")
Back to Journals » International Journal of Nanomedicine » Volume 13
Inhibitory effect of Au@Pt-NSs on proliferation, migration, and invasion of EJ bladder carcinoma cells: involvement of cell cycle regulators, signaling pathways, and transcription factor-mediated MMP-9 expression
Authors Shin SS, Noh DH, Hwang B, Lee JW, Park SL, Park SS , Moon B, Kim WJ , Moon SK
Received 30 November 2017
Accepted for publication 3 March 2018
Published 1 June 2018 Volume 2018:13 Pages 3295—3310
DOI https://doi.org/10.2147/IJN.S158463
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Seung-Shick Shin,1,* Dae-Hwa Noh,2,* Byungdoo Hwang,2 Jo-Won Lee,2 Sung Lyea Park,2 Sung-Soo Park,1 Bokyung Moon,2 Wun-Jae Kim,3 Sung-Kwon Moon2
1Department of Food Science and Nutrition, Jeju National University, Jeju, South Korea; 2Department of Food and Nutrition, Chung-Ang University, Anseong, South Korea; 3Department of Urology, Chungbuk National University, Cheongju, Chungbuk, South Korea
*These authors contributed equally to this work
Background: Although the diverse biological properties of nanoparticles have been studied intensively, research into their mechanism of action is relatively rare. In this study, we investigated the molecular mechanisms of the anticancer activity of heterometallic Au@Pt-nanoseeds (NSs) against bladder cancers.
Materials and methods: Mode of action of Au@Pt-NSs was investigated through MTT assay, flow cytometry analysis, Western immunoblots, real-time qPCR, wound-healing migration and invasion assays, zymography, and electrophoretic mobility shift assay (EMSA).
Results: Treatment with Au@Pt-NSs significantly inhibited the proliferation of EJ cells in a dose-dependent manner by inducing G1 phase cell cycle arrest. Among the regulators associated with the G1 cell cycle phase, CDK2, CDK4, cyclin D1, cyclin E, and p21WAF1 were shown to participate in the inhibitory pathways of Au@Pt-NSs. In addition, treatment with Au@Pt-NSs led to upregulation of phospho-p38 MAPK and downregulation of phospho-AKT in EJ cells. Interestingly, Au@Pt-NSs inhibited the migratory and invasive potential of the cells, which was attributed to the suppression of the enzymatic activity of matrix metalloproteinase-9 (MMP-9). Using MMP-9-specific oligonucleotides, we showed that transcription factors such as NF-κB and Sp-1 were responsible for the MMP-9-mediated metastatic potential of EJ cells.
Conclusion: Au@Pt-NSs significantly limited the progression, migration, and invasion of bladder cancer EJ cells. Our data represent a novel insight into developing cisplatin-like chemotherapeutic reagents with fewer side effects and provide useful information on molecular markers to monitor patients under Au@Pt-NSs-based chemotherapy.
Keywords: nanoseeds, nanomedicine, bladder cancer, molecular mechanism
Introduction
Urinary bladder cancer is one of the most common cancers in the US with an estimated 79,030 new cases in 2017.1 Approximately 90% of bladder cancers are transitional cell carcinomas (TCCs), comprising superficial and muscle-invasive lesions. Generally, superficial lesions are resectable and show favorable prognosis, while advanced-stage muscle-invasive tumors often have fatal consequences for the patients.2,3 Thus, identifying new therapeutic options is important to ensure the long-term survival of patients with bladder cancer.
Bladder cancer cells react to chemotherapeutic reagents through the activation or inhibition of key signaling pathways, including MAPKs and PI-3K/AKT.4–7 These molecular cues are transmitted to the nucleus by several steps of a signaling cascade, which redistributes the association of regulators of the cell cycle, including cyclin-dependent kinases (CDKs) and their binding partners, at certain cell cycle phases: G1, S, and G2/M.8–10 Transition from G1 phase to S phase is determined by interactions among key regulators, including cyclin D/E and CDK4/6, CDK inhibitors (p21WAF1 and p27KIP1), and retinoblastoma protein (pRB).11,12 When DNA is damaged, the kinase activity of individual cyclin/CDK complexes is negatively regulated by CDK inhibitors, which restrains cell cycle progression.13,14 Proteinases from cancerous cells are equally promising targets of chemotherapeutic reagents since it has been reported that the expression of matrix metalloproteinase-9 (MMP-9, also known as gelatinase B, a 92-kDa gelatinase) was closely related to the migration and invasion of bladder cancer cells via the activation of transcription factors, including AP-1, Sp-1, and NF-κB.15,16 Therefore, understanding the molecular mechanism of the inhibitory activity of chemotherapeutics and identifying effectors that participate in the process might increase our chances of preventing tumor proliferation and metastasis, consequently reducing the mortality of this fatal disease.
Along with the advancement in nanotechnologies, metal-based nanoparticles (NPs) comprising Au and Pt have attracted interest as a novel tool to detect, monitor, or even manage the progression of tumors. Although metal Au or Pt has little toxicity because of their inertness, NPs comprising them show very unique physicochemical properties, including catalytic activity,17–19 radical scavenging ability,20–22 and cytotoxicity.23,24 Therefore, NPs are considered as a new class of therapeutics to manage cancers. Recently, Au-NPs conjugated to anti-EGFR antibodies were used to treat early-stage bladder cancers.25 When integrating into EGFR-expressing bladder cancer cells, Au-NPs were heated using plasmon resonance with infrared light, which permitted highly targeted therapy.25 Another research group studied the efficacy of epigallocatechin gallate (EGCG) conjugated to Au-NPs in vivo, using C3H/HeN mice xenografted with MBT-2 murine bladder cancer cells.26 Interestingly, EGCG-Au-NPs significantly induced the apoptotic cell death of tumors compared with mice treated with free EGCG.26
Pt-based therapeutic reagents also have been used for decades. The most representative examples of Pt chemotherapeutics are the platins, such as cisplatin, carboplatin, oxaliplatin, and nedaplatin. In a recent preclinical study, Pt-NPs were reported to exhibit cytotoxicity by entering cell membranes and eventually disturbing the DNA integrity of cancer cells.27 Pelka et al28 demonstrated that the DNA-damaging toxicity of Pt-NP to human colon carcinoma HT29 cells was correlated with the particle size of platinum in an inverse manner, but was unrelated to the formation of reactive oxygen species. In a clinical setting, Pt-NPs have been used as a radiation-sensitizing reagent to enhance the efficacy of radiation therapy.29,30
Although individual monometallic NP has been reported as a promising therapeutic option for various cancers, still the discrepancy between preclinical results and clinical outcomes exists. Given that human tumors are intrinsically heterogeneous, heterometallic NPs may be more effective for the disease due to synergistic effects. Shmarakov et al31 have reported that the anticancer effect of bimetallic Ag@Au NP was dependent upon the size, shape, charge, composition, and molar ratio of metals distributed in the NPs and suggested that heterometallic Ag@Au NPs could be more effective than individual one. Wenzel et al32 also demonstrated that bimetallic titanocene–Au NPs displayed stronger cytotoxicity to human cancer cells than the corresponding monometallic NPs.
