")
Back to Journals » ImmunoTargets and Therapy » Volume 5
Insights into the pathogenesis of Langerhans cell histiocytosis: the development of targeted therapies
Received 20 April 2016
Accepted for publication 21 June 2016
Published 12 October 2016 Volume 2016:5 Pages 81—91
DOI https://doi.org/10.2147/ITT.S91058
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Caroline Hutter,1,2 Milen Minkov1–3
1International LCH Study Reference Center, Children’s Cancer Research Institute, Vienna, Austria; 2St. Anna Children's Hospital, University Clinic of Pediatrics and Adolescent Medicine, Medical University of Vienna, Vienna, Austria; 3Department of Pediatrics, Adolescent Medicine and Neonatology, Rudolfstiftung Hospital, Vienna, Austria
Abstract: More than a century after its first description, Langerhans cell histiocytosis (LCH) still remains an intriguing disease. Considerable progress in understanding its biology has been achieved recently. Description of the V600E BRAF mutation in samples of LCH tissue in 2010 was followed by description of additional mutations, all leading to constitutive ERK activation. Current experimental data suggest that LCH is a myeloid neoplasia with inflammatory properties, yet the exact pathophysiology remains poorly understood. Disease management paradigms have changed over time, closely reflecting the evolving view of the nature of the disease. The international Histiocyte Society have conducted three prospective clinical studies on multisystem LCH since the early 1990s. The standard frontline therapy for patients with multisystem LCH based on the cumulative knowledge of those trials consists of 6–12 weeks of initial therapy (daily oral steroids and weekly vinblastine injections), followed by pulses of prednisolone/vinblastine every 3 weeks, for a total treatment duration of 12 months. A currently ongoing study (LCH-IV) with a complex design (five interventional and two observational strata) targets further reduction of mortality and morbidity by tailoring treatment intensity depending on expected risk, as well as by exploring treatment regimens for special locations. Current knowledge on LCH pathobiology opens opportunities for improvement in the patient outcome. The activating BRAF and MAP2K1 mutations collectively accounting for about 75% of the LCH population as well as the resulting constitutive activation of downstream ERK offer an opportunity for targeted treatment. Related issues (eg, finding most effective and less toxic drugs or combinations, appropriate dosage, and optimal treatment duration) must be addressed in controlled prospective trials. Additional mechanisms, such as the interactions of the mutated dendritic cell clone with other inflammatory cells and key cytokines and chemokines, still remain attractive targets for therapeutic intervention, particularly in patients with localized, less aggressive disease.
Keywords: Langerhans cell histiocytosis, treatment, targeted therapy, BRAF, myeloid neoplasm
Introduction
The first ascertained report of Langerhans cell histiocytosis (LCH) dates back to the late 19th century.1 For many years, its description has been accompanied by the adjectives “enigmatic”, “poorly understood”, and “intriguing”, reflecting the uncertainty about its pathobiology. Many factors impeded progress in understanding LCH, the main obstacles being its rarity, difficulty in obtaining sufficient samples, lack of appropriate investigative technologies, and the extreme heterogeneity of its clinical spectrum and natural course.2 Therefore, the experience gathered from patient care and empirically developed treatment regimens was the main resource of knowledge until recently.
Finding a rational cure for LCH has been the driving force of the joined efforts of scientists, physicians, and patient/parent organizations (eg, Histiocyte Society, Histiocytosis Association, and Nikolas Symposium) during the past 3 decades. With the recently gained knowledge on LCH origin and pathobiology, this dream seems to be closer to reality.
Historical perspective
Because of characteristic morphological findings (tissue lesions containing large cells with histiocytic appearance), LCH has for a long time been categorized as a disease of the “monocyte-macrophage system”. Opinions on the origin and biology of LCH and the terminology used for its description have mirrored the main developments of the natural sciences and the advances of human biology over the past century (Figure 1).3–5 Two major hypotheses dominated the perception of LCH during the past 3 decades: that it is either a reactive disease (resulting of an inappropriate immune stimulation or intrinsic immune deregulation) or a neoplastic process. However, neither could be substantiated by unequivocal evidence.6,7
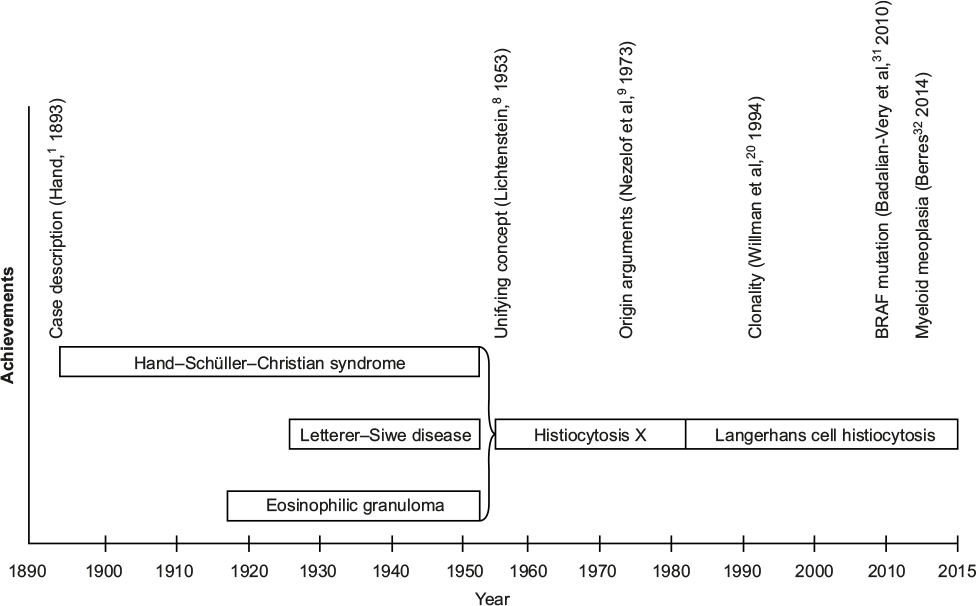 | Figure 1 LCH: historical timeline. Abbreviation: LCH, Langerhans cell histiocytosis. |
For decades, the disease had been referred to as “histiocytosis X,” stressing its unknown source.8 Identification of Birbeck granules (pentalaminar cytoplasmic bodies considered to be pathognomonic for the normal Langerhans cells [LC] of the human epidermis) in lesional histiocytes of patients with histiocytosis X led to the assumption of a histogenetic relation,9 and consequently to the current eponym “Langerhans cell histiocytosis”, as well as to the assignment of the entity to the histiocytoses of dendritic cell (DC) origin.10,11 Further evidence favoring this hypothesis was the shared surface phenotype between the normal LC and the lesional cells (named LCH cells).12 At that time, the epidermal LC have been thought to be of bone marrow origin, but it remains unclear what cause them to accumulate and form tissue granulomas (in cooperation with other inflammatory cells) in different organs. Dysregulation of chemokine production and/or chemokine receptor expression clearly plays a crucial role in LCH.13,14 Considering the role of the DC in antigen recognition and immune response initiation, inappropriate stimulation of the immune system or deregulated immune reaction is seemingly an appropriate assumptive explanation of a “reactive” disease process. This hypothesis was supported by the self-limiting course of LCH in a proportion of the patients, the inflammatory properties of the biopsy specimens (granuloma formation), and the abundance of local and systemic cytokines and chemokines.15–19
The finding of clonal LCH cells20 was the first solid challenge to the hypothesis of a “reactive” disease, but it was not sufficient to disprove it. Furthermore, though indirect, arguments in favor of a neoplastic origin were the observation of familial clustering,21 concurrent myelodysplasia,22 chromosomal instability,23 telomerase activity in some LCH lesions,24 and the increasing number of patients with malignancies preceding or following LCH.25 The description of a patient who has a common immunoglobulin gene rearrangement in LCH and B-cells26 and of two cases of LCH arising in the context of precursor T-lymphoblastic leukemia/lymphoma27 further supported the hypothesis that LCH might be associated with clonal malignant diseases of the hematopoietic stem cells.
