")
Back to Journals » The Application of Clinical Genetics » Volume 8
Insights into genetic susceptibility in the etiology of spontaneous preterm birth
Authors Parets S, Knight A, Smith A
Received 10 September 2015
Accepted for publication 12 November 2015
Published 14 December 2015 Volume 2015:8 Pages 283—290
DOI https://doi.org/10.2147/TACG.S58612
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Sasha E Parets,1 Anna K Knight,2 Alicia K Smith,1,2
1Department of Psychiatry and Behavioral Sciences, Emory University School of Medicine, Atlanta, GA, USA; 2Genetics and Molecular Biology Program, Emory University, Atlanta, GA, USA
Abstract: Preterm birth (PTB; <37 weeks of gestation) is a complex disorder, whose etiology is influenced by a variety of factors. A greater understanding of the biological mechanisms that contribute to PTB will facilitate identification of those at increased risk and may inform new treatments. To accomplish this, it is vital to elucidate the heritability patterns of this condition as well as the environment and lifestyle factors that increase risk for PTB. Identifying individual genes that contribute to the etiology of PTB presents particular challenges, and there has been little agreement among candidate gene and genome-wide studies performed to date. In this review we will evaluate recent genetic studies of spontaneous PTB, discuss common themes among their findings, and suggest approaches for future studies of PTB.
Keywords: PTB, GWAS, linkage, candidate gene, African–American, epigenetic
Introduction
Despite ongoing research and the advancement of health-care systems in the US, the rate of preterm birth (PTB) remains high. Prevention strategies have not been successful at reducing the PTB rate, and interventions for PTB after initiation of labor are only intermittently successful. Risk factors that increase the likelihood of a woman delivering preterm include her age, race, and current smoking status. Additionally, women who have had a previous preterm delivery are at higher risk of subsequent preterm deliveries.1–3
Over the past several years, clinical advancements have primarily occurred in managing the acute clinical needs of neonates who are born preterm.4 Despite this progress, PTB is the primary cause of mortality in the 1st year,5 and the morbidities of being born preterm can last throughout life as children born preterm are more likely to develop chronic diseases.2,6 The dire consequences of this condition cannot be fully addressed until the mechanisms that contribute to PTB are identified. This review will summarize the recent progress of genetic studies of PTB and discuss the insight they provide into its etiology.
PTB heritability
There are three primary categories of PTB. Approximately 30%–35% of all PTB are considered medically indicated and result from specific condition such as preeclampsia or fetal growth restriction affecting either the mother or fetus.2 Preterm premature rupture of the membranes (pPROMs) occurs in 25%–30% of PTB cases and is likely to occur in cases of infection, placental abruption, or if there are any anatomical abnormalities in the mother.2 Spontaneous PTB accounts for the remaining 35%–45% of PTB cases and has no known etiology.7 Genetic studies may be particularly useful for identifying biological pathways that contribute to spontaneous PTB.
Risk for PTB is not evenly distributed in the population. African–Americans are 1.5 times more likely to deliver preterm and more than twice as likely to deliver before 32 weeks gestation compared to Caucasians.2,8,9 A review by Anum et al10 highlighted other factors that could contribute to the increased risk of PTB among African–Americans including anemia during pregnancy, low serum folate levels, vitamin D deficiency, poor weight gain during pregnancy, or high prepregnancy body mass index (BMI). However, it is important to note that this increased risk of PTB among African–Americans appears to be independent of socioeconomic status or other social factors,11,12 as evidenced by the disparity remaining in military settings that have the same access to health care.10
One of the greatest risk factors for PTB is personal or familial history,2 and heritability studies have attempted to quantify how much of the variation in spontaneous PTB can be attributed to genetic differences. The estimates of PTB heritability in different populations vary substantially (17%–36%) depending on the types of PTB included and the nature of the cohort studied.1,6,13–16 For example, a study in a cohort of Australian twins estimated that the genetic contribution to PTB is 27% in any pregnancy and 17% for the first pregnancy.15 Another study specifically estimated the heritability of spontaneous PTB as high as 34%.17
A recent heritability study for PTB evaluated 1,784,172 noniatrogenic births from the Utah Population Database, of which 7.6% were spontaneous PTB.18 Overall, this comprehensive analysis conducted by Wu et al18 suggested that heritability of spontaneous PTB was less than 25%, which is relatively low when compared to other complex disorders.18 This study also emphasizes the profound role the environment plays in mediating the risk for PTB, as 75% of the variance could be attributed to the environment shared between a mother and offspring or to individual environmental factors that vary from one birth to another such as inflammation, stress, and socioeconomic status.18 They concluded that the relatively low heritability of spontaneous PTB suggests that studies of unrelated cases and controls would have relatively low power to detect causal variants. Given the likelihood that PTB risk results from multiple genes of small effect size that are involved in gene–gene and gene–environment interactions, Wu et al18 suggest that family studies would be more appropriate for mapping genes involved in spontaneous PTB.
