")
Back to Journals » International Journal of General Medicine » Volume 14
Integrated Analysis of Mutations, miRNA and mRNA Expression in Glioblastoma
Authors Wang S, Zhou H, Zhang R, Zhang Y
Received 2 September 2021
Accepted for publication 2 November 2021
Published 16 November 2021 Volume 2021:14 Pages 8281—8292
DOI https://doi.org/10.2147/IJGM.S336421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
ShiChao Wang,1 HuanMin Zhou,1 RuiJian Zhang,2 YanRu Zhang1
1College of Life Sciences, Inner Mongolia Agricultural University, Hohhot, Inner Mongolia, 010018, People’s Republic of China; 2Department of Neurosurgery, The People’s Hospital of Inner Mongolia, Hohhot, Inner Mongolia, 010017, People’s Republic of China
Correspondence: YanRu Zhang Tel +976 18047183314
Email [email protected]
Background: Glioblastoma multiforme (GBM) is a common, malignant brain tumor in adults, with a median survival of only 15– 23 months. Organisms respond to disease stress through sophisticated mechanisms at the physiological, transcriptional and metabolic levels. However, the molecular regulatory networks responsible for occurrence, progression and recurrence of glioma have yet to be elucidated.
Methods: In this study, we sought to determine the cause of gliomas by developing an RNA-seq technique that analyzes mRNA and small RNA (sRNA) with the aim of discovering potential methods for precisely blocking key signaling pathways in occurrence, progression, and recurrence. The explication of mechanisms leading to GBM formation has become a feasible and promising new therapeutic method.
Results: GBM-associated genes were identified based on their expression during the disease stress response. Analysis of the inverse correlations between microRNAs (miRNAs) and target mRNAs revealed 43 mRNA–miRNA interactions during disease progression. BOC-SMO and BOC-RAS were found to promote the malignant progression of glioma. A total of 3088 differentially expressed genes were identified as involved in several biological processes, such as amino acid metabolism, protein transport associated with immune response, cell proliferation, and cell apoptosis. Fifteen miRNAs were also identified as being differentially expressed in GBM and control groups.
Conclusion: The results of this study provide an important foundation for understanding the pathogenesis of glioma and discovering new therapeutic targets.
Keywords: glioblastoma multiforme, transcriptome, miRNA
Introduction
Glioblastoma multiforme (GBM) is the most common type of malignant primary central nervous system (CNS) tumor, and patient prognosis is usually poor.1 Based on histological classification, the World Health Organization (WHO) classified brain gliomas as I–IV grade. Astrocytoma (grades I–II) is classified as a low-grade glioma, and grades III and IV are considered high grade. The difference in histopathological features between high-grade and low-grade gliomas includes increased atypical cell density, enlarged necrotic areas, and abnormal vascular hyperplasia.2,3 Grade I has the best patient prognosis, and a cure can be achieved in clinical practice. GBM is classified as a grade IV tumor and is the most malignant tumor, accounting for 55% of intracranial tumors. Although strict treatment regimens currently exist for GBM, the survival rate of patients diagnosed with GBM remains very low, with a median overall survival rate of only 15–23 months.4
MicroRNAs (miRNAs) are a class of small (20–24 nucleotides in length) endogenous RNAs that play a variety of important regulatory roles in cells. miRNAs, as a family of small and evolutionarily conserved noncoding RNAs, are capable of regulating physiological and pathological processes via inhibiting downstream target mRNA translation. They can bind to the untranslated regions of target gene mRNAs, leading to mRNA cleavage and the negative regulation of post-transcriptional expression of target genes.5 Copathogenic gene expression is controlled by the up and down regulation of miRNAs, which contribute to cancer cell infection, proliferation, inbreak, angiogenesis, pro/anti inflammation, and metastasis in breast cancer, prostate cancer, nasopharyngeal carcinoma, and glioblastoma.6–9 Therefore, miRNAs can serve as important diagnostic and prognostic biomarkers for many cancers, including glioblastoma.10,11
Next-generation sequencing (NGS) technology provides an integrated understanding of gene status and has become a precise molecular analytical tool for transcriptional assays.12 With the development of NGS, changes to the genome, transcriptome and epigenome can be observed at the single-cell level, allowing disease occurrence and development to be monitored at the cellular level, including in tumors. Cell-cycle regulation is a fundamental feature of cancer pathogenesis and genes involved in cell growth and proliferation are expressed at high levels in almost all tumor microarray datasets. The sequencing approach has been used to identify the expression of cell growth and proliferation genes in gastric cancer,13 colorectal adenoma,14 and glioblastoma.15 Understanding the miRNA-mediated network of the cell-cycle response will provide a genetic basis for future studies.
In this study, we investigated miRNA and mRNA expression in glioblastoma cells, as well as the negative miRNA-mRNA correlation in a human astroglioma cell line. The technologies used in this study allowed interrogation of BOC gene expression and its relationship to GBM. Draft genome sequences of carcinoma and paracancerous tissues from four samples identified genes that provide a functional description of the disease and serve as a route for visual visualization management based on our research.
Materials and Methods
Ethics Statement
This study was approved by the local ethics committee of Hohhot First Hospital and Inner Mongolia People’s Hospital with the informed consent of all participants. And this study was conducted in accordance with the Declaration of Helsinki. All experimental protocols were in accordance with the approved guidelines for safety requirements of hospital department administrator. All information about the participants was anonymized.
Clinical Sample Collection
Study criteria were strictly aligned with the diagnostic guidelines in China for the diagnosis and treatment of glioma in the CNS and the classification rules of the WHO (2016) for tumors in the CNS.16 Tested samples, including the GBM focal zone, together with the corresponding adjacent non-involved tissue (ANIT) were collected from four patients (aged 35–60 years) at stages III and IV. ANIT samples were obtained at a distance from the edges of the tumor. GBM and ANIT were diagnosed using clinical histopathological approaches at the Department of Pathology and stages were determined according to Tumor-Node-Metastasis (TNM) classification. The TNM staging system has been imported globally for decades as a benchmark for staging cancers, which take into account the weight of tumor size, the category, and multiple distant metastases.17,18 All lesions were located in cerebral hemispheres, with three located in the frontal lobe and one in the temporal lobe.
