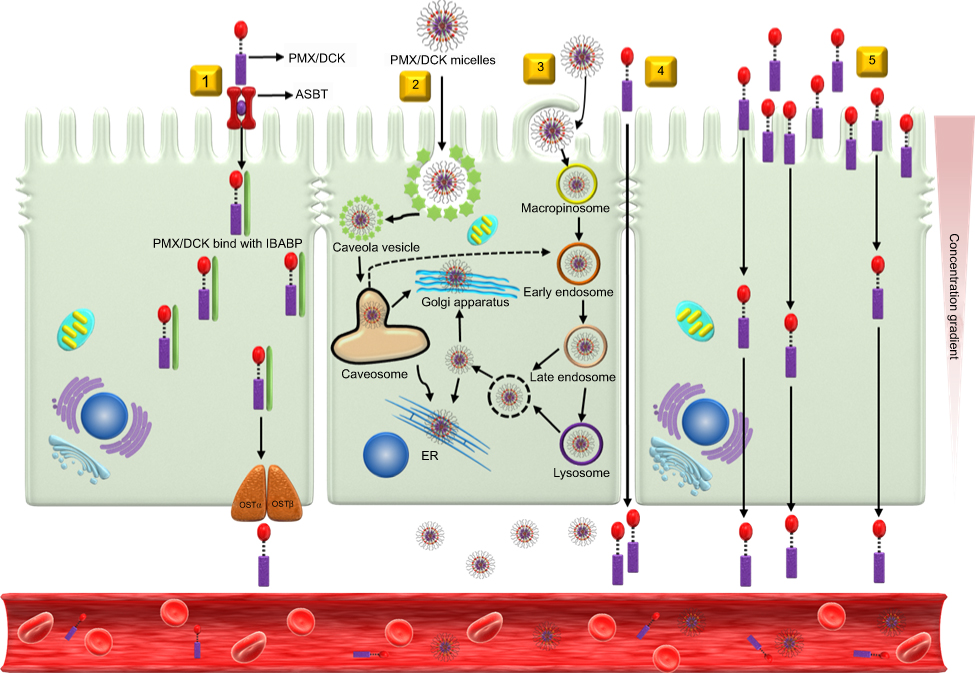 |
Figure 3 Schematic diagrams of possible specific and nonspecific transport pathways, and predicted intracellular trafficking and transcytosis of PMX/DCK-OP across an intestinal epithelium: (1) ASBT-mediated transport; (2) caveola/lipid raft-mediated endocytosis; (3) macropinocytosis; (4) paracellular transport; and (5) transcellular passive diffusion. Abbreviations: PMX, pemetrexed; DCK, Nα-deoxycholyl-L-lysyl-methylester; PMX/DCK, ion-pairing complex between PMX and DCK; PMX/DCK-OP, oral powder formulation of PMX/DCK; ASBT, apical sodium-dependent bile acid transporter; IBABP, intestinal bile acid-binding protein; OSTα, organic solute transporter alpha; OSTβ organic solute transporter beta; ER, endoplasmic reticulum. |
In addition to the ASBT-mediated transport, PMX/DCK-OP micelles themselves can be internalized by endocytosis, because they are the major route for nanoparticle transport across the intestinal epithelium by engulfment in membrane invaginations forming intracellular vesicles. First, we used inhibitors of both clathrin- and caveola/lipid raft-mediated endocytosis to explore their roles in the uptake of PMX/DCK-OP. After treatment with chlorpromazine, an inhibitor of clathrin-mediated endocytosis, the Papp of PMX/DCK-OP was only reduced by 7.95% in comparison to the control (without inhibitors), indicating the clathrin-mediated endocytosis plays a trivial role in the uptake of PMX/DCK-OP micelles (Figure 2A). On the other hand, genistein, a caveola/lipid raft-mediated endocytosis inhibitor, significantly decreased the Papp by 33.6% compared to the control (without inhibitors) (Figure 2A). Genistein is a commonly used broad-spectrum inhibitor of protein tyrosine kinase (PTK), which catalyzes tyrosine phosphorylation and activates multiple signal transduction pathways, including endocytosis regulation.45 Genistein has also been reported to be closely related with the actin network and recruitment of dynamin II, both of which are indispensable for caveola/lipid raft-mediated uptake.46 Therefore, the considerable decline in Papp in the presence of genistein confirmed the involvement of the caveola/lipid raft-mediated pathway in the uptake of PMX/DCK-OP. However, there was no significant difference in the Papp after treatment of Caco-2 cells with MβCD, a caveola/lipid raft-mediated endocytosis inhibitor (2.37% decrease in Papp of PMX/DCK-OP), which may be due to the difference in its mode of action. MβCD generally acts as a cholesterol-depleting agent and effectively inhibits caveola/lipid raft-mediated endocytosis.47 However, it was reported previously that the addition of cholesterol together with MβCD may exhaust its function due to its specific interaction with cholesterol, leading to a decrease in the inhibitory efficacy of MβCD.27 Therefore, in the present study, the inhibitory activity of MβCD may have decreased due to the presence of DOCA, a cholesterol derivative, in the DCK molecule. Macropinocytosis is another well-defined uptake mechanism that may be involved in the uptake of PMX/DCK-OP micelles. During this process, a large volume of fluid consisting of nanoparticles was engulfed by ruffling the plasma membrane to form cargo-loaded macropinosomes.48 Here, the inhibitor of macropinocytosis, amiloride, significantly decreased the uptake of PMX/DCK-OP (by 29.0%) compared to the control (without inhibitors), revealing the active role of macropinocytosis in the absorption of PMX/DCK-OP by the enterocytes (Figure 2A).
