")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Interleukin-16-producing NK cells and T-cells in the blood of tobacco smokers with and without COPD
Authors Andersson A, Malmhäll C, Houltz B, Tengvall S, Sjöstrand M, Qvarfordt I, Lindén A , Bossios A
Received 6 January 2016
Accepted for publication 31 May 2016
Published 15 September 2016 Volume 2016:11(1) Pages 2245—2258
DOI https://doi.org/10.2147/COPD.S103758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Anders Andersson,1,* Carina Malmhäll,2,* Birgitta Houltz,1 Sara Tengvall,1 Margareta Sjöstrand,2 Ingemar Qvarfordt,1 Anders Lindén,3 Apostolos Bossios2
1Respiratory Medicine and Allergology, Department of Internal Medicine and Clinical Nutrition, Institute of Medicine, Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden; 2Krefting Research Center, Department of Internal Medicine and Clinical Nutrition, Institute of Medicine, Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden; 3Unit for Lung and Airway Research, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden
*These authors contributed equally to this work
Background: Long-term exposure to tobacco smoke causes local inflammation in the airways that involves not only innate immune cells, including NK cells, but also adaptive immune cells such as cytotoxic (CD8+) and helper (CD4+) T-cells. We have previously demonstrated that long-term tobacco smoking increases extracellular concentration of the CD4+-recruiting cytokine interleukin (IL)-16 locally in the airways. Here, we hypothesized that tobacco smoking alters IL-16 biology at the systemic level and that this effect involves oxygen free radicals (OFR).
Methods: We quantified extracellular IL-16 protein (ELISA) and intracellular IL-16 in NK cells, T-cells, B-cells, and monocytes (flow cytometry) in blood samples from long-term tobacco smokers with and without chronic obstructive pulmonary disease (COPD) and in never-smokers. NK cells from healthy blood donors were stimulated with water-soluble tobacco smoke components (cigarette smoke extract) with or without an OFR scavenger (glutathione) in vitro and followed by quantification of IL-16 protein.
Results: The extracellular concentrations of IL-16 protein in blood did not display any substantial differences between groups. Notably, intracellular IL-16 protein was detected in all types of blood leukocytes. All long-term smokers displayed a decrease in this IL-16 among NK cells, irrespective of COPD status. Further, both NK and CD4+ T-cell concentrations displayed a negative correlation with pack-years. Moreover, cigarette smoke extract caused release of IL-16 protein from NK cells in vitro, and this was not affected by glutathione, in contrast to the decrease in intracellular IL-16, which was prevented by this drug.
Conclusion: Long-term exposure to tobacco smoke does not markedly alter extracellular concentrations of IL-16 protein in blood. However, it does decrease the intracellular IL-16 concentrations in blood NK cells, the latter effect involving OFR. Thus, long-term tobacco smoking exerts an impact at the systemic level that involves NK cells; innate immune cells that are critical for host defense against viruses and tumors – conditions that are overrepresented among smokers.
Keywords: COPD, interleukin-16, NK cells, CD4+ cells, CD8+ cells, CD69+
Background
Long-term tobacco smoking causes local inflammation in the airways. When the inflammatory disease chronic obstructive pulmonary disease (COPD) is established, there is a permanent loss of ventilatory function and capacity for gas exchange. It is known that the local inflammation present in COPD involves innate effector cells, including NK cells, which constitute first line of defense against foreign compounds from the environment,1,2 as well as adaptive immune cells, including cytotoxic (CD8+) T-cells3,4 and T helper (CD4+) cells.5–7 While the involvement of CD8+ and CD4+ T-cells in COPD is established, the corresponding involvement of NK cells remains to be established.
Notably, NK cells constitute the first line of defense against infection and are instrumental in tumor surveillance. A few preceding studies have indicated a reduction in the cytotoxic capacity of blood NK cells in smokers, suggesting a cell-specific immunosuppressive effect caused by tobacco smoke.8–10 It is now recognized that even NK cells are potent regulators of inflammation owing to their secretion of a wide range of cytokines and chemokines.11,12 Previous studies indicate that NK cells are also capable of regulating CD4+ T-cells, but the exact mechanism has not been clarified.13
It is known that interleukin (IL)-16 recruits CD4+ T-cells by cross-linking of the CD4 receptors.14 This cytokine has been highly conserved during evolution, possibly indicating its importance.15 In contrast to many other cytokines, IL-16 is synthesized as pro-IL-16, a form that is a regulator of T-cell growth.16 Although there is evidence from studies of immunoreactivity suggesting that IL-16 is produced by murine NK cells,17 the matching evidence from human NK cells is lacking.
We have previously demonstrated that tobacco smoke does affect the production and release of IL-16 from human T-cells in the airways in vivo and in isolated cells in vitro.18–20 Specifically, it seems as if tobacco smoke depletes IL-16 from local CD8+ T-cells in the airways.20 Moreover, we have shown that long-term exposure to tobacco smoke alters IL-16 protein in palatine tonsils, which is compatible with systemic effects.19 For the current study, we hypothesized that long-term exposure to tobacco smoke alters the biology of IL-16 at the systemic level by exerting effects on circulating blood leukocytes, including NK cells, CD8+ and CD4+ T-cells, B-cells, and monocytes.14,17,21
To address our hypothesis, we quantified extracellular IL-16 protein in blood from well-characterized tobacco smokers with COPD, asymptomatic smokers (AS), and never-smokers (NS), respectively. We related this extracellular IL-16 to intracellular IL-16 in circulating NK cells, CD8+ and CD4+ T-cells, B-cells, and monocytes. Given that we found altered expression of IL-16 in circulating NK cells, we isolated such cells from healthy donors and then cultured and exposed these cells to water-soluble tobacco smoke components in vitro for the study of IL-16 and the archetype NK cell cytokine IFN-γ. We also assessed the role of oxygen free radical scavenger’s (OFR’s) in this context.
Methods
Study population
The study was approved by the Regional Ethics Committee in Gothenburg (No 582-05). The subjects received oral and written information and provided both oral and written consent. Three different groups were included in this study. We included smokers with COPD, GOLD stages II and III (referred to as smokers with COPD; COPD) or smokers without COPD (referred to as AS). These two groups (ie, COPD + AS) were all long-term as well as current smokers with a matched tobacco load (≥10 pack-years). We utilized NS with normal lung function as a control group (Table 1). All subjects underwent an evaluation of medical history and physical findings by an experienced physician. Subjects with respiratory insufficiency or other serious conditions such as malignancy, ischemic heart disease, or rheumatoid arthritis were excluded. No subjects had a history of asthma. Sensitized subjects, as judged by Phadiatop, a screening test for inhalant allergy, were examined out of allergy season. For the definition of COPD by spirometry, a ratio of the forced expiratory volume in 1 second (FEV1) divided by the forced vital capacity lower than 0.70 was deemed abnormal according to GOLD criteria (www.gold.com). For subjects over 65 years, a ratio below 0.65 was deemed abnormal, in accordance with the National guidelines in Sweden.22,23 Furthermore, the COPD subjects were required to have a diffusion capacity for carbon monoxide (DLCO) reduced by >2 standard deviations (SD) from the predicted mean value. This criterion served as an indicator of emphysema. The NS and AS subjects were required to have a normal spirometry plus a normal DLCO (within 2 SD of the predicted mean value). All examinations were performed during a stable clinical phase without any clinical signs of infection. We also required the concentration of C-reactive protein (CRP) to be <10 mg/L on the sampling day. During the 4 weeks prior to the sampling, none of the subjects received corticosteroids, statins, or long-acting bronchodilators. One NS was excluded because the blood cell samples could not be analyzed with flow cytometry due to human error.