York et al19 questioned the conclusion that extended pedigrees would be informative for mapping genes related to PTB. In their extensive commentary, they highlight their work and the work of others using twin and family data to discuss methodological factors that should be considered by heritability studies of gestational age (GA) and PTB.19,20 Interestingly, these studies, which evaluated the heritability of GA in population-based records of primarily term deliveries, also support the conclusion that the environment plays the primary role in PTB risk while the genetic components contribute only modestly.21,22
Contributions of maternal and fetal genomes
Pregnancy involves a semi-allografic relationship between a mother and her fetus.23 The fetus is genetically distinct and recognized as such by the maternal immune system. Maternal physiology is adapted to tolerate and nurture the development of the fetus.24,25 Both the mother and fetus take an active role in the parturition process.6 However, the contribution of the mother and fetus toward the initiation of labor, or more generally toward PTB, remains an active debate.
Although there are undoubtedly multiple mechanisms that may contribute to the etiology of PTB, some argue that genetic studies of PTB should consider both the maternal and fetal genomes, recognizing that they may have both overlapping and independent contributions to PTB. Others argue that primary genetic contributions to PTB are inherent to the maternal genome.26,27 One such study arguing for the maternal genome’s contribution to PTB demonstrated that there was a recurrence risk of 1.85 for offspring of mothers who were born early preterm to be born preterm themselves, while there was a recurrence risk of 1.06 for offspring of fathers who were born early preterm to be born preterm themselves. Thus, they argue that risk is likely to be transmitted primarily through the maternal genome, with a minimal contribution from the fetal genome.26 Additionally, Plunkett et al27 found that a parent-of-origin model was the most likely mode of inheritance of PTB and that the maternal contribution to PTB is greater than that of the paternal contribution. However, they note that a maternal-only inheritance model did not outperform the model accounting for the contributions of both parents.27 This is consistent with the observations of Wu et al,18 who found that the heritability of GA estimated by comparing GA of fathers and their offspring was less than half of the estimate made by comparing GA of mothers and their offspring.18 Furthermore, when examining the heritability of GA, population-based studies also report that the maternal contribution is greater than the fetal contribution.21,22
The maternal and fetal genomes share approximately 50% sequence similarity, and it can be difficult to distinguish between the effects of maternal genes and the effects of maternally-inherited genes. This is further complicated by the potential contributions of the maternal genome in regulating the intrauterine environment in which the fetus develops and with which the fetal genome interacts. The biases inherent of different heritability study designs are summarized in other papers.19,20 Overall, it is likely that individual heritability estimates vary widely in different contexts and among different cohorts, suggesting that identification of genes that increase risk for PTB may be more productive from a public health standpoint than further refinement of heritability estimates in different cohorts. However, it remains important to consider which genome is being evaluated when trying to draw conclusions across published genetic studies of PTB.
Candidate gene studies
Candidate gene studies, which compare the frequency of a predetermined genetic variant in unrelated spontaneous PTB cases and term birth controls, were among the earliest genetic studies of PTB. Candidate genes are selected to test a specific hypothesis related to the biological pathways believed to contribute to a phenotype of interest, such as PTB, but are limited to the current understanding of the biological processes underlying that phenotype.28,29 Candidate gene studies for PTB have primarily focused on genes associated with inflammation or the transition of the uterus into a contractile state.30–39
Over time, candidate gene selection has become more comprehensive and has begun incorporating different hypotheses of PTB etiology. Romero et al40 examined 775 single nucleotide polymorphisms (SNPs) in 190 candidate genes to identify variants in maternal and fetal samples that associate with preterm delivery and PTB risk. The genes and variants were selected from a literature review of genes previously evaluated for association with spontaneous PTB, preterm labor, pPROM, small for GA, or preeclampsia. In maternal samples, they identified an association between preterm delivery and a SNP in tissue inhibitor of metalloproteinase 2 (TIMP2; rs2277698), a gene that is believed to regulate matrix-degrading enzyme activity and collagen IV metabolism. Higher TIMP2 levels have previously been associated with PROM and preeclampsia.40–42 Similarly, rs2277698 in TIMP2 has also been associated with increased risk for pPROM.43 In fetal