Current understanding of the LCH origin and biology
LCH is characterized by clonal proliferation and dissemination of cells that express CD1a and CD207, the hallmark features of the eponymous epidermal LC.28 However, gene expression profiling of LCH cells has revealed more than 2,000 differentially expressed transcripts compared to LC, showing that these two cell types differ from each other. Furthermore, the comparison of LCH cells with different DC subtypes has shown that the LCH cell is – at least at the transcriptomic level – similarly related to myeloid DC (mDC1) and to LC.29 Therefore, the bona fide progenitor cell of the CD207-positive LCH cell is still unknown. Perhaps in the future, comparative expression analysis with a larger set of reference cells or chromatin profiling could provide insight into the ontogeny of LCH.30
Badalian-Very et al who analyzed archival LCH samples using OncoMap, a high-throughput genotyping platform, achieved a breakthrough in the understanding of LCH biology in 2010. By using this technology, a gain-of-function mutation in the BRAF serine/threonine kinase (BRAF V600E) was found in 57% of the studied samples.31 This mutation, which causes the substitution of glutamate for valine at amino acid 600 (BRAF V600E), leads to the constitutive activation of the RAS/RAF/MEK/ERK pathway and is highly prevalent in various neoplasms. For this reason, LCH is now regarded as a neoplastic disease. In addition to the identification of a recurrent mutation, another important finding of that study was the universal ERK phosphorylation irrespective of mutational status, indicating that in the BRAF wild-type samples, other mutations or translocations could exist. Indeed, next-generation sequencing of LCH lesions subsequently identified mutations in MAP2K1, which occur in 50% of BRAF wild-type cases, and mutations in ARAF, in sporadic cases. Overall, the genomic landscape appears to be remarkably simple with just a few mutated genes that all seem to lie in the MAPK pathway. Because MAPK is active in all LCH lesions, it seems likely that the non-BRAF-, ARAF-, or MAP2K1-mutated LCH cells will have an acquired somatic mutations or translocations of other pathway members affecting the activation of MEK/ERK. Interestingly, the mutation status did not correlate with disease status and severity in the published series so far, although one study found an association with an increased risk of recurrence.32 In addition, this study detected BRAF V600E in bone marrow-derived cells in some multisystem LCH (MS-LCH) patients, whereas in single-system LCH (SS-LCH) patients, the mutation was only found in lesional LCH cells. The authors therefore propose a model of LCH in which the initiating cell defines the extent of the disease, ie, the presence of the mutated BRAF in a hematopoietic stem cell would cause high-risk LCH, whereas a mutation that is restricted to a more differentiated cell type would give rise to low-risk disease. The BRAF V600E mutation can also be detected in hematopoietic stem cells of almost all patients with hairy cell leukemia. How the same mutation in the same cell type can cause two clinically different diseases remains unclear to date. It is also unclear which additional factors can lead to the induction of the LCH-specific markers. One possibility might be the activation of the Notch pathway, because Notch and Notch receptors are highly expressed in LCH cells and because the stimulation of monocytes with the Notch ligand JAGGED2 can induce an LCH-like phenotype in vitro.29
A new classification of histiocytic disorders
On behalf of the Histiocyte Society, Emile et al33 recently proposed a revision of the classification of histiocytoses. The original classification took into account the assumed cell of origin and the morphological features of malignancy, and thus consisted of three categories: Class I:LCH; Class II: histiocytoses of mononuclear phagocytes other than LC; Class III: malignant histiocytic disorders.10 This classification of the Writing Group of the Histiocyte Society has been revised in 1997 in an attempt to refine diagnostic criteria and accommodate the varying biologic behavior of the diverse diseases with the original classification.11 In the currently proposed classification, Emile et al33 go a step further and integrate clinical presentation and pathology findings with molecular and genetic findings, therefore taking into account recent developments that shed new light onto LCH pathology. One example of a genetic finding guiding the classification of a disease is the presence of the BRAF V600E mutation in cases of both Erdheim–Chester disease (ECD) and LCH, which – together with the fact that almost 20% of ECD patients also have LCH lesions –hint at a close relationship between both diseases. Therefore, these two entities are now grouped together, which also makes sense in the view of the efficacy of RAF and MEK inhibitors in the treatment of both entities (see “New therapeutic approaches and tentative targets emerging from the (recent) insights into the LCH biology” section). Overall, the 2016 classification manages to place many different diseases into five groups, which do not just categorize but give a global perspective that helps the clinician to keep track in the jungle of more than 100 histiocytic disorders.