Although NPs have been studied intensively for many decades, research into the molecular mechanism of action of heterometallic Au@Pt-nanoseeds (NSs) is very rare. In this study, we observed that heterometallic Au@Pt-NSs inhibited the proliferation, migration, and invasion of bladder cancer cells significantly. We also demonstrated the molecular mechanisms of the inhibitory action in bladder cancer EJ cells. We believe that the use of NSs may potentiate antitumor efficacy against urothelial bladder cancers without negative side effects.
Materials and methods
Materials
Au@Pt-NSs were purchased from NanoSeedz™ (Hong Kong, China). The Pt:Au ratio was 1:4.19 Polyclonal antibodies against extracellular signal-regulated kinase (ERK), phospho-ERK, p38 MAPK, phospho-p38 MAPK, JNK, phospho-JNK, AKT, and phospho-AKT were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). Polyclonal antibodies against cyclin D1, cyclin E, CDK2, CDK4, p53, p21WAF1, p27KIP1, and GAPDH were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The nuclear extract kit and the electrophoretic mobility shift assay (EMSA) gel shift kit were obtained from Panomics (Fremont, CA, USA).
Cell culture
The EJ human bladder carcinoma cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a 5% CO2 humidified incubator. Normal human urothelial cells (HUCs) were obtained from ScienCell Research Laboratories (Carlsbad, CA, USA) and grown in the medium specific for HUCs with supplements according to the manufacturer’s protocol.
Au@Pt-NP (NSs) treatment and cell counting
EJ cells were plated in six-well plates and treated with NSs (0, 0.1, 0.3, and 0.5 μM) for 24 h. The cells were detached from the plates by treatment with 0.25% trypsin containing 0.2% EDTA (Corning Incorporated, Corning, NY, USA). Detached cells were mixed with 50 μL of 0.4% trypan blue (Sigma-Aldrich Co., St Louis, MO, USA) by gentle pipetting. Then, 20 μL of the mixture was loaded into each chamber of a hemocytometer and counted.
Cell viability assay
Cell viability was determined using a modification of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, cells were plated in 96-well plates (6 × 103 cells/well) followed by incubation with NSs (0, 0.1, 0.3, and 0.5 μM) for 24 h. After incubation, the medium was aspirated off, and fresh medium containing 10 μL of 5 mg/mL MTT was added. After 1 h, the medium was removed and replaced with 100 μL of dimethyl sulfoxide (DMSO). Absorbance at 540 nm was measured using a fluorescent plate reader. The cell morphology was photographed under a phase-contrast microscope.
Cell cycle analysis
Cells were harvested and fixed in 70% ethanol. After washing once with ice-cold phosphate-buffered saline (PBS), cells were incubated with RNase (1 mg/mL) followed by propidium iodide (50 mg/mL). The phase distribution of the cell cycle was measured using a flow cytometer (FACStar; BD Biosciences, San Jose, CA, USA) equipped with the BD Cell Fit software.
Immunoblots and immunoprecipitation
Cells were washed twice with cold PBS and freeze-thawed in 200 μL of lysis buffer (containing, in mmol/L, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) [pH 7.5] 50, NaCl 150, EDTA 1, dithiothreitol (DTT) 1, ethylene glycol tetraacetic acid (EGTA) 2.5, β-glycerophosphate 10, Na3VO4 0.1, NaF 1, phenylmethylsulfonyl fluoride (PMSF) 0.1, 10% glycerol, 0.1% Tween 20, 10 μg/mL of leupeptin, and 2 μg/mL of aprotinin). After the cells were scraped into 1.5 mL tubes, the lysates were incubated on ice for 10 min. The cells were then centrifuged at 10,000× g for 10 min at 4°C. The amount of protein was determined using a bicinchoninic acid (BCA) protein assay reagent kit (Thermo Fisher Scientific, Waltham, MA, USA). Protein (25 μg each) was loaded onto a 0.1% sodium dodecyl sulfate (SDS), 10% polyacrylamide gel, and separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) under denaturing conditions. The proteins were transferred onto nitrocellulose membranes (Hybond; GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). After blocking in 5% skim milk, the membranes were incubated with primary antibodies for 12 h followed by incubation with peroxidase-conjugated secondary antibodies for 90 min. The immunocomplexes were then detected using a chemiluminescence reagent kit (GE Healthcare Bio-Sciences Corp.). For immunoprecipitation analysis, equal amounts of cell lysates were incubated with the indicated antibodies at 4°C overnight. Protein A-Sepharose beads (Santa Cruz Biotechnology Inc.) were then added to the immunocomplexes followed by incubation at 4°C for 2 h. The immunoprecipitated complexes were washed with 1× lysis buffer three times, resuspended in SDS-PAGE sample buffer containing β-mercaptoethanol (Bio-Rad Laboratories Inc., Hercules, CA, USA), and separated by electrophoresis. Experiments were repeated at least three times.
Wound-healing migration assay
Exponentially grown cells (3 × 105/well) were plated in six-well plates. Cells were pretreated with mitomycin C (5 μg/mL, Sigma-Aldrich Co #M4287) for 2 h to inhibit cell proliferation. The cell surface area was then scratched with a 2-mm-wide pipette tip. After washing with PBS three times, the plate was incubated with culture media in the presence or absence of NSs (0, 0.1, 0.3, and 0.5 μM) for 24 h. The recovery capacity of the cells migrating into the scratched area was measured and compared with that of the control. Cellular images were photographed under an inverted microscope at 40× magnification.
Boyden chamber invasion assay
Invasiveness was assessed using an invasion assay kit (Cell Biolabs, San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, 2.5 × 104 cells were resuspended in serum-free culture medium and incubated with mitomycin C (5 μg/mL) for 2 h before being seeded in the upper chamber. Medium containing 10% FBS was added to the lower chamber as a chemoattractant. After 24 h, cells in the lower chamber were stained and photographed.
Zymography
Conditioned medium was collected and electrophoresed through a polyacrylamide gel containing 0.25% gelatin. The gel was washed twice for 15 min at room temperature with 2.5% Triton X-100. Subsequently, the gel was incubated at 37°C overnight in a buffer containing 150 mM NaCl, 50 mM Tris–HCl, and 10 mM CaCl2, pH 7.5. The gel was stained with 0.2% Coomassie blue and photographed on a light box. Proteolysis was detected as a white zone in a blue field.
