")
Back to Journals » International Journal of Nanomedicine » Volume 13
Insights into the antibacterial mechanism of PEGylated nano-bacitracin A against Streptococcus pneumonia: both penicillin-sensitive and penicillin-resistant strains
Authors Hong W, Liu L, Zhang Z, Zhao Y, Zhang D, Liu M
Received 28 June 2018
Accepted for publication 18 September 2018
Published 10 October 2018 Volume 2018:13 Pages 6297—6309
DOI https://doi.org/10.2147/IJN.S178596
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Wei Hong, Lipeng Liu, Zehui Zhang, Yining Zhao, Dexian Zhang, Mingchun Liu
Key Laboratory of Zoonosis of Liaoning Province, College of Animal Science and Veterinary Medicine, Shenyang Agricultural University, Shenyang, Liaoning Province 110866, People’s Republic of China
Background: Multidrug-resistant (MDR) Streptococcus pneumonia constitute a major worldwide public health concern.
Materials and methods: In our preliminary study, PEGylated nano-self-assemblies of bacitracin A (PEGylated Nano-BA12K) showed strong antibacterial potency against reference S. pneumonia strain (ATCC 49619). In this study, the possibility of applying PEGylated Nano-BA12K against penicillin-resistant S. pneumonia was further investigated. In addition, the underlying antibacterial mechanism of PEGylated Nano-BA12K against both sensitive and resistant S. pneumonia was also clarified systematically, since S. pneumonia was naturally resistant to its unassembled counterpart bacitracin A (BA).
Results: PEGylated Nano-BA12K showed strong antibacterial potency against 13 clinical isolates of S. pneumonia, including five penicillin-resistant strains. Structural changes, partial collapse, and even lysis of both penicillin-sensitive and penicillin-resistant bacteria were observed after incubation with PEGylated Nano-BA12K via transmission electron microscopy and atomic force microscopy. Thus, the cell wall or/and cell membrane might be the main target of PEGylated Nano-BA12K against S. pneumonia. PEGylated Nano-BA12K exhibited limited effect on the permeabilization and peptidoglycan content of cell wall. Surface pressure measurement suggested that PEGylated Nano-BA12K was much more tensioactive than BA, which was usually translated into a good membranolytic effect, and is helpful to permeabilize the cell membrane and damage membrane integrity, as evidenced by depolarization of the membrane potential, permeabilization of membrane and leakage of calcein from liposomes.
Conclusion: Collectively, great cell membrane permeability and formidable membrane disruption may work together for the strong antibacterial activity of PEGylated Nano-BA12K against S. pneumonia. Taken together, PEGylated Nano-BA12K has excellent potential against both penicillin-sensitive and penicillin-resistant S. pneumonia and might be suitable for the treatment of S. pneumonia infectious diseases.
Keywords: PEGylated Nano-BA12K, multidrug-resistant, Streptococcus pneumonia, penicillin-sensitive and penicillin-resistant
Corrigendum for this paper has been published
Corrigendum for this paper has been published
Introduction
The number of life-threatening bacterial infections has increased sharply in recent years because of a rapid increase of multidrug-resistant (MDR) bacteria together with a low rate of development of new antibacterial agents, which has become a serious threat to public health. Thus, the design and identification of alternative classes of antibacterial agents that can effectively overcome drug resistance is more pressing than ever.1 Streptococcus pneumonia is a major cause of many childhood diseases such as respiratory tract infections, acute otitis media, community-acquired pneumonia, meningitis, and sepsis. The treatment of these infections remains challenging because of the emergence of MDR phenotypes.2 The most important mechanisms of resistance described so for in S. pneumonia is β-lactams resistance, which is mediated by stepwise alternations of penicillin-binding proteins (PBPs), resulting in decreased affinity of PBP1a, PBP2x, and PBP2b. Resistant isolate PBPs are encoded by mosaic genes that contain sequence blocks highly divergent from those of sensitive strains. They have been recognized as the product of transformation events, resulting from horizontal gene transfer.3
Many strategies are being pursued to eradicate bacterial resistance. Using new generations of antibiotics, combination therapy, natural antibacterial substances, and targeted drug delivery systems are common approaches in this field. Recent developments of nanoparticles with suitable physicochemical properties for better drug delivery have been considered as distinct strategy for overcoming bacterial resistant organisms.4 It has been demonstrated that nanoparticles are able to 1) alter bacterial efflux pump activity, 2) anti-biofilm activity, 3) enhance penetration through biofilm structures, 4) protect against enzymatic degradation and inactivate via polyanionic compounds, 5) intracellular killing of bacteria, and 6) specific targeting. Numerous research studies about effectiveness of nanoparticles to combat bacterial resistance are recently published.5–7 Lipid-based nanoparticles, polymeric nanoparticles, nitric oxide releasing nanoparticles, and metal nanoparticles are the main system that have been developed and used in this field.8–10
The polypeptide antibiotics are promising agents for combating MDR bacterial pathogens. Among them, bacitracin is an attractive target for drug discovery since it possesses strong antibacterial activity against Gram-positive bacteria without triggering multidrug resistance. BA is target to its site of action by inhibiting the cell wall synthesis.11–13 In addition, BA can also disintegrate the bacterial cell membrane, leading to the loss of various ions and amino acids, resulting in the death of bacteria.14–16 However, BA showed limited antibacterial activity against Gram-negative bacteria and high nephrotoxicity, which prevents its direct use clinically, but only used for the treatment of some local infections caused by penicillin-resistant bacteria. The studies of structure–activity relation of peptides have shown that a hydrophobic modification of polypeptide antibiotics can promote their membrane adsorption, insertion, permeabilization, and disruption potency, which usually translates into a broad spectrum of antimicrobial activities and lower toxicity.17 Hydrophobic modification can also make the hydrophilic polypeptide antibiotics that have amphiphilic structure, which can spontaneously form nanoparticles, “nanoantibiotics,” with core-shell structure in water. Nanoantibiotics are also available to efficiently administer antibiotics by improving pharmacokinetics and accumulation, while reducing the adverse effects of antibiotics.18–20 In our previous study, the PEGylated Nano-BA12K based mainly on BA-PEG-PLGA12K-PEG-BA showed strong antibacterial activities against both Gram-positive and Gram-negative bacteria including S. pneumonia, yet induced relatively lower toxicity both in vitro and in vivo.21 The objective of the present research was to evaluate the possibility of using PEGylated Nano-BA12K to treat penicillin-resistant S. pneumonia. The antibacterial potency of PEGylated Nano-BA12K was first tested against 13 isolates of S. pneumonia. Consequently, fluorescent, spectrographic, transmission electron microscopy (TEM) and atomic force microscopy (AFM) assays were employed to investigate the potential mechanism of PEGylated Nano-BA12K against both penicillin-sensitive and penicillin-resistant strains.
Materials and methods
Materials
Reagents
HO-PEG(2K)-PLGA(12K)-PEG(2K)-OH was purchased from Xi’an ruixi Biological Technology Co., Ltd. (Xi’an, People’s Republic of China). Bacitracin A (BA), Penicillin G, sodium fluorescein, stannous 2-ethylhexanoate, t-butyldimethylsilanol, triphenylsilanol, N,N′-carbonyldiimidazole, o-nitrophenyl-β-D-galactopyranoside (ONPG), 3,3′-dipropylthiadicarbocynine iodide (diSC3-5), Triton X-100, phosphatidylglycerol (PTG), and caidiolipin (CL) were purchased from Sigma-Aldrich (Shanghai, People’s Republic of China). A LIVE/DEAD® BacLight™ Bacterial Viability Kit (L7012) was purchased from Thermo Fisher Scientific Inc. (Shanghai, People’s Republic of China). AKP activity colorimetric quantitative detection kit was purchased from Shanghai Haling Biotechnology Co., Ltd. (Shanghai, People’s Republic of China). All the other regents and chemicals were of analytical or chromatographic grade and were purchased from Concord Technology (Tianjing, People’s Republic of China).
Bacteria
S. pneumonia ATCC49619 was purchased from American Type Culture Collection (Manassas, VA, USA). The 12 additional strains for susceptibility testing are clinical isolates of S. pneumonia from cerebrospinal fluid of meningitis patients, which were obtained from the First Hospital of China Medical University (Shenyang, People’s Republic of China) and stored at −80°C in 40% (v/v) glycerol prior to use. These were part of the routine hospital laboratory procedure.