General approach and principles of treating LCH
The clinical spectrum of LCH ranges from a single skeletal lesion to disseminated disease affecting multiple organs. The clinical course and outcome are also quite variable and hardly predictable upon diagnosis.34 Therefore, the “one size fits all” treatment approach is not feasible. Different prognostic factors, clinical scores, and stratification systems have been elaborated over the years to assist therapeutic decisions and enable risk-adapted treatment.5,35–40
Clinical classification of LCH
It has been empirically established that patients presenting with involvement of one organ system (SS-LCH) have excellent prognosis.41 Patients with involvement of two or more organs (MS-LCH), particularly those with dysfunction of the liver and of hematopoiesis, may have progressive disease associated with considerable mortality.34 This empirical knowledge formed the basis of the clinical classification of LCH,42 which has been adopted for the clinical trials of the Histiocyte Society (Table 1). In addition to involvement of risk organs at the time of diagnosis, response after 6 weeks of standard treatment proved to be of independent prognostic value,38,43 providing guidance for early adjustment of treatment intensity. Risk organ involvement at diagnosis and response to initial treatment allow defining the following groups at risk for treatment failure and unfavorable outcome (Table 2). Appropriate classification of disease extent and severity and assignment of risk-adapted therapy mandate complete clinical evaluation of each patient at initial diagnosis and relapse.44–46
 | Table 1 Current clinical classification of LCHa Notes: aReprinted from Hematology/Oncology Clinics of North America, 12, Broadbent V, Gadner H. Current therapy for Langerhans cell histiocytosis, 327–338, Copyright 1998 with permission from Elsevier.42 bRisk organ involvement is defined as the presence of at least one of the following: hematopoietic system (bi- or pancytopenia), liver (hepatomegaly and/or dysfunction), and/or spleen (splenomegaly). Abbreviations: LCH, Langerhans cell histiocytosis; SS-LCH, single-system LCH; MS-LCH, multisystem LCH. |
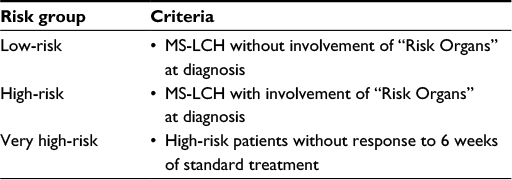 | Table 2 Stratification of MS-LCH Note: Risk organ involvement is defined as the presence of at least one of the following: hematopoietic system (bi- or pancytopenia), liver (hepatomegaly and/or dysfunction), and/or spleen (splenomegaly). Abbreviations: LCH, Langerhans cell histiocytosis; MS-LCH, multisystem Langerhans cell histiocytosis. |
Current treatment approach to LCH
Therapeutic principles in LCH have emerged from clinical experience despite lack of knowledge regarding its etiology and pathogenesis. Therefore, the evolution of LCH therapy over the years has closely reflected the paradigm and hypothesis shifts regarding LCH biology.3,5 Indeed, the difficulties in developing optimal therapies were, at least in part, due to the gaps in the understanding of the pathogenesis of LCH.
In view of the excellent prognosis, SS-LCH (the majority of the cases confined to skeleton or skin) is usually managed as conservatively as possible.
Approach to SS-LCH
Randomized trials for the treatment of SS-LCH were never carried out, and the treatment recommendations are based on experience gained from retrospective cohorts.41,47–53 Overall, the course of localized skeletal SS-LCH is benign, and no initial treatment strategy appears to have a clear advantage with respect to rate and speed of healing of the individual lesions. In contrast, in the study of Titgemeyer et al,41 systemic combination chemotherapy was given to all patients with multifocal skeletal disease as prescribed by the study protocol. The reactivation rate of 18% in patients with multifocal bone disease in that study contrasts to the 50%–80% reported in the literature and suggests a positive influence of systemic therapy on the disease course in multifocal bone disease. This finding underlines the biological difference between unifocal and multifocal skeletal LCH, postulated by a recent biological study.32
Given the very good prognosis, the treatment of unifocal bone lesions focuses on local control and prevention of reactivations and permanent consequences. Treatment options range from expectant attitude, through surgery or topical drug delivery, to systemic therapy in selected cases. Decisive for the treatment decision in unifocal bone lesion is the location (weight-bearing or compression of crucial adjacent structures), the size, the surgical accessibility, the presence of considerable adjacent soft-tissue mass, pain or functional impairment, and the risk of permanent sequelae. A wait-and-see approach is justified in small asymptomatic osseous or cutaneous lesions in view of the high likelihood for spontaneous healing. Surgical procedures such as biopsy, curettage, or resection are used to treat solitary bone lesions, solitary affected lymph nodes, or circumscribed nodular skin lesions. A biopsy is necessary to confirm the diagnosis and at the same time represents a healing stimulus. It is empirically well established that radical surgery is not necessary and usually not useful in localized LCH.44,46 Wide surgical resection is particularly harmful in skull vault and jawbone lesions, as it impedes bone reconstitutions and causes permanent defects, which are not observed in nonresected lesions. An intralesional application of crystalline methylprednisolone (100–150 mg) in symptomatic bone lesion can quickly bring about a reduction in symptoms and cure.54,55 Because of its potential to induce secondary malignancies, radiotherapy at a low dose (6–10 Gy) is limited to specific indications such as imminent compression of vital structures (eg, the spinal cord or the optic nerve). In case of large, symptomatic lesions, which are not easily accessible and bear high likelihood for pathologic fractures and permanent consequences, mild systemic treatment of short duration (3–6 months) may be the preferable option for local disease control. A best practice-based treatment approach to SS-LCH is depicted in Figure 2.
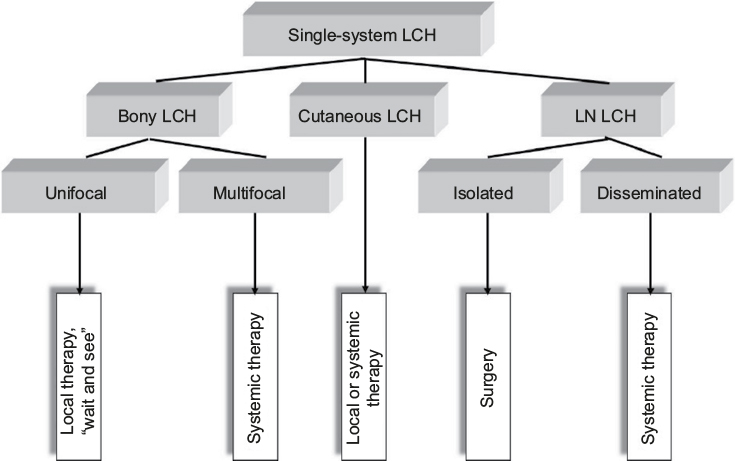 | Figure 2 Treatment approach to SS-LCH. Abbreviations: LCH, Langerhans cell histiocytosis; SS-LCH, single-system LCH; LN LCH, lymph node LCH. |
Approach to disseminated disease
There is a general agreement on the indication of systemic therapy for patients with MS-LCH.34,42,56–58 The value of systemic therapy for multifocal skeletal SS-LCH is less well documented and still needs evaluation in controlled prospective trials. The individual drugs, drug combinations, and regimens with established activity in MS-LCH have been extensively reviewed elsewhere.34,44,56,58,59 Therefore, only the results of the clinical trials of the Histiocyte Society43,60–64 and the current standard of care will be briefly summarized here.
