")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Integrating risk minimization planning throughout the clinical development and commercialization lifecycle: an opinion on how drug development could be improved
Received 26 November 2014
Accepted for publication 16 January 2015
Published 26 February 2015 Volume 2015:11 Pages 339—348
DOI https://doi.org/10.2147/TCRM.S78202
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Garry Walsh
Elaine H Morrato,1 Meredith Y Smith2
1Colorado School of Public Health, University of Colorado, Anschutz Medical Campus, Aurora, CO, 2EMD Serono, Inc, Rockland, MA, USA
Abstract: Pharmaceutical risk minimization programs are now an established requirement in the regulatory landscape. However, pharmaceutical companies have been slow to recognize and embrace the significant potential these programs offer in terms of enhancing trust with health care professionals and patients, and for providing a mechanism for bringing products to the market that might not otherwise have been approved. Pitfalls of the current drug development process include risk minimization programs that are not data driven; missed opportunities to incorporate pragmatic methods and market-based insights, outmoded tools and data sources, lack of rapid evaluative learning to support timely adaption, lack of systematic approaches for patient engagement, and questions on staffing and organizational infrastructure. We propose better integration of risk minimization with clinical drug development and commercialization work streams throughout the product lifecycle. We articulate a vision and propose broad adoption of organizational models for incorporating risk minimization expertise into the drug development process. Three organizational models are discussed and compared: outsource/external vendor, embedded risk management specialist model, and Center of Excellence.
Keywords: pharmaceuticals, drug development, risk management, organizational design
Introduction
Pharmaceutical drug safety is poised at the brink of a new era: the emergence of a 21st century pharmacovigilance – “More than just adverse drug reaction reporting – it must be about leadership in drug safety, in safe use, and in achieving better and more regular positive therapeutic clinical outcomes”.1 A signature feature of this transformation is the growing emphasis on proactive approaches to managing product-related risks via the use of formal risk minimization programs (also known as risk evaluation and mitigation strategies [REMS] in the US).2,3 Essentially, such efforts are public health interventions that seek to ensure safe and appropriate use of a specific drug product. Examples of risk minimization activities include special manufacturing and supply chain procedures (to reduce tampering and/or diversion), health care professional (HCP) and patient education, patient screening and monitoring, and specific restrictions on prescribing and/or dispensing procedures. Outside of REMS, the increased use of boxed warnings and patient-directed medication guides has also contributed to growing risk minimization efforts in the US.4
This transformation has been essentially codified in the recent publications by the European Medicines Agency (EMA) of the European Union Good Pharmacovigilance Practice (GVP), Module V Risk Management Systems5 and Module XVI Risk Minimization Measures: Selection of Tools and Effectiveness Measures,6 as well as specific commitments outlined in the Prescription Drug User Fee Act (PDUFA) V for the US Food and Drug Administration (FDA).7 Collectively, these initiatives pose heightened accountability for regulators and industry for developing risk minimization programs that are demonstrably effective. A particularly key element of that accountability is recognition of the need to incorporate the patient’s perspective in all phases of the risk minimization process (as acknowledged in EMA’s GVP Modules V and XVI, and in FDA’s REMS guidance.8)
In both the US and the EU, the ability to require risk minimization programs is statutorily defined. However, these statutes are still relatively recent, and much remains to be determined with regard to integrating these regulatory requirements into the drug development process. As a result, the pharmaceutical industry is grappling with the challenge of identifying organizational structures and team processes that can optimally support effective and efficient development and execution of risk management commitments, including avenues for patient engagement. In particular, successful execution and evaluation of these programs require pharmaceutical companies to invest in both new types of professional expertise in the form of social scientists, and new types of cross-functional collaboration across the drug safety, medical, regulatory, and commercial teams.9,10
As a point of comparison, drug development transformation triggered by the 1962 Kefauver-Harris Drug Amendments in the US took over a decade to evolve into the modern drug development framework we use today.11 The amendments required manufacturers to meet investigational new drug standards and demonstrate efficacy as a condition for drug approval. New preclinical and clinical trial methodologies (eg, Phase I, II, and III trial designs) had to be developed to support the new legal standard of “substantial evidence” and “adequate and well-controlled data”. Today, in the post-passage era of the 2007 FDA Amendment Act and the EMA GVP Modules V and XVI, industry and regulators are in an analogous situation – that is, how to best standardize and operationalize methodologies in support of enhancement and modernization of drug safety and risk management.