After entry into the cell, nanoparticles generally (i) are either captured by endosomes and consequently degraded by the lysosome, (ii) escape into the cytoplasm, or (iii) undergo recycling. In addition, nanoparticles can be localized to various organelles via endocytic vesicles and the complex intracellular trafficking machinery.49 Therefore, during the processes of endocytosis and macropinocytosis, PMX/DCK-OP micelles may be engulfed in membrane invaginations forming caveosomes and macropinosomes, and then delivered to various organelles, such as the early endosome, late endosome, and lysosome, after which they escape to the endoplasmic reticulum (ER) and Golgi apparatus, and enter the enterocytes.49,50 During this process, endosomal escape may be crucial for the intracellular delivery of PMX/DCK-OP micelles because of the digestive enzymes in the lysosome. The PMX/DCK-OP micelles escaping from the endosomes can enter the cytoplasm or exit the enterocytes via the ER and Golgi apparatus (Figure 3). However, the cytoplasmic route is usually not preferred because most drugs must enter the blood circulation to exert their pharmacological action. In addition, the nanomaterials with the required intracellular delivery density can utilize ER and Golgi routes, as well as the retrograde trafficking pathway.
It has been reported that both the Golgi complex and ER are important regulators of the secretory ER/Golgi pathway, as well as the endocytic recycling pathway.51 Therefore, to clarify whether the ER-to-Golgi apparatus is involved in the intracellular trafficking of PMX/DCK-OP micelles, we examined the effects of brefeldin A, a specific inhibitor of the secretory pathway from the ER to the Golgi apparatus,52 on the permeation of PMX/DCK-OP. The results indicated that the Papp of PMX/DCK-OP was significantly decreased (by 43.2%) compared to the control (without inhibitors) after treatment of the cells with brefeldin A, suggesting that the ER and Golgi apparatus are actively involved in the exocytosis of PMX/DCK-OP micelles (Figure 2A). Furthermore, the involvement of the ER-to-Golgi apparatus in the intracellular transport of PMX/DCK-OP micelles clarified the roles of endocytosis and macropinocytosis; lysosomal escape of PMX/DCK-OP micelles was also observed (Figure 3).
P-gp is responsible for the multidrug resistance phenotype of cancer, by mediating the ATP-dependent efflux of anticancer drugs out of the cell.53 To examine the effects of P-gp-mediated efflux on the transport of PMX/DCK-OP, we used cyclosporine A as an inhibitor of P-gp.54,55 The Papp of PMX/DCK-OP was increased by 16.0% compared to the control (without inhibitors), but the difference was not statistically significant (Figure 2A). Therefore, P-gp-mediated efflux does not have a significant influence on PMX/DCK-OP uptake.
The oral formulation can also facilitate passive diffusion and paracellular drug transport. To understand the involvement of passive diffusion and paracellular transport in the uptake of PMX/DCK-OP, we examined the effects of EGTA on the Papp of PMX/DCK-OP. EGTA is a selective calcium chelator, which is used to bind free extracellular Ca+2 ions and can reversibly open the intercellular TJs of the Caco-2 monolayer.28 As shown in Figure 2B, when the Caco-2 monolayer (without EGTA) was treated with all inhibitors except cyclosporine A, the Papp of PMX/DCK-OP decreased by 66.7% compared to the control (without inhibitors and EGTA), indicating that 33.3% of PMX/DCK-OP was permeated by passive diffusion and paracellular transport (Figure 2B). As described above, the passive diffusion of PMX through the PAMPA membrane was markedly improved after ion-pairing complex formation with DCK, followed by formulation with Labrasol and P188. Furthermore, the Papp of PMX/DCK-OP across the EGTA-treated Caco-2 