Most trials before the era of the Histiocyte Society have pooled patients with varying clinical presentation, course, and prognosis in order to collect higher numbers.65 Large-scale clinical trials in rare diseases such as LCH are possible only in the setting of international cooperation. The Histiocyte Society has conducted a series of randomized clinical trials that defined the current ground for the management of LCH.
The LCH-I trial (1991–1995) was the first international randomized trial for MS-LCH. It compared the effectiveness of vinblastine (VBL) and etoposide in the treatment of patients with MS-LCH, and the main conclusion was that these drugs have equivalent activity in children with MS-LCH.43 In addition, this trial has proven that based on age and organ involvement at diagnosis, it is possible to define a subgroup of MS-LCH with a survival probability of 100% (low-risk group) and a subgroup at risk of mortality (risk group).43
The LCH-II trial (1996–2000) explored the value of the addition of etoposide to a standard initial therapy combination of prednisolone (PRED) and VBL in patients with risk of MS-LCH.61 The continuation therapy included oral mercaptopurine and pulses of PRED and VBL for a total treatment duration of 6 months. The outcomes in the standard and the experimental arm were similar with respect to response at week 6 (63% vs 71%), 5-year survival (74% vs 79%), reactivation frequency (46% vs 46%), and permanent consequences (43% vs 37%).61 This trial proved that the stratification in low-risk and risk group based solely on organ involvement at the time of diagnosis (age did not prove to have an independent prognostic value) is feasible.
In the LCH-III (2001–2008) study, patients with MS-LCH were divided into two groups (low-risk and risk) depending on the risk of mortality. In the low-risk group, the value of the continuation therapy (6 vs 12 months) was studied with respect to reactivation rate and sequelae. In the risk group, the value of the addition of intermediate-dose methotrexate to the standard combination of PRED and VBL was studied with respect to early response and mortality. A second 6-week course of initial therapy was delivered in patients without optimal response in both groups. The final results of the risk group trial did not prove superiority of the experimental arm with respect to initial response, overall and reactivation-free survival, and toxicity. In the low-risk group, prolongation of the treatment duration resulted in a reduced risk of reactivation (0.50 in the 6-month vs 0.35 in the 12-month arm).62 Overall, the LCH-III study concluded that early intensification with a second induction phase for patients with slow responses and therapy prolongation result in significantly improved outcomes for patients with MS-LCH.
The cumulative experience of the prospective clinical trials conducted by the Histiocyte Society can be summarized as follows:
- Risk organ involvement at diagnosis (defined as at least one of the following: peripheral blood cytopenia and/or liver enlargement ± organ dysfunction and/or spleen enlargement) allows stratification of MS-LCH into low-risk (probability of survival of nearly 100%) and risk group (probability of survival of 80%–90%). Patients with risk organ involvement (particularly those with bi- and pancytopenia and liver dysfunction) who do not respond to 6 weeks of standard treatment have particularly dismal prognosis (survival less than 50%). This small subgroup categorized as “very high-risk” deserves treatment intensification. As continuing standard treatment usually fails to change the outcome for those patients, experimental approaches targeting improved survival seem justified.
- The standard frontline therapy for patients with MS-LCH treated outside of controlled clinical trials consists of 6–12 weeks of initial therapy (oral steroids and weekly VBL injections), followed by pulses of PRED / VBL every 3 weeks, for a total treatment duration of 12 months (Figure 3).34,44,59 The main advantages of this regimen are the solid evidence for its activity in LCH, its acceptable toxicity, its applicability in an outpatient setting, drug availability, and the relatively low price.65
- A standard of care for patients who could not be cured by the frontline therapy (suboptimal response, disease progression, or relapse) has not been established yet. Patients with low-risk disease, particularly those who have a relapse after complete resolution, can be successfully treated by repetition of the frontline regimen, or by application of a number of other single drugs or drug combinations.34,56,59,64,66–68 Controlled prospective trials with appropriate end points (prevention of subsequent relapses and permanent consequences, as well as improvement in the quality of life) are still lacking. To date, only few options have shown promising results in the treatment of severe progressive LCH in small series and pilot trials,60,69–71 but their applicability is limited by either high toxicity or availability of matched donors, as well as by the need for highly specialized expertise for treatment delivery.
- The same is true for some specific or rare clinical scenarios, ie, isolated destructive pulmonary LCH, sclerosing cholangitis, LCH reactivation presenting with isolated diabetes insipidus, and central nervous system-LCH of neurodegenerative type.