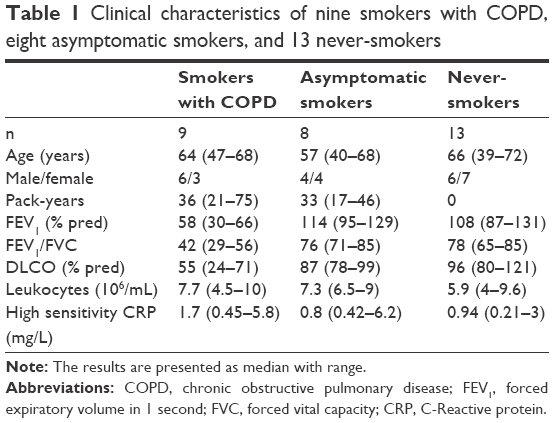 | Table 1 Clinical characteristics of nine smokers with COPD, eight asymptomatic smokers, and 13 never-smokers |
Spirometry
All subjects underwent dynamic flow spirometry (Jaeger Masterscope, Cardinal Health, Würzburg, Germany). FEV1 and forced vital capacity were measured according to the American Thoracic Society (ATS) and European Respiratory Society (ERS) Guidelines.24
Measurement of gas diffusion capacity
The diffusion capacity of the lung for carbon monoxide (DLCO) was assessed by the single-breath method (Sensor Medics 2200, Sensor Medics Corporation, the Netherlands) in accordance with ATS/ERS Guidelines and adjusted for the hemoglobin level.25 Both spirometry and DLCO measurements were performed after administration of bronchodilators (terbutaline 1.5 mg). FEV1 and DLCO values were expressed as percent predicted (% pred). The FEV1 was based on the reference values of the European Coal and Steel Community and the ERS.26 The reference value for DLCO was based on the reference equation by Salorinne.27
Whole blood samples
Peripheral venous blood samples were taken for flow cytometry analysis, preparation of plasma or serum, analysis of differential cell counts, as well as high-sensitivity (HS) CRP. Plasma and serum were stored frozen (−80°C) until analysis of IL-16 and IL-1β protein.
Enrichment of NK cells
Human peripheral blood mononuclear cells (PBMCs) were isolated from the buffy coat from healthy blood donors (Sahlgrenska University Hospital, Gothenburg, Sweden) by density centrifugation using Ficoll Paque PLUS (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) as previously described.19 The NK cells were then enriched using a negative magnetic separation technique (NK Cell Isolation Kit, Miltenyi Biotec, Bergisch Gladbach, Germany). Cell purity was >95% and viability >98% as determined by flow cytometry (see “Intracellular IL-16 and IFN-γ protein analysis” section).
Preparation of cigarette smoke extract
The water-soluble components of tobacco smoke were generated in the form of cigarette smoke extract (CSE) as described in detail previously.19,20 Briefly, CSE was generated by leading mainstream smoke from ten medium nicotine and tar commercial cigarettes (Marlboro™, Philip Morris Int, Richmond, VA, USA) through 15 mL of cell culture medium (HyClone RPMI1640 Media, GE Healthcare Bio-Sciences AB) by vacuum suction at room temperature. The burning time for each cigarette was 3 minutes. The prepared solution with CSE was thereafter passed through a sterile filter and divided into aliquots and stored frozen (−80°C). The chosen concentration of CSE (1:25) resulted in IL-16 response with well-preserved cell viability (see “Results” section).
Culture of NK cells
The enriched NK cells were cultured with or without CSE (dilution 1:25) and with or without an OFR scavenger (L-glutathione, 10−2 M) in complete medium (ie, cell culture media, supplemented with fetal calf serum [FCS: 10%] [both from Sigma-Aldrich, St Louis, MO, USA], L-glutamine [4 mmol/mL], and penicillin/streptomycin [100 units/100 μg/mL] [HyClone] as previously described19,20). The utilized concentration of L-glutathione (10−2 M) was chosen based on its effect on IL-16 in a previous study.20 The NK cells were cultured for 20 hours in a humidified incubator (5% CO2, 37°C). The viability for NK cells after 20 hours of culture was assessed using exclusion of trypan blue dye (see “Results” section). For the experiments on extracellular IL-16 protein, the conditioned medium was carefully collected and centrifuged (300 ×g, 4°C, 10 minutes) to achieve a cell-free supernatant that was stored frozen (−80°C). For those experiments intended for analysis of intracellular proteins (IL-16 & IFN-γ) by flow cytometry, monensin (Golgi Stop™; BD Biosciences, San Jose, CA, USA) was added 6 hours prior to harvest to enable detection of intracellular proteins.
Analysis of extracellular IL-16 and IL-1β protein
IL-16 concentrations in plasma, serum, and conditioned medium from cell culture were quantified using commercial ELISA kits (in vivo: BioSource; in vitro: R&D Systems, Minneapolis, MN, USA). Samples below the lowest concentration of the standard curve were assigned the mean value of 0 and the lowest value of standard curve. The IL-1β concentrations were quantified in plasma, 29 out of 30 were above the lowest concentration of the standard curve (Human IL-1β ELISA, Mabtech AB, Stockholm, Sweden).
Intracellular IL-16 and IFN-γ protein analysis
In essence, flow cytometry analysis was performed on fresh peripheral whole blood or enriched NK cells from buffy coat prior to and after cell culture as described earlier.20,28 The following antibodies were used to detect surface antigens: CD3 (clone SK7), CD4 (clone SK3), CD8 (clone RPA-T8), CD14 (clone MΦP9), CD16 (clone 3G8), CD19 (clone SJ25C1) and CD56 (clone N-CAM) purchased from BD Biosciences, Franklin Lakes, NJ, USA and CD69 (clone FN50; BioLegend, San Diego, CA, USA). 7-aminoactinomycin D (7AAD; BD Biosciences) was used to select viable cells. Additionally, blocking buffer (PBS supplemented with human IgG; Sigma Aldrich) and monensin (Golgistop™, BD Biosciences), red blood cell lysing solution (hypertonic saline), wash buffer (PBS with 1% FCS), fixative (PBS with 4% paraformaldehyde; Sigma Aldrich), and SAP buffer (HBSS with 0.1% saponin; Sigma Aldrich) were used. Intracellular antibodies were IL-16 (clone 14.1; BD Biosciences) and IFN-γ (clone 25723; R&D Systems, Minneapolis, MN, USA). Matched isotype controls (IC) and fluorescence minus one controls (FMO) were included in the analysis. All flow cytometry analyses were performed with a BD FACS Aria Flow Cytometer running FACS Diva version 6.0 Software (BD Biosciences) and analyzed with FlowJo software (Tree Star, Ashland, OR, USA). NK cells were classified as CD3−CD16+CD56+ cells, intact cells were classified as 7AAD−, and finally the signal for intracellular protein was calculated as relative mean fluorescence index (rMFI: sample MFI expression/MFI in the corresponding control).20
Statistical analysis
The statistical analysis was performed with StatView 4.01 (Abacus Concepts, Berkeley, CA, USA) using a nonparametric approach. The Kruskal–Wallis test for comparison between multiple groups, followed by the Mann–Whitney U-test, was applied. The latter was also applied for comparisons between two groups (smokers and NS). The correlation analysis was performed using the Spearman rank correlation test. P<0.05 were considered statistically significant in all analyses.
Results
Subject characteristics
Seventeen smokers, nine smokers with COPD (COPD) and eight AS plus 13 NS were included in the study and their clinical characteristics are shown in Table 1. The smokers with COPD were in GOLD stages II and III, all of them displaying pathological gas diffusion capacity and, as expected, hsCRP was slightly increased. The AS group displayed a normal spirometry and a normal diffusion capacity, just like the NS group. Moreover, the tobacco load was very similar in the AS and COPD groups. Table 2 shows the basic characteristics for the corresponding NK cell, CD8+ and CD4+ T-cell, B-cell, and monocyte concentrations in blood. We found no pronounced differences in the total or relative number of NK cells, CD8+ or CD4+ T-cells, or in monocytes between the study groups. For all samples, we first analyzed the three groups separately. However, since there was no substantial difference (with the exception of CD69+ CD8+ T-cells, see “The activation marker CD69 alone or together with intracellular IL-16 in NK cells, CD8+ and CD4+ T-cells, B-cells, and monocytes” section), we pooled the results from both groups of long-term smokers (COPD + AS) into one, from here on referred to as smokers. In the group of smokers, there was a slight increase in total number of blood B-cells (Table 2) (0.42 [0.24–1.24] ×106/mL) compared to NS (0.27 [0.13–0.8] ×106/mL) (Mann–Whitney U-test P<0.05).