monolayer (without inhibitors) increased by 34.0% in comparison to the control (without inhibitors and EGTA), suggesting the importance of opening the TJs to increase the uptake of PMX/DCK-OP (Figure 2B). On the other hand, the Papp of EGTA-treated Caco-2 monolayer (with all inhibitors except cyclosporine A) was similar to the control (without inhibitors and EGTA) but increased by 66.0% compared to the EGTA-untreated Caco-2 monolayer (with all inhibitors except cyclosporine A). This observation also supported a significant role of EGTA in increasing the transport of PMX/DCK-OP across the Caco-2 monolayer via reversibly opening the intracellular TJs. Therefore, it is clear that reversible opening of intracellular TJs can increase the transport of PMX/DCK-OP, and that the incorporation of components capable of opening TJs in the formulation of PMX/DCK-OP can enhance the intestinal permeation of PMX (Figure 3). The presence of Labrasol and bile acid, which open TJs, in PMX/DCK-OP may have favored the permeation of PMX/DCK-OP through the paracellular route.41,56,57
In vivo oral absorption in rats
The pharmacokinetic parameters in rats after intravenous administration of PMX, and oral administration of PMX, PMX/DCK, and PMX/DCK-OP, are shown in Figure 4A and B, respectively. The maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) achieved after oral administration of PMX/DCK (equivalent to 25 mg/kg PMX) were 4.58±4.63 µg/mL and 4.13±2.29 µg·h/mL, respectively, which were 3.57- and 1.76-fold higher than those of oral PMX alone, suggesting significant improvement of intestinal absorption of PMX and resulting in 75.3% greater oral bioavailability of PMX/DCK than PMX (Table 3). The improvement of oral bioavailability was supported by the results of the mechanistic study, which demonstrated significant reduction of Papp after inhibition of ASBT by Act D, suggesting active involvement of bile acid transporter in the uptake of PMX/DCK. Moreover, the significant increases in in vitro artificial membrane and Caco-2 cell monolayer permeability of PMX/DCK compared to PMX confirmed the major role of paracellular and transcellular transport in the enhancement of oral bioavailability. In addition, DOCA induced disruption of intestinal barrier function by dephosphorylation of occludin, and cytoskeletal rearrangement of epithelial TJs may have enhanced the oral absorption of PMX/DCK.37,56–58 Furthermore, the Cmax and AUClast values for PMX/DCK-OP were 1.29- and 2.25-fold higher than those of PMX/DCK, respectively. Therefore, the oral bioavailability of PMX/DCK-OP was 294% higher than that of PMX solution. The time to reach the maximum concentration (Tmax) was 0.63±0.25 and 0.50±0.00 hrs after oral administration of PMX and PMX/DCK-OP, respectively, suggesting the immediate dissolution of PMX/DCK from the powder formulation, followed by rapid absorption of PMX/DCK or PMX/DCK-OP micelles across the intestinal membrane. The enhanced oral absorption of PMX in PMX/DCK-OP proceeds as follows: (i) uptake of free PMX/DCK by the bile acid transporter, which was confirmed by inhibition of bile acid transporter; and (ii) significant decrease in Papp after inhibition of caveola/lipid raft-mediated endocytosis by genistein, suggesting internalization of PMX/DCK-OP micelles by endocytosis; 3) formation of PMX/DCK-OP macropinosomes that entered the enterocytes (which also revealed the prominent role of macropinocytosis in enhanced oral absorption); and 4) PMX/DCK-OP uptake via paracellular transport and passive diffusion, which was confirmed by reversible opening of TJs by EGTA and in vitro artificial membrane permeability.
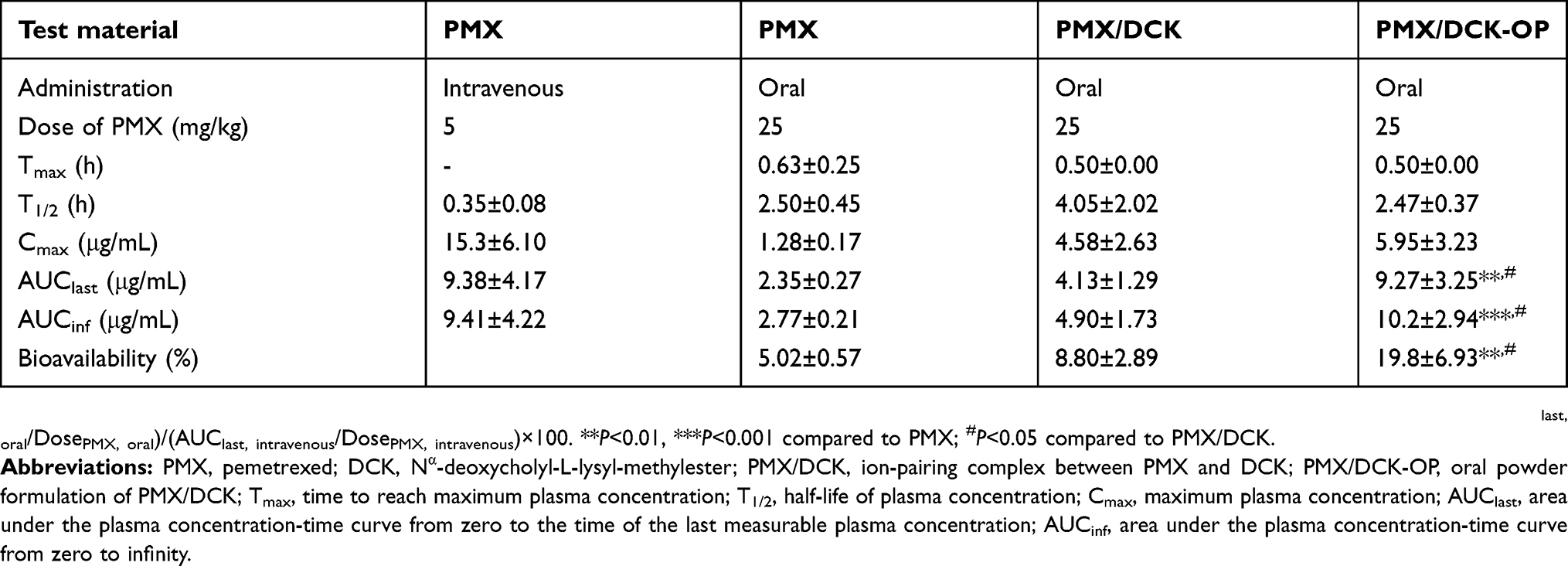 |
Table 3 Pharmacokinetic parameters of PMX in rats after intravenous injection of PMX and oral administration of PMX, PMX/DCK, or PMX/DCK-OP |