samples, the authors identified an association between PTB and a SNP in interleukin-6 receptor (IL6R; rs8192282), which binds IL6 and is associated with immune signaling.44 Genetic variation in IL6R has been associated with preterm labor.40 A recent study found that rs8192282 is part of a haplotype block that may influence soluble IL6R levels to modulate inflammatory disease risk.45 Finally, Romero et al40 identified haplotypes in four genes (IL6R; collagen, type IV, alpha 3 – COL4A3; lactotransferrin – LTF; and fibroblast growth factor 1 – FGF1) associated with delivering preterm in maternal samples and haplotypes in three genes (insulin-like growth factor 2 – IGF2, interleukin-2 – IL2, collagen, type IV, alpha 1 – COL4A1) associated with PTB in fetal samples. This study concludes that both the fetal and maternal genomes may contribute to PTB risk.40
A systemic review of the literature identified 189 SNPs in 84 genes that had been associated with PTB.46 A meta-analysis of these studies supported significant, albeit modest, associations between PTB and maternal SNPs in interleukin-1 receptor antagonist (IL1RN), beta-2 adrenergic receptor (ADRB2), interferon gamma (IFNG), and factor 2 (F2). Of these genes, IL1RN and IFNG are involved in immune response, ADRB2 is involved in G protein-coupled receptor, and F2 is involved in coagulation. In the fetal genome, the meta-analysis suggested a single SNP (rs1799963) in F2 also associated with PTB.46
Overall, candidate gene association studies in unrelated cases and controls have neither resulted in overwhelming reproducibility in maternal or fetal samples, nor have they provided substantial insight into the etiology of spontaneous PTB. Thus, investigators have begun focusing on genetic approaches that do not require an a priori understanding of the biological mechanisms underlying spontaneous PTB.
Genome-wide studies
Genome-wide association studies (GWASs) are valuable for examining the associations between genetic variants and diseases with complex etiologies, such as PTB.47 GWASs are unbiased and allow for generation of new hypotheses. However, they require large sample sizes to garner the statistical power necessary to account for the number of independent tests performed.
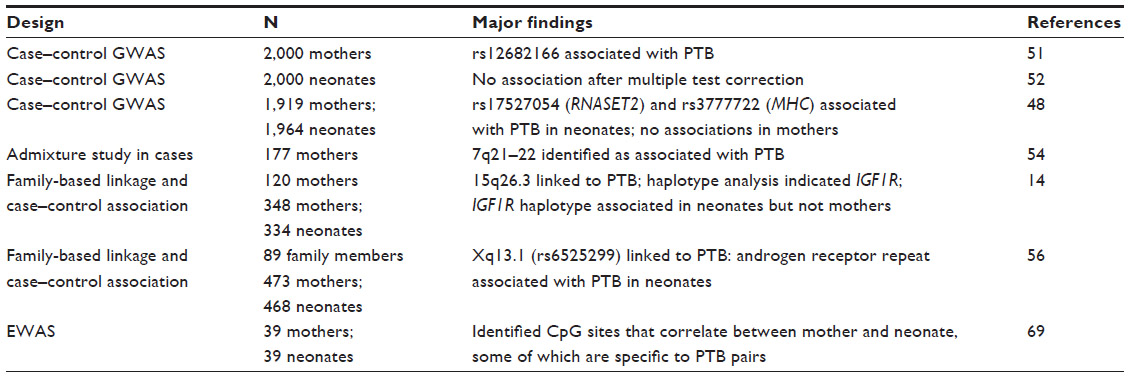
One such GWAS examined paired maternal–infant samples from early PTB in 959 mothers and 979 infants compared to term births in 960 mothers and 985 infants.48 No SNP in the analysis of the maternal genome remained associated with PTB after correction for multiple testing. However, SNPs from the fetal genome, located on chromosome 6 in ribonuclease T2 (RNASET2) and in the extended major histocompatibility complex, were associated with PTB at experiment-wide significance levels.48 Both genes are involved in regulation of the immune response,49,50 although their connection with PTB remains unclear. Despite this initial finding, the investigators were unable to replicate the association in an independent fetal cohort of 243 cases and 149 controls.48
Uzun et al51 nested a candidate gene study inside a GWAS. Using maternal GWAS data from the GENEVA (Gene Environment Association Studies Initiative) cohort, they first examined candidate genes that were previously associated with PTB.51 They found no association between PTB and genes from the literature after correction for multiple tests. They next evaluated all of the SNPs from the GWAS and identified one that remained associated after multiple test correction. However, that SNP was not located near any gene and has no known regulatory function. Using a curated SNP and gene list (N=515 genes), they then preformed gene set enrichment analysis for biological process, molecular function, and cellular components and reported gene enrichment for 30 pathways, compared to 39 pathways identified using the GWAS data. Many of these pathways were associated with inflammation and metabolic disorders, and the authors suggest that these pathways may provide clues as to the gene–gene interactions that may contribute to PTB.51
Another GWAS evaluated 1,000 PTBs and 1,000 term births from the Danish National Birth Cohort and found no association with any autosomal SNP in the fetal genome. The authors suggest that family-based linkage studies may be more informative of genetic variants associated with PTB risk.52 Overall, GWAS has had limited success in identifying potentially causal genetic variants associated with PTB, potentially due to study limitations including inadequate power, variations in sample collection and analyses methods, and the heterogeneous mechanism and definitions of PTB. It is not clear how best to improve the future likelihood of success in identifying PTB-associated variants. Some suggest that larger studies with careful phenotypic characterization will be required to identify genes of subtle effect sizes. However, it is also possible that studies of unrelated cases and controls may not be the ideal design for gene identification and that family-based approaches would be more fruitful. Indeed, the inconsistencies among findings from case–control studies performed to date suggest that other methods may prove more fruitful in identifying the genetic contribution to PTB.