Nuclear extracts and EMSA
EMSA was performed with the NSs (0, 0.3, and 0.5 μM) for 24 h. Nuclear extracts were prepared using a Nuclear Extraction Kit (Panomics). Briefly, cells were harvested by centrifugation, washed, and resuspended in a buffer containing 10 mM HEPES (pH 7.9), 10 mM KCl, 1 mM DTT, 0.5 mM PMSF, 0.1 mM EDTA, and 0.1 mM EGTA. After incubation on ice for 15 min, the cells were mixed vigorously with 0.5% NP-40. The nuclear pellet was collected by centrifugation followed by extraction in a buffer containing 20 mM HEPES (pH 7.9), 400 mM NaCl, 1 mM DTT, 1 mM PMSF, 1 mM EDTA, and 1 mM EGTA at 4°C for 15 min. The nuclear extract (10–20 μg) was preincubated at 4°C for 30 min with a 100-fold excess of an unlabeled oligonucleotide spanning the -79 position of the MMP-9 cis-acting element of interest. The sequences were as follows: AP-1, CTGACCCCTGAGTCAGCACTT; NF-κB, CAGTGGAATTCCCCAGCC; and Sp-1, GCCCATTCCTTCCGCCCCCAGATGAAGCAG. The reaction mixture was then incubated at 4°C for 20 min in a buffer (25 mM HEPES buffer [pH 7.9], 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, and 2.5% glycerol) with 2 μg of poly dI/dC and 5 fmol (2 × 104 cpm) of a Klenow end-labeled (32P adenosine triphosphate [ATP]) 30-mer oligonucleotide, which spanned the DNA-binding site of the MMP-9 promoter. The reaction mixture was separated by electrophoresis at 4°C using a 6% polyacrylamide gel. The gel was exposed to X-ray film overnight. The gray values of the blots were measured using the ImagePro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
Statistical analyses
Where appropriate, data are presented as mean ± SD. Data were evaluated by factorial analysis of variance (ANOVA) and a Fisher’s least significant difference test, where appropriate. Statistical significance was considered at P < 0.05.
Results
Au@Pt-NSs inhibit the proliferation of bladder cancer EJ cells via G1 phase cell cycle arrest
To investigate the possibility of Au@Pt-NSs as a therapeutic reagent for bladder cancer, we first examined whether the Au@Pt-NSs possess anti-proliferative activity. We treated both cancer EJ and normal HUCs with the Au@Pt-NSs at concentrations of 0, 0.1, 0.3, and 0.5 μM for 24 h. Cellular viability was then measured using the MTT assay. Figure 1A shows that Au@Pt-NSs inhibited the proliferation of EJ cells in a dose-dependent manner. The viable cell counting method, using trypan blue, gave a similar result to that of the MTT assay (Figure 1B). The morphological change to round shape and the accumulation of floating cells clearly indicated that EJ cells underwent cellular stress due to Au@Pt-NSs (Figure 1C). However, the cytotoxicity of Au@Pt-NSs to normal HUCs was very low, as assessed by both the MTT and viable cell counting methods, compared with EJ cancer cells (Figure 1D and E). The morphology of HUC cells was unchanged by Au@Pt-NS treatment (Figure 1F). After verifying the anti-proliferative effect, we examined the distribution of the cell cycle to determine which cell cycle phase is involved in the inhibitory effect of Au@Pt-NSs. Cells were incubated with 0, 0.1, 0.3, and 0.5 μM of the Au@Pt-NSs for 24 h followed by flow cytometry analysis. Fluorescence-activated cell sorting (FACS) histograms showed that the EJ cells were accumulated dose dependently in the G1 phase and, accordingly, the number of cells in the S and G2/M phases was reduced in the presence of Au@Pt-NSs (Figure 2A–E). However, the cell cycle distribution of the HUCs was almost unchanged (Figure 2F–J). These results suggested that Au@Pt-NSs cause growth inhibition of EJ cells through G1 phase cell cycle arrest.