 |
Figure 4 Venous plasma concentration-time profiles of PMX after (A) single intravenous administration of PMX (5 mg/kg) and (B) oral administration of PMX (25 mg/kg), PMX/DCK (equivalent to 25 mg/kg of PMX), or PMX/DCK-OP (equivalent to 25 mg/kg of PMX) in aqueous solution to rats. Notes: Each value represents the mean ± standard deviation (n=4 for each group). Abbreviations: PMX, pemetrexed; DCK, Nα-deoxycholyl-L-lysyl-methylester; PMX/DCK, ion-pairing complex between PMX and DCK; PMX/DCK-OP, oral powder formulation of PMX/DCK. |
In vivo tumor growth inhibitory effect of orally administered PMX/DCK-OP
Based on the above findings, we used the B16F10 cell-bearing mouse tumor model to investigate the efficacy of combined oral administration of PMX/DCK-OP with intraperitoneal anti-PD1 in terms of inhibiting tumor progression. Compared to the untreated control group, the tumor growth rates were significantly delayed in mice treated with PMX/DCK-OP or PMX-IV alone (maximally suppressed by 35.5% and 38.4%, respectively; Figure 5A). The improved tumor growth inhibition efficacy may have been due to the metronomic scheduling of PMX/DCK-OP and PMX-IV, which can activate both innate and adaptive immunity, induce tumor dormancy, suppress tumor angiogenesis, and render tumor cells more sensitive to T cell-mediated lysis.1 However, PMX/DCK-OP showed similar antitumor efficacy as PMX-IV, although its bioavailability was approximately 20% that of PMX-IV. This may be due to the dosing schedule of PMX/DCK-OP, which allows the drug concentration in plasma to remain constant, resulting in continuous exposure of tumor endothelial cells to the chemotherapeutic agent and allowing them to proliferate slowly, and preventing them from repairing and recovering. PMX-IV dosing (twice-weekly intravenous administration of 20 mg/kg PMX) may not be sufficient to maintain the drug concentration around the tumor tissue, resulting in a drug-free period and allowing the tumor cells to regrow and mobilize circulating endothelial progenitor cells, thus eliciting tumor neovascularization. However, further mechanistic investigations accompanied by dosing studies for metronomic PMX/DCK-OP are required to explore antitumor activity according to anti-angiogenic and immunomodulation effects. On the other hand, after intraperitoneal administration of anti-PD1, the tumor volume was significantly suppressed, by 1.35- and 2.08-fold, compared to the PMX/DCK-OP and control groups, respectively. In addition, the tumor volume showed a more significant reduction after combined PMX/DCK-OP and intraperitoneal anti-PD1 treatment, which resulted in maximal reduction of tumor volume by 104% and 51.5% relative to PMX/DCK-OP and Anti-PD1, respectively. This synergistic tumor growth-inhibiting activity of chemotherapy with immunotherapy may have been due to enhancement of protective T cell responses and the antigen-presenting function of dendritic cells.59 However, oral administration of PMX/DCK-OP or combined treatment with PMX/DCK-OP and anti-PD1 did not affect the body weight of tumor-bearing mice (Figure 5B), suggesting that the administered drugs do not have systemic toxicity. The tumors isolated on day 14 after combined treatment with oral PMX/DCK-OP and intraperitoneal anti-PD1 also showed significant decreases in tumor mass, by 69.4% and 44.2% compared to the control and oral PMX/DCK-OP groups, respectively (Figure 5C and D). Langer et al performed an open-label phase 2 cohort study using anti-PD1 (pembrolizumab) along with intravenous carboplatin and PMX, and the results showed significantly improved anticancer efficacy compared to chemotherapy alone.60 Therefore, the metronomic combination of PMX/DCK-OP and intraperitoneal anti-PD1 was expected to play a synergistic role in cancer treatment. However, anticancer efficacy of intravenously administered PMX alone, or in combination with anti-PD1, must be demonstrated to confirm that the oral metronomic PMX/DCK-OP allows for a more favorable dosing schedule for synergistic anticancer effects with anti-PD1, via continuous exposure to the tumor endothelial cells, thus decreasing neoangiogenesis and boosting immune activity.