In vitro antibacterial activity assays
The minimal inhibitory concentrations (MICs) of PEGylated Nano-BA12K and BA against 13 isolates of S. pneumonia strains were determined using a modified standard micro-dilution method as previously reported.22,23 Briefly, the initial concentration of PEGylated Nano-BA12K was 256 μM and was serially diluted to 0.5 μM for use. About 100 μL of bacterial suspension (106 CFU/mL) form a log-phase bacterial culture and was added into 96-well microtiter plates, while 100 μL of PEGylated Nano-BA12K was also added to each well with a final volume of 200 μL. The final concentrations of the PEGylated Nano-BA12K range from 0.25 to 128 μM. Inhibition of bacterial growth was determined by measuring the absorbance at 600 nm with a multifunctional microplate reader (Tecan, Austria) after an incubation of 18 hours at 37°C. The MIC was defined as the lowest concentration that completely inhibited bacterial growth. Penicillin G was selected as positive control, while Mueller-Hinton Broth was used as the negative control. The tests were repeated at least three times.
Fluorescence microscope (FM)
The changes in viability of penicillin-sensitive S. pneumonia ATCC49619 and penicillin-resistant S. pneumonia 16167 after incubation with PEGylated Nano-BA12K were further assessed using LIVE/DEAD® BacLight™ Bacterial Viability Kit (L7012) after treatment of PEGylated Nano-BA12K, BA, and Penicillin G. Briefly, S. pneumonia cells (~107 CFU/mL) were incubated with tested formulations at 1× MIC for 0.5, 1, 2, 4, 8, and 12 hours. Then the bacterial cells were washed three times with PBS with centrifugation at 3,000 rpm for 10 minutes. Combination of equal volumes of SYTO 9 dye (component A) and propidium iodide (component B) in a microfuge tube was mixed thoroughly. Add 3 μL of the dye mixture for each milliliter of the bacterial suspension, mix thoroughly, and incubate at room temperature in the dark for 15 minutes. After staining, the bacterial cells were rinsed with PBS twice. Subsequently, trap 5 μL of the stained bacterial suspension between a slide and a 20 mm2 coverslip, and the microscope images were captured using an FM (Leica DM4 B; Leica Microsystems, Wetzlar, Germany) at λex (488 nm)/λem (590 nm) for green fluorescence or λex (568 nm)/λem (630 nm) for red fluorescence.
TEM observations
Bacterial cells of S. pneumonia ATCC49619 and S. pneumonia 16167 were grown to an exponential phase in MHB at 37°C under constant shaking at 220 rpm. After centrifugation at 3,000 rpm for 10 minutes, the cell pellets were harvested, washed twice with 10 mM PBS, and re-suspended to an OD600 of 0.2. The cells were incubated with PEGylated Nano-BA12K at its 0.5, 1, and 2× MICs for 2 hours at 37°C, respectively. Penicillin G was used as positive control. After incubation, the cells were centrifuged at 3,000 rpm for 10 minutes. The cell pellets were harvested, washed three times with PBS, and subjected to fixation with 2.5% glutaraldehyde at 4°C overnight followed by washing with PBS twice. Then the bacterial cells were post-fixed with 2% osmium tetroxide for 70 minutes. After dehydrated with a graded ethanol series (50%, 70%, 90%, and 100%) for 8 minutes each, bacterial samples were transferred to 100% ethanol, a mixture (1:1) of 100% ethanol and acetone, and 100% absolute acetone for 10 minutes each. Then, the specimens were transferred to 1:1 mixtures of absolute acetone and epoxy resin for another 30 minutes and to pure epoxy resin and incubated overnight at a constant temperature. Finally, the specimens were sectioned with ultramicrotome, stained by uranyl acetate and lead citrate, and examined using a H-7700 TEM (Hitachi Ltd., Tokyo, Japan).
AFM
S. pneumonia ATCC49619 cells and S. pneumonia 16167 cells were grown in MHB at 37°C under constant shaking at 220 rpm, respectively. The cultures were collected during the exponential phase by centrifugation at 3,000 rpm for 10 minutes and were resuspended in peptone water. Bacterial cells were incubated with 1× MIC of PEGylated Nano-BA12K at 37°C. The negative control was run with MHB, whereas Penicillin G was used as a positive control. After incubation, the bacterial cells were collected by centrifugation at 3,000 rpm for 10 minutes. The cell pellets were then harvested, washed thrice with 10 mM PBS, and resuspended in PBS. Images of the cells were visualized directly in air with a tapping mode AFM (Cypher™ ES; Asylum Research, Abingdon, UK). The height and size information were acquired using Imaging software.
Peptidoglycan (PG) content assay
The PG content of S. pneumonia ATCC49619 and S. pneumonia 16167 after incubation with different formulations was determined using the bacterial PG ELISA Kit. Briefly, bacterial cells were incubated to mid-log phase in MHB, washed thrice with 10 mM PBS, and diluted to an OD600 of 0.2 in the same buffer. Subsequently, 2 mL of cell suspension was added to a quartz cuvette and mixed with 1/4× MIC, 1/2× MIC, and 1× MIC of PEGylated Nano-BA12K, 128 μg/mL of BA, and 1× MIC of Penicillin G at 37°C for 1, 2, 5, 10, 20, and 30 minutes, respectively. Then, the bacterial suspension was washed thrice with 10 mM PBS and a bacterial suspension (100 μL, 107 CFU/mL) was added to 96-well microtiter plates. The PG content was determined according to the protocols provided by the vendor through a multifunctional microplate reader (Tecan) at 650 nm.
Assessment of PEGylated Nano-BA12K adsorption by surface tension measurements
PEGylated Nano-BA12K adsorption at the free air/buffer interface was assessed from surface tension measurements using Auto Surface Tensionmeter (A201; Shanghai Solon Information Technology Co. Ltd., Shanghai, China) based on Wilhelmy Plate method. Aliquots of PEGylated Nano-BA12K and BA solution were injected into the buffer subphase through the side arm of the measurement cell by means of a Hamilton syringe. The final concentration of the PEGylated Nano-BA12K and BA solution in the subphase ranged from 0 up to 1 mM. The surface tension of the solution was measured after equilibration for 24 hours. Each measurement was performed at least twice.
Cytoplasmic membrane electrical potential measurement
The ability of the PEGylated Nano-BA12K to alter cytoplasmic membrane electrical potential of bacteria was determined using membrane potential-sensitive dye diSC3-5 as previously described.23 Briefly, S. pneumonia ATCC49619 and S. pneumonia 16167 cells in the mid-log phase in MHB were harvested by centrifugation at 3,000 rpm for 10 minutes, washed thrice, and diluted to an OD600 of 0.05 with 5 mM N′-a-hydroxythylpiperazine-N′-ethanesulfanic acid (HEPES) buffer (pH 7.4, containing 20 mM glucose) containing 0.1 M KCL to equilibrate the cytoplasmic and external K+. Subsequently, the cell suspensions were incubated with 0.4 μM diSC3-5, and the bacteria were allowed to take up the dye for 60 minutes. Then, 2 mL of cell suspension was placed in a quartz cuvette and mixed with PEGylated Nano-BA12K at their 1× MICs to show the cytoplasmic membrane depolarization. The fluorescence intensity change due to the disruption of the membrane potential gradient across the cytoplasmic membrane was measured using an F-4500 fluorescence spectrophotometer (Hitachi Ltd.) with an excitation and emission wavelength of 622 and 670 nm from 0 to 300 seconds.
Cytoplasmic membrane permeability assay
Cytoplasmic membrane permeability of PEGylated Nano-BA12K was determined by measuring the release of cytoplasmic β-galactosidase from S. pneumonia cells (ATCC49619 and 16167) with ONPG as the substrate.22 S. pneumonia cells were grown to mid-log phase in MHB medium containing 2% lactose at 37°C. The cells were harvested by centrifugation, washed thrice, and diluted to an OD600 of 0.1 with 10 mM PBS (pH 7.4) containing 1.5 mM ONPG. Subsequently, 2 mL of the S. pneumonia cells were added to a quartz cuvette and incubated with 1× MICs of PEGylated Nano-BA12K, BA solution and Penicillin G at 37°C, respectively. OD420 measurements were recorded every 2 minutes for 30 minutes, which were taken as indicators of the permeability of the inner membrane.