A 2013 US government audit made several recommendations to FDA for improving the effectiveness of risk management efforts, including the need to: identify, develop, validate, and assess REMS components; clarify expectations for assessments and work with sponsors to obtain missing information; identify REMS that were not meeting their goals and take action to protect the public health; and seek legislative authority to enforce FDA assessment plans.12 Despite these challenges, progress is being made by regulators and the pharmaceutical industry. For example, the FDA has convened public workshops on social science and REMS standardization,13 and expert workshops on strengthening REMS through systematic analysis and evidence-based assessment,14 and has issued a report on standardizing REMS.15 Risk management programs have been successful in mitigating serious risks, for example, the THALOMID® (thalidomide) and iPLEDGE (isotretinoin) REMS programs. Some companies have implemented organizational change to incorporate risk minimization expertise into their drug development programs.
We believe more work is needed to accelerate and broaden the adoption of risk minimization practices into the drug development paradigm. This opinion article (1) highlights several shortcomings in the current approach taken by regulators and industry regarding risk minimization development and discusses how risk minimization development activities could be better integrated into existing clinical development and commercialization efforts for greater synergy, and (2) proposes possible organizational models for incorporating risk minimization expertise into the drug development enterprise.
Shortcomings of the current system
Risk minimization programs that are not data driven
Most risk minimization programs have been developed in reaction to heightened safety concerns from regulators and the public. Such a reactive approach can result in hastily designed programs that meet the minimum necessary requirements for approval or for remaining on the market. As a result, little evidence was generated to inform and support the design of the risk management program.2 This approach is equivalent to jumping straight into a Phase III clinical program without the benefit of Phase I and II results.
To illustrate this gap, let us consider a scenario when a company submits a new drug application with a risk that might potentially be minimized or mitigated by a formal risk minimization program. During the course of deliberations with the regulatory agency, it becomes clear that a risk management plan will be required in order to gain product approval. In the US, this drug application might come before an FDA advisory committee. Briefing materials might summarize numerous clinical studies and hundreds of pages of data in support of the unmet medical need and the efficacy and safety of the product. However, fewer than ten pages might be dedicated to describing the risk management program, and no data are provided to support its strategy. No protocols are provided in support of a post-marketing evaluation plan. Many times, companies will say at the advisory meeting that they have actively engaged patients in the development of educational materials; however, no evidence is provided, no draft materials are presented, and this is within a few months of potential market launch.
If this scenario involved a pre-Investigational New Drug meeting and no preclinical evidence was provided and no clinical plan detailed, the company would not receive go-ahead for human testing. Why should we hold risk management programs to a different evidentiary standard? Safety risks do not generally emerge overnight. There are opportunities for collecting data pertinent to risk management program design, including during the conduct of Phase III/IIIb studies and when promotional materials are being developed for clinicians and educational materials for patients prior to regulatory submissions.
Missed opportunities to incorporate pragmatic methods and market-based insights
Risk minimization programs are intended to promote safe use under real-world conditions. Registration trials are generally conducted in advanced clinical research centers with many resources, support staff, and services that may not be available to the average practitioner in day-to-day practice.16 To date, few risk minimization programs have explicitly incorporated pragmatic research methods, measures, or frameworks into their design. Risk management interventions should be pragmatic and adaptable for real-world health care settings if they are to be widely adopted and translated into clinical practice.17
Although companies vary in their approach, regulatory guidance implies that the leadership for the design and evaluation of risk minimization programs lies within the product safety team (or its equivalent), which typically includes drug safety, regulatory, clinical, and biostatistics team members. However, understanding of patient–provider needs and the constraints these stakeholders face in the real-world health care systems is typically housed within medical affairs and the commercial organization, thus representing a potential internal organizational barrier.
Outmoded tools and data sources
To date, the number and type of risk minimization tools and data sources employed have been both limited and non-evidence based.9 For example, web-based approaches have been confined to static websites and have not leveraged evidence-based, interactive, tailored methods that have been shown to be more engaging and effective in modifying behavior.18 For educational tools, there is a need to incorporate principles of plain language design and format, including pilot testing in the target audiences. Similarly, the types of data sources used to demonstrate effectiveness have been very limited, and are of low research quality.19
Lack of rapid evaluative learning to support timely adaption
The timetable for formal evaluation of risk management programs has been largely based on minimal regulatory requirements rather than on market-based learning cycles. For example, the standard milestones for submitting REMS assessments in the US are by 18 months and 3 years, and in the seventh year after the strategy was approved.20 This timetable contrasts with rapid market evaluation (as frequently as weekly/monthly) of commercial launch milestones and sales performance and quarterly pharmacovigilance drug safety updates for the first 2 years post-approval.21 Given the pace of health care change, health care research (such as risk management evaluations) must be more rapid22 and contribute toward a learning health care system.23
A separate issue is incomplete and late evaluations. A 2013 US government audit found that nearly half of the sponsor assessments for the 49 REMS reviewed did not include all the information requested in FDA assessment plans, and ten were not submitted to FDA within required timeframes.12 FDA was also slow in its REMS assessment review times and exceeded the 60-day goal for all but one sponsor assessment.12
Lack of systematic approaches for patient engagement
While FDA and EMA can be seen as being at an “inflection point” in regard to meaningful patient engagement in the drug development and review process, so, too, is the pharmaceutical industry.24 Broadly speaking, engaging patients can “influence research to be more patient centered, useful, and trustworthy and ultimately lead to greater use and uptake of research results by the patient and broader healthcare community”.25 From the pharmaceutical industry’s perspective, patient engagement should help optimize its ability to 1) address unmet medical need, 2) conduct patient benefit–risk assessment and optimization, 3) accelerate the product development and approval process, and 4) achieve rapid market penetration and product uptake.26 In instances where patient input has been obtained (eg, human factors testing conducted by product manufacturing; patient focus groups conducted by marketing), the information is often not widely shared nor conceived as part of a coherent, systematic, and comprehensive approach to pharmaceutical risk minimization.