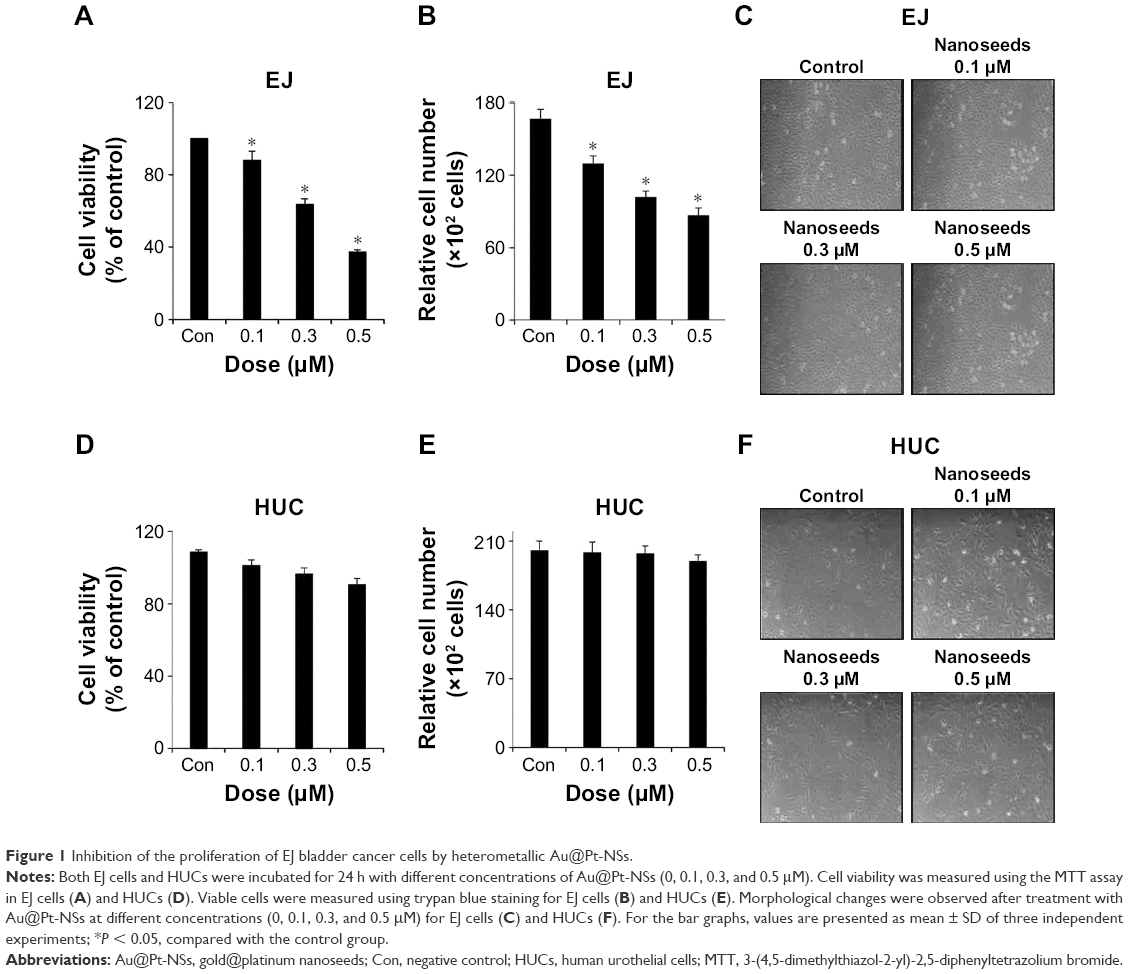 | Figure 1 Inhibition of the proliferation of EJ bladder cancer cells by heterometallic Au@Pt-NSs. |
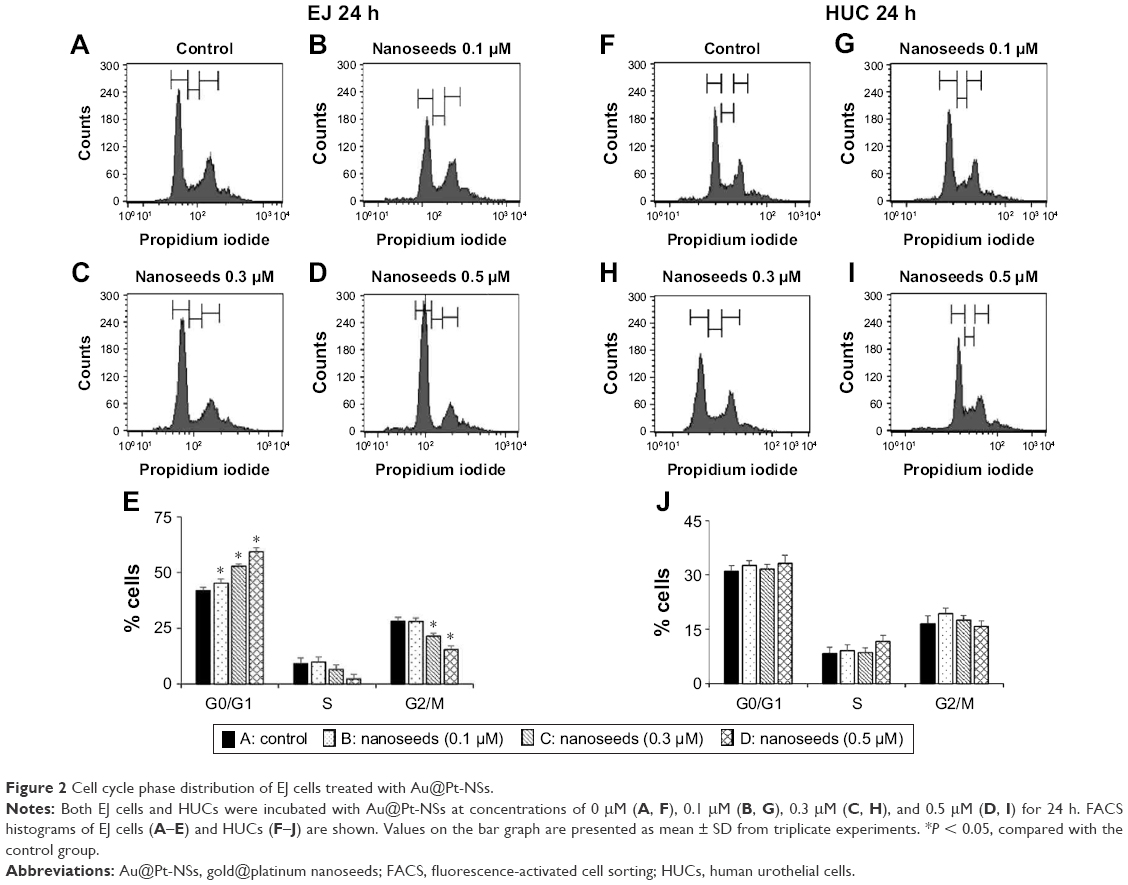 | Figure 2 Cell cycle phase distribution of EJ cells treated with Au@Pt-NSs. |
Cyclin/CDK complexes and p21WAF1 are involved in Au@Pt-NSs-mediated G1 phase cell cycle arrest
Division of mammalian cells is tightly controlled by the formation of complexes between cyclins and CDKs. When the progression of the cell cycle is limited, the kinase activities of CDKs are inhibited by cyclin-dependent kinase inhibitors (CKIs), including p21WAF1, p27KIP1, and p53. To understand the molecular mechanism of the Au@Pt-NSs, we incubated EJ cancer cells and normal HUCs with Au@Pt-NSs (0, 0.1, 0.3, and 0.5 μM) for 12 and 24 h and examined the levels of effectors that regulate the G1 to S phase transition. Among regulators of the G1 phase of EJ cells, the levels of CDK2, CDK4, cyclin D1, and cyclin E were downregulated significantly by treatment with Au@Pt-NSs. The levels of these proteins were measured as fold changes compared with the control (Figure 3A and C). As an example, at 0.5 μM of Au@Pt-NS treatment for 12 h, the relative protein levels of CDK2, CDK4, cyclin E, and cyclin D1 were 0.72, 0.60, 0.62, and 0.61, respectively. After 24 h, these levels had changed to 0.60, 0.33, 0.55, and 0.65, respectively. Among cell cycle inhibitors, the level of p21WAF1 was increased markedly, by approximately threefold, compared with the control after treatment with Au@Pt-NSs for 12 h. However, the levels of p27KIP1 and p53 were unaffected by the treatment (Figure 3B and D). To ensure the results of Western immunoblots, we performed real-time quantitative polymerase chain reaction (qPCR) for targets tested (Figures S1 and S2; Table S1). In agreement with the protein levels, mRNA expressions were equally modulated by the treatment with Au@Pt-NSs. p21WAF1 is one of the key regulators that control the activity of CDKs; therefore, we performed immunoprecipitation using anti-CDK2 and CDK4 antibodies. After 24 h of incubation of EJ cells with Au@Pt-NSs at 0.5 μM, cell lysates were prepared and pulled down with either anti-CDK2 or anti-CDK4 antibodies followed by immunobinding with an anti-p21WAF1 antibody. As shown in Figure 3E, the amount of p21WAF1 bound to CDK2 and CDK4 was increased significantly by treatment with Au@Pt-NSs (Figure 3E). However, in HUCs treated with Au@Pt-NSs (0, 0.1, 0.3, and 0.5 μM) for either 12 or 24 h, the protein levels of CDK2, CDK4, cyclin D1, cyclin E, p53, and CKIs, including p21WAF1 and p27KIP1, remained almost unchanged (Figure 4A–D). These data unequivocally suggested that the inhibition of EJ cell proliferation from Au@Pt-NSs was attributed to the upregulation of p21WAF1, which disturbed the formation of cyclin/CDK complexes.