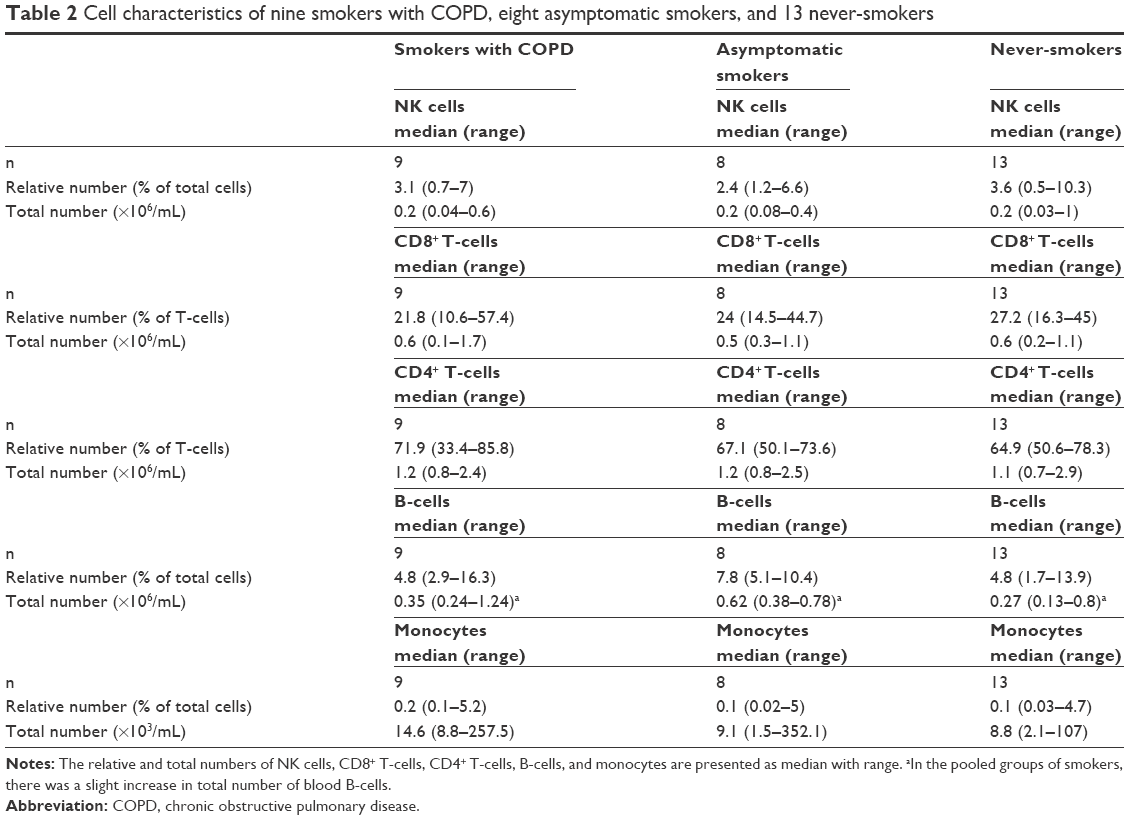 | Table 2 Cell characteristics of nine smokers with COPD, eight asymptomatic smokers, and 13 never-smokers |
Extracellular IL-16 and IL-1β protein in plasma
There were no statistically significant differences in the plasma concentrations of IL-16 between the COPD, AS, and NS groups or between the pooled group of smokers versus NS (Figure 1A), and the same was true for IL-1β (Figure 1B). The concentrations of these two interleukins did not display any statistically significant correlation (data not shown).
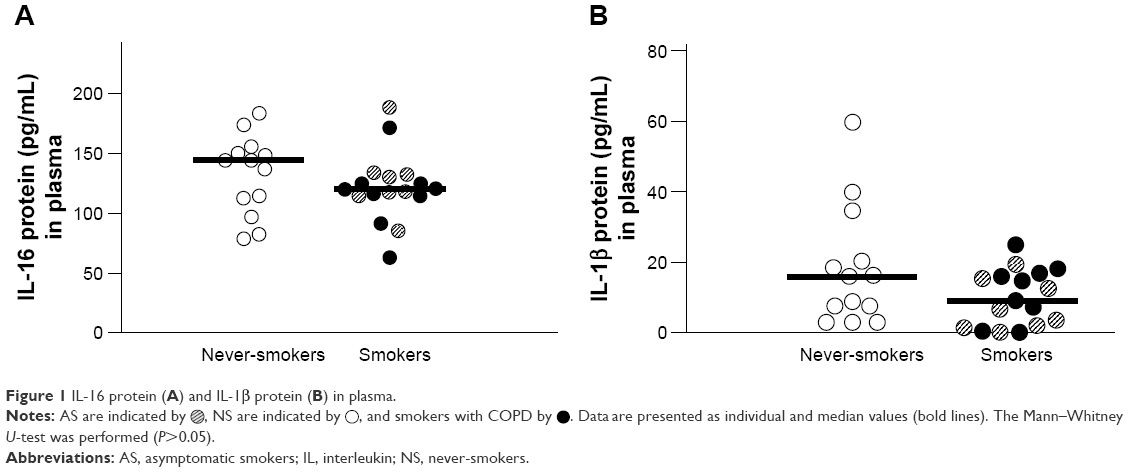 | Figure 1 IL-16 protein (A) and IL-1β protein (B) in plasma. |
 , NS are indicated by
, NS are indicated by  , and smokers with COPD by
, and smokers with COPD by  . Data are presented as individual and median values (bold lines). The Mann–Whitney U-test was performed (P>0.05).
. Data are presented as individual and median values (bold lines). The Mann–Whitney U-test was performed (P>0.05).Extracellular IL-16 protein in serum
To ascertain that coagulation did not affect the IL-16 protein, we also analyzed IL-16 protein in serum. However, just like for plasma samples, there were no statistically significant differences (median [range]) in the serum concentrations of IL-16 for COPD (127 [67–254] pg/mL), AS (157 [97–225] pg/mL), or NS subjects (150 [95–195] pg/mL) or for smokers (136 [67–254] pg/mL) versus NS.
Intracellular IL-16 protein in blood NK cells, CD8+ and CD4+ T-cells, B-cells, and monocytes
We also examined intracellular IL-16, since IL-16 protein is produced and stored intracellularly. Here, the pooled group of smokers exhibited a lower relative number of IL-16+ NK cells than did NS subjects (Figure 2A and B). Moreover, the total number of IL-16+ NK cells was lower in the group of smokers compared with NS group, but this difference was not statistically significant. Despite this, we could not detect any statistically significant difference in the intensity of intracellular IL-16 expression (rMFI) between the subject groups (Table 3). In addition, we detected no correlations between, on one hand, IL-16+ NK cells, and, on the other, extracellular concentrations of IL-16 protein in plasma, FEV1 (% pred), or pack-years (data not shown), respectively. However, importantly, all subsets of NK cells displayed expression of intracellular IL-16 protein (Figure 2A). Intracellular IL-16 protein was also examined in CD8+ and CD4+ T-cells in addition to B-cells and monocytes. No magnitudinal differences in IL-16 expression between smokers and NS were found in these cells (Table 3).