Questions on staffing and organizational infrastructure
From the industry’s perspective, several key questions need to be addressed before optimum organizational processes and structures can be identified. These include:
- What is the product portfolio (both pre- and post-marketing) – do we have enough critical mass to warrant building a risk minimization organization?
- What are the key internal barriers and facilitators to improving how risk minimization strategies are set and activities conducted?
- Can existing organizational structures and processes be modified in order to support better cross-functional communication and collaboration in regard to risk minimization program development, implementation, and evaluation (ie, is there internal, political support for such an initiative)?
- What potential synergies could result internally from a more comprehensive and coordinated approach to interacting with patients (eg, patient access programs, patient adherence initiatives, patient registries) for the purposes of risk minimization?
- Do effective change agents exist within and outside the organization in terms of structuring a more patient-centered approach to risk minimization? If not, can they be developed?
A proposal
We propose two approaches for operationalizing risk minimization into the drug development process: 1) bridging risk minimization research and practice with existing drug development and commercialization processes for greater synergy and cross-fertilization; and 2) fostering access to behavioral science expertise through formalized organizational structures.
Better integration of risk minimization with clinical drug development and commercialization work streams throughout the product lifecycle
Figure 1 provides a conceptual framework that illustrates how a proposed risk minimization development work stream could parallel, and serve as a bridge between the clinical development process27,28 (which provides the biological basis and regulatory requirements for risk minimization) and the commercialization process (which provides information regarding the health care system context and the target patients and prescriber groups).
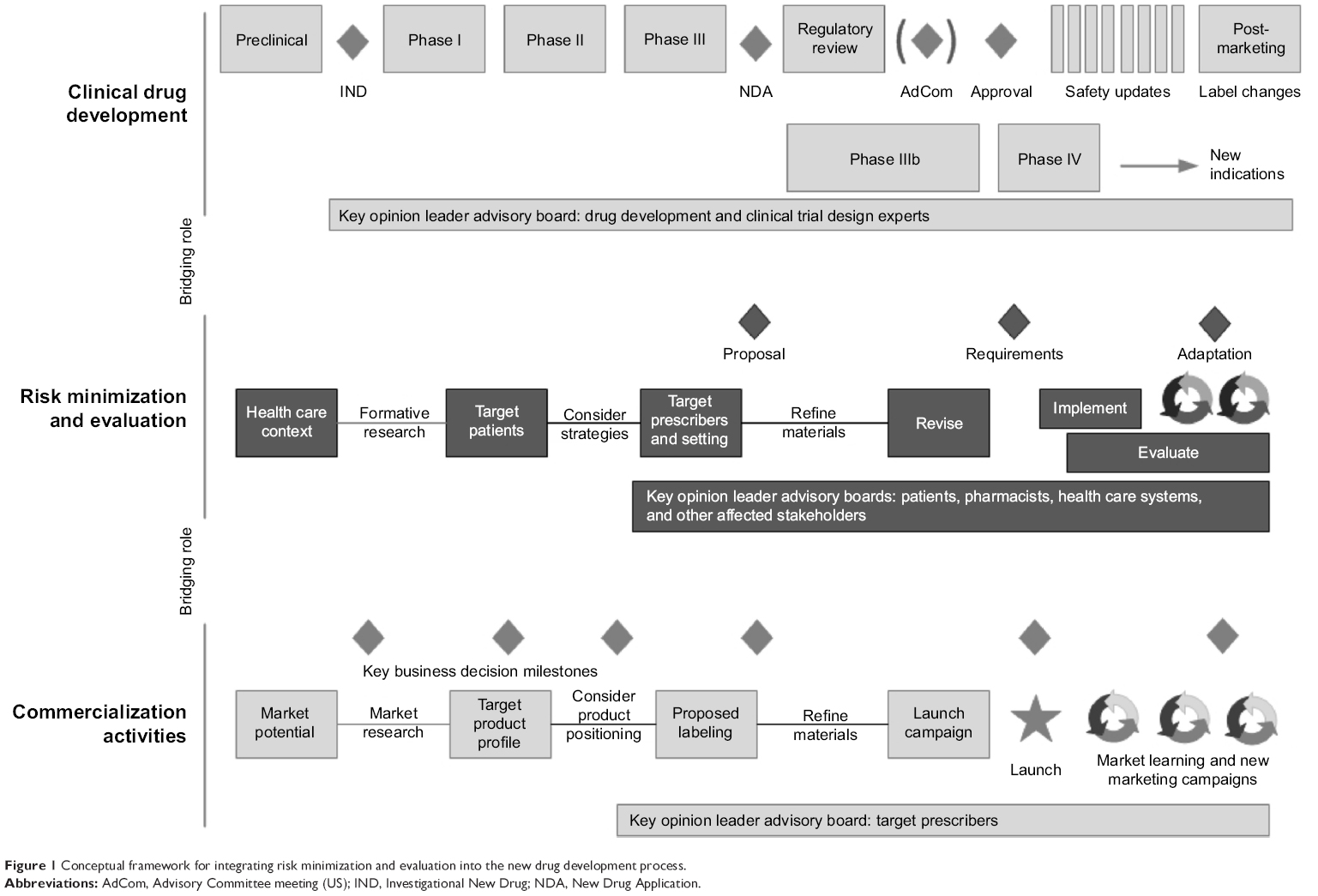 | Figure 1 Conceptual framework for integrating risk minimization and evaluation into the new drug development process. |
During the early phases of human testing, Phase I studies focus on the safety and pharmacology of a drug compound. Pharmacokinetic and pharmacodynamic studies provide support for mechanisms of drug action and the relationship between drug concentration and effect in humans. The commercial or new business development team continues to evaluate the drug’s market potential based on unmet market need and projected costs to develop and manufacture. The risk management work stream synthesizes the available clinical and market data to identify what risk minimization efforts might be necessary, given the health care context in which the drug would be used. This can include understanding anticipated treatment settings, typical habits and practices involving treatment within those settings, and the available resources and expertise of both patients and providers to manage potential risks.
The risk minimization plan draws upon internal market knowledge residing within the medical affairs and commercial organizations. While integrating insights from these two functional areas into the planning and implementation process is vital to the successful design and execution of a risk minimization program, it introduces a tension in that risk minimization programs, interventions, and materials are not, by definition, intended to be promotional in nature. One solution is to establish a new type of team member (a “risk minimization specialist”) who has the knowledge and applied expertise in designing and developing communication campaigns but who resides on the medical (non-promotional) side of the organization.