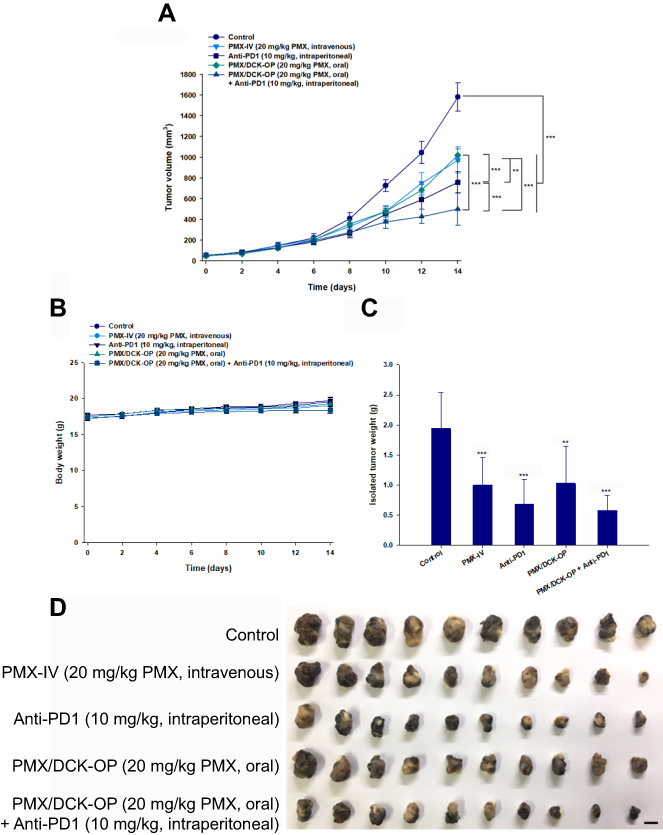 |
Figure 5 Efficacy of in vivo tumor growth inhibitory effects in B16F10 tumor-bearing mice after biweekly intravenous administration of 20 mg/kg PMX (PMX-IV), intraperitoneal administration of 10 mg/kg anti-PD1 (Anti-PD1) once every 3 days, once-daily oral administration of PMX/DCK-OP as 20 mg/kg PMX (PMX/DCK-OP), and combined treatment with once-daily oral administration of PMX/DCK-OP as 20 mg/kg PMX and intraperitoneal administration of 10 mg/kg anti-PD1 (PMX/DCK-OP+Anti-PD1) once every 3 days for 14 days. (A) Tumor volume in mice (**P<0.01, ***P<0.001). (B) Variation of body weight in mice during treatment. (C) Tumor weight in B16F10 tumor-bearing mice (**P<0.01, ***P<0.001 compared to the control group). (D) Photographs of tumors isolated from each group on day 15. Scale bar represents 10 mm.Notes: Statistics: one-way ANOVA followed by Tukey’s multiple-comparison test. Each value represents the mean ± standard deviation (n=10 for each group). Abbreviations: PMX, pemetrexed; DCK, Nα-deoxycholyl-L-lysyl-methylester; PMX/DCK, ion-pairing complex between PMX and DCK; PMX/DCK-OP, oral powder formulation of PMX/DCK. |
Histological evaluation of the tumor tissues from mice treated with PMX/DCK-OP, alone or in combination with anti-PD1 stained with PCNA, anti-CD31 antibody, and TUNEL (Figure 6) also showed similar trends. Tumors from mice treated with the combination of PMX/DCK-OP and anti-PD1 showed reduced numbers of PCNA-positive proliferative cells compared to the control and oral PMX/DCK-OP groups. In addition, the number of CD31-positive microvessels was markedly decreased in the combined treatment group compared to the control and intravenous PMX groups. In addition, TUNEL assay showed a similar tendency, with a significantly greater number of apoptotic cells in the combined treatment group compared to the oral PMX/DCK-OP and control groups.
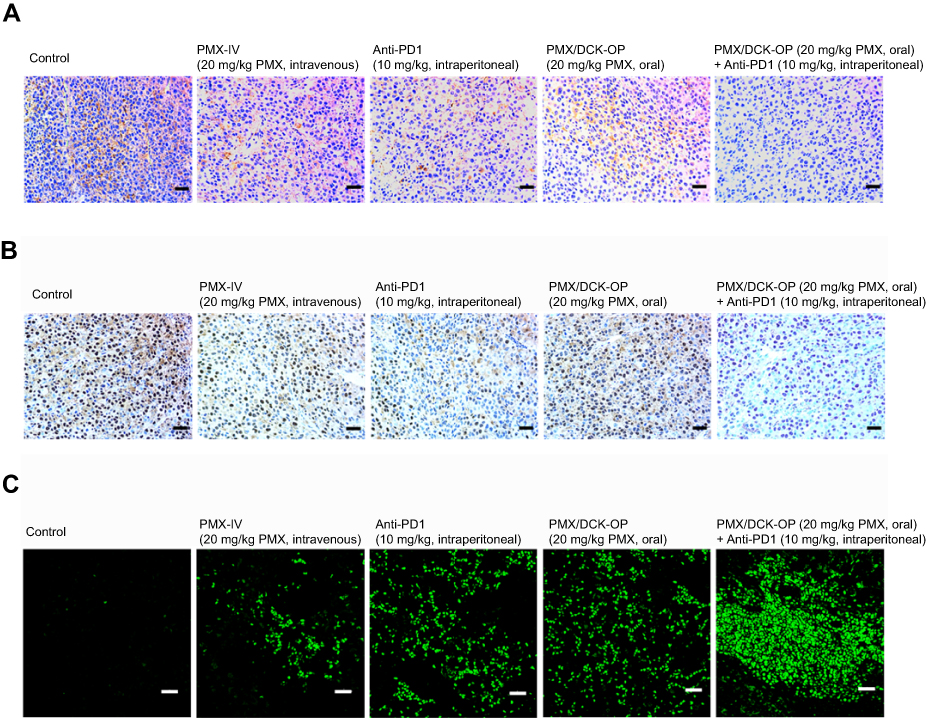 |
Figure 6 Representative cross-sectional images of isolated tumor tissues stained with (A) anti-CD31 antibody for microvessels (brown), (B) proliferating cell nuclear antigen (PCNA) for proliferating cells (brown), and (C) fluorescent terminal deoxynucleotidyl transferase-mediated dUPT nick end labeling (TUNEL) for apoptosis (green fluorescence) in the tumor tissues taken 14 days after treatment with various modes. Notes: The treatment modes were biweekly intravenous administration of 20 mg/kg PMX (PMX-IV), intraperitoneal administration of 10 mg/kg anti-PD1 (Anti-PD1) once every 3 days, once-daily oral administration of PMX/DCK-OP as 20 mg/kg of PMX (PMX/DCK-OP), and combined once-daily oral administration of PMX/DCK-OP as 20 mg/kg of PMX with once every 3 days intraperitoneal administration of 10 mg/kg anti-PD1 (PMX/DCK-OP + Anti-PD1). Scale bars represent 50 μm for anti-CD31 antibody and PCNA staining, and 20 μm for TUNEL staining, respectively. Abbreviations: PMX, pemetrexed; DCK, Nα-deoxycholyl-L-lysyl-methylester; PMX/DCK, ion-pairing complex between PMX and DCK; PMX/DCK-OP, oral powder formulation of PMX/DCK. |
Based on the findings of the present study, PMX/DCK-OP enhanced oral absorption, confirming the improvement in oral bioavailability of PMX with significant tumor growth suppression in combination with intraperitoneal anti-PD1 compared to PMX/DCK and anti-PD1. Therefore, PMX/DCK-OP may be suitable as a drug delivery carrier for PMX and exhibit synergistic activity with immunotherapy.