Admixture mapping uses the higher prevalence of PTB among those of African descent to identify regions of potential disease association that have higher frequencies of African alleles compared to European alleles. On average, individuals of African descent have less linkage disequilibrium and shorter haplotype blocks compared to individuals of European descent.53 A recent study used admixture to map a region on chromosome 7 (7q21–7q22) that may be associated with PTB in African–Americans.54 This region contains genes involved in metabolism, inflammation, calcium regulation, and collagen, all of which may be relevant to PTB. Admixture mapping in families may provide even more power to detect susceptibility loci.
Family-based studies
In contrast to GWASs, linkage studies use families to attempt to identify genomic regions that segregate with a trait or disorder of interests. Linkage refers to the tendency of two genomic loci to be inherited together, at a greater a frequency than random chance.55 A recent linkage study by Haataja et al14 evaluated 120 women from 20 large families with at least two recurrent spontaneous preterm deliveries. Seven large families were selected for linkage analysis from northern Finland, a region characterized by relative genetic homogeneity that facilitates linkage mapping. This analysis suggested that a region of 15q26.3 was linked to PTB in the infants of these families. No region was independently linked to PTB in the maternal samples, but the authors noted some evidence of linkage to 15q26.3, potentially supporting the linkage signal in the infants. This region includes the gene that encodes the insulin-like growth factor receptor 1, which the authors hypothesize may be involved in the signaling pathways related to parturition.14
Another linkage study performed by the same group examined the X chromosome in relation to spontaneous PTB in the same Finnish cohort.56 In fetal samples, Karjalainen et al56 reported linkage between PTB and Xq13.1, a region containing genes that encode the androgen receptor (AR) and interleukin-2 receptor gamma subunit (IL2RG). In a case–control analysis in an independent population they examined the association between spontaneous PTB and SNPs in AR and IL2RG; however, no SNP or haplotype associated with PTB status. However, upon examination of the CAG repeat of AR in fetal samples, they discovered that longer CAG repeats (≥26) associate with a greater risk of spontaneous PTB when compared to those with the shortest repeats (≤19). The authors suggest that repeat length variation may associate with length of gestation and pregnancy complications by altering the regulation of AR, which plays vital roles in pregnancy and development. However, an association between AR repeat length and PTB was not observed in maternal samples.56
Epigenetics
Since genetic studies have not yet identified genetic variants that increase risk of PTB or uncovered genes that inform its etiology, epigenetics has been proposed as a possible mechanism that incorporates environmental risk as well as the intergenerational risk.57 Epigenetics refers to the regulation of gene expression without changes in the underlying sequence of DNA. One such regulatory mark is DNA methylation at the 5′ position of cytosine. Recent studies have demonstrated that methylation may depend, in part, on the underlying genotype as methylation patterns from different tissue types are correlated within the same individual, and monozygotic twins show a higher rate of correlation than dizygotic twins.58,59 These areas of genetic–epigenetic correlation may underlie gene–environment interactions and have been associated with numerous complex traits and disorders.60–63 Thus, epigenetic regulation cannot be fully separated from the genomic sequence, and epigenetic differences have been associated with early life events, such as PTB and maternal stress, thus strengthening the hypothesis that changes in the epigenome may associate with, or even play a causal role in PTB.64–67
Epigenetic studies in murine models provide key insight into the intergenerational transmission of PTB. A recent study has examined the effects of maternal and grandmaternal stress on GA by exposing pregnant mice to a stressor for up to three generations. This study concluded that stress in previous generations may associate with shorter gestation, potentially through altered miRNA expression.68 This finding, if translated to human populations, would have enormous implications on how individual behaviors may contribute to the risk of PTB for subsequent generations.