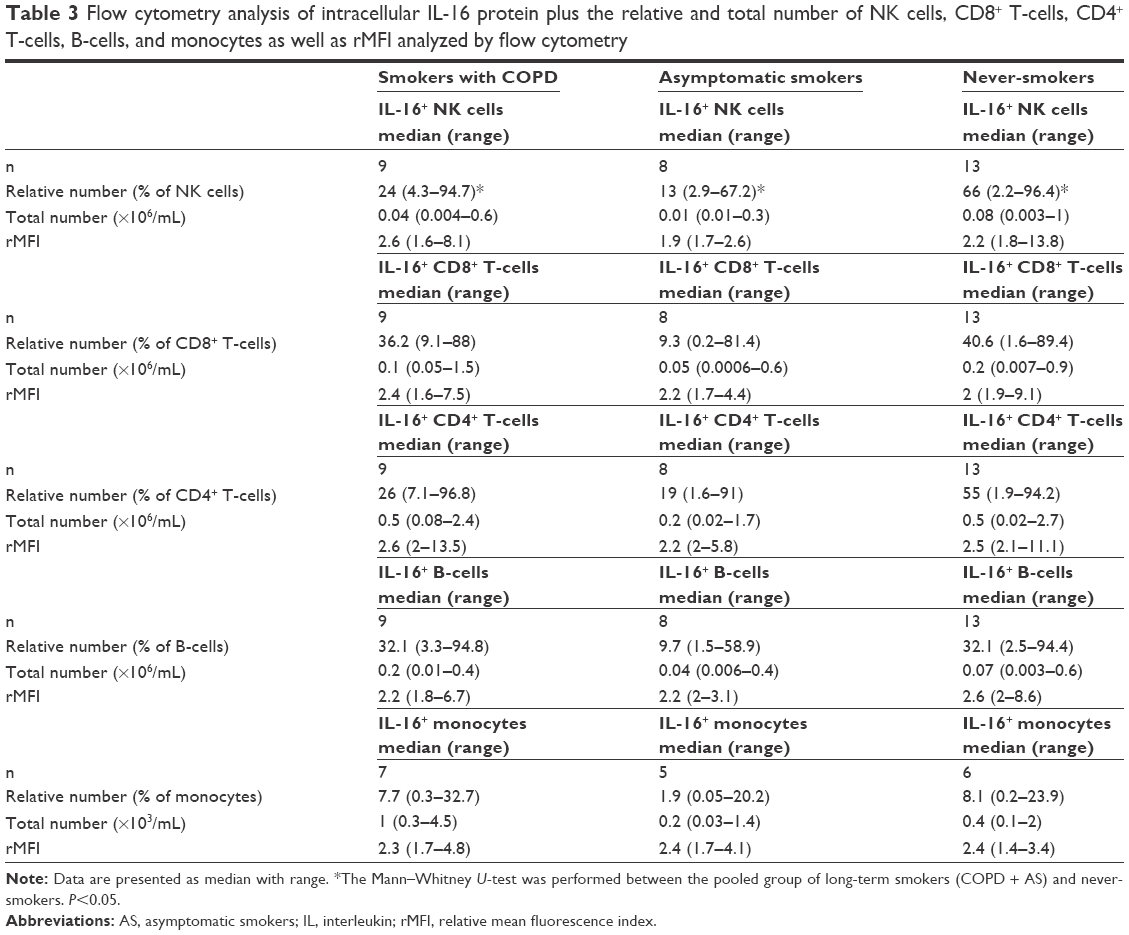 | Table 3 Flow cytometry analysis of intracellular IL-16 protein plus the relative and total number of NK cells, CD8+ T-cells, CD4+ T-cells, B-cells, and monocytes as well as rMFI analyzed by flow cytometry |
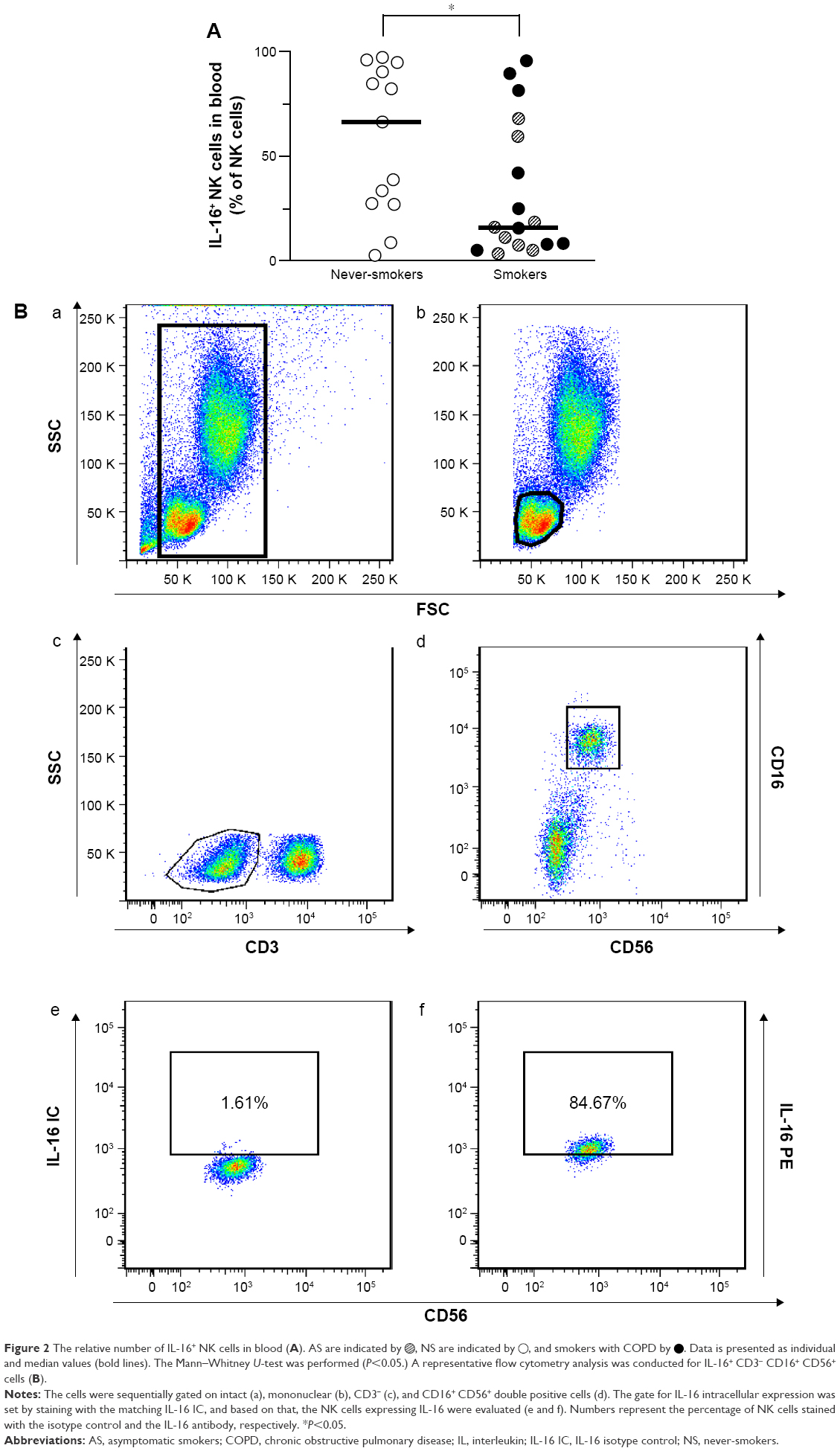 | Figure 2 The relative number of IL-16+ NK cells in blood (A). AS are indicated by |
The number of NK cells and CD4+ cells versus pack-years
A negative correlation was detected between the relative number of NK cells and the total number of NK cells and CD4+ cells, on one hand, versus pack-years, on the other hand, for the pooled group of smokers (Figure 3A–C).