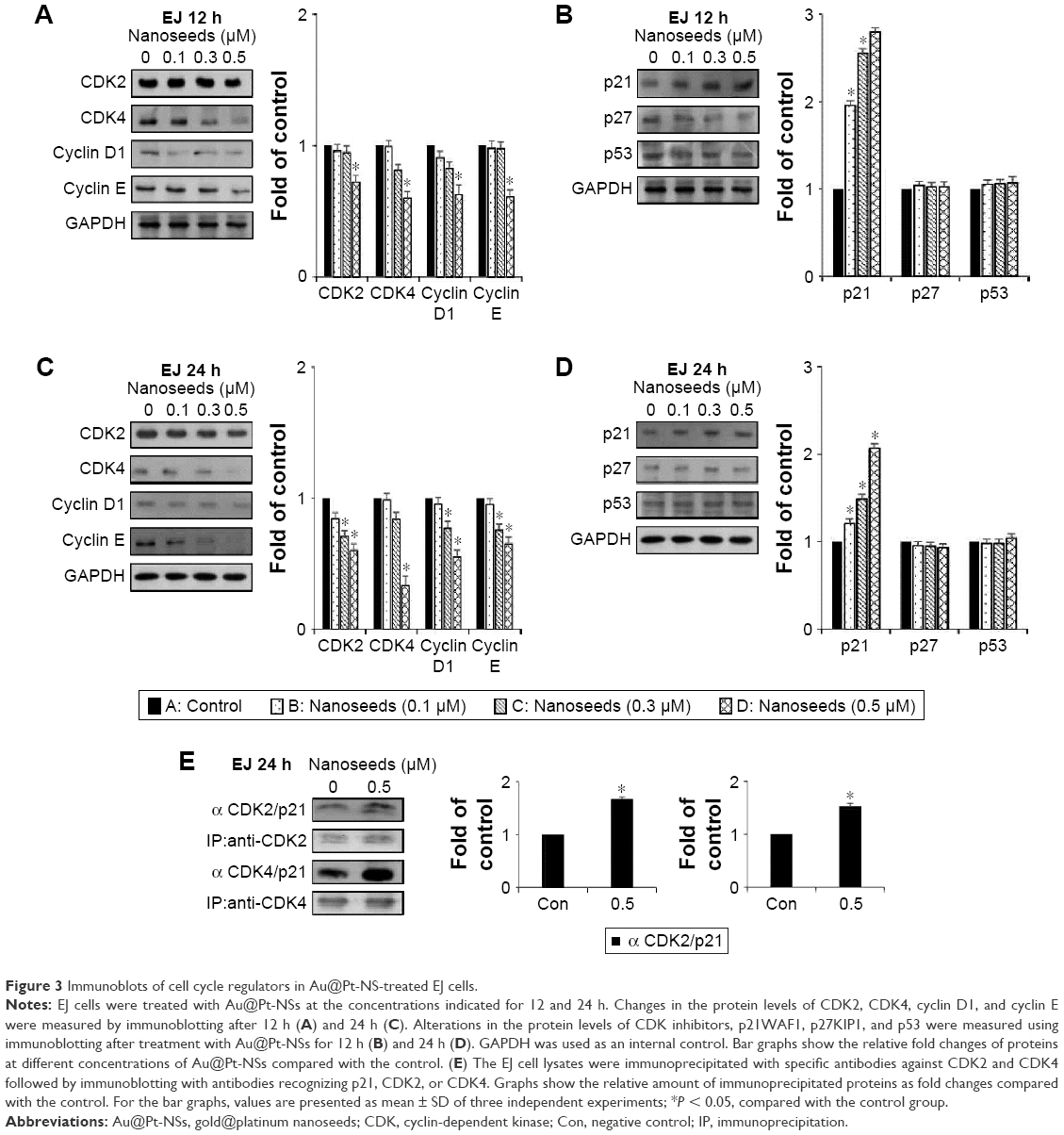 | Figure 3 Immunoblots of cell cycle regulators in Au@Pt-NS-treated EJ cells. |
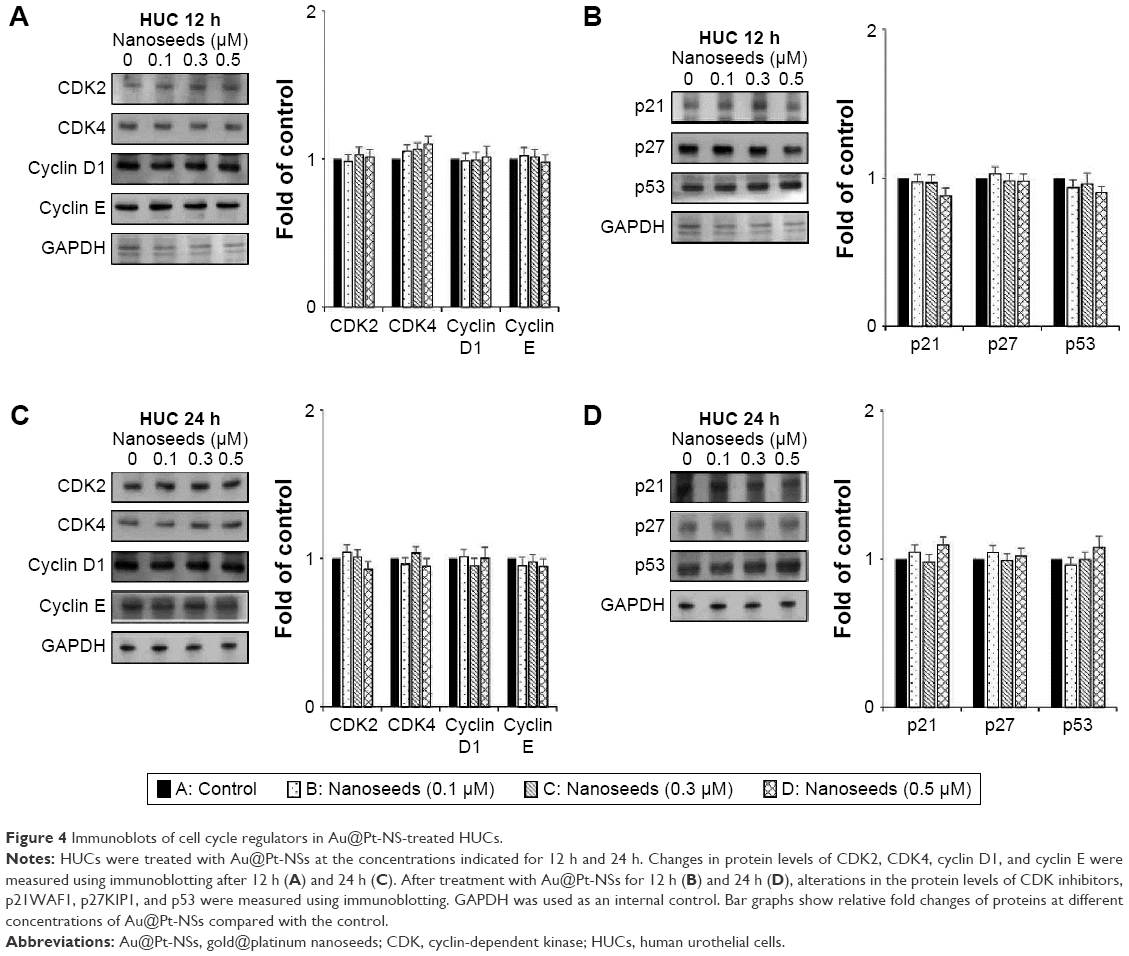 | Figure 4 Immunoblots of cell cycle regulators in Au@Pt-NS-treated HUCs. |
Au@Pt-NSs induce the phosphorylation of p38 and inhibit the phosphorylation of AKT
The MAPKs (ERK1/2, JNK1/2, and p38) and AKT signaling pathways have been reported as key regulators in the progression of bladder cancer.4,5 Thus, through signal induction experiments, we investigated whether Au@Pt-NSs influenced the phosphorylation of ERK1/2, JNK1/2, p38, or AKT. Both EJ and HUCs were treated with Au@Pt-NSs at different concentrations (0, 0.1, 0.3, and 0.5 μM) for different periods (1, 6, 12, and 24 h) followed by immunoblotting. As shown in Figure 5A–D, phosphorylation of p38 was increased significantly by treatment with Au@Pt-NSs in a dose-dependent manner. However, no significant changes in the phosphorylated form of either ERK1/2 or JNK1/2 were observed. Notably, phosphorylation of AKT was decreased significantly by treatment with Au@Pt-NSs. In particular, after 24 h of treatment with 0.5 μM Au@Pt-NSs, AKT phosphorylation in the EJ cells was almost completely suppressed. In comparison, normal HUCs did not show any significant changes in the phosphorylation levels of the signaling effectors (Figure 6A–D). These data suggested clearly that Au@Pt-NSs target the p38 and AKT signaling pathways in EJ bladder cancer cells.