A study by Parets et al69 evaluated the correlation of maternal and fetal DNA methylation patterns in African–Americans from a spontaneous PTB cohort. In this study, 5,171 CpG sites correlated between maternal and fetal samples, although most of the correlated CpG sites could be attributed to genetic variation. Furthermore, methylation of CpG sites in 57 genes was unique to PTB pairs.69 CpG sites that correlate between maternal and fetal pairs are enriched in pathways associated with metabolic, cardiovascular, and immune function.69 This study suggests that both genetic and epigenetic factors contribute to PTB, an observation that should be considered in the design of future studies.
Conclusion
Although the precise patterns of PTB heritability have yet to be determined, studies overwhelmingly suggest that genetic factors contribute to PTB risk. Candidate gene studies have provided an initial platform for exploring genetic variation associated with PTB, but have yielded few convincing contributions to explain PTB etiology. As summarized in Table 1, GWASs have generated some promising new leads. Studies that incorporate family-based designs versus unrelated spontaneous cases and controls are particularly appealing. Linkage studies in families have produced promising initial results, and further studies may help to identify genes involved in the etiology of spontaneous PTB. Similarly, epigenetic studies of families may be useful for incorporating the influence of a shared environment into ongoing studies of PTB. In time, such studies may facilitate biomarker development. Clearly, implementation of early preventative measures for women most at risk would have enormous positive implications for both acute and long-term maternal and fetal health.
 | Table 1 Summary of genome-wide studies of PTB |
The genetic studies presented in this review each have a different set of limitations, and further research in this area is clearly required. It is, however, tempting to consider the similarities across the results of the studies published to date. Overall, studies using linkage,14 admixture,54 and epigenome-wide69 approaches independently identify genes involved in metabolism and immune function. Indeed, women who deliver preterm are at increased risk to develop chronic disorders as they age,70–79 suggesting that spontaneous PTB may result from subsyndromic metabolic or immune dysregulation or may be an early indicator of future risk for mothers and their fetuses.
Acknowledgments
This work was supported by grants from the National Institutes of Minority and Health Disparities (MD009064 to AKS).
Disclosure
Salary support for SEP and AKK was provided, in part, by National Institute of Environmental Health Sciences (T32ES012870) and National Institute of General Medical Sciences (T32GM008490), respectively. The authors report no other conflicts of interest in this work.
References
Parets SE, Bedient CE, Menon R, Smith AK. Preterm birth and its long-term effects: methylation to mechanisms. Biology. 2014;3(3):498–513. | |
Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371(9606):75–84. | |
Cnattingius S, Granath F, Petersson G, Harlow BL. The influence of gestational age and smoking habits on the risk of subsequent preterm deliveries. N Eng J Med. 1999;341(13):943–948. | |
Simmons LE, Rubens CE, Darmstadt GL, Gravett MG. Preventing preterm birth and neonatal mortality: exploring the epidemiology, causes, and interventions. Semin Perinatol. 2010;34(6):408–415. | |
Callaghan WM, MacDorman MF, Rasmussen SA, Qin C, Lackritz EM. The contribution of preterm birth to infant mortality rates in the United States. Pediatrics. 2006;118(4):1566–1573. | |
Institute of Medicine (US) Committee on Understanding Premature Birth and Assuring Healthy Outcomes; Behrman RE, Butler AS, editors. Preterm Birth Causes, Consequences, and Prevention. Washington, DC: Institute of Medicine (US) Committee on Understanding Premature Birth and Assuring Healthy Outcomes; 2007. | |
Moutquin JM. Classification and heterogeneity of preterm birth. BJOG. 2003;110(Suppl 20):30–33. | |