Dye leakage assay
Liposomes with a PG/CL mass ratio of 3/1 were prepared to mimic the S. pneumonia membrane.24–26 The small unilamellar vesicles (SUVs) were prepared by a modified thin-film hydration method.27 The lipids were dissolved in 10 mL of dichloromethane and sonicated for 10 minutes. The solvent was removed by rotary evaporation at 45°C for 1 hour to obtain a thin film. Residual solvent remaining in the film was further evaporated under vacuum for another 24 hours at room temperature. Calcein solution was prepared by dissolving 62 mg of calcein in 1 mL of HEPES buffer (pH 7.4). The NaOH was added in small aliquots until calcein was dissolved, to yield a dark orange solution. The resultant thin film was hydrated with calcein solution at 35°C for 1 hour to obtain a calcein-entrapped liposome solution. The calcein-entrapped liposome solution was then separated from free calcein through a Sephadex G50 column.
The calcein release was performed by transferring 2 mL of HEPES buffer solution (pH 7.4) and 4 mL of calcein-entrapped liposomes in a beaker, under gentle stirring. Membrane permeation was detected by an increase in fluorescence at λex (490 nm)/λem (520 nm), following the addition of 1× MICs of PEGylated Nano-BA12K, BA solution, and Penicillin G. To induce 100% dye release, 10% (v/v) Triton X-100 was added to dissolve the vesicles. The percentage of fluorescence intensity recovery, Ft was calculated by using Equation 1:
|
|
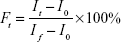
where I0 is the initial fluorescence intensity, If is the total fluorescence intensity with Triton X-100, and It is the fluorescence intensity observed at equilibrium after the addition of tested formulations.
Statistical analysis
All experiments were performed at least three times. Quantitative data are presented as mean ± SD. Statistical comparisons were determined by using Student’s t-test between two groups. P<0.05 and P<0.01 were considered statistically significant.
Results
In vitro antibacterial activities
Antibacterial activities of the PEGylated Nano-BA12K were determined against 13 isolates of S. pneumonia (Table 1). The growth of all the isolates was efficiently inhibited at a slightly lower concentration of PEGylated Nano-BA12K than those of Penicillin G. Furthermore, five penicillin-resistant strains, S. pneumonia 16033, 16092, 16121, 16129, and 16167 (MIC ≥4 μg/mL) were also susceptible to PEGylated Nano-BA12K with much lower MICs representing as 1, 0.5, 1, 2, and 1 μM, respectively.
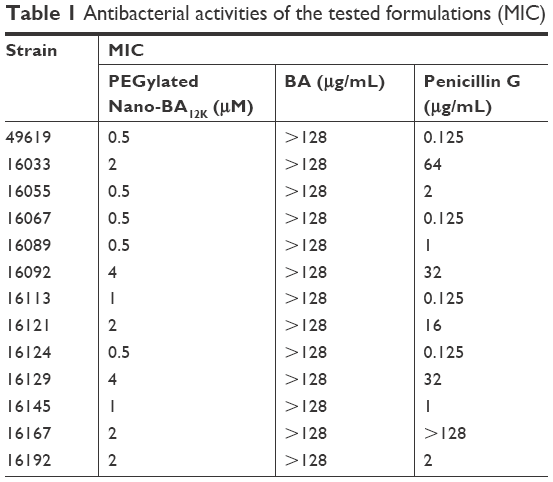 | Table 1 Antibacterial activities of the tested formulations (MIC) |
The change in viability of S. pneumonia ATCC 49619 and S. pneumonia 16167 after incubation with PEGylated Nano-BA12K was further assessed using FM. A red fluorescent nucleic acid dye (PI) was selected to detect the dead cell population, and a green fluorescent nucleic acid dye (SYTO 9) was applied to detect the viable cell population. Thus, bacteria with intact cell membranes appeared green fluorescence, whereas membrane-compromised bacteria appeared red. As illustrated in Figure 1, both the penicillin-sensitive and penicillin-resistant S. pneumonia cells exhibited low viability after treatment with PEGylated Nano-BA12K during the incubation time as displaying strong red fluorescence and weak green fluorescence in a time-dependent manner. The red fluorescence intensity increased from 0.5 to 8 h, and no viable green fluorescence was detected after 8 hours of incubation, indicating S. pneumonia cells are completely destroyed. BA solution had limited effect on the viability of S. pneumonia ATCC 49619 and S. pneumonia 16167 cells as observed by the bright green fluorescence intensity and minimal red fluorescence intensity. Penicillin G was only effective against sensitive S. pneumonia cells.
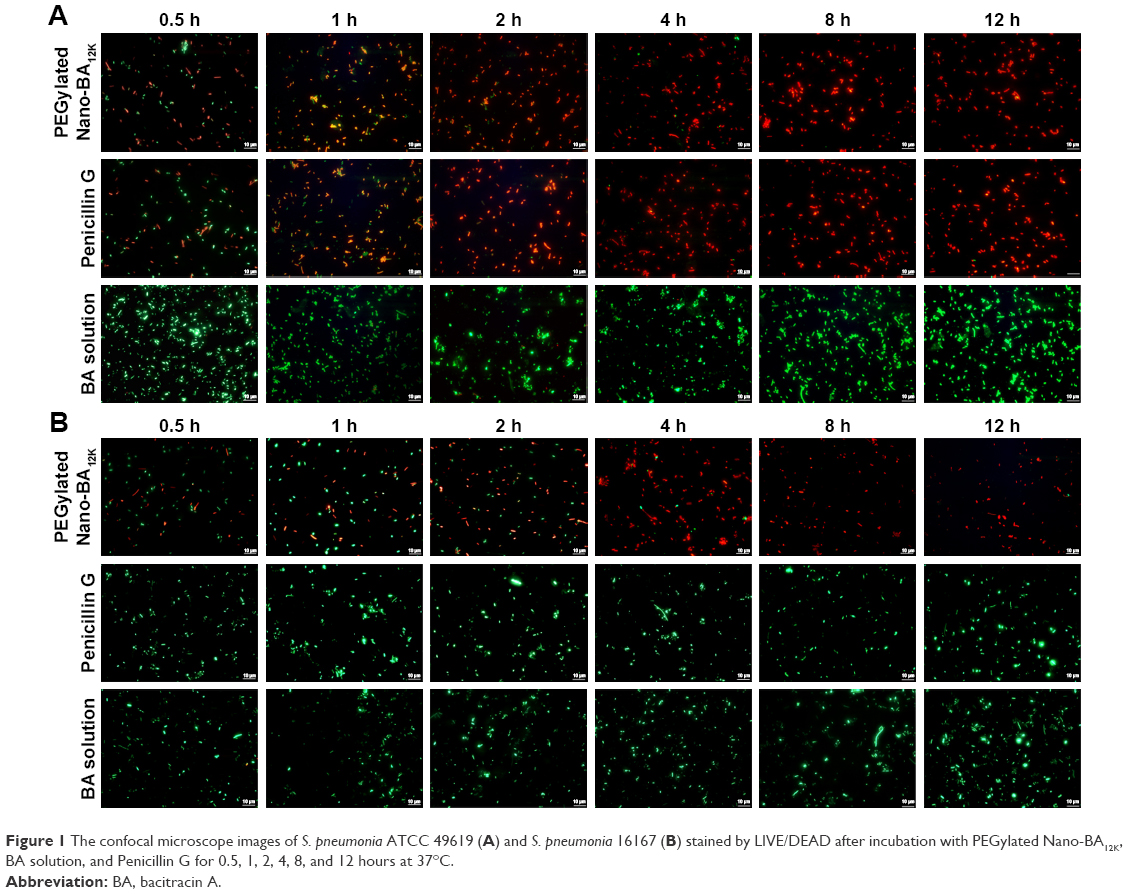 | Figure 1 The confocal microscope images of S. pneumonia ATCC 49619 (A) and S. pneumonia 16167 (B) stained by LIVE/DEAD after incubation with PEGylated Nano-BA12K, BA solution, and Penicillin G for 0.5, 1, 2, 4, 8, and 12 hours at 37°C. |
Nanoscale imaging of S. pneumonia cells
TEM was selected to study the morphological alternation of bacterial cells after treated with PEGylated Nano-BA12K. For S. pneumonia ATCC 49619, the bacterial cells had a normal smooth surface in the negative control group (Figure 2A). The cells treated with 0.5× MIC of PEGylated Nano-BA12K showed a partial surface disruption with a slight leakage of cellular cytoplasmic content (Figure 2B). Further addition of 1× MIC PEGylated Nano-BA12K to the bacteria caused significant morphological changes, but still retained their cytoplasmic membrane integrity (Figure 2C). When the concentration of PEGylated Nano-BA12K to 2× MIC was continuously increased, the cell wall/membrane was completely disrupted, and the detachment of cell wall from the cytoplasmic membrane could be observed, some were observed to even become ghost with a large amount of leakage of intracellular contents, and only fragments of the cells remained (Figure 2D). In contrast, the cells experienced no morphological changes after incubation with BA solution (Figure 2E). For penicillin-resistant strain S. pneumonia 16167 (Figure 3), similar results were obtained in the morphological changes after incubation with PEGylated Nano-BA12K. Vacuolar changes, breakdown of cell wall, and releasing of cytoplasm were clearly observed and exhibited a concentration-dependent profile (Figure 3B–D). For Penicillin G, only sensitive strain experienced morphological changes (Figure 2F), while resistant strain exhibited smooth surface (Figure 3F). BA solution showed limited effect on the morphological changes of both S. pneumonia ATCC 49619 (Figure 2E) and S. pneumonia 16167 (Figure 3E).