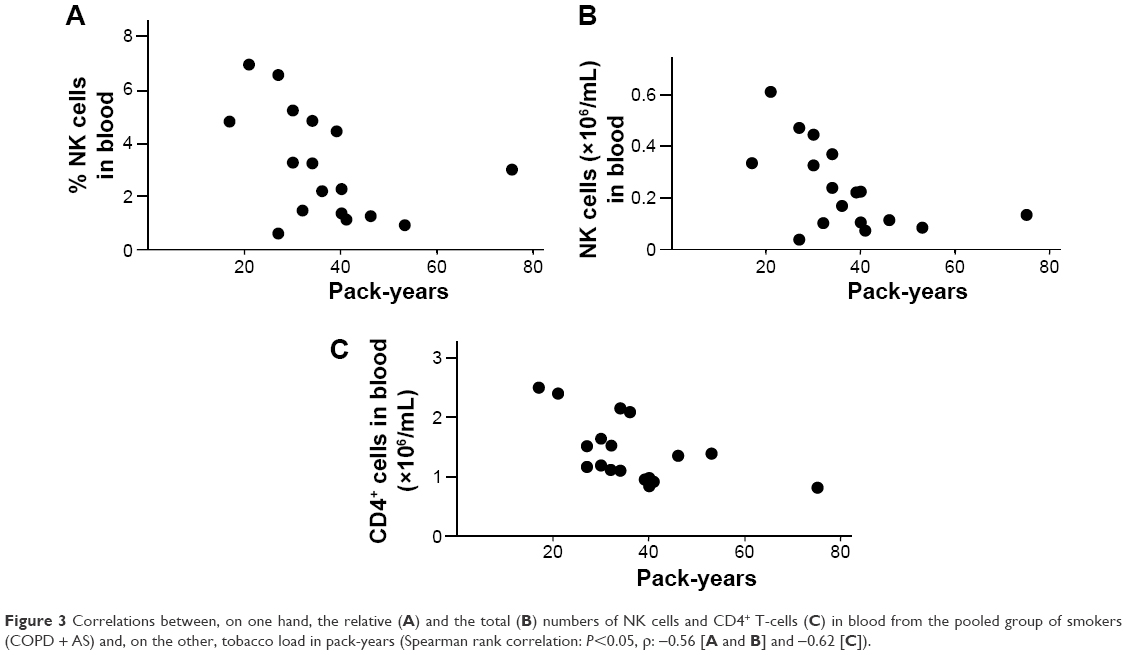 | Figure 3 Correlations between, on one hand, the relative (A) and the total (B) numbers of NK cells and CD4+ T-cells (C) in blood from the pooled group of smokers (COPD + AS) and, on the other, tobacco load in pack-years (Spearman rank correlation: P<0.05, ρ: −0.56 [A and B] and −0.62 [C]). |
The activation marker CD69 alone or together with intracellular IL-16 in NK cells, CD8+ and CD4+ T-cells, B-cells, and monocytes
We found no smoking-related group difference for the number of CD69+ or IL-16+CD69+ NK cells (COPD, AS, and NS, or the pooled group of smokers versus NS). Neither the relative nor the total number exhibited any statistically significant group-related differences (data not shown). However, smokers with COPD were found to have a modest increase in the total number of CD69+ CD8+ T-cells compared to AS, and AS had a slight but statistically significant decrease compared with NS (Figure 4). There was also a modest increase in the total number of CD69+ monocytes in the pooled group of smokers (2.4 [0–5.4] ×103/mL) versus NS (0.8 [0–3.5] ×103/mL) (Mann–Whitney U-test P<0.05).
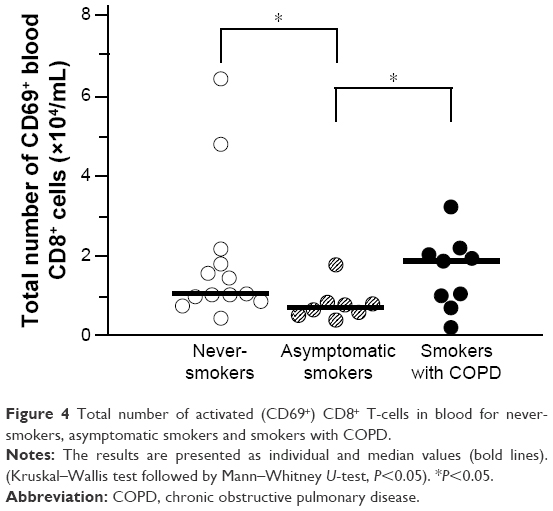 | Figure 4 Total number of activated (CD69+) CD8+ T-cells in blood for never-smokers, asymptomatic smokers and smokers with COPD. |
Extracellular IL-16 protein from NK cells in vitro
Exposure to CSE markedly increased the concentrations of extracellular IL-16 protein in the conditioned medium (Figure 5). Treatment with the OFR scavenger glutathione increased the concentrations of extracellular IL-16 protein in unstimulated NK cells, and there was no clear difference between unexposed NK cells treated with glutathione and those exposed to CSE and treated with glutathione. The viability (median percentage with range; n=3) was as follows for the different treatment groups: 100% (na) for vehicle alone, 97% (96–99) for CSE-treated cells, 99% (97–100) for vehicle and glutathione and, finally, 97% (91–98) for CSE and glutathione.
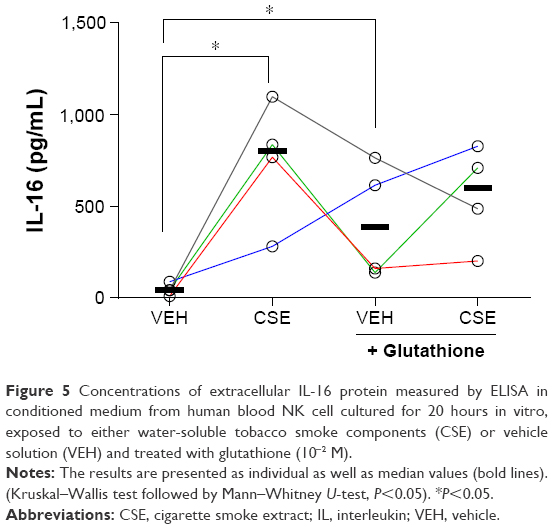 | Figure 5 Concentrations of extracellular IL-16 protein measured by ELISA in conditioned medium from human blood NK cell cultured for 20 hours in vitro, exposed to either water-soluble tobacco smoke components (CSE) or vehicle solution (VEH) and treated with glutathione (10−2 M). |
Intracellular IL-16 protein in vitro
The relative number of intact (7AAD−) NK cells was markedly decreased by exposure to CSE. Glutathione per se caused a slight decrease in 7AAD− cells, and treatment with this OFR scavenger prevented a further decrease in intact NK cells exposed to CSE (Figure 6A). The relative number of IL-16+ NK cells was markedly decreased by CSE. Glutathione per se caused a slight decrease in IL-16+ cells, which was, however, not statistically significant. Notably, treatment with glutathione prevented a further decrease in IL-16+ cells exposed to CSE (Figure 6B). The very same pattern was observed when assessing the intracellular content of IL-16 protein as rMFI (Figure 6C).