Study Design
Samples, including neoplastic and paracancerous tissues, were collected and fixed with formalin for pathological detection, as well as stained with hematoxylin and eosin (H&E). The adjacent cancer tissues and tumor tissues were differentiated and confirmed by optical microscopy. The enhanced MRI images of enrolled patients were provided (Figure S1). The remaining tissue samples were cut into 0.5 × 0.5 × 0.5-cm pathological slices and immediately frozen with liquid nitrogen for the subsequent sequencing experiments. Detailed sample information is listed in Table 1.
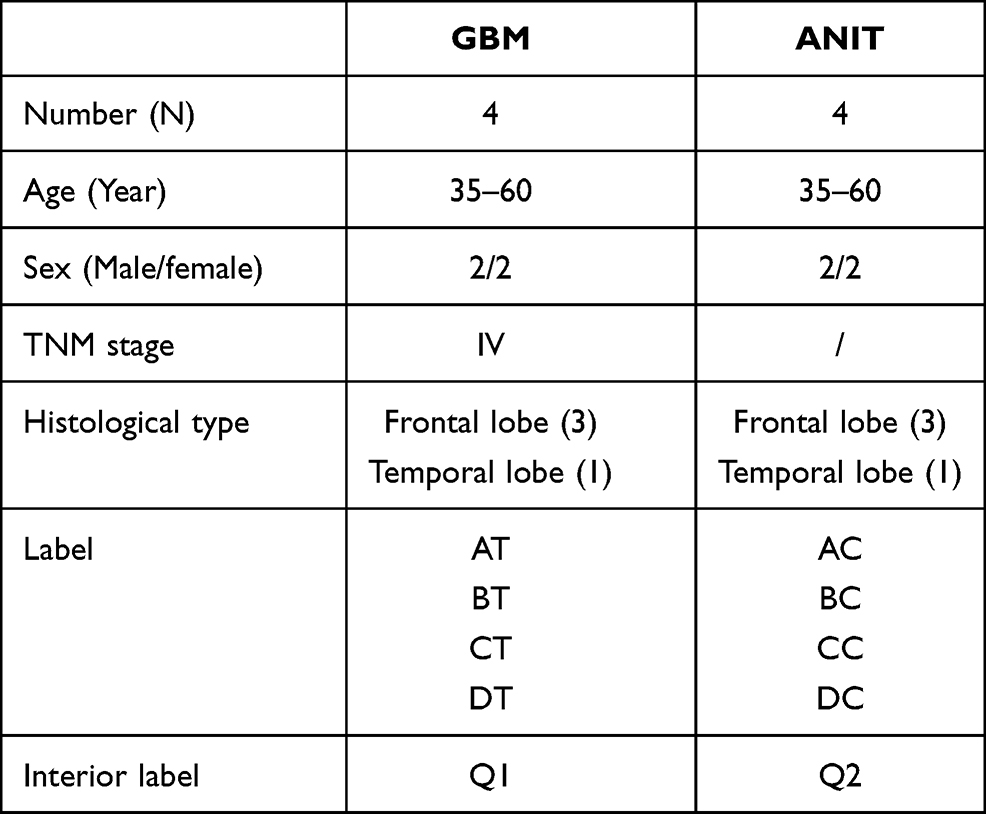 |
Table 1 Sample Grouping Schedule of GBM Tissue and Corresponding Paracancerous Enrolled in This Study |
Tissue RNA Extraction and Quality Determination
Appropriate amounts of tissue were thoroughly ground in liquid nitrogen. TRIzol reagent mixture (1 mL) was added, and the sample was thoroughly mixed prior to a 10-min incubation at room temperature. Chloroform (200 μL) was added, and the samples were thoroughly shaken before centrifugation at 12,000 rpm for 10 min at 4 °C. The upper aqueous layer with RNA was separated using an equal volume of phenol:chloroform (25:24) prior to centrifugation at 12,000 rpm at 4°C. The upper aqueous phase was then added to an equal volume of chloroform and centrifuged at 12, 000 rpm for 10 min at 4°C. Finally, the upper aqueous phase was mixed with an equal volume of isopropyl alcohol and centrifuged at 12,000 rpm for 10 min at 4°C and the mixture was purified using ethanol. Total plasma RNA was eluted using 20–50 μL of RNase-free water and stored at –80°C until subsequent analysis. Quality control was performed by RT-q PCR using four control and four GBM tissue samples and the quality and quantity of RNA extracted from tissue samples were determined using a nanodrop assay. RNA integrity number (RIN) and the percentage of RNA fragments > 200 nt were monitored as quality indicators.
Sequencing Library Preparation and Data Analysis
Three micrograms of total RNA per sample was used for RNA preparation. Ribosomal RNA was first removed using the Epicentre Ribo-zero rRNA Removal Kit (Epicentre, USA) and the rRNA-free residue was cleaned by ethanol precipitation. Sequencing libraries were subsequently generated using the rRNA-depleted RNA and NEBNext Ultra Directional RNA Library Prep Kits for Illumina (NEB, USA) following manufacturer’s recommendations. Briefly, fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X). First strand cDNA was synthesized using random hexamer primers and M-MuLV Reverse Transcriptase (RNaseH-). Second strand cDNA synthesis was performed using DNA Polymerase I and RNase H. In the reaction buffer, dNTPs with dTTP were replaced with dUTP. The remaining overhanging ends were converted to blunt ends using exonuclease and polymerase activities. Following the adenylation of the 3ʹ ends of DNA fragments, they were ligated with the EBNext Adaptor with a hairpin loop structure in preparation for hybridization. cDNA fragments that were approximately 150–200 bp in length were purified for the library using the AMPure XP system (Beckman Coulter, Beverly, USA). USER Enzyme (3 μL; NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C prior to PCR analysis. PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. At last, products were purified (AMPure XP system), and library quality was assessed on the Agilent Bioanalyzer 2100 system.
Clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. The libraries were then sequenced on an Illumina HiSeq 4000 platform and 150-bp paired-end reads were generated. Raw data in FastQ format (raw read) was initially processed with an internal Perl Script. This step included obtaining clean data by deleting reads that contained the adapter, the ploy-N, and the low-quality reads from the original data (clean reads). The Q20, Q30 and GC content in the clean data were calculated, and all subsequent analyses were based on the processed clean data.
Cuffdiff was used to evaluate fragments per kilobase of exon per million reads mapped (FPKMs) for both long non-coding RNAs (lncRNAs) and coding genes in each sample.19 Gene FPKMs were computed by adding the transcript FPKMs for each group. FPKMs were numbered based on the length of the fragment and read count mapped to the fragment.
The Ballgown suite includes functions for interactive data exploration of the transcriptome assembly, visualization of transcript structures, feature-specific abundances for each locus, and post-hoc annotation of assembled features to annotated features. Transcripts with a P-adjust <0.05 were determined to be differentially expressed transcripts. Cuffdiff provides statistical routines for determining different expressions in digital transcript and gene expression data using a model based on the negative binomial distribution and transcripts with an P-adjusted were assigned as differentially expressed.20
Gene Ontology (Go) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
GO enrichment analysis of differentially expressed genes or lncRNA target genes was implemented using the Goseq R package with gene length bias correction.21 GO terms with corrected P-values <0.05 were considered to be significantly enriched by differentially expressed genes.
The KEGG database is a widely used resource for annotation at high levels, as well as identifying relevant functions of the biological system. Molecular-level information of the cell, the organism, and the ecosystem can be determined, especially when using large-scale molecular datasets generated by genome sequencing and high-throughput experimental technologies. We used KOBAS software to test the statistical enrichment of differentially expressed genes and lncRNA target genes in KEGG pathways.22
Transduction with Lentiviruses
The significance of the BOC gene in glioblastoma was verified using a BOC gene knockdown in human glioblastoma U251 cell lines using lentivirus transduction. A knock-down (KD) group and negative control (NC) group were established to evaluate the knockout effect. The process of transfection was as follows: Lentivirus (MOI = 10) was inoculated into 6-well plates and the cell density was set to 30%. Polybrene was added to the plates 10 min prior to the addition of the lentivirus to enhance the transfection efficiency. The plates were incubated at 37°C with 5% CO2 for 16 h before replacing the medium with normal culture medium. Following incubation for 24 h, 1 μg/mL puromycin was added to the medium to select for stably transfected cells. After 72 h of incubation under the above conditions, the infection effect was verified using fluorescence photography (Table S1).
Verification of BOC Knockdown Efficiency by qRT-PCR
mRNA expression levels of BOC and downstream genes HRAS, MRAS, and SMO in KD-BOC cell strains and the control group using qRT-PCR. Trizol reagent was used to extract total RNA from cells according to manufacturer’s introduction. A Promega M-MLV kit was used to synthesize cDNA by reverse transcription, and the cDNA was amplified using the SYBR Premix Ex Taq RT-PCR kit. Finally, mRNA levels were detected by quantitative PCR using a Roche LightCycler 480 II instrument. GAPDH was used as a reference gene and the mRNA expression levels of BOC and downstream genes were calculated using 2−ΔΔCT method. Primer information is shown in Table S2.
Results
Stained Pathological Sections
Tumor areas were darker than the surrounding normal brain tissue in pathological sections stained with H&E. Glioma cells and palisade necrosis were also visible and are demonstrated indicators of glioma. Figure 1 shows long fusiform and stellate tumor cells, with large, round hyperchromatic nuclei. Pathological mitosis is common, with occasional binuclear cells. Tumor tissues were hemorrhagic, necrotic, and contained an abundance of microvessels. Tumor tissues were densely arranged around vessels with focal infiltration. Tumor cells also displayed characteristics of active growth with bundle arrangement in small clumps. Other pathology reports were shown in Figure S2.
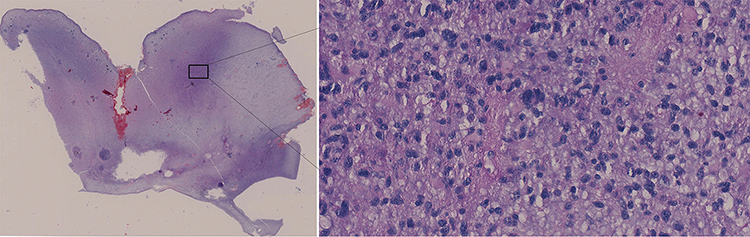 |
Figure 1 H&E staining result. Note: The left image is the tissue image at 10 × 10 low power, and the right image is the tissue image at 40 × 10 power of the selected area. |
miRNA Sequencing Data Analysis
Eight qualified libraries from the GBM illness group, along with corresponding paracarcinoma tissue, were sequenced. An overview of the sequencing and assembly data is provided in Table 2. Raw data were cleaned by removing low-quality reads. GLAT, GLBT, GLCT and GLDT from the four samples in group Q1 were constructed using the small RNA Sample Prep Kit, resulting in 14,491,255, 15,478,540, 15,161,494 and 14,660,621 raw reads, respectively. After removal of the low-quality reads from the raw data, 14,107,719, 15,019,165, 14,746,657 and 14,205,763 clean reads were obtained, respectively. The paracarcinoma tissues corresponding to the above samples produced, 15,495,417, 13,323,123, 14,247,033 and 14,370,467 raw reads, respectively.