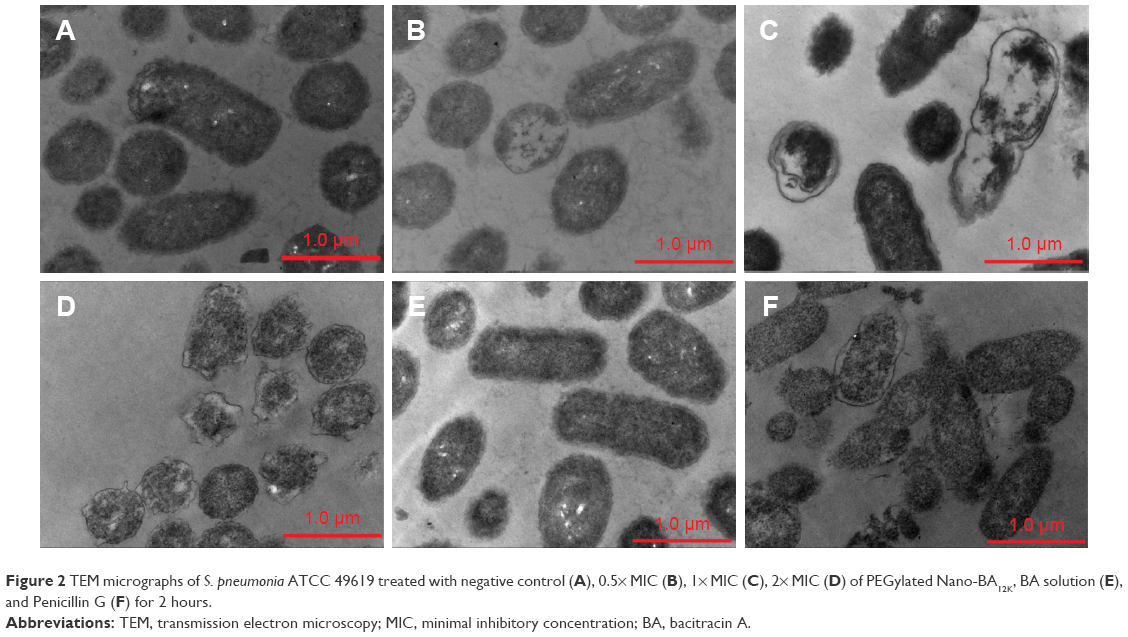 | Figure 2 TEM micrographs of S. pneumonia ATCC 49619 treated with negative control (A), 0.5× MIC (B), 1× MIC (C), 2× MIC (D) of PEGylated Nano-BA12K, BA solution (E), and Penicillin G (F) for 2 hours. |
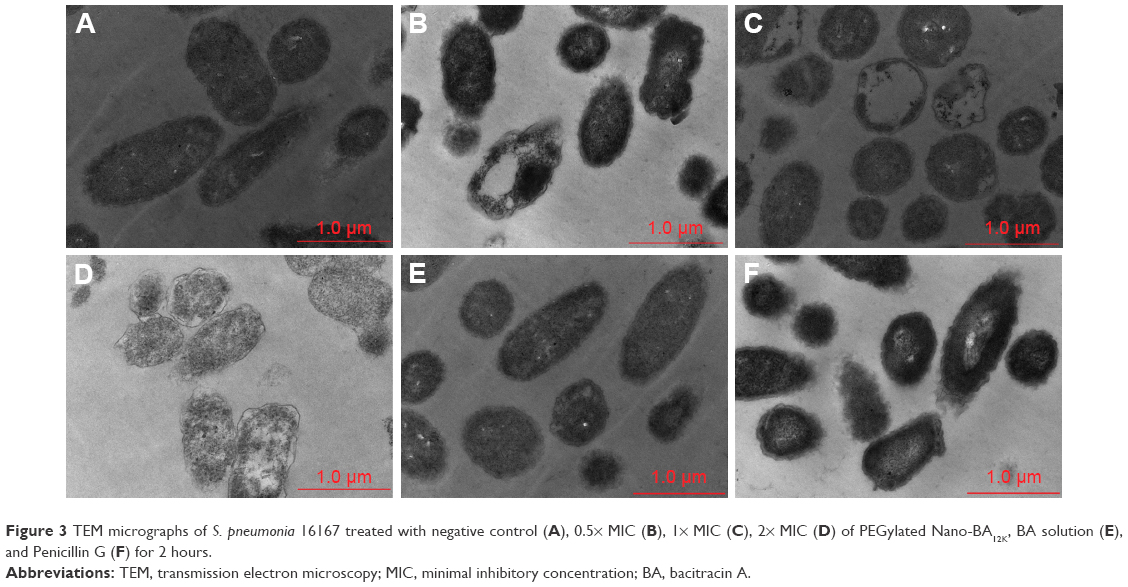 | Figure 3 TEM micrographs of S. pneumonia 16167 treated with negative control (A), 0.5× MIC (B), 1× MIC (C), 2× MIC (D) of PEGylated Nano-BA12K, BA solution (E), and Penicillin G (F) for 2 hours. |
The effect of PEGylated Nano-BA12K on the surface of S. pneumonia was further investigated using in situ AFM imaging (Figure 4). As shown in the vertical cross-sections, both penicillin-sensitive and penicillin-resistant S. pneumonia bacteria exhibited a typical catenoid shape in air. After treated with PEGylated Nano-BA12K, the bacterium progressively flattened over the course of the experiment. It is totally resulting in a drastic decrease of the bacterial kurtosis and average size. We interpret such swelling as evidence for the alteration of the cell wall/membrane leading to the discharge of most of the intracellular content, and no more well-organized membrane structure was exhibited. Moreover, the root mean square of bacteria was significantly increased, and debris was observed on the mica substrate in the vicinity of bacterial aggregates (Table 2). Such fibrous materials, already observed as a result of cell surface perturbation28 are indicative of cellular leakages.