A desired future state would be to solicit patient input systematically throughout drug development. A patient-driven approach to health research is consistent with engagement strategies recommended by the Patient-Centered Outcomes Research Institute29 in the US and in legislation under development to reform FDA regulation.30 Patient perspective can provide valuable input on benefit–risk trade-offs that ultimately can inform safety/efficacy success criteria, the interpretation of clinical findings, and subsequent drug development decisions. For example, patient perspective on desired product attributes could be ascertained starting at proof of principle. At planning and execution of First-in-Human studies, patient preferences and trade-offs concerning the product’s benefit–risk profile could be assessed to inform development choices. During the confirmatory phases of drug development, understanding the patient’s journey through the health care delivery system could help determine feasible and acceptable ways to preventing or mitigating specific risks in the post-marketing context.
Phase II studies examine the effectiveness of a compound and determine dosing. Phase III studies confirm previous findings in a larger patient population based on the drug’s proposed indications for use. It is during these confirmatory phases of drug development that the conditions for establishing safety and efficacy are set. The risk minimization specialist would assess how well these conditions reflect real-world practice and whether the protocols and materials used in the pivotal program can be easily translated into practice. This will inform whether parallel research is necessary (eg, to develop and pilot education materials in language and format accessible for typical patients and prescribers). Qualitative and quantitative research may be combined in a variety of ways to achieve a deeper level of understanding than can be achieved by one method alone.31
Following successful completion of the pivotal trial program, it becomes evident what degree of risk minimization is required, ranging from: product labeling (package insert and patient-directed medication guides), enhanced communication programs, required education and training, or more restrictive distribution procedures. The risk management specialist produces the scientific justification for the risk minimization plan to regulators based on implementation science best practices,9 and they develop synergies with commercial activities to increase the likelihood of effective implementation once approved. In the US, the Veterans Administration serves as a model for fostering adoption of quality improvement requirements within its health care system using implementation science frameworks.32 The regulatory landscape regarding risk minimization requirements is evolving, and so the risk management specialist also contributes by actively anticipating regulatory needs. For example, the FDA is currently developing standards for when REMS are required as part of its PDUFA V commitments.8
At the time of regulatory submission, the organizational focus is on speed to submission. It is critical that the preliminary work to defend the risk minimization plan be completed by the end of the Phase III program. Ill-supported plans can delay approval.2 Postlaunch, the risk minimization specialist synthesizes pharmacovigilance findings with market understanding regarding drug use to determine whether goals are being met and whether adaptation is necessary.