Conclusion
This study confirmed that PMX/DCK-OP enhances the intestinal membrane permeability and oral bioavailability of PMX. PMX/DCK-OP was prepared by ionic complex formation of PMX with DCK at a 1:1 molar ratio, along with Labrasol and P188 as dispersants. The optimum oral powder formulation improved the Papp of PMX by 8.26-fold compared to free PMX. The results also indicated that inhibition of bile acid transporters on a Caco-2 cell monolayer significantly curtailed the Papp of PMX/DCK-OP, suggesting that an ASBT-mediated pathway is the predominant mechanism underlying intestinal absorption of PMX/DCK-OP, followed by caveola/lipid raft-mediated endocytosis, macropinocytosis, passive diffusion, and paracellular transport. For these reasons, oral PMX/DCK-OP showed a 294% increase in oral bioavailability in comparison to free PMX, further confirming the role of bile acid derivative and dispersants used in the formulation. Moreover, the tumor growth was maximally suppressed after combined treatment with PMX/DCK-OP and intraperitoneal anti-PD1. These observations suggested the therapeutic potential of combined oral metronomic chemotherapy and immunotherapy for synergistic anticancer efficacy.
Acknowledgment
This study was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean Government Ministry of Science and ICT (MSIT; NRF-2017M3A9F5029656).
Disclosure
The authors report no conflicts of interest in relation to this work.
References
1. Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. 2015;3(5):436–443. doi:10.1158/2326-6066.CIR-15-0064
2. Maiti R. Metronomic chemotherapy. J Pharmacol Pharmacother. 2014;5(3):186–192. doi:10.4103/0976-500X.136098
3. Guengerich FP. Characterization of human cytochrome P450 enzymes. Faseb J. 1992;6(2):745–748. doi:10.1096/fasebj.6.11.1644261
4. Kuppens IE, Bosch TM, van Maanen MJ, et al. Oral bioavailability of docetaxel in combination with OC144-093 (ONT-093). Cancer Chemother Pharmacol. 2005;55(1):72–78. doi:10.1007/s00280-004-0864-4
5. Denies S, Cicchelero L, Van Audenhove I, Sanders NN. Combination of interleukin-12 gene therapy, metronomic cyclophosphamide and DNA cancer vaccination directs all arms of the immune system towards tumor eradication. J Control Release. 2014;187:175–182. doi:10.1016/j.jconrel.2014.05.045
6. Chen CA, Ho CM, Chang MC, et al. Metronomic chemotherapy enhances antitumor effects of cancer vaccine by depleting regulatory T lymphocytes and inhibiting tumor angiogenesis. Mol Ther. 2010;18(6):1233–1243. doi:10.1038/mt.2010.34
7. Shan F, Zhang B, Sun L, Xie L, Shen M, Ruan S. The role of combination maintenance with pemetrexed and bevacizumab for advanced stage nonsquamous non-small cell lung cancer: a systematic review and meta-analysis. Biomed Res Int. 2018;2018:5839081. doi:10.1155/2018/5839081
8. Rinaldi DA, Kuhn JG, Burris HA, et al. A phase I evaluation of multitargeted antifolate (MTA, LY231514), administered every 21 days, utilizing the modified continual reassessment method for dose escalation. Cancer Chemother Pharmacol. 1999;44(5):372–380. doi:10.1007/s002800050947
9. Hanauske AR, Chen V, Paoletti P, Niyikiza C. Pemetrexed disodium: a novel antifolate clinically active against multiple solid tumors. Oncologist. 2001;6(4):363–373. doi:10.1634/theoncologist.6-4-363
10. Thanki K, Gangwal RP, Sangamwar AT, Jain S. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170(1):15–40. doi:10.1016/j.jconrel.2013.04.020
11. Bansal T, Akhtar N, Jaggi M, Khar RK, Talegaonkar S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov Today. 2009;14(21–22):1067–1074. doi:10.1016/j.drudis.2009.07.010
12. Aungst BJ. Absorption enhancers: applications and advances. Aaps J. 2012;14(1):10–18. doi:10.1208/s12248-011-9307-4
13. Rahman MA, Harwansh R, Mirza MA, Hussain S, Hussain A. Oral lipid based drug delivery system (LBDDS): formulation, characterization and application: a review. Curr Drug Deliv. 2011;8(4):330–345. doi:10.2174/156720111795767906
14. Franke K, Kettering M, Lange K, Kaiser WA, Hilger I. The exposure of cancer cells to hyperthermia, iron oxide nanoparticles, and mitomycin C influences membrane multidrug resistance protein expression levels. Int J Nanomedicine. 2013;8:351–363. doi:10.2147/IJN.S37465
15. Balakrishnan A, Polli JE. Apical sodium dependent bile acid transporter (ASBT, SLC10A2): a potential prodrug target. Mol Pharm. 2006;3(3):223–230. doi:10.1021/mp060022d
16. Pangeni R, Choi JU, Panthi VK, Byun Y, Park JW. Enhanced oral absorption of pemetrexed by ion-pairing complex formation with deoxycholic acid derivative and multiple nanoemulsion formulations: preparation, characterization, and in vivo oral bioavailability and anticancer effect. Int J Nanomedicine. 2018;13:3329–3351. doi:10.2147/IJN.S177627
17. Bonina F, Puglia C, Rimoli MG, et al. Glycosyl derivatives of dopamine and L-dopa as anti-Parkinson prodrugs: synthesis, pharmacological activity and in vitro stability studies. J Drug Target. 2003;11(1):25–36.