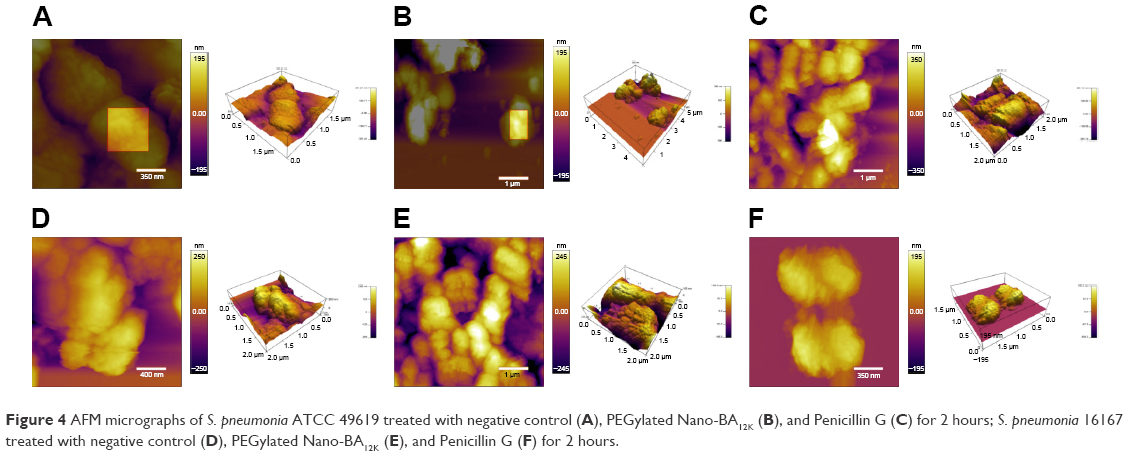 | Figure 4 AFM micrographs of S. pneumonia ATCC 49619 treated with negative control (A), PEGylated Nano-BA12K (B), and Penicillin G (C) for 2 hours; S. pneumonia 16167 treated with negative control (D), PEGylated Nano-BA12K (E), and Penicillin G (F) for 2 hours. |
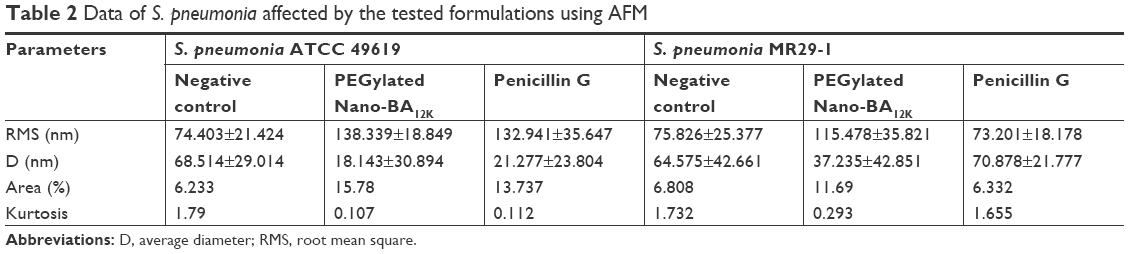 | Table 2 Data of S. pneumonia affected by the tested formulations using AFM |
PG content assay
PG is thought to be the major structural component in the cell wall of Gram-positive bacteria, forming a protective wall to resist a variety of host defense molecules.29 The fluctuation of PG content after incubation with PEGylated Nano-BA12K was further examined to evaluate its effect on cell wall integrity. As shown in Figure 5, for both S. pneumonia ATCC 49619 (Figure 5A) and S. pneumonia 16167 (Figure 5B), the PG content remained constant and did not response to the change of drug concentration and incubation time. Penicillin G could significantly reduce the PG content of S. pneumonia ATCC 49619 and totally lost its activities against S. pneumonia 16167.
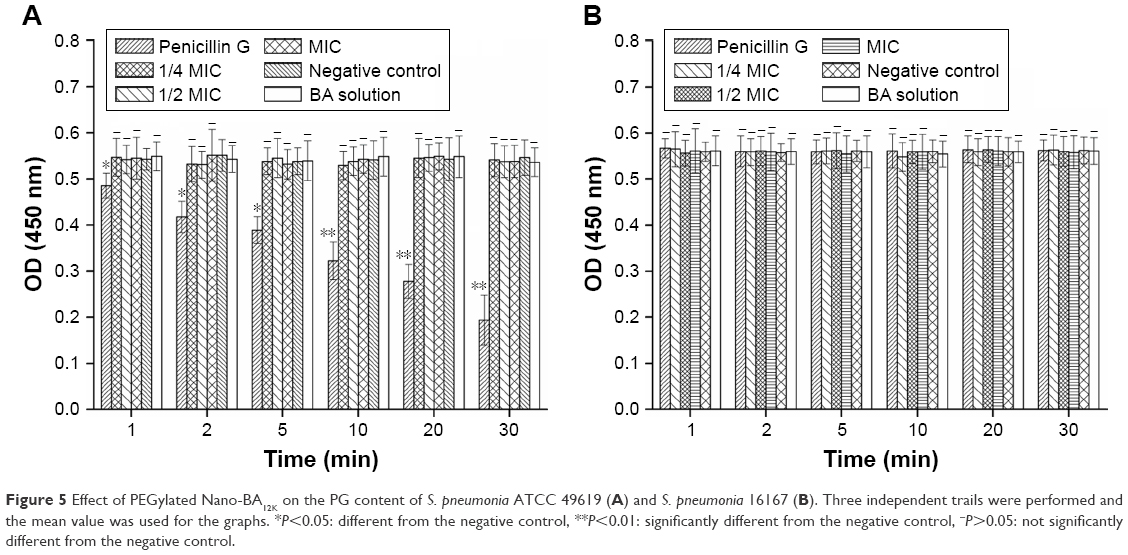 | Figure 5 Effect of PEGylated Nano-BA12K on the PG content of S. pneumonia ATCC 49619 (A) and S. pneumonia 16167 (B). Three independent trails were performed and the mean value was used for the graphs. *P<0.05: different from the negative control, **P<0.01: significantly different from the negative control, −P>0.05: not significantly different from the negative control. |
Behavior of BA-PEG-PLGA12K-PEG-BA at the air/buffer interface
The adsorption of BA-PEG-PLGA12K-PEG-BA and BA at the air/solution interface was detected by the surface tension measurement. As shown in Figure 6, at the beginning, the surface tension of the buffer without copolymer was measured to be 57.45 mN/m. At a low copolymer concentration, the surface tension decreased with increasing concentration, which was in accordance with Gibbs’ adsorption isotherm. The decrease in surface tension was due to the BA-PEG-PLGA12K-PEG-BA unimers distributing to the air/water interface, indicating that BA-PEG-PLGA12K-PEG-BA unimers were free within this concentration. It could be noticed that a change in slope (“a” first break) was observed at a characteristic concentration (~1×10−3 g/L). Then the surface tension values continued to decrease until a plateau was reached (“b” second break), after which the surface tension values remained approximately constant with further increasing the concentration of BA-PEG-PLGA12K-PEG-BA unimers until “c.” At the break at the high-concentration part of the surface tension curve (“b”) corresponding to the CMC value (~1.43×10−2 g/L), the BA-PEG-PLGA12K-PEG-BA unimers began to form micelles (PEGylated Nano-BA12K) due to the physical interactions between the hydrophobic blocks, leading to a reduction of BA-PEG-PLGA12K-PEG-BA unimers at the air/water interface. While, the origin of the low-concentration break (“a”) was less clear and has been attributed to the formation of “monomolecular” micelles, the presence of impurities, and the arrangement of the copolymer molecules adsorbed on the air/water interface at complete surface coverage. When further increasing the concentration of BA-PEG-PLGA12K-PEG-BA from “c” to “d,” the surface tension began to decrease. This was because PEGylated Nano-BA12K was saturated in buffer and the excessive BA-PEG-PLGA12K-PEG-BA unimers were distributed to the air/water interface again, leading to a decrease in the surface tension. After the concentration passed “d,” the surface tension remained constant, due to the limited solubility of BA-PEG-PLGA12K-PEG-BA unimers in the buffer. BA was chosen for comparison and appeared to be much less tensioactive than the PEGylated Nano-BA12K, which was in good correlation with the previous study.30 Overall, the modified BA (BA-PEG-PLGA12K-PEG-BA) was highly surface active, which usually translated into a good membranolytic effect.
 | Figure 6 The surface tension variation of addition of BA-PEG-PLGA12K-PEG-BA and BA with various concentrations (n=6). Data are expressed as the mean ± SD (error bars). |
PEGylated Nano-BA12K induced membrane depolarization
The ability of the PEGylated Nano-BA12K to depolarize the S. pneumonia cytoplasmic membrane was first determined using a membrane potential-sensitive dye diSC3-5. Under the influence of a membrane potential, the dye can concentrate in the cytoplasmic membrane, resulting in a self-quenching of fluorescence. Upon permeabilization and disruption of the cytoplasmic membrane, the diSC3-5 will release into the medium, causing a consequent increase in fluorescence intensity. As shown in Figure 7A and B, the addition of PEGylated Nano-BA12K caused a rapid increase in fluorescence intensity in both sensitive and resistant S. pneumonia due to the collapse of the ion gradients that generate the membrane potential. Penicillin G only showed moderate effect on sensitive S. pneumonia strain.