Another point worth noting is the use of advisory boards throughout the product’s lifecycle. To inform the clinical development program, key opinion leaders with expertise in trial design are convened to guide what is necessary for market registration. To inform market launch, key opinion leaders respected for their medical expertise are convened to provide advice on product positioning. To inform the risk minimization plan, additional advisory boards engaging patients, pharmacists, health care systems, and/or other affected stakeholders may be highly beneficial. Engagement supports the scientific justification for the proposed plan and increases the likelihood for successful implementation.
In summary, the risk management specialist serves as a bridge between what is known clinically, what is required by regulators, and what is known about how clinical care is provided. By incorporating risk minimization thinking throughout the development and commercialization process, companies can be more proactive and nimble in their response should serious events requiring management emerge.
Articulate a vision and promote adoption of organizational models for incorporating risk minimization expertise into drug development
In order to achieve a viable integration of the risk minimization function with the clinical and commercialization work streams, a clear vision of the unique responsibilities and activities associated with the risk minimization function must be articulated and communicated across the organization. While several companies already have a “risk management” function or position, the positions typically reside in the pharmacovigilance/safety departments and their responsibilities are to write, manage, and coordinate the various risk management plan requirements across regulators. Instead, we argue that the risk minimization function should also be accountable for a broader set of strategic activities:
- leading strategic planning for risk minimization activities for the research portfolio as a whole as well as for individual products;
- executing or overseeing the execution of “best-in-class” risk minimization program design, implementation, and evaluation using knowledge from implementation science in health;
- conducting targeted research to develop improved risk minimization tools, methodologies, and evaluation approaches that support the company’s pipeline and marketed products’ portfolio;
- establishing a knowledge management system that: a) documents both internal and external “lessons learned” and the evolving risk minimization requirements and practices of regulatory authorities worldwide, and b) promulgates best practices in risk minimization science to internal teams;
- optimizing operational and cost efficiencies of risk minimization processes by standardizing processes where appropriate and leveraging preferred supplier and service provider arrangements;
- publishing risk minimization evaluations and research findings in order to advance the science in a “pre-competitive” context; and
- achieving a sustained level of compliance globally with regard to risk minimization commitments through standard setting, monitoring, and ongoing technical support to company affiliate offices.
Making the business case
So what is the business case for more proactive integration of risk minimization efforts into the drug development process? Some argue that changing the current development paradigm adds extra cost and requires additional communication coordination which will ultimately be transferred to the patients who would have to absorb the cost. On the other hand, many drugs would not otherwise be approved, or allowed to remain on the market, without effective risk minimization programs.2
Lessons from the business literature on the value of corporate responsibility and sustainability can shed light on this question. Broadly speaking, corporations have a societal responsibility to those groups and individuals that they affect, that is, their stakeholders. For pharmaceutical corporations, stakeholders include patients and caregivers, health care providers, health care organizations, and communities. Ensuring safe and appropriate use of medicines could be considered one form of social responsibility.
Practical examples of the business value generated by the allocation of resources in socially responsible activities include: reducing exposure to corporate risk and costs, enhancing corporate reputation and legitimacy, and gaining competitive advantage through shared value creation.33 Based on what the business literature argues, patients and other health care stakeholders who participate in the development of risk minimization programs should develop shared value with the pharmaceutical manufacturer and greater commitment to ensuring the appropriate use of the drug. Over time, this should lead to greater market sustainability and increased sales.
Organizational design
Several organizational structures for incorporating risk minimization expertise into the drug development enterprise are possible. Each requires senior management support and some degree of dedicated resources in order to be successful. We focus on three that are adaptable to differences in a company’s size, structure, resources, and level of organizational commitment to risk management. These three models include: 1) outsourcing of risk minimization design, implementation, and evaluation to one or more contract research organizations or vendors; 2) the hiring of one or more dedicated “risk management specialists; and 3) the establishment of an in-house risk management “Center of Excellence” (CoE).
Figure 2 summarizes the strengths and weaknesses of each model and provides example companies adopting a given model. Over time, there will be great value in learning about the respective successes and failures of these different organizational models with regard to directing risk minimization activities and enhancing safe use.