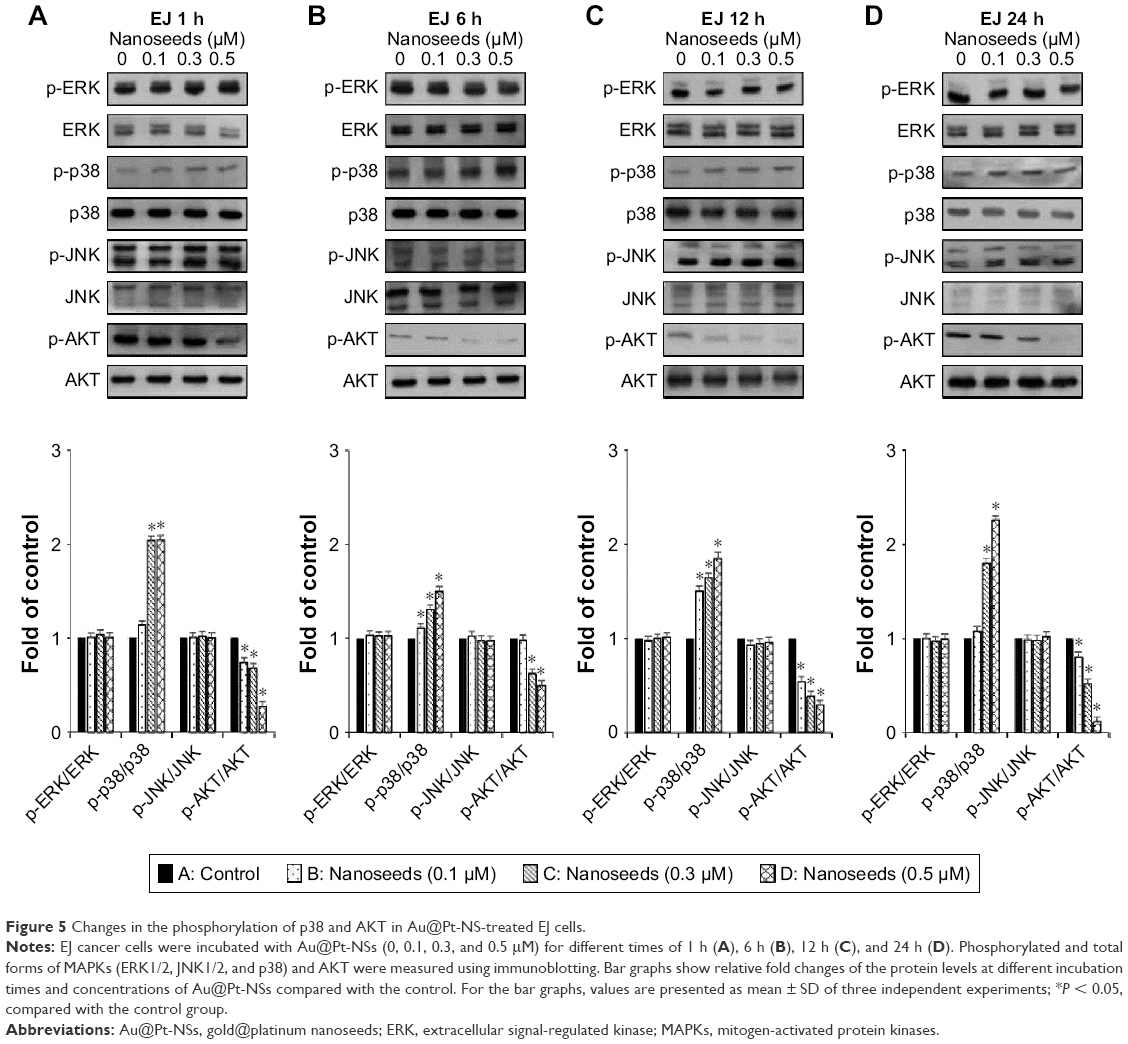 | Figure 5 Changes in the phosphorylation of p38 and AKT in Au@Pt-NS-treated EJ cells. |
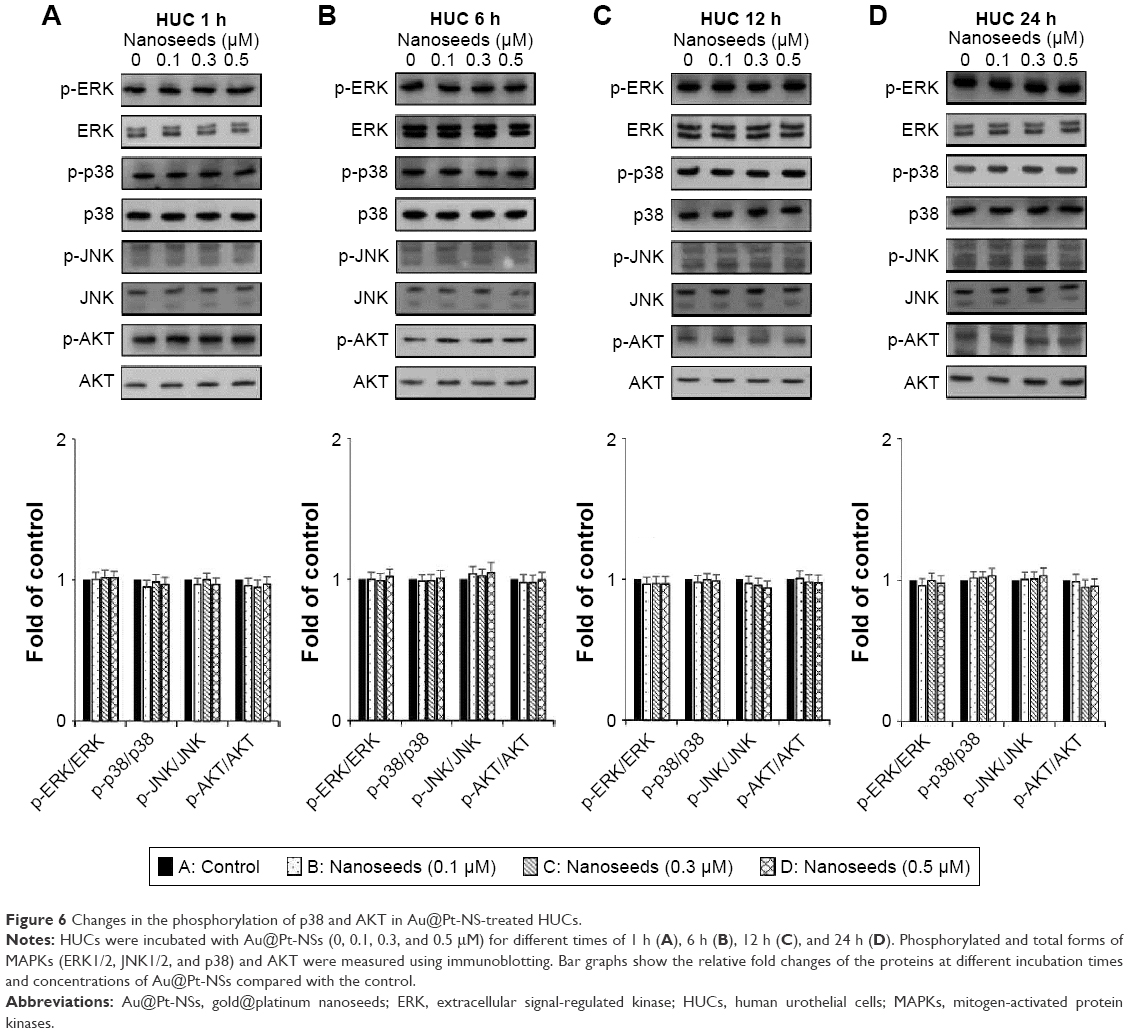 | Figure 6 Changes in the phosphorylation of p38 and AKT in Au@Pt-NS-treated HUCs. |
Au@Pt-NSs reduce the migration and invasion of EJ bladder cancer cells
When transformed, urothelial bladder cells frequently acquire migratory and invasive phenotypes, which might cause fatal consequences to patients. Thus, we investigated whether Au@Pt-NSs influenced the migration and invasion of EJ bladder cancer cells. To distinguish cellular migration or invasion from proliferation, cells were preincubated with mitomycin C for 2 h. These cells were then scratched with a pipette tip and incubated with medium in the presence or absence of the Au@Pt-NSs for 24 h. As shown in Figure 7, the migratory potential of the EJ cells was inhibited significantly and dose dependently. The migration of EJ cells in 0.5 μM Au@Pt-NSs was reduced by ~50% compared with that of the control (Figure 7A). However, migratory potential of normal HUCs was unaffected even at a concentration of 0.5 μM (Figure 7B). Subsequently, we examined the potential anti-invasive capacity of 0.5 μM Au@Pt-NSs using Boyden chamber assays. As shown in Figure 7C, the invasiveness of EJ cancer cells was diminished significantly by treatment with Au@Pt-NSs in a dose-dependent manner. Compared with the control, treatment with 0.5 μM Au@Pt-NSs inhibited the invasion of ~70% of the cells through the transwell membrane (Figure 7C). However, the invasiveness of HUCs was unchanged by treatment with Au@Pt-NSs (Figure 7D). Overall, these results demonstrated clearly that Au@Pt-NSs inhibit the metastatic potential of bladder EJ cancer cells effectively.
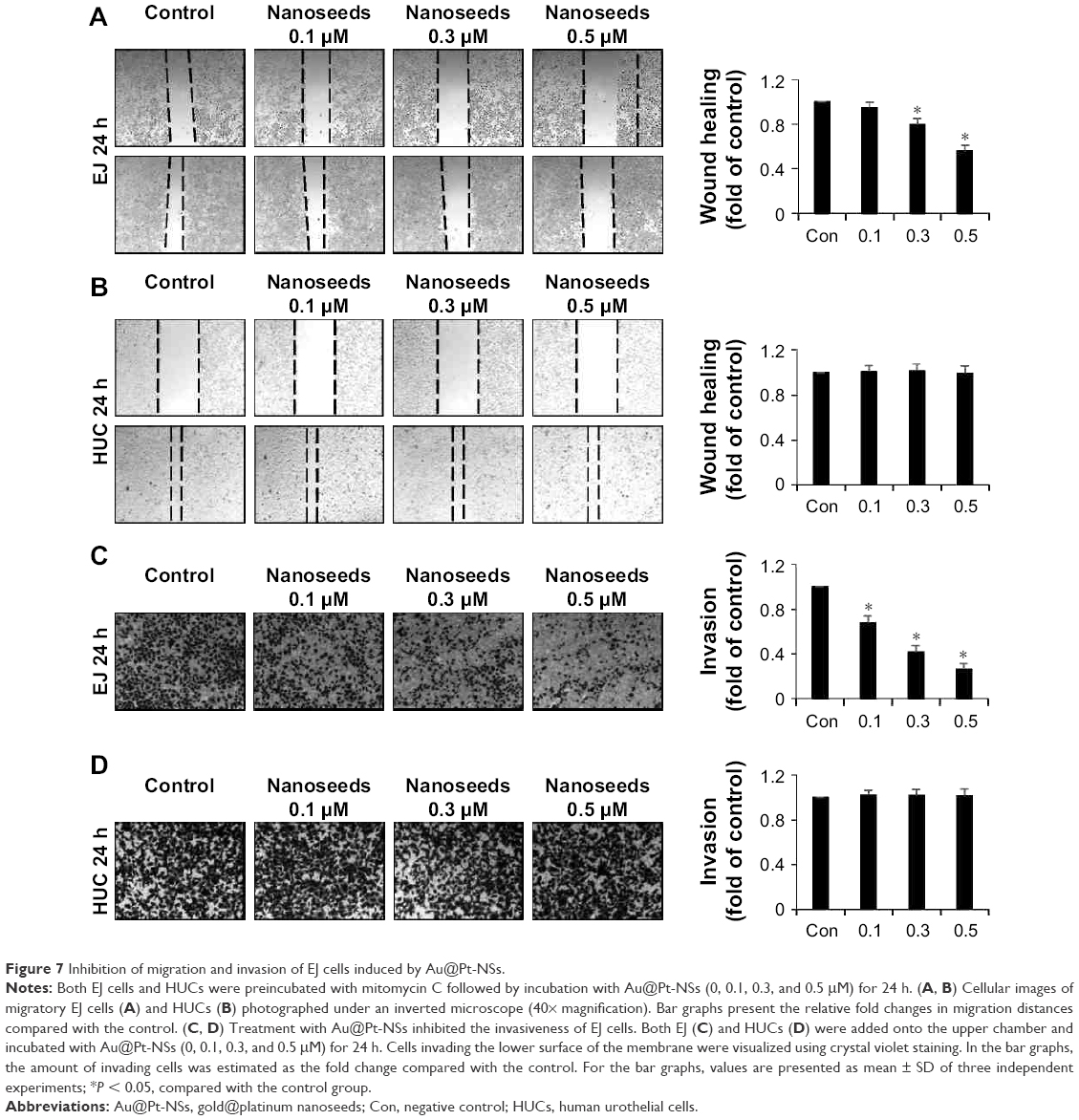 | Figure 7 Inhibition of migration and invasion of EJ cells induced by Au@Pt-NSs. |