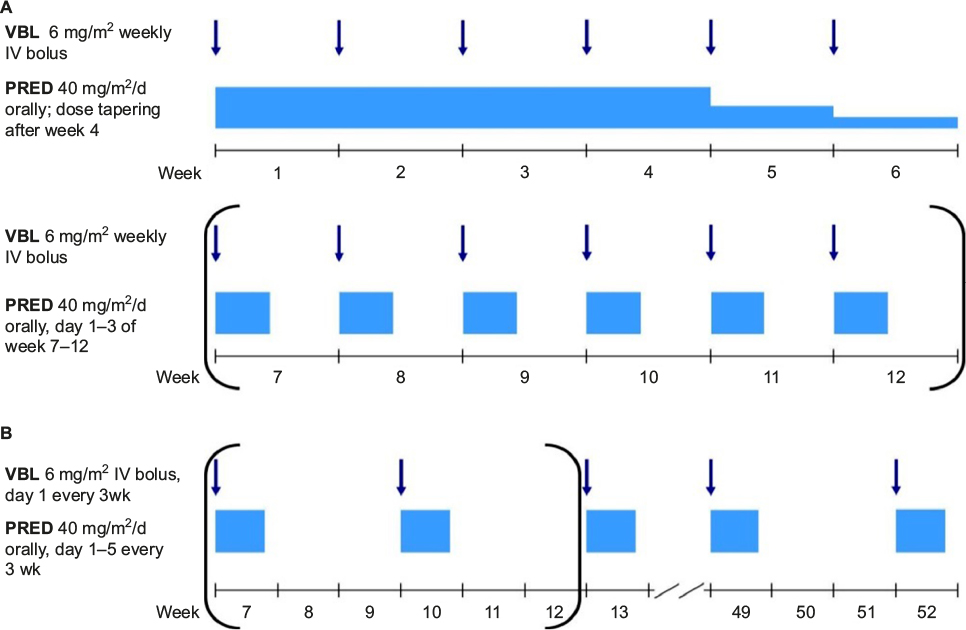 | Figure 3 Standard frontline treatment of MS-LCH. Notes: (A) Initial treatment, parentheses indicate optional for patients with an incomplete response; (B) continuation treatment, parentheses indicate for patients who have received second initial course start this continuation treatment at week 13. Abbreviations: LCH, Langerhans cell histiocytosis; MS-LCH, multisystem LCH; VBL, vinblastine; PRED, prednisolone. IV, intravenous. |
A current, ongoing international trial of the Histiocyte Society (LCH-IV International Collaborative Treatment Protocol for Children and Adolescents with Langerhans Cell Histiocytosis; NCT02205762) with a complex design (five interventional and two observational strata) is looking for improvement of relapse-free survival and quality of life by targeting the unsolved issues listed earlier.59,65
New therapeutic approaches and tentative targets emerging from the (recent) insights into the LCH biology
The detection of the mutations in the MAPK pathway has revolutionized our understanding of LCH biology. The opportunities offered by this knowledge and the spectrum of its clinical implications reach far beyond the rationale for targeted RAF and MEK inhibition, which has been extensively discussed elsewhere.2,32,34,44,59,65,72–77
The explosion of basic knowledge on LCH poses a huge challenge how to translate it into clinical practice and incorporate it into the patient care. LCH has been recently redefined as a myeloid neoplasia.2,44,75 Besides the difficulty in providing a sound answer to the patient’s question “is it a cancer?”, the most obvious question is “are we going to treat it as a cancer?”. New insights have changed the view on LCH and help in partly explaining its pathomechanisms, but it does not change anything on its mostly benign behavior. In contrast to most malignancies, mortality is very low. The major challenges are chronic recurrent course over years or multiple reactivations, which could both result in acute morbidity (eg, pain, motion deficits, and itchy rashes) and permanent disabilities (hormone deficits, lung honeycombing, liver cirrhosis, neurocognitive deficits, being the most severe, but fortunately rare). What is the price a patient should pay for alleviation of acute morbidity and assumed, but unproven, prevention of permanent consequences in terms of acute and long-term treatment side effect?
As there are no biological markers, treatment decisions solely rely on clinical presentation. In face of the currently unpredictable course of LCH in patients with multisystem disease and the high reactivation rate of LCH, the possibility to monitor treatment response with more quantifiable and objective parameters would be of immense clinical value. The documented mutations in the MAPK pathway are appealing biomarkers. According to the data from Berres et al,32 BRAF V600E expression in circulating cells is associated with disease severity. Translated into clinical practice, this could provide a more objective way for clinical classification and therapeutic stratification than the current one that is solely based on conventional imaging of involved organs. For example, multifocal skeletal LCH is diagnosed predominantly in preschool children and is frequently accompanied by clinical or laboratory signs of systemic inflammation. It is currently categorized as SS-LCH, but is biologically closer to MS-LCH. Quantification of mutation-bearing cells could perhaps enable more precise clinical classification and possibly guide treatment stratification. Therefore, evaluation of cell-free DNA in accessible body fluids (blood and urine) could provide a measure both for the disease burden and for therapeutic success, but this needs a careful proof in a prospective study.78
The MAPK signaling pathway plays a key role in the regulation of gene expression, cellular growth, and survival. A number of activating mutations affecting this pathway result in overactive downstream ERK, which proves to be the ultimate driving event in LCH. Both specific inhibition of the mutated kinases and downstream ERK inhibition (Figure 4) are undoubtedly appealing treatment options.2,44,75,79 The clinical experience available to date confirmed at least two essential expectations to BRAF inhibitors, namely in vivo activity in patients with LCH and ECD and rapid clinical response.80–84 As there is no curative therapy for patients with advanced ECD, these findings are likely to change the treatment approach in this setting. In patients with severe life-threatening LCH, rapid clinical response is of particular importance. The series of Haroche et al82 show impressively rapid response to vemurafenib and prove that sustainable treatment effect is achievable. However, although MAPK inhibition might lead to clinical improvement, it is currently unclear whether treatment with a single inhibitor can eradicate the disease. The major tasks to be addressed in controlled prospective trials are, therefore, finding the most effective and less toxic specific inhibitors, establishing downstream inhibition for patients without known mutations, defining appropriate pediatric dosages, and establishing how long and in which combinations (if any) the drugs should be given. There are few ongoing trials listed on https://clinicaltrials.gov/ (eg, NCT02281760, NCT02649972, NCT02089724, and NCT01677741), evaluating the role of MAPK inhibitors in patients with histiocytic disorders. Because of concerns of potentially severe toxicity, pediatric trials should initially be restricted to patients with severe diseases who could not be rescued with less toxic treatments.44,85 In view of low expected numbers of eligible patients and the need to address multiple issues, a meaningful study design will require multi-institutional cooperation. The currently ongoing LCH-IV trial offers an appropriate setting for piloting new drugs.59 Patients with high-risk MS-LCH who fail first-line therapy are offered either repeated cladribine/cytarabine courses or hematopoietic stem cell transplantation after reduced-intensity conditioning at the discretion of the treating physician (as soon as strict criteria of disease severity are fulfilled). Although effective, both salvage options are toxic and associated with treatment-related mortality.60,70 Therefore, therapeutic window piloting specific MAPK pathway inhibitors would be an ethically justifiable intervention. The limited available experience with these drugs suggests that they are able to induce rapid response and thus expected to reduce septic complications because of prolonged immunosuppression or rapid progression of the disease.