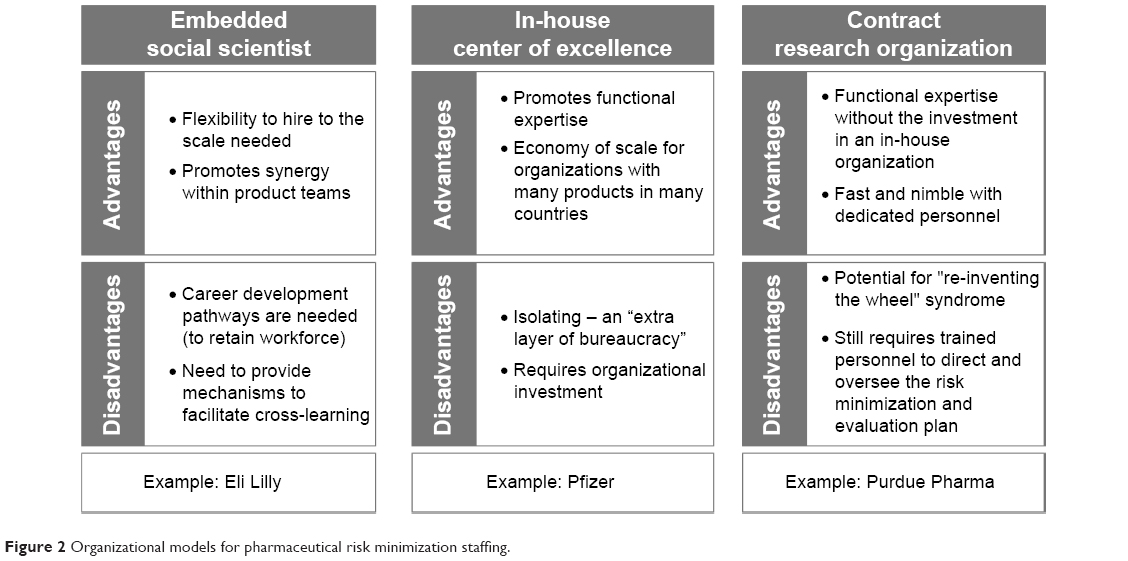 | Figure 2 Organizational models for pharmaceutical risk minimization staffing. |
Outsource/external vendor model
In this model, the organization relies exclusively on the resources and expertise of an outside vendor to fulfill risk management commitments, thus minimizing the need to redirect internal resources. Drawbacks of this approach, however, are multiple. These include the fact that: a) it provides limited opportunities for the development of internal expertise; b) costs can be higher, and program coordination can be compromised, especially if multiple vendors are hired for different tasks (eg, design, implementation, and evaluation); c) the lack of organizational investment in the risk management function can serve to undermine the efforts of those internally delegated to coordinate the planning, implementation, and evaluation of risk minimization and hamper their ability to gain cross-functional cooperation; and d) it can lead to a “reinventing the wheel” syndrome in which the learnings gained from one risk minimization program are not shared widely within the organization.
Embedded “risk management specialist” model
The risk management specialist model involves the hiring of a new type of pharmaceutical professional who would be responsible for risk management activities and be located on the medical side of the organization. This individual should have specific training in the behavioral/social sciences and applied experience in developing, implementing, and evaluating public health/health care interventions in real world settings. The risk management specialist could function in one of the two ways: 1) as an in-house “consultant”, circulating from team to team and cross-therapeutic areas to provide guidance and advice on how to plan, implement, and evaluate risk minimization programs on an “as-needed” basis; or 2) could be assigned to a specific team/product/therapeutic area for the lifecycle of the product. The key limitation of this approach is that the effectiveness of these new professionals, if lacking the institutional support offered by a dedicated functional department and adequate funding, may be quite constrained.
In-house CoE model
The CoE concept is both a familiar and widely applied concept within the pharmaceutical industry. It has been used to enhance organizational capacity in various aspects of the drug development process (eg, translational research) or to promote adoption of new methodologies (eg, benefit–risk assessment). Staffing for a risk minimization CoE should consist of a multidisciplinary team of individuals with training and/or expertise in one or more of the following areas: behavioral/social science, program evaluation, benefit–risk communication and social marketing techniques, social and other digital media design and content, human factors usability specialist, qualitative and quantitative methods (“mixed methods”), and other types of data analytics, and patient/customer engagement knowledge. In particular, the increasing demand for mixed methods approaches to evaluating risk minimization programs necessitates broader methodological training and deepened collaboration between medical, clinical, and social scientists, a collaboration that would be fostered in the context of a CoE.31
Establishing a CoE for risk management, with designated staff and budget, enables the development of internal expertise in risk minimization, identification, and sharing of “best practices” across teams and therapeutic areas, and signals a level of organizational commitment to both key internal and external stakeholders (eg, regulatory authorities, HCPs, and patients), that can enhance the credibility and acceptability of any proposed risk minimization activities. The main potential downside to this approach is cost, since it requires more infrastructure and staff to operate than the previous two models. Costs, however, might be offset to some significant extent by savings from a reduction or more efficient use of outsourcing of risk minimization deliverables.
Summary
Pharmaceutical risk minimization programs are now an established requirement in the regulatory landscape. However, pharmaceutical companies have been slow to recognize and embrace the significant potential these programs offer in terms of enhancing trust with HCPs and patients, and for providing a mechanism for bringing products to the market that would not otherwise have been approved.