Au@Pt-NSs inhibit MMP-9 activity by suppressing the binding capacities of transcription factors such as NF-κB and Sp-1 in EJ cells
One of the intrinsic characteristics of transformed cells, particularly in bladder cancer, is the expression of proteolytic enzymes, including matrix metalloproteinases (MMPs), which are used tactically to invade adjacent tissue or the extracellular matrix.15,33 Thus, we investigated whether Au@Pt-NSs could inhibit the activities of MMP-2 and MMP-9 using gelatin zymography. As shown in Figure 8, compared with the control, the Au@Pt-NSs suppressed the activities of MMP-2 and MMP-9 in EJ cells significantly. The enzymatic activities of both MMP-2 and MMP-9 were reduced by >50% after treatment with 0.5 μM Au@Pt-NSs (Figure 8A). However, these anti-MMP activities were not observed in normal HUCs at the same concentration of Au@Pt-NSs (Figure 8B). To understand the mechanism of the Au@Pt-NSs-mediated anti-MMP-9 activity, we performed EMSAs using oligonucleotides from transcription factors such as NF-κB, AP-1, and Sp-1, spanning the sequence of the MMP-9 cis-element. Both cancer EJ cells and normal HUCs were treated with 0.3 and 0.5 μM of the Au@Pt-NSs for 24 h followed by EMSA. Interestingly, the binding activities of both NF-κB and Sp-1 to MMP-9 in the EJ cells were decreased by up to 24% and 67%, respectively, compared with each control (Figure 8C). However, the binding activity of AP-1 was unchanged in the presence of Au@Pt-NSs (Figure 8C). The binding activities of the transcription factors in HUCs were unaffected by treatment with Au@Pt-NSs (Figure 8D). Overall, these results suggested that Au@Pt-NSs inhibit the migration and invasion of EJ cells effectively by suppressing MMP-9 activity, which was demonstrated by the reduced binding activities of NF-κB and Sp-1 motifs.