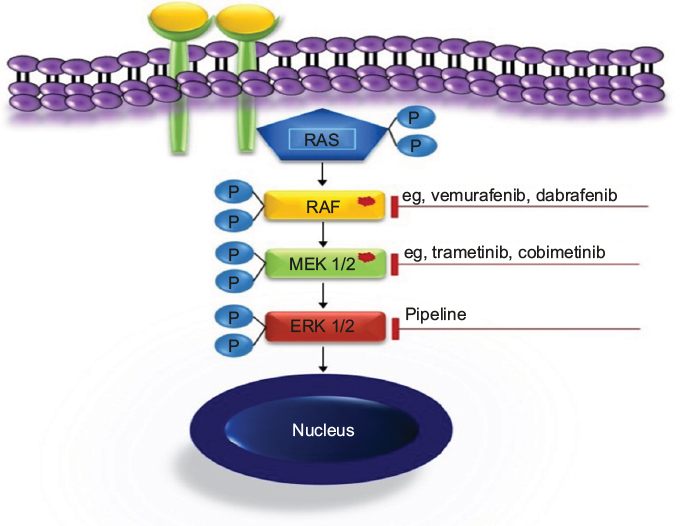 | Figure 4 MAPK targets and drugs. |
The dream of targeted treatment and preventing LCH mortality seems to become a reality. Nevertheless, it is very unlikely that the currently available MAPK pathway inhibitors will become the standard of care for all patients of LCH, both for reasons of high costs and worrisome side effects.
Should the search for cure be restricted to inhibiting the MAPK pathway? Pathology findings (granuloma formation), cytokine studies,15–19,86 and clinical observations suggest that mortality and permanent organ damage in LCH are most probably due to deleterious effects of an unbridled local and systemic inflammatory response, rather than to an uncontrolled proliferation of the misguided myeloid clone. Therefore, different aspects of the inflammatory response (eg, cellular cross talk, specific cytokines, and chemokines) offer further potential therapeutic targets. Both experimental data (ie, morphoproteomic analysis of LCH lesions)87 and limited clinical experience with bisphosphonates, etanercept, and indomethacin88–93 provide rationale for targeting the inflammation evoked by the aberrant myeloid clone in LCH. The recently approved RANKL inhibitor denosumab for the treatment of giant-cell tumor is another attractive option for patients with skeletal LCH.94–97 Granuloma formation in LCH reflects a complex microenvironment comprising a number of cells and key molecules,16,28 and gives rationale for further exploration of the communicative tumor system reprogramming approach, which has been reported in anecdotal cases of LCH.98 Expansion of regulatory T-cells in LCH and the evidence that the tissue infiltration by the DC clone in LCH is due to accumulation and better survival, rather than to uncontrolled proliferation of CD1a/CD207-positive cells, let to the speculation that targeting T-regs, or overcoming tolerance, may be beneficial.99 Interestingly, a recent immunohistochemical study demonstrated significant expression of the PD-1 ligand in a number of histiocytic disorders, including LCH.100 If validated by experimental data, this pathway could provide a further therapeutic target.
Fortunately, the armamentarium available in the fight with LCH is getting diverse and more precise. Optimizing patient stratification and tailoring treatment to the need of the individual patient will remain a major task in the upcoming decades.
Disclosure
The authors report no conflicts of interest in this work.
References
Hand A. Polyuria and tuberculosis. Arch Pediatr. 1893;10:673–675. | ||
Rollins BJ. Genomic alterations in Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):839–851. | ||
Komp DM. Historical perspectives of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1987;1(1):9–21. | ||
Komp DM. Therapeutic strategies for Langerhans cell histiocytosis. J Pediatr. 1991;119(2):274–275. | ||
Ladisch S. Histiocytosis. In: Willoughby ML, Siegel SE, editors. Butterworth’s International Medical Reviews: Pediatrics. Vol 1. London, UK: Butterworth Scientific; 1982:95–109. | ||
Egeler RM, Annels NE, Hogendoorn PC. Langerhans cell histiocytosis: a pathologic combination of oncogenesis and immune dysregulation. Pediatr Blood Cancer. 2004;42(5):401–403. | ||
Fadeel B, Henter JI. Langerhans-cell histiocytosis: neoplasia or unbridled inflammation? Trends Immunol. 2003;24(8):409–410; author reply 410–411. | ||
Lichtenstein L. Histiocytosis X; integration of eosinophilic granuloma of bone, Letterer-Siwe disease, and Schuller-Christian disease as related manifestations of a single nosologic entity. AMA Arch Pathol. 1953;56(1):84–102. | ||
Nezelof C, Basset F, Rousseau MF. Histiocytosis X histogenetic arguments for a Langerhans cell origin. Biomedicine. 1973;18(5):365–371. | ||
Histiocytosis syndromes in children. Writing Group of the Histiocyte Society. Lancet. 1987;1(8526):208–209. | ||
Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee on histiocytic/reticulum cell proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol. 1997;29(3):157–166. | ||
Chu T, Jaffe R. The normal Langerhans cell and the LCH cell. Br J Cancer Suppl. 1994;23:S4–S10. | ||
Coury F, Annels N, Rivollier A, et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med. 2008;14(1):81–87. | ||
Ikawa Y, Nishimura R, Araki R, et al. Pathognomonic serum cytokine profiles identify life-threatening langerhans cell histiocytosis. Br J Haematol. Epub April 7, 2016. | ||
Kannourakis G, Abbas A. The role of cytokines in the pathogenesis of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994;23:S37–S40. | ||
Egeler RM, Favara BE, van Meurs M, Laman JD, Claassen E. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94(12):4195–4201. | ||
Rosso DA, Karis J, Braier JL, Henter JI, Fadeel B. Elevated serum levels of the decoy receptor osteoprotegerin in children with langerhans cell histiocytosis. Pediatr Res. 2006;59(2):281–286. | ||
Rosso DA, Ripoli MF, Roy A, Diez RA, Zelazko ME, Braier JL. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003;25(6):480–483. | ||
Ishii R, Morimoto A, Ikushima S, et al. High serum values of soluble CD154, IL-2 receptor, RANKL and osteoprotegerin in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2006;47(2):194–199. | ||
Willman CL, Busque L, Griffith BB, et al. Langerhans’-cell histiocytosis (histiocytosis X) – a clonal proliferative disease. N Engl J Med. 1994;331(3):154–160. | ||
Arico M, Nichols K, Whitlock JA, et al. Familial clustering of Langerhans cell histiocytosis. Br J Haematol. 1999;107(4):883–888. | ||
Surico G, Muggeo P, Rigillo N, Gadner H. Concurrent Langerhans cell histiocytosis and myelodysplasia in children. Med Pediatr Oncol. 2000;35(4):421–425. | ||
Betts DR, Leibundgut KE, Feldges A, Pluss HJ, Niggli FK. Cytogenetic abnormalities in Langerhans cell histiocytosis. Br J Cancer. 1998;77(4):552–555. | ||