18. Bilsky EJ, Egleton RD, Mitchell SA, et al. Enkephalin glycopeptide analogues produce analgesia with reduced dependence liability. J Med Chem. 2000;43(13):2586–2590. doi:10.1021/jm000018k
19. Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50(12):2340–2357. doi:10.1194/jlr.R900012-JLR200
20. Tolle-Sander S, Lentz KA, Maeda DY, Coop A, Polli JE. Increased acyclovir oral bioavailability via a bile acid conjugate. Mol Pharm. 2004;1(1):40–48. doi:10.1021/mp034010t
21. Jeon OC, Hwang SR, Al-Hilal TA, et al. Oral delivery of ionic complex of ceftriaxone with bile acid derivative in non-human primates. Pharm Res. 2013;30(4):959–967. doi:10.1007/s11095-013-1045-0
22. Jeon OC, Seo DH, Kim HS, Byun Y, Park JW. Oral delivery of zoledronic acid by non-covalent conjugation with lysine-deoxycholic acid: in vitro characterization and in vivo anti-osteoporotic efficacy in ovariectomized rats. Eur J Pharm Sci. 2016;82:1–10. doi:10.1016/j.ejps.2015.11.004
23. Dave VS, Gupta D, Yu M, Nguyen P, Gupta SV. Current and evolving approaches for improving the oral permeability of BCS Class III or analogous molecules. Drug Dev Ind Pharm. 2017;43(2):177–189. doi:10.1080/03639045.2016.1269122
24. Park JW, Kim SJ, Kwag DS, et al. Multifunctional delivery systems for advanced oral uptake of peptide/protein drugs. Curr Pharm Des. 2015;21(22):3097–3110. doi:10.2174/1381612821666150531163944
25. Swaan PW. Recent advances in intestinal macromolecular drug delivery via receptor-mediated transport pathways. Pharm Res. 1998;15:826–834. doi:10.1023/A:1011908128045
26. Qu X, Zou Y, He C, et al. Improved intestinal absorption of paclitaxel by mixed micelles self-assembled from vitamin E succinate-based amphiphilic polymers and their transcellular transport mechanism and intracellular trafficking routes. Drug Deliv. 2018;25(1):210–225. doi:10.1080/10717544.2018.1474967
27. He B, Lin P, Jia Z, et al. The transport mechanisms of polymer nanoparticles in Caco-2 epithelial cells. Biomaterials. 2013;34(25):6082–6098. doi:10.1016/j.biomaterials.2013.04.053
28. Artursson P, Magnusson C. Epithelial transport of drugs in cell culture. II: effect of extracellular calcium concentration on the paracellular transport of drugs of different lipophilicities across monolayers of intestinal epithelial (Caco-2) cells. J Pharm Sci. 1990;79(7):595–600. doi:10.1002/jps.2600790710
29. Bobin-Dubigeon C, Amiand MB, Herrenknecht C, Bard JM. Development and validation of an improved liquid chromatography-mass spectrometry method for the determination of pemetrexed in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(24):2451–2456. doi:10.1016/j.jchromb.2009.06.020
30. Prasad YV, Puthli SP, Eaimtrakarn S, et al. Enhanced intestinal absorption of vancomycin with Labrasol and D-alpha-tocopheryl PEG 1000 succinate in rats. Int J Pharm. 2003;250(1):181–190. doi:10.1016/S0378-5173(02)00544-6
31. Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212(2):233–246. doi:10.1016/S0378-5173(00)00614-1
32. Fischer SM, Parmentier J, Buckley ST, Reimold I, Brandl M, Fricker G. Oral bioavailability of ketoprofen in suspension and solution formulations in rats: the influence of poloxamer 188. J Pharm Pharmacol. 2012;64(11):1631–1637. doi:10.1111/j.2042-7158.2012.01541.x
33. Newa M, Bhandari KH, Li DX, et al. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int J Pharm. 2007;343(1–2):228–237. doi:10.1016/j.ijpharm.2007.05.031
34. Sun S, Liang N, Kawashima Y, Xia D, Cui F. Hydrophobic ion pairing of an insulin-sodium deoxycholate complex for oral delivery of insulin. Int J Nanomedicine. 2011;6:3049–3056. doi:10.2147/IJN.S25646
35. Mrestani Y, Bretschneider B, Hartl A, Neubert RH. In-vitro and in-vivo studies of cefpirom using bile salts as absorption enhancers. J Pharm Pharmacol. 2003;55(12):1601–1606. doi:10.1211/0022357022214
36. Moghimipour E, Ameri A, Handali S. Absorption-enhancing effects of bile salts. Molecules. 2015;20(8):14451–14473. doi:10.3390/molecules200814451
37. Raimondi F, Santoro P, Barone MV, et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294(4):G906–913. doi:10.1152/ajpgi.00043.2007
38. Pavlovic N, Golocorbin-Kon S, Ethanic M, et al. Bile acids and their derivatives as potential modifiers of drug release and pharmacokinetic profiles. Front Pharmacol. 2018;9:1–23. doi:10.3389/fphar.2018.01283
39. Sahay G, Batrakova EV, Kabanov AV. Different internalization pathways of polymeric micelles and unimers and their effects on vesicular transport. Bioconjug Chem. 2008;19(10):2023–2029. doi:10.1021/bc8002315