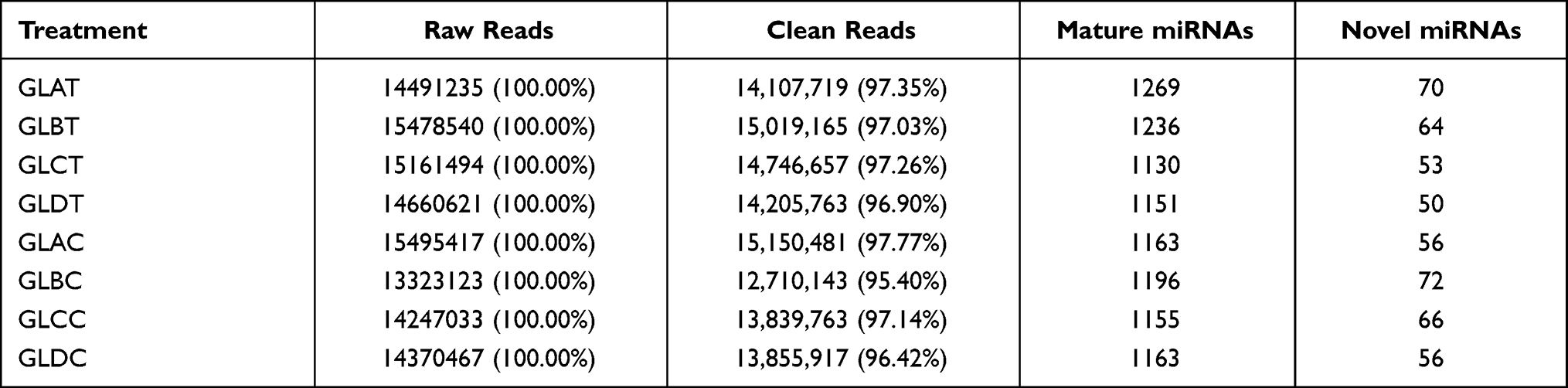 |
Table 2 Summary of Small RNA (sRNA) Sequencing Datasets |
After removing low-quality data, there were 15,150,481, 12,710,143, 13,839,763, 13,855,917 corresponding clean reads, respectively. A majority of the sRNAs were 21–24 nt in length for all eight libraries, with 24 nt being the most frequent length (Figure S3). Based on the described method, a total of 1730 miRNAs and 120 novel miRNAs were identified in the eight sRNA libraries (Table S3). Details about the distributions of the sRNAs are provided in Table S4.
Differential Expression of miRNAs in GBM
The miRNA-seq data analysis identified a total of 15 differentially expressed miRNAs (DE miRNAs) in GBM samples and their corresponding paracarcinoma tissue, which fit the criteria | log2 FC | > 0.5 and P-value <0.05. Two of these were identified as novel miRNAs (Table 3). The conserved (n=13) and novel (n=2) miRNAs were significantly expressed in GBM, with eight being upregulated and seven being downregulated. The novel miRNAs (novel_912 and novel_328) were similarly regulated in GBM tissues.
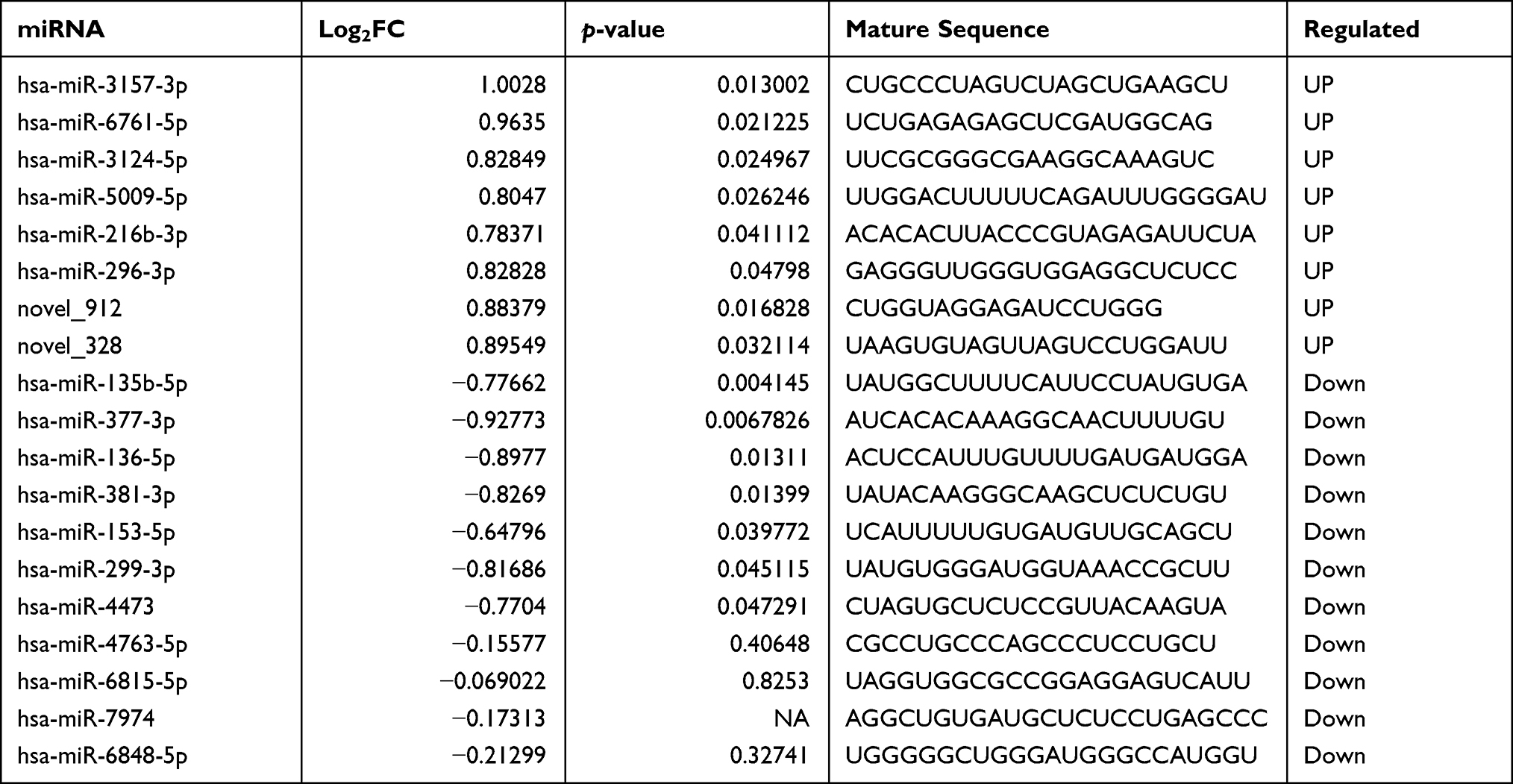 |
Table 3 List of Differentially Expressed microRNAs (DE miRNAs) in Response to GBM |
mRNA Sequencing Data
Eight qualified libraries from the GBM illness group and the corresponding paracarcinoma tissue were sequenced. An overview of the sequencing and assembly data is shown in Table 4. The raw data from the carcinoma tissue were cleaned by removing low-quality reads, resulting in 100,911,602, 100,277,916, 112,758,002 and 82,991,704 clean reads, respectively. More than 94.28% of the clean reads had Q-scores at the Q30 level (an error threshold of < 0.01%). The reliability between the replicates of Q1 and Q2 samples was evaluated using Pearson correlation analysis. Figure 2 shows the results of the correlation analysis indicating the similarity between tumor tissues was >69% and para-carcinoma tissue was >60%.