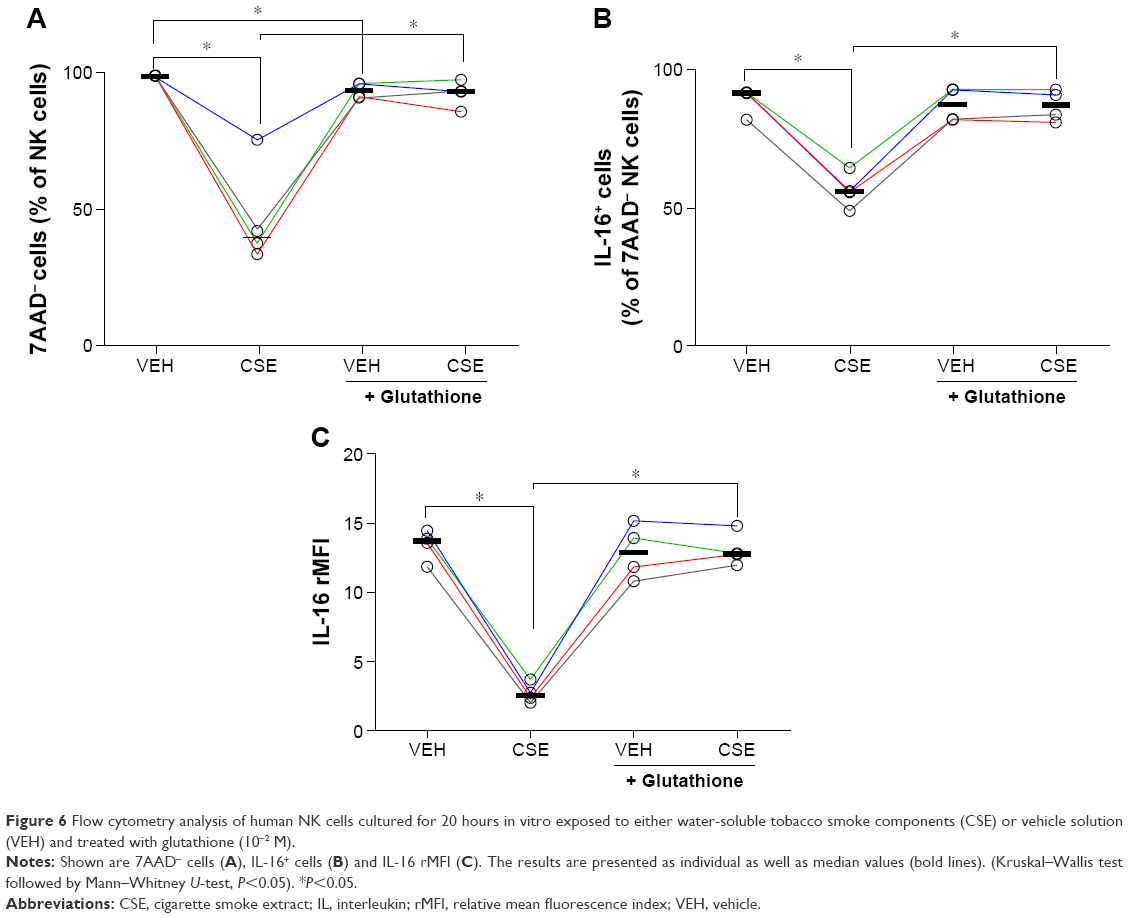 | Figure 6 Flow cytometry analysis of human NK cells cultured for 20 hours in vitro exposed to either water-soluble tobacco smoke components (CSE) or vehicle solution (VEH) and treated with glutathione (10−2 M). |
Intracellular IFN-γ protein in vitro
To investigate if exposure to CSE also leads to protein synthesis and not only release of IL-16 protein, we analyzed the archetype NK cell cytokine IFN-γ. A representative plot of flow cytometry analysis for IFN-γ proteins in NK cells cultivated with and without glutathione treatment is shown in Figure 7A. The relative number of IFN-γ+ NK cells was markedly increased by exposure to CSE, an increase that was fully prevented by treatment with glutathione (Figure 7B). When measuring the intracellular content of IFN-γ protein as rMFI, no statistically significant difference was found. However, there was a trend toward an increase in IFN-γ protein in NK cells exposed to CSE (Figure 7C).
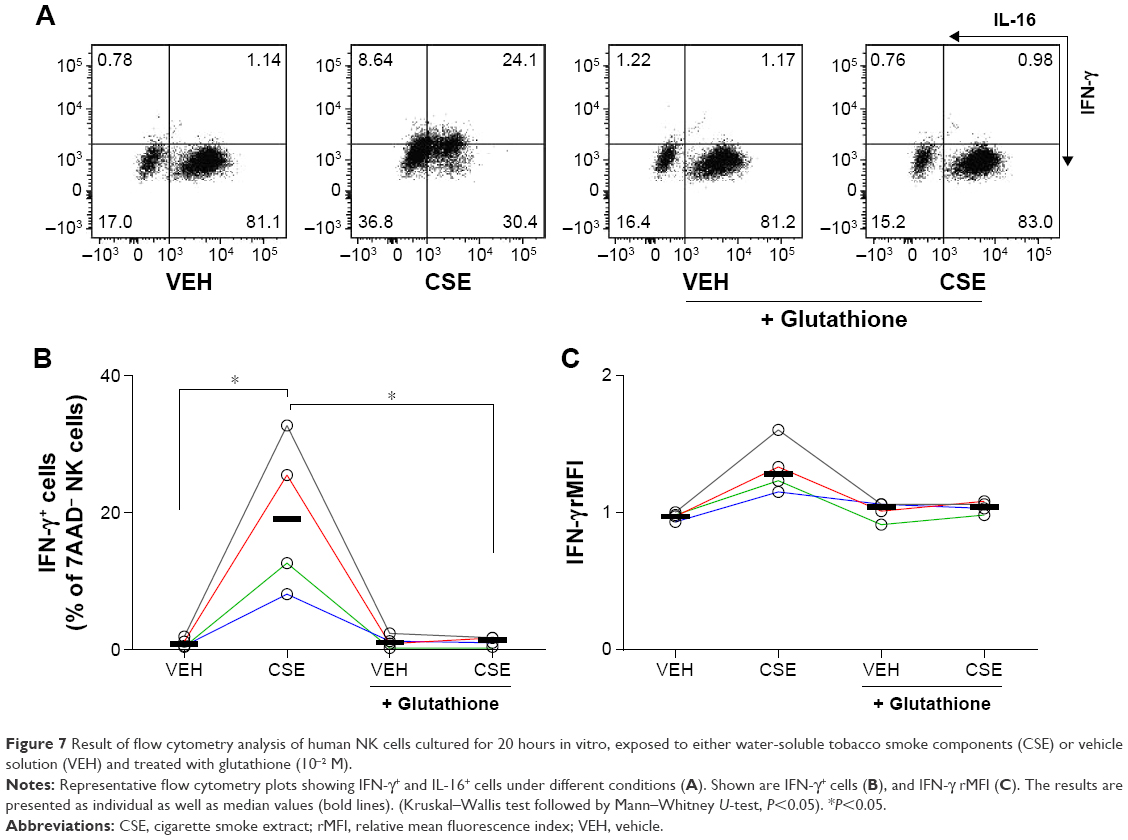 | Figure 7 Result of flow cytometry analysis of human NK cells cultured for 20 hours in vitro, exposed to either water-soluble tobacco smoke components (CSE) or vehicle solution (VEH) and treated with glutathione (10−2 M). |
Discussion
The present study shows that the CD4+-recruiting cytokine IL-16 can be produced by human NK cells and released from these cells in a selective manner by exposure to CSE. The present study also demonstrates that the number of blood IL-16+ NK cells is decreased in long-term tobacco smokers. Notably, the effect of exposure to CSE on intracellular IL-16 in NK cells in vitro was prevented by the OFR scavenger glutathione. However, the present study did not reveal any substantial difference in soluble IL-16 protein or in CD8+ and CD4+ T-cells, B-cells, and monocytes at the systemic level, as assessed in blood. Taken together, our data suggests that long-term exposure to tobacco smoke may clearly alter the systemic biology of IL-16, even though without major impact on IL-16 in the extracellular compartment.
We think that the most significant aspect of our study is the demonstration of a decrease in the relative number of blood IL-16+ NK cells in the pooled group of long-term smokers (COPD + AS), a finding that seems to be related to smoking irrespectively of COPD. In line with this observation in long-term smokers who are also current smokers, we found a clear, negative correlation between tobacco load (ie, pack-years) and the absolute and relative number of blood NK cells. In addition, we verified that this may be a causative relationship, by demonstrating that tobacco smoke components (ie, CSE) exert an impact on IL-16-producing NK cells in vitro. Moreover, to the best of our knowledge, this study is the first to identify the population of IL-16-producing NK cells (ie, CD3−CD16+CD56+ cells) in humans, regardless of smoking and COPD.
It is of mechanistic interest that the release of IL-16 from human NK cells was not inhibited by the OFR scavenger glutathione, in contrast to what was the case for CD8+ cells in our previous study.20 In our current study on NK cells, the water-soluble components of tobacco smoke decreased intracellular IL-16 according to two analytical approaches, affecting the number of IL-16+ NK cells as well as the intracellular content of IL-16 protein measured as rMFI. However, this decrease was prevented by glutathione, which argues for the involvement of OFR in setting intracellular IL-16 concentrations.
The mechanisms by which tobacco smoke components deplete IL-16 from lymphocytes are not yet fully elucidated. Since we found a decrease in intracellular IL-16 in 7AAD− NK cells (ie, cells with intact membranes) caused by the water-soluble components of tobacco smoke, we suggest that the release mechanism is at least partially an active process. To further evaluate this idea, we assessed intact NK cells in vitro and found that tobacco smoke components increased the number of IFN-γ+ NK cells. However, the addition of the OFR scavenger glutathione extinguished this response to tobacco smoke components.