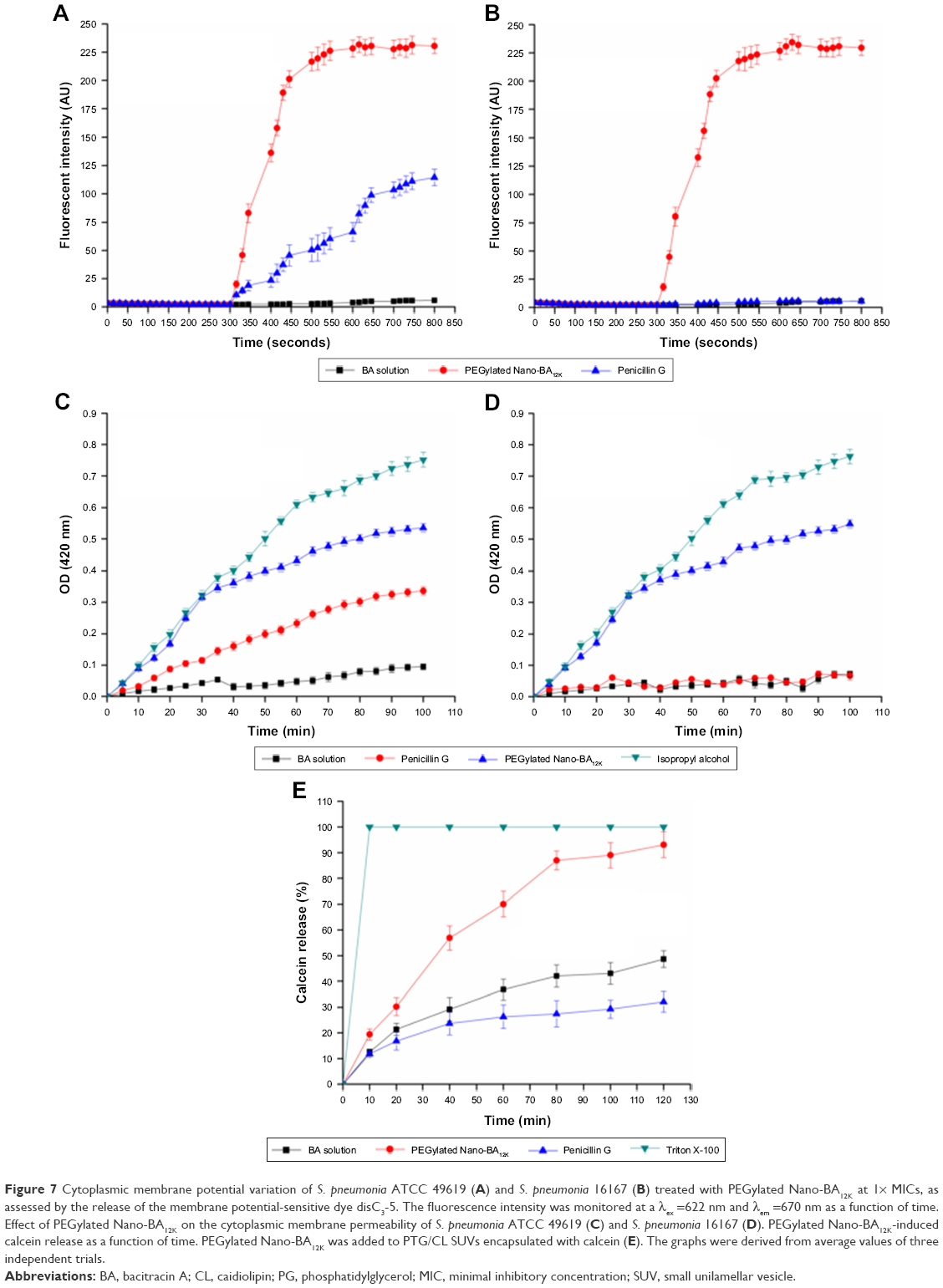 | Figure 7 Cytoplasmic membrane potential variation of S. pneumonia ATCC 49619 (A) and S. pneumonia 16167 (B) treated with PEGylated Nano-BA12K at 1× MICs, as assessed by the release of the membrane potential-sensitive dye disC3-5. The fluorescence intensity was monitored at a λex =622 nm and λem =670 nm as a function of time. Effect of PEGylated Nano-BA12K on the cytoplasmic membrane permeability of S. pneumonia ATCC 49619 (C) and S. pneumonia 16167 (D). PEGylated Nano-BA12K-induced calcein release as a function of time. PEGylated Nano-BA12K was added to PTG/CL SUVs encapsulated with calcein (E). The graphs were derived from average values of three independent trials. |
Cytoplasmic membrane permeability
The ability of PEGylated Nano-BA12K permeabilizing the cytoplasmic membrane of bacterial cells was also evaluated. The altered cytoplasmic membrane permeability could let ONPG enter into the cytoplasm and be degraded by β-galactosidase, producing o-nitrophenol with a certain absorbance at 420 nm. As illustrated in Figure 7C and D, PEGylated Nano-BA12K induced a rapid increase in the permeability of the cytoplasmic membrane at 1× MIC for both penicillin-sensitive and penicillin-resistant S. pneumonia. Unlike its cell wall permeability, PEGylated Nano-BA12K revealed greater cytoplasmic membrane permeability. Thus, PEGylated Nano-BA12K is believed to act on the cytoplasmic membrane more than the cell wall.
Lipid membrane permeability
The above experiments proposed a hypothesis that the PEGylated Nano-BA12K might kill the bacteria through disruption of the cell membrane, which could cause the leakage of the intracellular contents and ultimately lead to cell death. To determine whether bacterial membrane can be preferentially targeted by PEGylated Nano-BA12K, membrane perturbation was further examined by measuring calcein release from liposomes resembling the lipid composition of S. pneumonia cytoplasmic membranes (PTG/CL).31 Calcein was entrapped within these vesicles at a self-quenching concentration. The membrane disruption was indicated by the increase of fluorescence due to the release of calcein. As shown in Figure 7E, PEGylated Nano-BA12K rapidly induced calcein leakage from the PTG/CL liposomes in a time-dependent manner. Finally, PEGylated Nano-BA12K induced 93.1% calcein leakage from the PTG/CL liposomes, implying that S. pneumonia cytoplasmic membrane was susceptible to PEGylated Nano-BA12K. The Penicillin G and BA were less effective than PEGylated Nano-BA12K in terms of causing calcein leakage from the PTG/CL liposomes (32.1% and 48.7%). Further addition of the membrane-disruptive agent of Triton X-100 could induce a complete leakage of calcein.
Discussion
MDR S. pneumonia constitute a major worldwide public health concern. In our preliminary study, PEGylated nano-self-assemblies of bacitracin A (PEGylated Nano-BA12K) demonstrated strong antibacterial potency against reference S. pneumonia strain (ATCC 49619). In this study, the possibility of applying PEGylated Nano-BA12K against penicillin-resistant S. pneumonia was further investigated. In addition, the underlying antibacterial mechanism of PEGylated Nano-BA12K against both sensitive and resistant S. pneumonia was also clarified systematically, since S. pneumonia was naturally resistant to its unassembled counterpart BA. The in vitro antibacterial activity assay revealed a comprehensive result that PEGylated Nano-BA12K was effective against both penicillin-sensitive and penicillin-resistant S. pneumonia stains. Vacuolar changes, breakdown of cell wall/membrane, and releasing of cytoplasm were clearly observed in S. pneumonia ATCC 49619 and S. pneumonia 16167 after being treated with PEGylated Nano-BA12K for 2 hours through TEM and AFM. We presumed that PEGylated Nano-BA12K might kill S. pneumonia bacteria via cell wall/membrane disruption and content leakage. To verify this hypothesis, a series of molecular and cell experiments with cell wall/membrane were conducted.
Previous studies have proposed that cell wall/membrane damage occurs via wall/membrane destabilization and/or transmembrane pore formation.32 PEGylated Nano-BA12K exhibited limited effect on PG-outer cell wall barrier. The formation of PEGylated Nano-BA12K did not endow the micelles with the cell wall damage capability either, supported by no change in peptidoglycan content of S. pneumonia after treatment with various concentrations and incubation time of PEGylated Nano-BA12K. Previous studies indicated that S. pneumonia was naturally resistant to bacitracin. An activated CiaRH system capacitate S. pneumonia highly resistant to lysis induced by a wide variety of early and late cell wall inhibitors, such as cycloserine, bacitracin, and vancomycin.33–36 Impressively, surface pressure measurement suggested that PEGylated Nano-BA12K was much more tensioactive than BA, which was usually translated into a good membranolytic effect, and is helpful to permeabilize the cell membrane and damage membrane integrity. PEGylated Nano-BA12K could permeabilize the cytoplasmic membrane and collapse cell membrane potential rapidly and in a concentration-dependent manner. PEGylated Nano-BA12K nearly span the cell wall and thus could form channels, and their antimicrobial activity could arise from the ability to alter cell wall characteristics and/or cross the cell wall spontaneously and act on intracellular targets or perturb the coordination of multienzymatic complexes located in the vicinity of the membrane.37–39 PEGylated Nano-BA12K was further carried out in membrane mimetic conditions to better understand its interaction with the lipid. Calcein leakage assay monitored through fluorescence is well documented as a technique for probing antibacterial drugs activity.40,41 Calcein is a water soluble relatively large molecule and its release from the liposomes is assumed to involve in the formation of some type of pores in the liposomes.42 The data obtained from fluorescence experiment indicated that PEGylated Nano-BA12K could cause calcein leakage from PTG/CL SUVs (representing the cytoplasmic membrane of S. pneumonia) within the tested range of time, indicating significant perturbation of membrane mimetics. Thus, previously noted increased permeability of the cytoplasmic membrane of S. pneumonia after the introduction of PEGylated Nano-BA12K would appear to arise from the direct action with the phospholipids.