da Costa CE, Egeler RM, Hoogeboom M, et al. Differences in telomerase expression by the CD1a+ cells in Langerhans cell histiocytosis reflect the diverse clinical presentation of the disease. J Pathol. 2007;212(2):188–197. | ||
Egeler RM, Neglia JP, Arico M, et al. The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH-Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am. 1998;12(2):369–378. | ||
Magni M, Di Nicola M, Carlo-Stella C, et al. Identical rearrangement of immunoglobulin heavy chain gene in neoplastic Langerhans cells and B-lymphocytes: evidence for a common precursor. Leuk Res. 2002;26(12):1131–1133. | ||
Feldman AL, Berthold F, Arceci RJ, et al. Clonal relationship between precursor T-lymphoblastic leukaemia/lymphoma and Langerhans-cell histiocytosis. Lancet Oncol. 2005;6(6):435–437. | ||
Laman JD, Leenen PJ, Annels NE, Hogendoorn PC, Egeler RM. Langerhans-cell histiocytosis “insight into DC biology”. Trends Immunol. 2003;24(4):190–196. | ||
Hutter C, Kauer M, Simonitsch-Klupp I, et al. Notch is active in Langerhans cell histiocytosis and confers pathognomonic features on dendritic cells. Blood. 2012;120(26):5199–5208. | ||
Winter DR, Jung S, Amit I. Making the case for chromatin profiling: a new tool to investigate the immune-regulatory landscape. Nat Rev Immunol. 2015;15(9):585–594. | ||
Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–1923. | ||
Berres ML, Lim KP, Peters T, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014;211(4):669–683. | ||
Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681. | ||
Minkov M. Multisystem Langerhans cell histiocytosis in children: current treatment and future directions. Paediatr Drugs. 2011;13(2):75–86. | ||
Komp DM. Concepts in staging and clinical studies for treatment of Langerhans’ cell histiocytosis. Semin Oncol. 1991;18(1):18–23. | ||
Lahey E. Histiocytosis X – an analysis of prognostic factors. J Pediatr. 1975;87(2):184–189. | ||
Lahey ME. Prognostic factors in histiocytosis X. Am J Pediatr Hematol Oncol. 1981;3(1):57–60. | ||
Minkov M, Grois N, Heitger A, Potschger U, Westermeier T, Gadner H. Response to initial treatment of multisystem Langerhans cell histiocytosis: an important prognostic indicator. Med Pediatr Oncol. 2002;39(6):581–585. | ||
Donadieu J, Piguet C, Bernard F, et al. A new clinical score for disease activity in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2004;43(7):770–776. | ||
Lavin PT, Osband ME. Evaluating the role of therapy in histiocytosis-X. Clinical studies, staging, and scoring. Hematol Oncol Clin North Am. 1987;1(1):35–47. | ||
Titgemeyer C, Grois N, Minkov M, Flucher-Wolfram B, Gatterer-Menz I, Gadner H. Pattern and course of single-system disease in Langerhans cell histiocytosis data from the DAL-HX 83- and 90-study. Med Pediatr Oncol. 2001;37(2):108–114. | ||
Broadbent V, Gadner H. Current therapy for Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998;12(2):327–338. | ||
Gadner H, Grois N, Arico M, et al. A randomized trial of treatment for multisystem Langerhans’ cell histiocytosis. J Pediatr. 2001;138(5):728–734. | ||
Allen CE, Ladisch S, McClain KL. How I treat Langerhans cell histiocytosis. Blood. 2015;126(1):26–35. | ||
Broadbent V, Gadner H, Komp DM, Ladisch S. Histiocytosis syndromes in children: II. Approach to the clinical and laboratory evaluation of children with Langerhans cell histiocytosis. Clinical Writing Group of the Histiocyte Society. Med Pediatr Oncol. 1989;17(6):492–495. | ||
Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60(2):175–184. | ||
Berry DH, Gresik M, Maybee D, Marcus R. Histiocytosis X in bone only. Med Pediatr Oncol. 1990;18(4):292–294. | ||
Bollini G, Jouve JL, Gentet JC, Jacquemier M, Bouyala JM. Bone lesions in histiocytosis X. J Pediatr Orthop. 1991;11(4):469–477. | ||
Fiorillo A, Sadile F, De Chiara C, et al. Bone lesions in Langerhans cell histiocytosis. Clin Pediatr (Phila). 1993;32(2):118–120. | ||
Kilpatrick SE, Wenger DE, Gilchrist GS, Shives TC, Wollan PC, Unni KK. Langerhans’ cell histiocytosis (histiocytosis X) of bone. A clinicopathologic analysis of 263 pediatric and adult cases. Cancer. 1995;76(12):2471–2484. | ||
Lau LM, Stuurman K, Weitzman S. Skeletal Langerhans cell histiocytosis in children: permanent consequences and health-related quality of life in long-term survivors. Pediatr Blood Cancer. 2008;50(3):607–612. | ||
Womer RB, Raney RB Jr, D’Angio GJ. Healing rates of treated and untreated bone lesions in histiocytosis X. Pediatrics. 1985;76(2):286–288. | ||
Morimoto A, Ishida Y, Suzuki N, et al. Nationwide survey of single-system single site Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2010;54(1):98–102. | ||
Cohen M, Zornoza J, Cangir A, Murray JA, Wallace S. Direct injection of methylprednisolone sodium succinate in the treatment of solitary eosinophilic granuloma of bone: a report of 9 cases. Radiology. 1980;136(2):289–293. | ||
Egeler RM, Thompson RC Jr, Voute PA, Nesbit ME Jr. Intralesional infiltration of corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr Orthop. 1992;12(6):811–814. | ||
Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: current concepts and treatments. Cancer Treat Rev. 2010;36(4):354–359. | ||
Arceci RJ, Brenner MK, Pritchard J. Controversies and new approaches to treatment of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998;12(2):339–357. | ||
McClain KL. Drug therapy for the treatment of Langerhans cell histiocytosis. Expert Opin Pharmacother. 2005;6(14):2435–2441. | ||
Monsereenusorn C, Rodriguez-Galindo C. Clinical characteristics and treatment of Langerhans Cell histiocytosis. Hematol Oncol Clin North Am. 2015;29(5):853–873. | ||
Donadieu J, Bernard F, van Noesel M, et al. Cladribine and cytarabine in refractory multisystem Langerhans cell histiocytosis: results of an international phase 2 study. Blood. 2015;126(12):1415–1423. | ||
Gadner H, Grois N, Potschger U, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood. 2008;111(5):2556–2562. | ||
Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121(25):5006–5014. | ||
Minkov M, Grois N, Broadbent V, Ceci A, Jakobson A, Ladisch S. Cyclosporine A therapy for multisystem langerhans cell histiocytosis. Med Pediatr Oncol. 1999;33(5):482–485. | ||
Weitzman S, Braier J, Donadieu J, et al. 2′-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH): results of the LCH-S-98 protocol of the histiocyte society. Pediatr Blood Cancer. 2009;53(7):1271–1276. | ||
Minkov M, Rodriguez-Galindo C. Treatment of Langerhans cell histiocytosis: it is time to learn from the past. Br J Haematol. 2015;171(1):148–149. | ||
Minkov M, Steiner M, Potschger U, et al. Reactivations in multisystem Langerhans cell histiocytosis: data of the international LCH registry. J Pediatr. 2008;153(5):700–705, 705.e701–e702. | ||
Morimoto A, Kobayashi R, Maeda M, Asami K, Bessho F, Imashuku S. Impact of reactivation on the sequelae of multi-system Langerhans cell histiocytosis patients. Pediatr Blood Cancer. 2008;50(4):931–932; author reply 932. | ||
Pollono D, Rey G, Latella A, Rosso D, Chantada G, Braier J. Reactivation and risk of sequelae in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2007;48(7):696–699. | ||
Bernard F, Thomas C, Bertrand Y, et al. Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur J Cancer. 2005;41(17):2682–2689. | ||
Steiner M, Matthes-Martin S, Attarbaschi A, et al. Improved outcome of treatment-resistant high-risk Langerhans cell histiocytosis after allogeneic stem cell transplantation with reduced-intensity conditioning. Bone Marrow Transplant. 2005;36(3):215–225. | ||
Akkari V, Donadieu J, Piguet C, et al. Hematopoietic stem cell transplantation in patients with severe Langerhans cell histiocytosis and hematological dysfunction: experience of the French Langerhans Cell Study Group. Bone Marrow Transplant. 2003;31(12):1097–1103. | ||
Abla O, Weitzman S. Treatment of Langerhans cell histiocytosis: role of BRAF/MAPK inhibition. Hematol Am Soc Hematol Educ Program. 2015;2015(1):565–570. | ||
Allen CE, Parsons DW. Biological and clinical significance of somatic mutations in Langerhans cell histiocytosis and related histiocytic neoplastic disorders. Hematol Am Soc Hematol Educ Program. 2015;2015:559–564. | ||
Brown NA, Furtado LV, Betz BL, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood. 2014;124(10):1655–1658. | ||
Chakraborty R, Hampton OA, Shen X, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;124(19):3007–3015. | ||
Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6(2):154–165. | ||
Nelson DS, van Halteren A, Quispel WT, et al. MAP2K1 and MAP3K1 mutations in Langerhans cell histiocytosis. Genes Chromosomes Cancer. 2015;54(6):361–368. | ||
Hyman DM, Diamond EL, Vibat CR, et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov. 2015;5(1):64–71. | ||
Bubolz AM, Weissinger SE, Stenzinger A, et al. Potential clinical implications of BRAF mutations in histiocytic proliferations. Oncotarget. 2014;5(12):4060–4070. | ||
Charles J, Beani JC, Fiandrino G, Busser B. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71(3):e97–e99. | ||
Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121(9):1495–1500. | ||
Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol. 2015;33(5):411–418. | ||
Heritier S, Jehanne M, Leverger G, et al. Vemurafenib use in an infant for high-risk Langerhans cell histiocytosis. JAMA Oncol. 2015;1(6):836–838. | ||
Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–736. | ||
Haroche J, Cohen-Aubart F, Emile JF, Donadieu J, Amoura Z. Vemurafenib as first line therapy in BRAF-mutated Langerhans cell histiocytosis. J Am Acad Dermatol. 2015;73(1):e29–e30. | ||
Brown RE. Cyclo-oxygenase-2 in the osteolytic lesions of Langerhans cell histiocytosis. Med Pediatr Oncol. 2000;34(4):311. | ||
Brown RE. Brief communication: morphoproteomic analysis of osteolytic Langerhans cell histiocytosis with therapeutic implications. Ann Clin Lab Sci. 2005;35(2):131–136. | ||
Arzoo K, Sadeghi S, Pullarkat V. Pamidronate for bone pain from osteolytic lesions in Langerhans’-cell histiocytosis. N Engl J Med. 2001;345(3):225. | ||
Brown RE. More on pamidronate in Langerhans’-cell histiocytosis. N Engl J Med. 2001;345(20):1503. | ||
Elomaa I, Blomqvist C, Porkka L, Holmstrom T. Experiences of clodronate treatment of multifocal eosinophilic granuloma of bone. J Intern Med. 1989;225(1):59–61. | ||
Kamizono J, Okada Y, Shirahata A, Tanaka Y. Bisphosphonate induces remission of refractory osteolysis in langerhans cell histiocytosis. J Bone Miner Res. 2002;17(11):1926–1928. | ||
Munn SE, Olliver L, Broadbent V, Pritchard J. Use of indomethacin in Langerhans cell histiocytosis. Med Pediatr Oncol. 1999;32(4):247–249. | ||
Henter JI, Karlen J, Calming U, Bernstrand C, Andersson U, Fadeel B. Successful treatment of Langerhans’-cell histiocytosis with etanercept. N Engl J Med. 2001;345(21):1577–1578. | ||
Brodowicz T, Hemetsberger M, Windhager R. Denosumab for the treatment of giant cell tumor of the bone. Future Oncol. 2015;11(13):1881–1894. | ||
Dahiya N, Khadka A, Sharma AK, Gupta AK, Singh N, Brashier DB. Denosumab: a bone antiresorptive drug. Med J Armed Forces India. 2015;71(1):71–75. | ||
Goldenberg MM. Pharmaceutical approval update. P T. 2013;38(8):443–445. | ||
Vaishya R, Agarwal AK, Vijay V. “Salvage Treatment” of aggressive giant cell tumor of bones with denosumab. Cureus. 2015;7(7):e291. | ||
Hart C, Vogelhuber M, Wolff D, et al. Anakoinosis: communicative reprogramming of tumor systems – for rescuing from chemorefractory neoplasia. Cancer Microenviron. 2015;8(2):75–92. | ||
Senechal B, Elain G, Jeziorski E, et al. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med. 2007;4(8):e253. | ||
Gatalica Z, Bilalovic N, Palazzo JP, et al. Disseminated histiocytoses biomarkers beyond BRAFV600E: frequent expression of PD-L1. Oncotarget. 2015;6(23):19819–19825. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.