Although we argue that our in vitro system for studying CSE is a model that elucidates potential cellular mechanisms caused by tobacco smoke, clearly, this system has limitations. First, it employs only the water-soluble components of tobacco smoke. Second, in vivo, the immunological alterations among tobacco smokers seem to arise from repeated exposure to tobacco smoke. However, hypothetically, it is possible that the total dose of CSE generated in vitro is only reached in vivo after long-time exposure to tobacco smoke.
The population of NK cells constitutes a first line of defense against viral infections29,30 and against tumors.31,32 Previous studies have also revealed an increase in NK cells locally in the airways of tobacco smokers.33 From a theoretical point of view, it seems plausible that these NK cells may contribute to the increased concentration of IL-16 protein in that compartment – a possibility that deserves further study.18–20
The effect of tobacco smoking on the IL-16+ subset of NK cells may reflect a suppressive effect of tobacco smoke on NK cells directly as well as indirectly. In addition to the suppressive effect of tobacco smoke components on NK cells, we did find that the number of NK cells, as well as CD4+ T-cells, correlates negatively with tobacco load (ie, pack-years). Evidently, this could be due to a suppressive effect of tobacco smoke on these cells but it could also be due to aging, since aging inevitably follows the course of tobacco smoking. One other possible mechanism might be cellular senescence caused by tobacco smoke.34,35 However, the fact that both viral infections and tumors are clearly overrepresented among tobacco smokers is in support of a true immunosuppressive effect.36,37 This is also in line with the reported reduction in cytotoxic capacity of blood NK cells caused by tobacco smoke.8,9 Moreover, several studies demonstrate that COPD exacerbations can be triggered by viral infections, and, hypothetically, the systemic impact of long-term tobacco smoking demonstrated in the current study could contribute to viral infections by both reducing the total number and the cytotoxic capacity of each NK cell.38 The decrease in intracellular IL-16 expression could also be a sign of a transient, recent cell activation and release of this protein. The negative outcome for the activation marker in blood NK cells could thus be due to a postactivation state, and this is supported by a recent study.39
We did not find any difference in relative or absolute numbers of blood NK cells between our study groups, whereas Tollerud et al40 previously claimed that the relative number of NK cells is decreased in smokers. However, they did not investigate the IL-16 expression among these cells. Tollerud et al40 observed an interesting phenomenon that the decrease in the relative number of NK cells lasts at least 10 years after smoking cessation, and this observation was in contrast to the case for other lymphocyte subsets, subsets which were normalized during the same observation period. Fully in line with these previously published results, we detected a negative correlation between tobacco load as well as the total and relative numbers of NK cells for the smokers in our current study. However, we obtained no evidence that the total number of NK cells in blood markedly differ between the NS, AS, and COPD group or in all pooled long-term smokers versus NS, and, in fact, neither did Tollerud et al.40
The total number of CD8+ and CD4+ T-cells did not differ between groups, which may reflect that the samples were harvested from subjects during a clinically stable phase, in contrast to another study conducted during exacerbations in which the investigators found a decrease in peripheral CD8+ and CD4+ T-cells.41 We did not find any difference in IL-16+ CD8 or CD4+ T-cells in contrast to the NK cells. IL-16 has also been found to regulate CD4+ cells by inducing apoptosis, arguing for a more complex role in regulation of these cells.42 We also found a modest increase in B-cells and in activated monocytes in blood from the pooled group of smokers. Another interesting finding in our study is that AS, ie, smokers without COPD, showed a decrease in activated CD8+ T-cells in comparison with smokers who did develop COPD. However, these observations are beyond the scope of our current study and deserve further study in the future.
Clearly, the lack of a substantial impact of long-term smoking on extracellular IL-16 protein in the blood, as demonstrated in our current study, is in contrast to the corresponding impact on IL-16 protein locally in the airways.18–20 This is also in contrast to a report from Bade et al43 who showed an increase in serum IL-16 among COPD subjects (current smokers and ex-smokers) compared with nonsmokers and AS.43 In our study, the COPD group had a decreased gas diffusion capacity, indirectly suggesting emphysema, whereas the AS group, had a normal gas diffusion capacity, arguing against emphysema. However, we did not detect any correlation between IL-16 in blood versus lung function (FEV1% of pred) or diffusion capacity (data not shown). This is also in contrast to a study by Bowler et al44 who found an association between a decrease in IL-16 levels in the blood and emphysema. However, in this study, the emphysema was estimated with a CT scan.44 Taken together, our findings of an increase in soluble IL-16 protein in the airways of tobacco smokers is in contrast to blood we found no changes in IL-16 among smokers. Hence, we think that soluble IL-16 protein could be considered more as a pathogenic mediator causing recruitment of CD4+-bearing cells than as a biomarker for COPD.
We have previously shown that long-term smoking is associated with a clear-cut increase in extracellular IL-16 protein locally in the airways.18–20 From a statistical point of view, it can be argued that the size of our current study was too small to detect more modest differences between the study groups (type II error). We therefore cannot rule out the possibility of overlooking limited alterations of IL-16 in the extracellular compartment of the blood. However, the current study allowed us to prove other group-related differences, and therefore it seems unlikely that long-term tobacco smoking causes substantial alterations of extracellular IL-16 at the systemic level. It is therefore probable that the impact of long-term tobacco smoking on extracellular IL-16 protein is mainly local.
In our current study, we included a few sensitized subjects (ie, atopic nonasthmatics) in all groups. We find it unlikely that this influenced IL-16 concentrations since a previous study clearly showed that neither IL-16 mRNA nor the protein in the lung of atopic nonasthmatics differ from controls.45 In addition, we harvested all samples out of allergy season.45 In our study, the difference was mostly between smokers and NS and not between smokers with COPD and AS. One explanation could be that the samples from the subjects were collected during a clinically stable phase.
Conclusion
This study demonstrates for the first time that human NK cells in the blood produce the CD4+-recruiting cytokine IL-16 cytokine and suggest that long-term exposure to tobacco smoke decreases IL-16+ NK cells at the systemic level without substantially altering IL-16 protein in the extracellular compartment of the blood. Moreover, tobacco smoke components seem to deplete the NK cells of IL-16 protein in a process linked to oxidative stress. Thus, we provide evidence for a previously unknown impact of long-term tobacco smoking at the systemic level that involves NK cells; innate immune cells that play an important role in host defense against viruses and tumors – conditions that are overrepresented among smokers. Whether the negative correlation between tobacco load and specific leukocyte concentrations represents an impact of tobacco smoke or age or both deserves further study.
Acknowledgment
We gratefully acknowledge Dorota Persson for her skillful lung physiology measurements, and Eric Fortes Lähdet and Lory Fägerström for their assistance with the study. The study was funded by the Swedish Research Council, the Swedish Heart-Lung Foundation and federal funding in accordance with the ALF/LUA agreement. No direct or indirect financial support was obtained from the tobacco industry.