 |
Table 4 Summary of mRNA Sequencing Datasets |
Differentially Expressed Genes in GBM
The differences in gene expression between glioma and carcinoma tissues during disease progression were examined by differential expression analysis using DESeq software with the strict criteria of |log 2 FC|> 2 and P-value <0.05 as the screen difference standard. A total of 3088 genes were differentially expressed in response to GBM, with 1375 upregulated and 1713 downregulated (Table S5, S6).
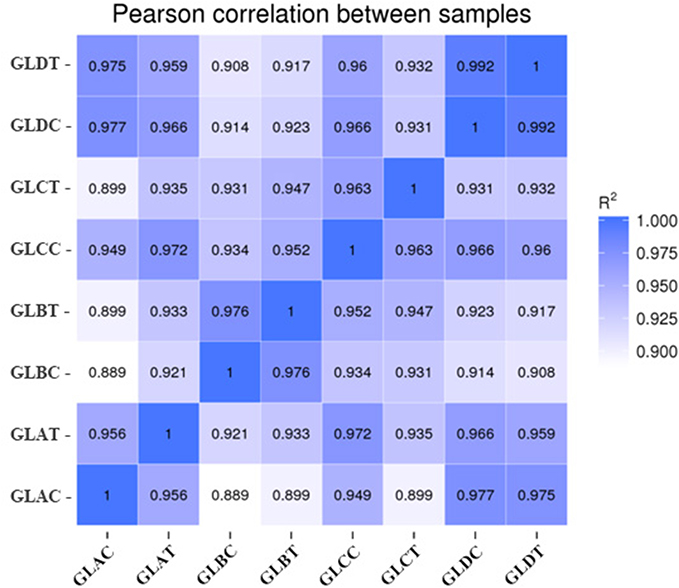 |
Figure 2 Heatmap of Pearson correlations of the miRNA expression levels among samples. |
Functional Classification of GBM Responsive Genes
All responsive genes were mapped to the KEGG database for a comprehensive understanding of the DEG functions. Differentially expressed mRNA transcripts were screened in tumor tissues and paracancerous tissues, and 19 significant pathways were obtained by co-enrichment. Significantly enriched KEGG terms were identified for 27 upregulated and 18 downregulated genes. The enriched KEGG pathways were related to immune response, amino acid metabolism, cell proliferation, protein transport, as well as other physiological and biochemical reactions. The enrichment analysis is shown in Figure 3 and mainly involves the hedgehog (Hh) signaling pathway (hsa04340), glioma signaling pathway (hsa05214), pancreatic cancer signaling pathway (hsa05212), axon guidance (hsa04360), TNF signaling pathway (hsa04668), among others.
 |
Figure 3 Enriched KEGG pathway scatterplot. |
Integrated Analysis of miRNA and mRNA
The interaction between DE miRNAs and their targets was investigated using Cytoscape version 3.6.1 software. This analytical process identified miRNA–mRNA interactions with the involvement of DE miRNAs and DE mRNAs (Figure 4). A single miRNA can regulate multiple mRNAs, and a single mRNA can be targeted by more than one miRNA. These results suggest that the miRNA and mRNA interaction network involved in disease progression is highly complex. Based on network regulation, a deep analysis of the interactions between miRNA and mRNA was performed to identify the location distribution and sequence correlation. Different RNA molecules with potential crossover effects were systematically and comprehensively analyzed. As a whole, these findings have important scientific value in revealing the complex mechanism of tumor genesis.
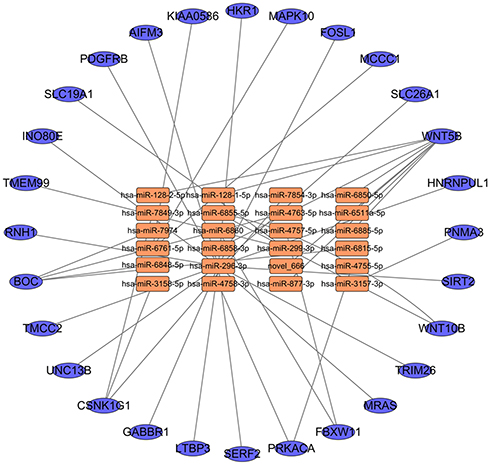 |
Figure 4 miRNA-mRNA correlation network. The circles in the network indicate the target mRNAs. The orange squares and blue ellipse in the network indicate the DE miRNAs and mRNAs. |
Results of Lentivirus Transfection
Cells exhibited marked green fluorescence, after selection with puromycin, and the adherent cells displayed integral morphologies, which is an indicator to evaluate the effect of virus transfection. Dead cells were observed in the experimental group and the empty carrier group. Many dead cells were detected in the untreated control group. Adherent cells lacking intact cell morphology were sparse. Puromycin-resistant cells were successfully structured after virus transduction, and the stably transfected cells were identified by screening with puromycin (Figure 5).