Kistka ZA, Palomar L, Lee KA, et al. Racial disparity in the frequency of recurrence of preterm birth. Am J Obstet Gynecol. 2007;196(2):131. e131–e136. | |
Hamilton BE, Martin JA, Ventura SJ. Births: preliminary data for 2011. National Vital Statistics Reports: From the Centers for Disease Control and Prevention, National Center for Health Statistics, National Vital Statistics System. Vol 61, No 5. Hyattsville, MD: National Center for Health Statistics. 2012:1–18. | |
Anum EA, Springel EH, Shriver MD, Strauss JF 3rd. Genetic contributions to disparities in preterm birth. Pediatr Res. 2009;65(1):1–9. | |
Goldenberg RL, Cliver SP, Mulvihill FX, et al. Medical, psychosocial, and behavioral risk factors do not explain the increased risk for low birth weight among black women. Am J Obstet Gynecol. 1996;175(5):1317–1324. | |
McGrady GA, Sung JF, Rowley DL, Hogue CJ. Preterm delivery and low birth weight among first-born infants of black and white college graduates. Am J Epidemiol. 1992;136(3):266–276. | |
Ward K, Argyle V, Meade M, Nelson L. The heritability of preterm delivery. Obstet Gynecol. 2005;106(6):1235–1239. | |
Haataja R, Karjalainen MK, Luukkonen A, et al. Mapping a new spontaneous preterm birth susceptibility gene, IGF1R, using linkage, haplotype sharing, and association analysis. PLoS Genet. 2011;7(2):e1001293. | |
Treloar SA, Macones GA, Mitchell LE, Martin NG. Genetic influences on premature parturition in an Australian twin sample. Twin Res. 2000; 3(2):80–82. | |
Committee on Practice Bulletins-Obstetrics TACoO, Gynecologists. Practice bulletin no 130: prediction and prevention of preterm birth. Obstet Gynecol. 2012;120(4):964–973. | |
Clausson B, Lichtenstein P, Cnattingius S. Genetic influence on birthweight and gestational length determined by studies in offspring of twins. BJOG. 2000;107(3):375–381. | |
Wu W, Witherspoon DJ, Fraser A, et al. The heritability of gestational age in a two-million member cohort: implications for spontaneous preterm birth. Hum Genet. 2015;134(7):803–808. | |
York TP, Strauss JF 3rd, Eaves LJ. A narrow heritability evaluation of gestational age at birth. Hum Genet. 2015;134(7):809–811. | |
Svensson AC, Sandin S, Cnattingius S, et al. Svensson et al respond to “Maternal genes and environment in preterm birth”. Am J Epidemiol. 2009;170(11):1386–1387. | |
Lunde A, Melve KK, Gjessing HK, Skjaerven R, Irgens LM. Genetic and environmental influences on birth weight, birth length, head circumference, and gestational age by use of population-based parent-offspring data. Am J Epidemiol. 2007;165(7):734–741. | |
York TP, Eaves LJ, Lichtenstein P, et al. Fetal and maternal genes’ influence on gestational age in a quantitative genetic analysis of 244,000 Swedish births. Am J Epidemiol. 2013;178(4):543–550. | |
Lee J, Romero R, Xu Y, et al. A signature of maternal anti-fetal rejection in spontaneous preterm birth: chronic chorioamnionitis, anti-human leukocyte antigen antibodies, and C4d. PloS One. 2011;6(2):e16806. | |
Trowsdale J, Betz AG. Mother’s little helpers: mechanisms of maternal-fetal tolerance. Nat Immunol. 2006;7(3):241–246. | |
Veenstra van Nieuwenhoven AL, Heineman MJ, Faas MM. The immunology of successful pregnancy. Hum Reprod Update. 2003;9(4):347–357. | |
Wilcox AJ, Skjaerven R, Lie RT. Familial patterns of preterm delivery: maternal and fetal contributions. Am J Epidemiol. 2008;167(4):474–479. | |
Plunkett J, Feitosa MF, Trusgnich M, et al. Mother’s genome or maternally-inherited genes acting in the fetus influence gestational age in familial preterm birth. Hum Hered. 2009;68(3):209–219. | |
Kwon JM, Goate AM. The candidate gene approach. Alcohol Res Health. 2000;24(3):164–168. | |
Tabor HK, Risch NJ, Myers RM. Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet. 2002;3(5):391–397. | |
Dizon-Townson DS, Major H, Varner M, Ward K. A promoter mutation that increases transcription of the tumor necrosis factor-alpha gene is not associated with preterm delivery. Am J Obstet Gynecol. 1997;177(4):810–813. | |
Annells MF, Hart PH, Mullighan CG, et al. Interleukins-1, -4, -6, -10, tumor necrosis factor, transforming growth factor-beta, FAS, and mannose-binding protein C gene polymorphisms in Australian women: risk of preterm birth. Am J Obstet Gynecol. 2004;191(6):2056–2067. | |