Both the quality and credibility of risk minimization programs can be significantly enhanced or impeded by the organizational structure within individual pharmaceutical companies. No one single blueprint for an optimal risk management organizational structure exists. Instead, multiple factors, including the size and depth of the company’s pipeline, budgetary and staffing constraints, and organizational commitment to patient centeredness, dictate which particular approach might work best in which context. Further investigation is warranted to learn more about the respective successes and failures of the different organizational structures with regard to enhancing safe and appropriate drug use. In theory, companies that are able to establish an organizational structure that features superior cross-functional communication and collaboration, have appropriate expertise and adequate funding, and systematically solicit patient input will be able to develop and execute more effective and high-quality risk minimization programs. Such ability offers a significant competitive advantage for pharmaceutical and biotechnology companies and places them in an optimal position to fully embrace the new “21st century” model of pharmacovigilance.
Disclosure
Dr Morrato received no financial support for the conduct of this study or for the preparation of this manuscript. She has received consulting fees from the Consumer Healthcare Products Association, Merck & Company, and Janssen Pharmaceuticals. She has also received travel support from the Consumer Healthcare Products Association and Merck & Company. She has received research grant support from Janssen Pharmaceuticals. She is an advisor to the FDA and a former member of its Drug Safety and Risk Management Advisory Committee and current member of its Pulmonary-Allergy Drugs Advisory Committee. She has been an invited expert at the FDA REMS Assessments Public Workshop (June 2012), the Brookings Institution’s Expert Workshop “Strengthening Risk Evaluation and Mitigation Strategies” (September 2013 – paid travel), the CTTI-FDA Opioid Workshop (paid travel), and the Conjoint Committee on Continuing Education, Opioid REMS (no financial interest). She is a member of Benefit-Risk Assessment, Communication and Evaluation (BRACE) Special Interest Group (SIG) and International Society for Pharmacoepidemiology (ISPE) (no financial interest).
Dr Smith is a full-time employee of EMD Serono, Inc. and owns stock in Abbott Laboratories and AbbVie, Inc. Other than salary support, she received no funding for the conduct of this study or for the preparation of this manuscript. She is a former member of the Council on International Organizations of Medical Sciences Working Group VIII “Signal Detection in Pharmacovigilance” and Working Group IX “Practical Considerations for the Development and Application of a Toolkit for Medicinal Product Risk Management”. She has been an invited expert speaker at the US FDA Monthly Staff Forum, the FDA REMS Assessments Public Workshop (June 2012), and the Brookings Institution’s Expert Workshop “Strengthening Risk Evaluation and Mitigation Strategies” (September 2013). She is a member of the BRACE SIG and ISPE (no financial interest). She is a co-inventor of the US patent 12/237,853, title: System and methods for management of risk data and analytics (reference #: 241957.000019; Purdue reference: 07-BM-0014US02).
This manuscript represents the views of the authors and does not represent the views of EMD Serono, Inc. nor FDA policy or opinion.
References
Pitts PJ. openFDA: An Open Question. Ther Innov Regul Sci. 2014:1–2. | ||
Morrato EH, Ling SB. The Drug Safety and Risk Management Advisory Committee: a case study of meeting frequency, content, and outcomes before and after FDAAA. Med Care. 2012;50(11):970–986. | ||
U.S. Food and Drug Administration. Approved Risk Evaluation and Mitigation Strategies (REMS). [Updated July 7, 2014]. Available from: http://www.fda.gov/Drugs/DrugSafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm111350.htm. Accessed February 4, 2015. | ||
Frank C, Himmelstein DU, Woolhandler S, et al. Era of faster FDA drug approval has also seen increased black-box warnings and market withdrawals. Health Aff (Millwood). 2014;33(8):1453–1459. | ||
Guideline on Good Pharmacovigilance Practices (GVP) Module V – Risk Management Systems (Rev 1). United Kingdom: European Medicines Agency and Heads of Medicines Agencies; 2014. | ||
Guideline on Good Pharmacovigilance Practices (GVP) Module XVI– Risk Minimisation Measures: Selection of Tools and Effectiveness Indicators (Rev 1). United Kingdom: European Medicines Agency and Heads of Medicines Agencies; 2014. | ||
U.S. Food and Drug Administration. PDUFA Reauthorization Performance Goals and Procedures. Fiscal Years 2013 through 2017. | ||