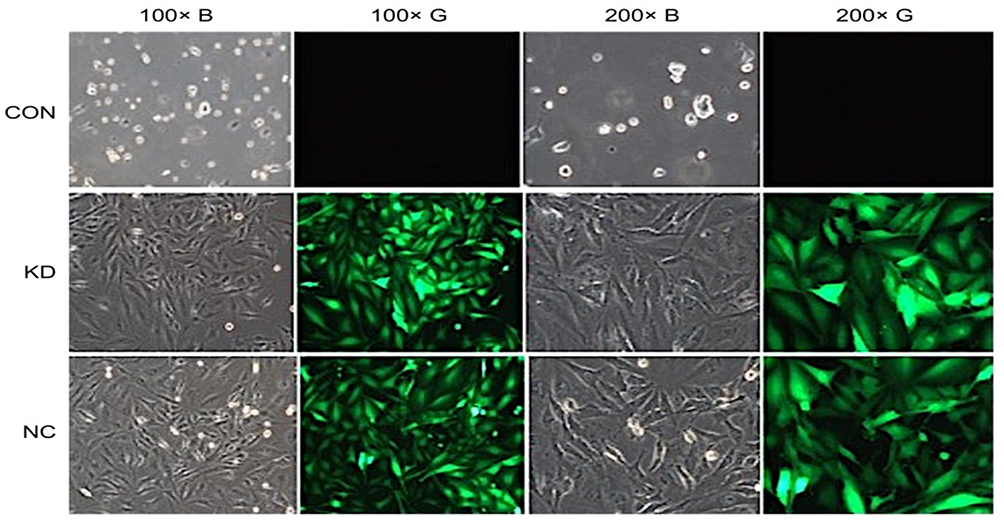 |
Figure 5 Cell pattern after puromycin screening. |
Verification Results of qRT-PCR
BOC is a receptor-like protein in the Ig/ FNIII repeating family, which can directly bind to Hh ligand. Abnormal expression of BOC can amplify Hh signal by increasing local Hh concentration or prolonging the binding time of Hh ligand, thus affecting the response of the whole nervous central system to Hh. Therefore, we performed quantitative analysis based on BOC and its downstream genes. The mechanism of interaction between BOC and downstream genes in glioma cells is shown in Figure S3. Expression levels of BOC, SMO, HRAS, and MRAS in the KD-BOC group of U251 cells were markedly reduced relative to the control group (P<0.05), indicating the successful construction of an ideal model using knockdown technology. BOC knockdown impacted downstream gene expression, resulting in reduced expression levels, which affected the occurrence and development of glioblastoma (Figure 6).
 |
Figure 6 Verification of qRT-PCR results. Notes: BOC (A); SMO (B); HRAS (C); MRAS (D). *P < 0.05, **P < 0.01, ***P < 0.001. |
Discussion
Gliomas are the most common primary tumors of the CNS and are a significant threat to human health. The accelerated development of NGS technologies has enabled the investigation of a wide range of physiological and metabolic changes at the molecular level. In this study, we concurrently analyzed mRNA and miRNA expression levels in GBM disease progression. NGS technology not only provides comprehensive analytical results for RNA, it also effectively narrows the target gene range and captures key genes by associating multiple analyses. Using integrative analysis, we obtained a set of mRNAs and miRNAs associated with the occurrence and development of GBM and identified interactions and potential roles of these mRNAs and miRNAs during disease progression. These data provide a systematic understanding and allow for an in-depth exploration of the relevant molecular mechanism.
During disease stress, humans can accumulate essential substances as an effect of a tumor, as well as regulate gene expression to maintain metabolic homeostasis.23 The Hh signaling pathway was first reported in the common fruit fly and plays a vital role in normal embryonic development. When its function is disrupted, Hh has been associated with a variety of neoplasms.24 Acting through a signaling cascade, Hh can cause a change in the balance between activator and inhibitor forms of glioma-associated oncogene (GLi) transcription factors. Elements of the Hh signaling pathway involved in the signal transfer to the GLi transcription factors include hedgehog ligands, patched receptors (Ptch1, Ptch2), smoothened receptor (Smo), protein kinase A (PKA), and cyclic adenosine monophosphate (cAMP). GLi activation targets it to the nucleus, where it modifies the transcription of target genes by binding to their promoters. The primary genes targets of the Hh signaling pathway contribute to many important pathways including those associated with PTCH1,25 Rab23,26 RAS, BRAF,27 VEGF, and MAPK.28 Indeed, Rab 23 mRNA and its associated protein are highly expressed in glioma tissues and contribute to the pathological grade.29 Likewise, the RAS-mitogen-activated protein kinase (RAS-MAPK) pathway, a common feature of tumors, is important in the development of targeted therapeutics aimed at mitigating patient morbidity.30 In this study, mRNA expression levels of RAS family proteins were significantly upregulated in tumor tissues compared with adjacent tissues, which is consistent with previous studies. This includes the protein encoded by the BRAF gene, a serine/threonine protein kinase of Raf/mil, which is involved in the regulation of the MAP/ERKs signaling pathway and plays an important role in the process of cell division, differentiation and secretion. Mutations in the BRAF gene have been associated with a variety of cancers, including colorectal cancer, malignant melanoma, non-small cell lung cancer, and lung adenocarcinoma. Mitogen-activated protein kinases (MAPKs) are highly conserved serine/threonine protein kinases in eukaryotes, which are involved in biological processes, such as cell growth, proliferation, differentiation, movement and apoptosis. In our study, an increased abundance of BRAF and MAPKs was identified in tumor tissues (Table S5), suggesting a potential role for this mRNA in malignant proliferation and invasion of glioma.