Chen D, Hu Y, Wu B, et al. Tumor necrosis factor-alpha gene G308A polymorphism is associated with the risk of preterm delivery. Beijing Da Xue Xue Bao. 2003;35(4):377–381. | |
Engel SA, Erichsen HC, Savitz DA, Thorp J, Chanock SJ, Olshan AF. Risk of spontaneous preterm birth is associated with common proinflammatory cytokine polymorphisms. Epidemiology. 2005;16(4):469–477. | |
Simhan HN, Krohn MA, Roberts JM, Zeevi A, Caritis SN. Interleukin-6 promoter -174 polymorphism and spontaneous preterm birth. Am J Obstet Gynecol. 2003;189(4):915–918. | |
Hartel C, Finas D, Ahrens P, et al. Polymorphisms of genes involved in innate immunity: association with preterm delivery. Mol Hum Reprod. 2004;10(12):911–915. | |
Landau R, Xie HG, Dishy V, et al. beta2-Adrenergic receptor genotype and preterm delivery. Am J Obstet Gynecol. 2002;187(5):1294–1298. | |
Maltier JP, Benghan-Eyene Y, Legrand C. Regulation of myometrial beta 2-adrenergic receptors by progesterone and estradiol-17 beta in late pregnant rats. Biol Reprod. 1989;40(3):531–540. | |
Bezold KY, Karjalainen MK, Hallman M, Teramo K, Muglia LJ. The genomics of preterm birth: from animal models to human studies. Genome Med. 2013;5(4):34. | |
Hao K, Wang X, Niu T, et al. A candidate gene association study on preterm delivery: application of high-throughput genotyping technology and advanced statistical methods. Hum Mol Genet. 2004;13(7):683–691. | |
Romero R, Velez Edwards DR, Kusanovic JP, et al. Identification of fetal and maternal single nucleotide polymorphisms in candidate genes that predispose to spontaneous preterm labor with intact membranes. Am J Obstet Gynecol. 2010;202(5):431. e431–e434. | |
Eleuterio NM, Palei AC, Rangel Machado JS, Tanus-Santos JE, Cavalli RC, Sandrim VC. Positive correlations between circulating adiponectin and MMP2 in preeclampsia pregnant. Pregnancy Hypertens. 2015;5(2):205–208. | |
Vadillo-Ortega F, Hernandez A, Gonzalez-Avila G, Bermejo L, Iwata K, Strauss JF 3rd. Increased matrix metalloproteinase activity and reduced tissue inhibitor of metalloproteinases-1 levels in amniotic fluids from pregnancies complicated by premature rupture of membranes. Am J Obstet Gynecol. 1996;174(4):1371–1376. | |
Romero R, Friel LA, Velez Edwards DR, et al. A genetic association study of maternal and fetal candidate genes that predispose to preterm prelabor rupture of membranes (PROM). Am J Obstet Gynecol. 2010; 203(4):361. e361–e361, e330. | |
Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70(1):11–20. | |
Gigante B, Strawbridge RJ, Velasquez IM, et al. Analysis of the role of interleukin 6 receptor haplotypes in the regulation of circulating levels of inflammatory biomarkers and risk of coronary heart disease. PloS One. 2015;10(3):e0119980. | |
Dolan SM, Hollegaard MV, Merialdi M, et al. Synopsis of preterm birth genetic association studies: the preterm birth genetics knowledge base (PTBGene). Public Health Genomics. 2010;13(7–8):514–523. | |
McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9(5):356–369. | |
Zhang H, Baldwin DA, Bukowski RK, et al. A genome-wide association study of early spontaneous preterm delivery. Genet Epidemiol. 2015; 39(3):217–226. | |
Azizia M, Lloyd J, Allen M, Klein N, Peebles D. Immune status in very preterm neonates. Pediatrics. 2012;129(4):e967–e974. | |
Luhtala N, Parker R. T2 family ribonucleases: ancient enzymes with diverse roles. Trends Biochem Sci. 2010;35(5):253–259. | |
Uzun A, Dewan AT, Istrail S, Padbury JF. Pathway-based genetic analysis of preterm birth. Genomics. 2013;101(3):163–170. | |
Wu Easc W, Manuck TA, Esplin MS, Varner MW, Jorde LB. A Genome-Wide Association Study of spontaneous preterm birth in a European population. F1000Research. 2013;2:255. | |
Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–2229. | |
Manuck TA, Lai Y, Meis PJ, et al. Admixture mapping to identify spontaneous preterm birth susceptibility loci in African Americans. Obstet Gynecol. 2011;117(5):1078–1084. | |
Dawn Teare M, Barrett JH. Genetic linkage studies. Lancet. 2005; 366(9490):1036–1044. | |