U.S. Food and Drug Administration. REPORT: Standardizing and Evaluating Risk Evaluation and Mitigation Strategies (REMS); 2014. | ||
Smith MY, Morrato E. Advancing the field of pharmaceutical risk minimization through application of implementation science best practices. Drug Saf. 2014;37(8):569–580. | ||
Practical Approaches to Risk Minimisation for Medicinal Products: Report of CIOMS Working Group IX. Geneva, Switzerland: Council for International Organizations of Medical Sciences; 2014. | ||
Junod SW. FDA and Clinical Drug Trials: A Short History. [Updated July 7, 2014]. Available from: http://www.fda.gov/AboutFDA/WhatWeDo/History/Overviews/ucm304485.htm. Accessed February 4, 2015. | ||
Levinson DR. FDA Lacks Comprehensive Data To Determine Whether Risk Evaluation and Mitigation Strategies Improve Drug Safety (OEI-04-11-00510). Washington, DC: Health and Human Services, Office of Inspector General; 2013. | ||
Department of Health and Human Services, Food and Drug Administration. Risk Evaluation and Mitigation Strategy Assessments: Social Science Methodologies to Assess Goals Related to Knowledge: Public Workshop. Fed Regist. 2012;77(86):26292–26294. | ||
Engelberg Center for Health Care Reform at Brookings. Strengthening Risk Evaluation and Mitigation Strategies (REMS) Through Systematic Analysis, Standardized Design, and Evidence-Based Assessment: Meeting Summary. Washington DC; 2013. | ||
Department of Health and Human Services, Food and Drug Administration. REPORT: Standardizing and Evaluating Risk Evaluation and Mitigation Strategies (REMS); September 2014. | ||
Jazieh AR. Future of translational research: why go pragmatic? Pragmatic Obs Res. 2011;2011:1–4. | ||
Glasgow RE, Riley WT. Pragmatic measures: what they are and why we need them. Am J Prev Med. 2013;45(2):237–243. | ||
Lustria ML, Noar SM, Cortese J, Van Stee SK, Glueckauf RL, Lee J. A meta-analysis of web-delivered tailored health behavior change interventions. J Health Commun. 2013;18(9):1039–1069. | ||
Gridchyna I, Cloutier AM, Nkeng L, Craig C, Frise S, Moride Y. Methodological gaps in the assessment of risk minimization interventions: a systematic review. Pharmacoepidemiol Drug Saf. 2014;23(6): 572–579. | ||
Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for Industry Format and Content of Proposed Risk Evaluation and Mitigation Strategies (REMS), REMS Assessments, and Proposed REMS Modifications 2009 (Draft Guidance). Silver Spring, MD: U.S. Department of Health and Human Services, Food and Drug Administration; 2009. | ||
U.S. Food and Drug Administration. Guidance for Industry: Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment; 2005. Available from: http://www.fda.gov/cber/guidelines.htm#pharma. Accessed April 7, 2005. | ||
Peek CJ, Glasgow RE, Stange KC, Klesges LM, Purcell EP, Kessler RS. The 5 R’s: an emerging bold standard for conducting relevant research in a changing world. Ann Fam Med. 2014;12(5):447–455. | ||
Gluck ME, Radomski L. The AcademyHealth Listening Project Report: Improving the Evidence Base for Medicare Policymaking. Washington, DC: AcademyHealth; 2014. | ||
Looney W. FDA’s patient outreach: finding ties that bind. Pink Sheet. 2014 July 01. | ||
Institute P-COR. What We Mean by Engagement; 2014. Available from: http://www.pcori.org/content/what-we-mean-engagement. Accessed October 30, 2014. | ||
Milligan PA, Brown MJ, Marchant B, et al. Model-based drug development: a rational approach to efficiently accelerate drug development. Clin Pharmacol Ther. 2013;93(6):502–514. | ||
Lipsky MS, Sharp LK. From idea to market: the drug approval process. J Am Board Fam Pract. 2001;14(5):362–367. | ||
Hughes M, Inglese J, Kurtz A, et al. Early drug discovery and development guidelines: for academic researchers, collaborators, and start-up companies. In: Sittampalam GS, Coussens NP, Nelson H, editors. Assay Guidance Manual [Internet]. Bethesda, MD: Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2012:2004. | ||
Patient-Centered Outcomes Research Institute. PCORI Engagement Rubric with Table; 2014. Available from: http://www.pcori.org/assets/2014/08/PCORI-Engagement-Rubric-with-Table.pdf. Accessed October 12, 2014. | ||
FDA. FDA and patient groups take center stage as part of effort to accelerate #Path2Cures [press release]. July 11, 2014;2014. | ||
Albright K, Gechter K, Kempe A. Importance of mixed methods in pragmatic trials and dissemination and implementation research. Acad Pediatr. 2013;13(5):400–407. | ||
Damschroder LJ, Aron DC, Keith RE, Kirsh SR, Alexander JA, Lowery JC. Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4:50. | ||
Tonello M. The Business Case for Corporate Social Responsibility. The Harvard Law School Forum on Corporate Governance and Financial Regulation; 2011. Available from: http://blogs.law.harvard.edu/corpgov/2011/06/26/the-business-case-for-corporate-social-responsibility/. Accessed February 17, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.