BOC is a transmembrane protein located upstream of the Hh signaling pathway. BOC mRNA expression levels were altered in tumor tissues when compared to adjacent tissues, although the change was not significant. The small number of participants enrolled in this study may contribute to the variability of expression levels for related genes. The main reason our study focused on BOC and the Hh signaling pathway is the novelty and reliability of the Hh pathway. An increasing number of studies are indicating that BOC expression is significantly elevated in glioma tissue compared to normal brain tissue. In addition, the survival time of glioma patients with high BOC levels is significantly reduced, suggesting BOC may be a potential drug target due to its activation of the Hh signaling pathway, which promotes the progression of GBM.31,32 The Ivy Glioblastoma Atlas Project (Ivy GAP) is an anatomical transcription map of human glioblastoma, which links individual histological characteristics with genomic and gene expression patterns. It allows researchers to explore the anatomical and genetic mechanisms of glioblastoma at the cellular and molecular levels. The database creates a comprehensive collection of suspected molecular variants in glioblastoma cells, allowing for a deeper understanding of tumors.33 Utilizing this tool, we focused our research on the regulated genes of the Hh signaling pathway. As part of the study, we added a cell line validation assay to further verify the relationship between the BOC gene and the malignancy, invasion, and migration of the tumor. q-PCR technology was used extensively for the quantitative analysis of BOC expression levels in glioma tumor tissues and adjacent tissues to verify the accuracy of the whole transcriptome sequencing results. Based on this, we selected U251 cell lines for our studies and BOC-targeted silencing was performed using RNA interference technology to knockdown BOC expression. We then observed changes to the expression of the downstream genes and pathways affected by reduced BOC expression to understand the molecular mechanisms leading to BOC-mediated promotion of glioma proliferation, invasion, and migration.
Numerous studies have recently confirmed the crucial role of miRNAs in the occurrence and development of disease. Our studies utilized sRNA sequencing to identify 13 known miRNAs and 2 novel miRNAs in GBM samples. Consistent with a previous sRNA sequencing study of cancer samples, our results indicate that the most abundant transcripts across all libraries were has-miR-4763-5P, has-miR-6848-5p, and has-miR-6860. miRNA expression is correlated with cancer type, stage, and other clinical variables and miRNA profiling is a tool for cancer diagnosis and prognosis. miRNAs function in nearly all stages of cancer biology, including proliferation, apoptosis, invasion/metastasis, and angiogenesis.34–36 Our study identified 13 known and 2 novel miRNAs that correspond to the occurrence and development of disease. Significant regulation of the miR135 family was observed in GBM samples and tissues adjacent to GBM. The conserved miR135 family is a well-studied family that plays a vital role in both diffuse and intestinal gastric cancer subtypes. In cancer, antisense oligonucleotides (anti-miRs) were transfected to inhibit the activity of specific microRNAs. In prostate carcinoma, transfection of antimiRs against miR-375 leads to tumor cell inhibition in 2D models, which further verifies its regulatory role in the carcinogenesis process.37 AntimiRs transfection significantly reduced the expression of hsa-miR-135b-5p, resulting in regulation of the target APC genes.38 An investigation of miR299 in papillary thyroid carcinoma (PTC) found it to be significantly dysregulated in PTC tissues compared to matched normal thyroid tissues.39 Our experimental data further confirmed the significance of miR135 and miR299 in cancer development, including cellular proliferation, apoptosis, and migration. These results also provide valuable information on the role of miR135 and miR299 in the cancer development. Four additional miRNAs (hsa-miR-4763-5p, hsa-miR-6815-5p, hsa-miR-7974, and hsa-miR-6848-5p) were found to be dysregulated and became the focus genes in our study because of their intimate connection with the BOC gene. BOC contains potential target binding sites for hsa-miR-4763-5P, hsa-miR-6848-5P, has-miR-6860. Additionally, the BOC downstream genes POU2F1 and TP53 are associated with tumor gene transcripts, which could explain the ability of BOC to affect the tumor formation process by regulating downstream genes through miRNA binding. A similar effect was previously observed with hsa-miR-3157-3p, where the downstream gene PNMA was identified as a Bax-associating protein that induced apoptosis in cultured mammalian cells.40
In general, miRNAs negatively regulate gene transcription by targeting complementary RNA sequences in gene targets for cleavage. Our integrated analysis of miRNA and mRNA transcript levels revealed inverse regulation of 20 and 28 interactions in GBM group and control group. For instance, the miRNA 196 was upregulated in GBM, and the mRNA HNRNPUL1 showed high mutation frequencies, and HNRNPs participated in cancer-related pathways including protein secretion and mitotic spindle.41,42 However, the miRNA 196 target HNRNPs was not identified in our study. Recently, a role for the miR128/WNT5B network in the regulation of depression susceptibility was identified in rats model.43 The miR-299-3P target TVF4 in the regulation of inhibiting proliferation and invasion of cervical cell via targeting TCF4 was also identified in our study, and SLC19A1 serve as a potential biomarker for diagnosis and prognosis in osteosarcoma.43 As expected, nt-miR128 targeted WNT5B exhibited strict negative regulation in GBM, suggesting that nt-miR128 play an important role in GBM by regulating the expressions of WNT5B.
Additionally, a large number of tumor-responsive miRNAs and their targets, such as miR-3157/PNMA3, miR-6860/FBXW11, miR-6858-3p/WNT5B the target–action relationship has been found, and there have been separate reports on mRNA and miRNA in tumor development (Figure 6); however, the interactions of this regulatory network need to be further studied.
Conclusions
To conclude, the sequencing analysis provides an overview of dysregulated miRNAs and mRNAs that might have potential for diagnosis and treatment of glioma and may be useful excavating new potential biomarkers. Further research on the function of these RNAs will help us understand the mechanisms of occurrence and progression of glioblastoma.
Funding
This study was funded by grants from the Science and Technology Planning Project of Hohhot, Inner Mongolia (No.2019-1-5).
Disclosure
The authors have declared that no competing interests exist.
References
1. Nagaraja S, Vitanza NA, Woo PJ, et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell. 2017;31:635–652.e6.
2. Shergalis A, Bankhead A
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19:v1–v88.
4. Lim KY, Won JK, Park CK, et al. H3 G34-mutant high-grade glioma. Brain Tumor Pathol. 2021;38:4–13.
5. Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252–263.
6. Que T, Song Y, Liu Z, et al. Decreased miRNA-637 is an unfavorable prognosis marker and promotes glioma cell growth, migration and invasion via direct targeting Akt1. Oncogene. 2015;34:4952–4963.
7. Hu N, Zhang J, Cui W, et al. miR-520b regulates migration of breast cancer cells by targeting hepatitis B X-interacting protein and interleukin-8. J Biol Chem. 2011;286:13714–13722.