Collectively, it could be concluded that the emergence antibacterial activity of PEGylated Nano-BA12K against S. pneumonia might be due to the cytoplasmic membrane permeabilization and disruption. Self-promoted uptake has been proposed for the interaction between peptides and cell wall/membranes.43 PG is thought to be the major structural component in the cell wall of Gram-positive bacteria and forms a protective wall to resist a variety of host defence molecules.29 The addition of PLGA was sufficient to endow the resulting PEGylated Nano-BA12K with strong hydrophobic interactions and van der Waals with the PG, which let PEGylated Nano-BA12K probably inserted more deeply into adjacent PG molecules and acted as a spacer in the plane of the bilayer, reducing the short-range attractive forces between PG saccharide cores, consequently altering the lipid packing and the supermolecular structure of PG. The compromised cell wall may allow an increased influx of PEGylated Nano-BA12K into the periplasmic space. In addition, the formation of nanoparticles was expected to increase the local density of BA mass on the surface. Previous studies indicated that BA could disintegrate the bacterial cytoplasmic membrane of bacteria. We believed that the high local density of BA molecules on the surface could strengthen the interactions of PEGylated Nano-BA12K with cytoplasmic membrane of S. pneumonia cells, resulting in cytoplasmic damage. Additionally to the possible cytoplasmic membrane disintegration, PEGylated Nano-BA12K may permeate through the cytoplasmic membrane of the organisms due to the relatively large volume compared to the thickness of the cell membrane, as a consequence, loss of important metabolites and cellular components which usually culminated in cell shrinkage and ultimately cell death (Figure 8).
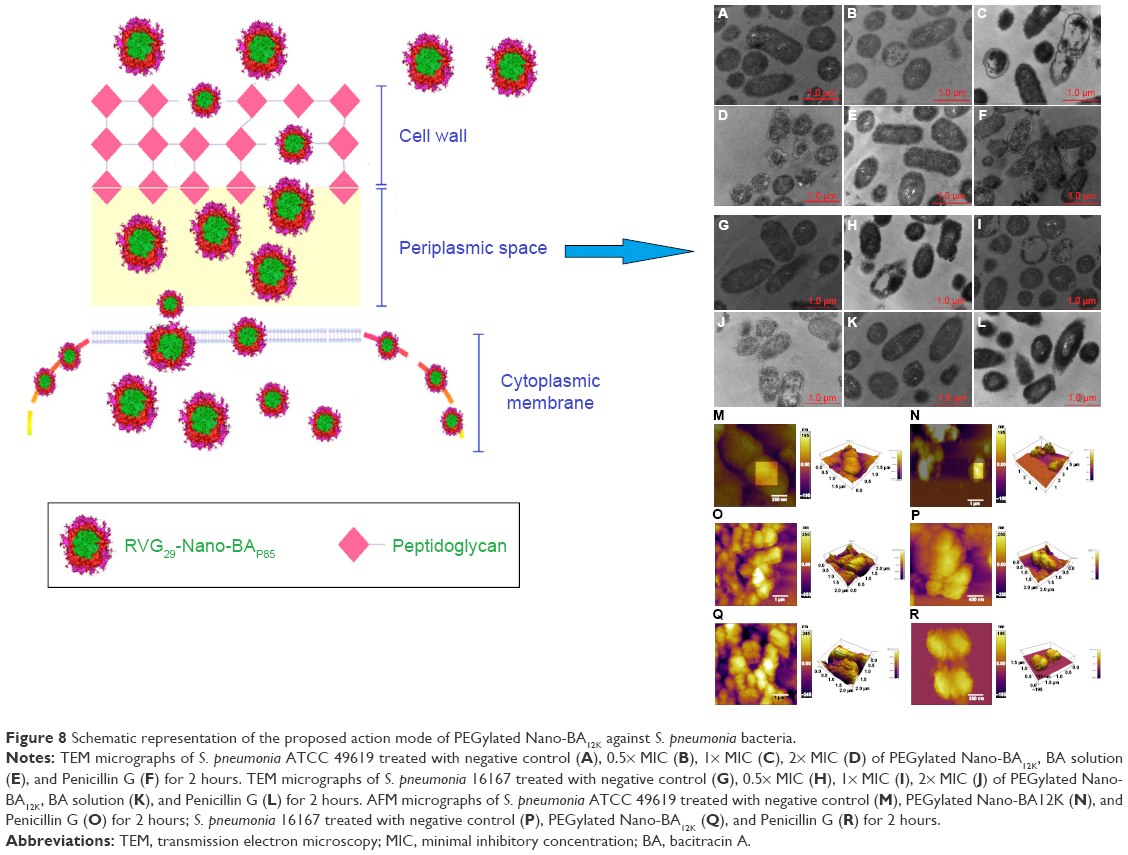 | Figure 8 Schematic representation of the proposed action mode of PEGylated Nano-BA12K against S. pneumonia bacteria. |
Currently, an increasing number of studies have given evidence that membrane permeabilization alone appears insufficient to cause cell death and therefore other complementary or novel mechanisms have been recently been reported.13,44 The recent work of Ciesiołka et al indicated that bacitracin also has the ability to degrade nucleic acids, being especially active against RNA molecules and single-stranded DNA.14 Thus, the interaction with the phospholipids present in the bacterial membrane may also act as a means of promoting internalization of the PEGylated Nano-BA12K. Nano-BA5K might cause cell death not only by disrupting cell membrane but also by targeting intracellular components such as DNAs and RNAs. Thus, the antibacterial mechanism of PEGylated Nano-BA12K will be further investigated in the future study.
Conclusion
In summary, mechanism studies indicated that PEGylated Nano-BA12K could come across the cell wall of bacteria, and then disrupted the cytoplasmic membrane due to the high local density of BA mass on the surface and high surface activity, ultimately leading to cell death. Nowadays, the field of nanoantibiotics is barely in its infancy, compared to caner- and cardiovascular diseases-targeted nanomedicine, while there are very little data on the clinical applications, toxicity, and antibacterial mechanism of nanoantibiotics. In this study, all of the data we presented are helpful in the design of self-assembly nano-polypeptide antibiotic candidates for future therapeutic purposes with clear antibacterial mechanism, and PEGylated Nano-BA12K could be a potential anti-infective agent for S. pneumonia infection treatment.
Acknowledgment
The authors are grateful for the financial support by the PhD Start-up Fund of Natural Science Foundation of Liaoning Province (Grant No 201601101), the National Science Foundation for Young Scientists of China (Grant No 31602108), the National Key Research and Development Program of China (Grant No 2016YFD0501309), and the Key Laboratory of Zoonosis of Liaoning Province.
Disclosure
The authors report no conflicts of interest in this work.