Karjalainen MK, Huusko JM, Ulvila J, et al. A potential novel spontaneous preterm birth gene, AR, identified by linkage and association analysis of X chromosomal markers. PloS One. 2012;7(12):e51378. | |
Menon R, Conneely KN, Smith AK. DNA methylation: an epigenetic risk factor in preterm birth. Reprod Sci. 19(1):6–13. | |
Yang HH, Hu N, Wang C, et al. Influence of genetic background and tissue types on global DNA methylation patterns. PLoS One. 2010;5(2):e9355. | |
Kaminsky ZA, Tang T, Wang SC, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41(2):240–245. | |
Drong AW, Nicholson G, Hedman AK, et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PloS One. 2013;8(2):e55923. | |
Smith AK, Kilaru V, Kocak M, et al. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics. 2014;15:145. | |
Almli LM, Stevens JS, Smith AK, et al. A genome-wide identified risk variant for PTSD is a methylation quantitative trait locus and confers decreased cortical activation to fearful faces. Am J Med Genet B Neuropsychiatr Genet. 2015;168(5):327–336. | |
Dayeh TA, Olsson AH, Volkov P, Almgren P, Ronn T, Ling C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia. 2013;56(5):1036–1046. | |
Parets SE, Conneely KN, Kilaru V, et al. Fetal DNA methylation associates with early spontaneous preterm birth and gestational age. PloS One. 2013;8(6):e67489. | |
Behnia F, Parets SE, Kechichian T, et al. Fetal DNA methylation of autism spectrum disorders candidate genes: association with spontaneous preterm birth. Am J Obstet Gynecol. 2015;212(4):533. e531–e539. | |
Vidal AC, Benjamin Neelon SE, Liu Y, et al. Maternal stress, preterm birth, and DNA methylation at imprint regulatory sequences in humans. Genet Epigenet. 2014;6:37–44. | |
Nemoda Z, Massart R, Suderman M, et al. Maternal depression is associated with DNA methylation changes in cord blood T lymphocytes and adult hippocampi. Transl Psychiatry. 2015;5:e545. | |
Yao Y, Robinson AM, Zucchi FC, et al. Ancestral exposure to stress epigenetically programs preterm birth risk and adverse maternal and newborn outcomes. BMC Med. 2014;12:121. | |
Parets SE, Conneely KN, Kilaru V, Menon R, Smith AK. DNA methylation provides insight into intergenerational risk for preterm birth in African Americans. Epigenetics. 2015:10(9):784–792. | |
York TP, Eaves LJ, Neale MC, Strauss JF 3rd. The contribution of genetic and environmental factors to the duration of pregnancy. Am J Obstet Gynecol. 2014;210(5):398–405. | |
Kessous R, Shoham-Vardi I, Pariente G, Holcberg G, Sheiner E. An association between preterm delivery and long-term maternal cardiovascular morbidity. Am J Obstet Gynecol. 2013;209(4):368. e361–e368. | |
Catov JM, Newman AB, Roberts JM, et al. Preterm delivery and later maternal cardiovascular disease risk. Epidemiology. 2007;18(6):733–739. | |
Hastie CE, Smith GC, Mackay DF, Pell JP. Maternal risk of ischaemic heart disease following elective and spontaneous pre-term delivery: retrospective cohort study of 750 350 singleton pregnancies. Int J Epidemiol. 2011;40(4):914–919. | |
Smith GC, Pell JP, Walsh D. Pregnancy complications and maternal risk of ischaemic heart disease: a retrospective cohort study of 129,290 births. Lancet. 2001;357(9273):2002–2006. | |
James-Todd T, Wise L, Boggs D, Rich-Edwards J, Rosenberg L, Palmer J. Preterm birth and subsequent risk of type 2 diabetes in black women. Epidemiology. 2014;25(6):805–810. | |
Lykke JA, Paidas MJ, Damm P, Triche EW, Kuczynski E, Langhoff-Roos J. Preterm delivery and risk of subsequent cardiovascular morbidity and type-II diabetes in the mother. BJOG. 2010;117(3):274–281. | |
Catov JM, Dodge R, Yamal JM, Roberts JM, Piller LB, Ness RB. Prior preterm or small-for-gestational-age birth related to maternal metabolic syndrome. Obstet Gynecol. 2011;117(2 Pt 1):225–232. | |
Robbins CL, Hutchings Y, Dietz PM, Kuklina EV, Callaghan WM. History of preterm birth and subsequent cardiovascular disease: a systematic review. Am J Obstet Gynecol. 2014;210(4):285–297. | |
Melbye M, Wohlfahrt J, Andersen AM, Westergaard T, Andersen PK. Preterm delivery and risk of breast cancer. Br J Cancer. 1999;80(3–4):609–613. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.