40. Gupta S, Kesarla R, Omri A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013;2013:1–16.
41. DiMarco RL, Hunt DR, Dewi RE, Heilshorn SC. Improvement of paracellular transport in the Caco-2 drug screening model using protein-engineered substrates. Biomaterials. 2017;129:152–162. doi:10.1016/j.biomaterials.2017.03.023
42. Shneider BL. Intestinal bile acid transport: biology, physiology, and pathophysiology. J Pediatr Gastroenterol Nutr. 2001;32(4):407–417. doi:10.1097/00005176-200104000-00002
43. Kanda T, Foucand L, Nakamura Y, et al. Regulation of expression of human intestinal bile acid-binding protein in Caco-2 cells. Biochem J. 1998;330(Pt 1):261–265. doi:10.1042/bj3300261
44. Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56(6):1085–1099. doi:10.1194/jlr.R054114
45. Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–398. doi:10.1146/annurev.biochem.69.1.373
46. Orlandi PA, Fishman PH. Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J Cell Biol. 1998;141(4):905–915. doi:10.1083/jcb.141.4.905
47. Dutta D, Donaldson JG. Search for inhibitors of endocytosis: intended specificity and unintended consequences. Cell Logist. 2012;2(4):203–208. doi:10.4161/cl.23967
48. Kerr MC, Teasdale RD. Defining macropinocytosis. Traffic. 2009;10(4):364–371. doi:10.1111/tra.2009.10.issue-4
49. Azevedo C, Macedo MH, Sarmento B. Strategies for the enhanced intracellular delivery of nanomaterials. Drug Discov Today. 2018;23(5):944–959. doi:10.1016/j.drudis.2017.08.011
50. Singh S, Kumar A, Karakoti A, Seal S, Self WT. Unveiling the mechanism of uptake and sub-cellular distribution of cerium oxide nanoparticles. Mol Biosyst. 2010;6(10):1813–1820. doi:10.1039/c0mb00014k
51. Burd CG. Physiology and pathology of endosome-to-Golgi retrograde sorting. Traffic. 2011;12(8):948–955. doi:10.1111/j.1600-0854.2011.01188.x
52. Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116(5):1071–1080. doi:10.1083/jcb.116.5.1071
53. Lee SD, Osei-Twum JA, Wasan KM. Dose-dependent targeted suppression of P-glycoprotein expression and function in Caco-2 cells. Mol Pharm. 2013;10(6):2323–2330. doi:10.1021/mp300668e
54. Tang F, Horie K, Borchardt RT. Are MDCK cells transfected with the human MRP2 gene a good model of the human intestinal mucosa? Pharm Res. 2002;19(6):773–779. doi:10.1023/A:1016192413308
55. Hu K, Cao S, Hu F, Feng J. Enhanced oral bioavailability of docetaxel by lecithin nanoparticles: preparation, in vitro, and in vivo evaluation. Int J Nanomedicine. 2012;7:3537–3545. doi:10.2147/IJN.S30631
56. Forsgard RA, Korpela R, Stenman LK, Osterlund P, Holma R. Deoxycholic acid induced changes in electrophysiological parameters and macromolecular permeability in murine small intestine with and without functional enteric nervous system plexuses. Neurogastroenterol Motil. 2014;26(8):1179–1187. doi:10.1111/nmo.12383
57. Mithani SD, Bakatselou V, TenHoor CN, Dressman JB. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharm Res. 1996;13(1):163–167. doi:10.1023/A:1016062224568
58. Tian C, Asghar S, Wu Y, et al. Improving intestinal absorption and oral bioavailability of curcumin via taurocholic acid-modified nanostructured lipid carriers. Int J Nanomedicine. 2017;12:7897–7911. doi:10.2147/IJN.S145988
59. Chen YL, Chang MC, Cheng WF. Metronomic chemotherapy and immunotherapy in cancer treatment. Cancer Lett. 2017;400:282–292. doi:10.1016/j.canlet.2017.01.040
60. Langer CJ, Gadgeel SM, Borghaei H, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17(11):1497–1508. doi:10.1016/S1470-2045(16)30498-3
Supplementary material
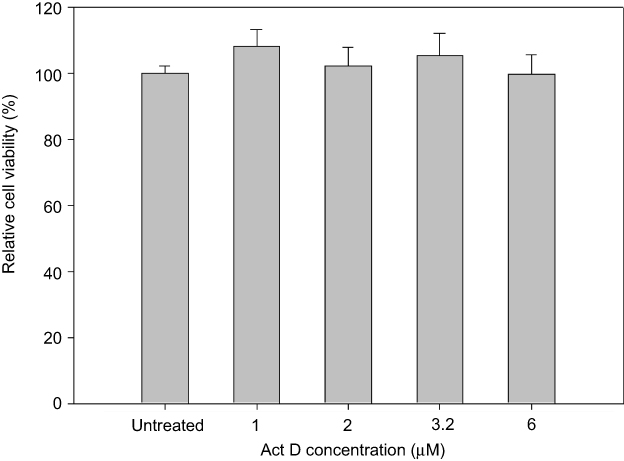 |
Figure S1 In vitro cytotoxic effect of Act D on Caco-2 cells. Notes: Cell viability was measured using WST-8 and the growth of Caco-2 cells compared to the untreated control group. All data are expressed as means ± standard deviation (n=6). Abbreviation: Act D, actinomycin D. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.