8. LeBlanc VC, Morin P. Exploring miRNA-associated signatures with diagnostic relevance in glioblastoma multiforme and breast cancer patients. J Clin Med. 2015;4:1612–1630.
9. Chiosea S, Jelezcova E, Chandran U, et al. Up-regulation of dicer, a component of the MicroRNA machinery, in prostate adenocarcinoma. Am J Pathol. 2006;169:1812–1820.
10. Deng B, Wang M, Liu Z. A panel of 8 miRNAs as a novel diagnostic biomarker in pancreatic cancer. Medicine (Baltimore). 2020;99:e22261.
11. Rezaei O, Honarmand K, Nateghinia S, et al. miRNA signature in glioblastoma: potential biomarkers and therapeutic targets. Exp Mol Pathol. 2020;117:104550.
12. Behjati S, Tarpey PS. What is next generation sequencing?. Arch Dis Child Educ Pract Ed 2013;98:236–238.
13. Manicum T, Ni F, Ye Y, et al. Prognostic values of E2F mRNA expression in human gastric cancer. Biosci Rep. 2018;38:BSR20181264.
14. Xue VW, Cheung MT, Chan PT, et al. Non-invasive potential circulating mRNA markers for colorectal adenoma using targeted sequencing. Sci Rep. 2019;9:12943.
15. Liu G, Pan Y, Li Y, et al. lncRNA and mRNA signature for prognosis prediction of glioblastoma. Future Oncol. 2020;16:837–848.
16. Jiang T, Nam DH, Ram Z, et al. Clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. 2021;499:60–72.
17. Denaro N, Russi EG, Merlano MC. Pros and cons of the new edition of TNM classification of head and neck squamous cell carcinoma. Oncology. 2018;95:202–210.
18. Paner GP, Stadler WM, Hansel DE, et al. Updates in the eighth edition of the tumor-node-metastasis staging classification for urologic cancers. Eur Urol. 2018;73:560–569.
19. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515.
20. Frazee AC, Pertea G, Jaffe AE, et al. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol. 2015;33:243–246.
21. Young MD, Wakefield MJ, Smyth GK, et al. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14.
22. Mao X, Cai T, Olyarchuk JG, et al. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–3793.
23. Sjodahl G, Eriksson P, Liedberg F, et al. Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J Pathol. 2017;242:113–125.
24. Girardi D, Barrichello A, Fernandes G, et al. Targeting the Hedgehog pathway in cancer: current evidence and future perspectives. Cells. 2019;8:135.
25. Agren M, Kogerman P, Kleman MI, et al. Expression of the PTCH1 tumor suppressor gene is regulated by alternative promoters and a single functional Gli-binding site. Gene. 2004;330:101–114.
26. Wang M, Dong Q, Wang Y. Rab23 is overexpressed in human astrocytoma and promotes cell migration and invasion through regulation of Rac1. Tumour Biol. 2016;37:11049–11055.
27. Chiang J, Li X, Liu APY, et al. Tectal glioma harbors high rates of KRAS G12R and concomitant KRAS and BRAF alterations. Acta Neuropathol. 2020;139:601–602.
28. Chen X, Hao A, Li X, et al. Activation of JNK and p38 MAPK mediated by ZDHHC17 drives glioblastoma multiforme development and malignant progression. Theranostics. 2020;10:998–1015.
29. Dong J, Liu Y, Hao Y, et al. Expression and correlation of Rab23 with pathological grades in human glioma cells. J Biol Regul Homeost Agents. 2019;33:1885–1889.
30. Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun. 2020;8:30.
31. Nitta Y, Miyachi M, Tomida A, et al. Identification of a novel BOC-PLAG1 fusion gene in a case of lipoblastoma. Biochem Biophys Res Commun. 2019;512:49–52.
32. Jiang C, Qu X, Ke H, et al. Association between the HMGB1/TLR4 signaling pathway and the clinicopathological features of ovarian cancer. Mol Med Rep. 2018;18:3093–3098.
33. Zheng S, Tao W. Identification of novel transcriptome signature as a potential prognostic biomarker for anti-angiogenic therapy in glioblastoma multiforme. Cancers (Basel). 2020;12:2368.
34. Sun Z, Shi K, Yang S, et al. Effect of exosomal miRNA on cancer biology and clinical applications. Mol Cancer. 2018;17:147.
35. Xu B, Liu J, Xiang X, et al. Expression of miRNA-143 in pancreatic cancer and its clinical significance. Cancer Biother Radiopharm. 2018;33:373–379.
36. Zhang B, Pan X, Cobb GP, et al. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12.
37. Kunz M, Brandl M, Bhattacharya A, et al. Nanoparticle-complexed antimiRs for inhibiting tumor growth and metastasis in prostate carcinoma and melanoma. J Nanobiotechnology. 2020;18:173.
38. Magalhaes L, Quintana LG, Lopes DCF, et al. APC gene is modulated by hsa-miR-135b-5p in both diffuse and intestinal gastric cancer subtypes. BMC Cancer. 2018;18:1055.
39. Liu X, He M, Hou Y, et al. Expression profiles of microRNAs and their target genes in papillary thyroid carcinoma. Oncol Rep. 2013;29:1415–1420.
40. Schuller M, Jenne D, Voltz R. The human PNMA family: novel neuronal proteins implicated in paraneoplastic neurological disease. J Neuroimmunol. 2005;169:172–176.
41. Li H, Liu J, Shen S, et al. Pan‐cancer analysis of alternative splicing regulator heterogeneous nuclear ribonucleoproteins (hnRNPs) family and their prognostic potential. J Cell Mol Med. 2020;24:11111–11119.
42. Guan Y, Mizoguchi M, Yoshimoto K, et al. MiRNA-196 is upregulated in glioblastoma but not in anaplastic astrocytoma and has prognostic significance. Clin Cancer Res. 2010;16:4289–4297.
43. Roy B, Dunbar M, Agrawal J, et al. Amygdala-based altered mirnome and epigenetic contribution of miR-128-3p in conferring susceptibility to depression-like behavior via Wnt signaling. Int J Neuropsychopharmacol. 2020;23:165–177.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.