Author contributions
Anders Andersson, Carina Malmhäll, and Apostolos Bossios had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Anders Andersson and Carina Malmhäll contributed with the principal study design, sample collection, laboratory analyses, and data interpretation and were primary authors. Birgitta Houltz contributed with expertise on lung physiology measurements, data interpretation, and critical reading of the manuscript. Sara Tengvall contributed with laboratory analyses, data interpretation, and critical reading of the manuscript. Margareta Sjöstrand contributed to the study design, in particular with reference to laboratory analyses, and with critical reading of the manuscript. Ingemar Qvarfordt contributed to the study design, as well as with critical review and finalizing the manuscript. Anders Lindén contributed with the principal study design, data interpretation, funding, as well as the development and finalization of the manuscript. Apostolos Bossios contributed with the principal study design, sample collection, laboratory analyses, data interpretation, funding, as well as development and finalization of the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Tetley TD. Macrophages and the pathogenesis of COPD. Chest. 2002;121(5 Suppl):156S–159S. | ||
Burgel PR, Bourdin A, Pilette C, Garcia G, Chanez P, Tillie-Leblond I. Modifications structurales et inflammation dans la BPCO: importance des voies aériennes distales [Structural abnormalities and inflammation in COPD: a focus on small airways]. Rev Mal Respir. 2011;28(6):749–760. French. | ||
Saetta M, Baraldo S, Corbino L, et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(2):711–717. | ||
Ekberg-Jansson A, Arva E, Nilsson O, Lofdahl CG, Andersson B. A comparison of the expression of lymphocyte activation markers in blood, bronchial biopsies and bronchoalveolar lavage: evidence for an enrichment of activated T lymphocytes in the bronchoalveolar space. Respir Med. 1999;93(8):563–570. | ||
Turato G, Zuin R, Miniati M, et al. Airway inflammation in severe chronic obstructive pulmonary disease: relationship with lung function and radiologic emphysema. Am J Respir Crit Care Med. 2002;166(1):105–110. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Lofdahl MJ, Roos-Engstrand E, Pourazar J, et al. Increased intraepithelial T-cells in stable COPD. Respir Med. 2008;102(12):1812–1818. | ||
Urbanowicz RA, Lamb JR, Todd I, Corne JM, Fairclough LC. Altered effector function of peripheral cytotoxic cells in COPD. Respir Res. 2009;10:53. | ||
Ferson M, Edwards A, Lind A, Milton GW, Hersey P. Low natural killer-cell activity and immunoglobulin levels associated with smoking in human subjects. Int J Cancer. 1979;23(5):603–609. | ||
Mian MF, Lauzon NM, Stampfli MR, Mossman KL, Ashkar AA. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J Leukoc Biol. 2008;83(3):774–784. | ||
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510. | ||
Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115(11):2167–2176. | ||
Lu L, Ikizawa K, Hu D, Werneck MB, Wucherpfennig KW, Cantor H. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity. 2007;26(5):593–604. | ||
Cruikshank WW, Kornfeld H, Center DM. Interleukin-16. J Leukoc Biol. 2000;67(6):757–766. | ||
Keane J, Nicoll J, Kim S, et al. Conservation of structure and function between human and murine IL-16. J Immunol. 1998;160(12):5945–5954. | ||
Wilson KC, Cattel DJ, Wan Z, et al. Regulation of nuclear Prointerleukin-16 and p27(Kip1) in primary human T lymphocytes. Cell Immunol. 2005;237(1):17–27. | ||
Meagher C, Beilke J, Arreaza G, et al. Neutralization of interleukin-16 protects nonobese diabetic mice from autoimmune type 1 diabetes by a CCL4-dependent mechanism. Diabetes. 2010;59(11):2862–2871. | ||
Laan M, Qvarfordt I, Riise GC, Andersson BA, Larsson S, Linden A. Increased levels of interleukin-16 in the airways of tobacco smokers: relationship with peripheral blood T lymphocytes. Thorax. 1999;54(10):911–916. | ||
Andersson A, Qvarfordt I, Laan M, et al. Impact of tobacco smoke on interleukin-16 protein in human airways, lymphoid tissue and T lymphocytes. Clin Exp Immunol. 2004;138(1):75–82. | ||
Andersson A, Bossios A, Malmhall C, et al. Effects of tobacco smoke on IL-16 in CD8+ cells from human airways and blood: a key role for oxygen free radicals? Am J Physiol Lung Cell Mol Physiol. 2011;300(1):L43–L55. | ||
Laberge S, Cruikshank WW, Kornfeld H, Center DM. Histamine-induced secretion of lymphocyte chemoattractant factor from CD8+ T cells is independent of transcription and translation. Evidence for constitutive protein synthesis and storage. J Immunol. 1995;155(6):2902–2910. | ||
Swedish Respiratory Society. Swedish National Guidelines for the Treatment of the COPD, 2013. Available from: www.slmf.se/äldre-vårdprogram | ||
Swedish Medical Products Agency. Treatment of Chronic Obstructive Pulmonary Disease, 2009. Available from: https://lakemedelsverket.se/upload/om-lakemedelsverket/publikationer/information-fran-lakemedelsverket/Info_fr_LV_2009-2.pdf | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Macintyre N, Crapo RO, Viegi G, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. 2005;26(4):720–735. | ||
Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC. Lung volumes and forced ventilatory flows. Report working party standardization of lung function tests, European community for steel and coal. Official statement of the European Respiratory Society. Eur Respir J Suppl. 1993;16:5–40. | ||
Salorinne Y. Single-breath pulmonary diffusing capacity. Reference values and application in connective tissue diseases and in various lung diseases. Scand J Respir Dis Suppl. 1976;96:1–84. | ||
Shimonkevitz R, Northrop J, Harris L, Craun M, Bar-Or D. Interleukin-16 expression in the peripheral blood and CD8 T lymphocytes after traumatic injury. J Trauma. 2005;58(2):252–258. | ||
Lanier LL. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol. 2008;8(4):259–268. | ||
Culley FJ. Natural killer cells in infection and inflammation of the lung. Immunology. 2009;128(2):151–163. | ||
Srivastava S, Lundqvist A, Childs RW. Natural killer cell immunotherapy for cancer: a new hope. Cytotherapy. 2008;10(8):775–783. | ||
Yang Q, Goding SR, Hokland ME, Basse PH. Antitumor activity of NK cells. Immunol Res. 2006;36(1–3):13–25. | ||
Wang J, Urbanowicz RA, Tighe PJ, Todd I, Corne JM, Fairclough LC. Differential activation of killer cells in the circulation and the lung: a study of current smoking status and chronic obstructive pulmonary disease (COPD). PloS One. 2013;8(3):e58556. | ||
Boyer L, Savale L, Boczkowski J, Adnot S. Sénescence cellulaire et pathologies pulmonaires: exemple de la BPCO [Cellular senescence and pulmonary disease: COPD as an example]. Rev Mal Respir. 2014;31(10):893–902. French. | ||
Kerbrat S, Baskara I, Dagouassat M, et al. Rôle du stress oxydant dans la sénescence des lymphocytes T induite par l’exposition à la fumée de cigarette. Rev Mal Respir. 2015;32(3):318. French. | ||
Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164(20):2206–2216. | ||
Quoix E, Lemarie E. Épidémiologie du cancer bronchique primitif: aspects classiques et nouveautés [Epidemiological novelties in lung cancer]. Rev Mal Respir. 2011;28(8):1048–1058. French. | ||
Wilkinson TM, Donaldson GC, Johnston SL, Openshaw PJ, Wedzicha JA. Respiratory syncytial virus, airway inflammation, and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(8):871–876. | ||
Hodge G, Mukaro V, Holmes M, Reynolds PN, Hodge S. Enhanced cytotoxic function of natural killer and natural killer T-like cells associated with decreased CD94 (Kp43) in the chronic obstructive pulmonary disease airway. Respirology. 2013;18(2):369–376. | ||
Tollerud DJ, Clark JW, Brown LM, et al. Association of cigarette smoking with decreased numbers of circulating natural killer cells. Am Rev Respir Dis. 1989;139(1):194–198. | ||
Freeman CM, Martinez CH, Todt JC, et al. Acute exacerbations of chronic obstructive pulmonary disease are associated with decreased CD4+ & CD8+ T cells and increased growth & differentiation factor-15 (GDF-15) in peripheral blood. Respir Res. 2015;16(1):94. | ||
Zhang XM, Xu YH. The associated regulators and signal pathway in rIL-16/CD4 mediated growth regulation in Jurkat cells. Cell Res. 2002;12(5–6):363–372. | ||
Bade G, Khan MA, Srivastava AK, et al. Serum cytokine profiling and enrichment analysis reveal the involvement of immunological and inflammatory pathways in stable patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2014;9:759–773. | ||
Bowler RP, Bahr TM, Hughes G, et al. Integrative omics approach identifies interleukin-16 as a biomarker of emphysema. OMICS. 2013;17(12):619–626. | ||
Laberge S, Ernst P, Ghaffar O, et al. Increased expression of interleukin-16 in bronchial mucosa of subjects with atopic asthma. Am J Respir Cell Mol Biol. 1997;17(2):193–202. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.