References
Musher DM. Infections caused by Streptococcus pneumoniae: clinical spectrum, pathogenesis, immunity, and treatment. Clin Infect Dis. 1992;14(4):801–809. | ||
Lynch JP, Zhanel GG. Escalation of antimicrobial resistance among Streptococcus pneumoniae: implications for therapy. Semin Respir Crit Care Med. 2005;26(6):575–616. | ||
Hakenbeck R, Briese T, Chalkley L, et al. Antigenic variation of penicillin-binding proteins from penicillin-resistant clinical strains of Streptococcus pneumoniae. J Infect Dis. 1991;164(2):313–319. | ||
Singh R, Smitha MS, Singh SP. The role of nanotechnology in combating multi-drug resistant bacteria. J Nanosci Nanotechnol. 2014;14(7):4745–4756. | ||
Diab R, Khameneh B, Joubert O, Duval R. Insights in nanoparticle-bacterium interactions: new frontiers to bypass bacterial resistance to antibiotics. Curr Pharm Des. 2015;21(28):4095–4105. | ||
Drulis-Kawa Z, Dorotkiewicz-Jach A. Liposomes as delivery systems for antibiotics. Int J Pharm. 2010;387(1–2):187–198. | ||
Huang CM, Chen CH, Pornpattananangkul D, et al. Eradication of drug resistant Staphylococcus aureus by liposomal oleic acids. Biomaterials. 2011;32(1):214–221. | ||
Catuogno C, Jones MN. The antibacterial properties of solid supported liposomes on Streptococcus oralis biofilms. Int J Pharm. 2003;257(1–2):125–140. | ||
Moghadas-Sharif N, Fazly Bazzaz BS, Khameneh B, Malaekeh-Nikouei B. The effect of nanoliposomal formulations on Staphylococcus epidermidis biofilm. Drug Dev Ind Pharm. 2015;41(3):445–450. | ||
Homhuan A, Kogure K, Akaza H, et al. New packaging method of mycobacterial cell wall using octaarginine-modified liposomes: enhanced uptake by and immunostimulatory activity of dendritic cells. J Control Release. 2007;120(1–2):60–69. | ||
Chapnick EK, Gradon JD, Kreiswirth B, et al. Comparative killing kinetics of methicillin-resistant Staphylococcus aureus by bacitracin or mupirocin. Infect Control Hosp Epidemiol. 1996;17(3):178–180. | ||
Tsuji K, Robertson JH. Improved high-performance liquid chromatographic method for polypeptide antibiotics and its application to study the effects of treatments to reduce microbial levels in bacitracin powder. J Chromatogr. 1975;112:663–672. | ||
Ming LJ, Epperson JD. Metal binding and structure-activity relationship of the metalloantibiotic peptide bacitracin. J Inorg Biochem. 2002;91(1):46–58. | ||
Ciesiołka J, Jeżowska-Bojczuk M, Wrzesiński J, et al. Antibiotic bacitracin induces hydrolytic degradation of nucleic acids. Biochim Biophys Acta. 2014;1840(6):1782–1789. | ||
Joy SR, Li X, Snow DD, Gilley JE, Woodbury B, Bartelt-Hunt SL. Fate of antimicrobials and antimicrobial resistance genes in simulated swine manure storage. Sci Total Environ. 2014;481:69–74. | ||
Thevenard B, Besset C, Choinard S, et al. Response of S. thermophilus LMD-9 to bacitracin: involvement of a BceRS/AB-like module and of the rhamnose-glucose polysaccharide synthesis pathway. Int J Food Microbiol. 2014;177:89–97. | ||
Tamaki M, Fujinuma K, Harada T, et al. Fatty acyl-gramicidin S derivatives with both high antibiotic activity and low hemolytic activity. Bioorg Med Chem Lett. 2012;22(1):106–109. | ||
Salehi B, Mehrabian S, Ahmadi M. Investigation of antibacterial effect of cadmium oxide nanoparticles on Staphylococcus aureus bacteria. J Nanobiotechnology. 2014;12:26. | ||
Chockalingam AM, Babu HK, Chittor R, Tiwari JP. Gum arabic modified Fe3O4 nanoparticles cross linked with collagen for isolation of bacteria. J Nanobiotechnology. 2010;8:30. | ||
Cavassin ED, de Figueiredo LF, Otoch JP, et al. Comparison of methods to detect the in vitro activity of silver nanoparticles (AgNP) against multidrug resistant bacteria. J Nanobiotechnology. 2015;13:64. | ||
Hong W, Zhao Y, Guo Y, et al. PEGylated self-assembled nano-bacitracin A: probing the antibacterial mechanism and real-time tracing of target delivery in vivo. ACS Appl Mater Interfaces. 2018;10(13):10688–10705. | ||
Ma QQ, Dong N, Shan AS, et al. Biochemical property and membrane-peptide interactions of de novo antimicrobial peptides designed by helix-forming units. Amino Acids. 2012;43(6):2527–2536. | ||
Dong N, Ma Q, Shan A, et al. Strand length-dependent antimicrobial activity and membrane-active mechanism of arginine- and valine-rich β-hairpin-like antimicrobial peptides. Antimicrob Agents Chemother. 2012;56(6):2994–3003. | ||
Chen Y, Mant CT, Hodges RS. Preparative reversed-phase high-performance liquid chromatography collection efficiency for an antimicrobial peptide on columns of varying diameters (1mm to 9.4mm I.D.). J Chromatogr A. 2007;1140(1–2):112–120. | ||
Epand RF, Schmitt MA, Gellman SH, Epand RM. Role of membrane lipids in the mechanism of bacterial species selective toxicity by two alpha/beta-antimicrobial peptides. Biochim Biophys Acta. 2006;1758(9):1343–1350. | ||
Ishibashi J, Saido-Sakanaka H, Yang J, Sagisaka A, Yamakawa M. Purification, cDNA cloning and modification of a defensin from the coconut rhinoceros beetle, Oryctes rhinoceros. Eur J Biochem. 1999;266(2):616–623. | ||
Dzoyem JP, Hamamoto H, Ngameni B, Ngadjui BT, Sekimizu K. Antimicrobial action mechanism of flavonoids from Dorstenia species. Drug Discov Ther. 2013;7(2):66–72. | ||
Alves CS, Melo MN, Franquelim HG, et al. Escherichia coli cell surface perturbation and disruption induced by antimicrobial peptides BP100 and pepR. J Biol Chem. 2010;285(36):27536–27544. | ||
Papo N, Shai Y. A molecular mechanism for lipopolysaccharide protection of Gram-negative bacteria from antimicrobial peptides. J Biol Chem. 2005;280(11):10378–10387. | ||
Rodrigues JC, Caseli L, Jefferson Carnevalle Rodrigues LC. Incorporation of bacitracin in Langmuir films of phospholipids at the air-water interface. Thin Solid Films. 2017;622:95–103. | ||
Cronan JE. Bacterial membrane lipids: where do we stand? Annu Rev Microbiol. 2003;57:203–224. | ||
Aquila M, Benedusi M, Koch KW, dell’orco D, Rispoli G. Divalent cations modulate membrane binding and pore formation of a potent antibiotic peptide analog of alamethicin. Cell Calcium. 2013;53(3):180–186. | ||
Mascher T, Heintz M, Zähner D, Merai M, Hakenbeck R. The CiaRH system of Streptococcus pneumoniae prevents lysis during stress induced by treatment with cell wall inhibitors and by mutations in pbp2x involved in beta-lactam resistance. J Bacteriol. 2006;188(5):1959–1968. | ||
Gómez-Mejia A, Gámez G, Hammerschmidt S. Streptococcus pneumoniae two-component regulatory systems: The interplay of the pneumococcus with its environment. Int J Med Microbiol. 2018;308(6):722–737. | ||
Li J, Tan C, Zhou Y, et al. The two-component regulatory system CiaRH contributes to the virulence of Streptococcus suis 2. Vet Microbiol. 2011;148(1):99–104. | ||
Hakenbeck R. Transformation in Streptococcus pneumoniae: mosaic genes and the regulation of competence. Res Microbiol. 2000;151(6):453–456. | ||
Pag U, Oedenkoven M, Sass V, et al. Analysis of in vitro activities and modes of action of synthetic antimicrobial peptides derived from an alpha-helical ‘sequence template’. J Antimicrob Chemother. 2008;61(2):341–352. | ||
Joanne P, Falord M, Chesneau O, et al. Comparative study of two plasticins: specificity, interfacial behavior, and bactericidal activity. Biochemistry. 2009;48(40):9372–9383. | ||
Deshayes S, Decaffmeyer M, Brasseur R, Thomas A. Structural polymorphism of two CPP: an important parameter of activity. Biochim Biophys Acta. 2008;1778(5):1197–1205. | ||
Medina ML, Chapman BS, Bolender JP, Plesniak LA. Transient vesicle leakage initiated by a synthetic apoptotic peptide derived from the death domain of neurotrophin receptor, p75NTR. J Pept Res. 2002;59(4):149–158. | ||
Russell AL, Kennedy AM, Spuches AM, Venugopal D, Bhonsle JB, Hicks RP. Spectroscopic and thermodynamic evidence for antimicrobial peptide membrane selectivity. Chem Phys Lipids. 2010;163(6):488–497. | ||
Sobko AA, Kotova EA, Antonenko YN, Zakharov SD, Cramer WA. Effect of lipids with different spontaneous curvature on the channel activity of colicin E1: evidence in favor of a toroidal pore. FEBS Lett. 2004;576(1–2):205–210. | ||
Hancock RE. Peptide antibiotics. Lancet. 1997;349(9049):418–422. | ||
Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66(4):236–248. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.