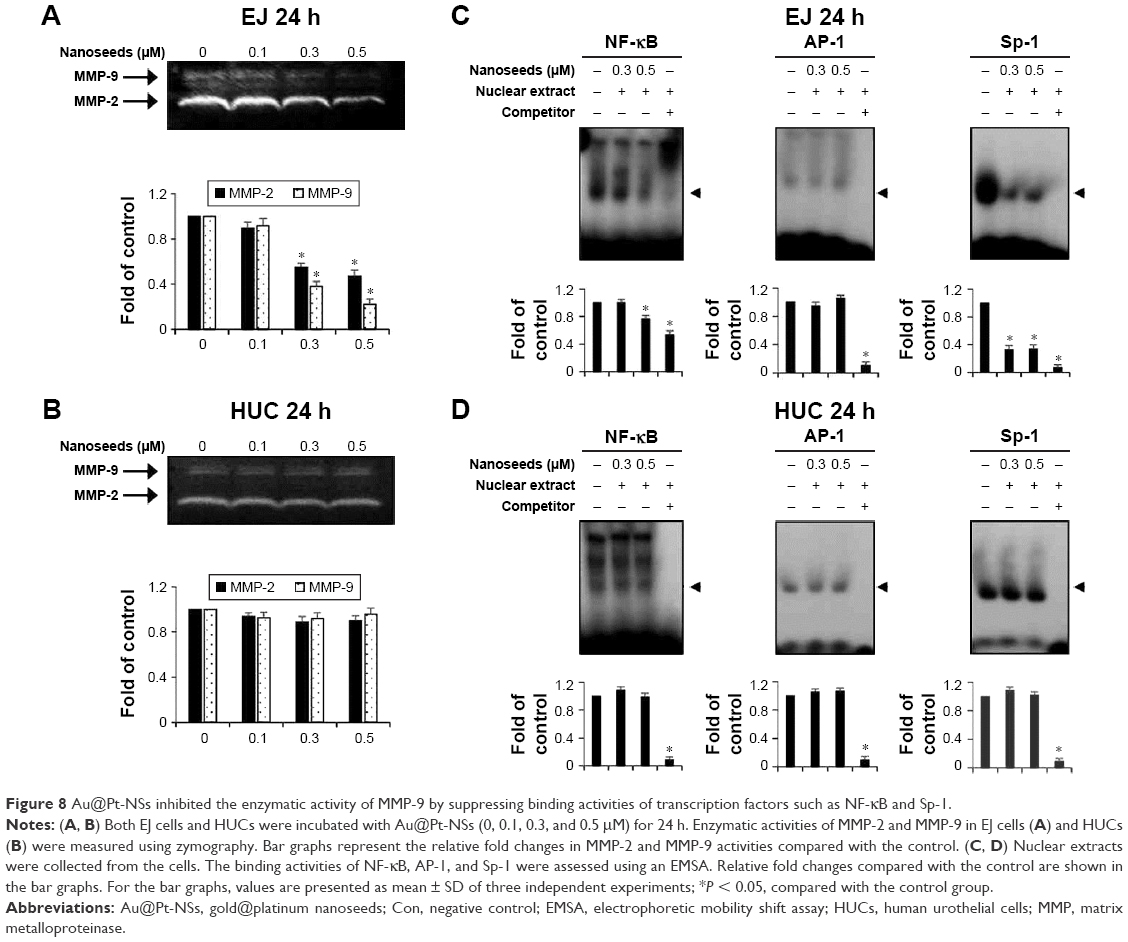 | Figure 8 Au@Pt-NSs inhibited the enzymatic activity of MMP-9 by suppressing binding activities of transcription factors such as NF-κB and Sp-1. |
Discussion
During recent decades, it has been noted that substances display unpredictable traits when examined at the nanoscale, which has led to the advent of metal NPs. Metal NPs have been reported to possess diverse biological activities, and NPs based on different types of metals, including gold, silver, platinum, and copper, have been developed as novel therapeutics to manage cancer. However, although there are many studies related to the efficacy of NPs, very few have investigated the mechanism of antitumor effects of the Au@Pt-NSs. In this study, we investigated the molecular mechanism of the anticancer efficacy of Au@Pt-NSs using EJ bladder carcinoma cells. First, we observed that Au@Pt-NSs showed a strong and dose-dependent anti-proliferative effect against EJ cells. This agreed with the results of previous studies, for example, Saha et al34 reported that Au NPs inhibited the proliferation of both pancreatic cancer cells (PCCs) and normal pancreatic stellate cells (PSCs) efficiently by hampering the bidirectional communication between PCCs and PSCs. Given that NPs are intrinsically genotoxic because of their high affinity for cellular DNA, assessing the cytotoxicity of NPs toward normal counterparts is a critical step in the development of chemotherapeutic reagents. In our study, the cytotoxicity of Au@Pt-NSs was limited to EJ cancer cells: cytotoxicity toward normal HUCs was negligible up to 0.5 μM. Similar to our results, Qu and Lu35 reported that citrate-coated Au NPs at a size of 10–50 nm were nontoxic to embryonic fibroblast cells. Our results indicated that certain dose ranges of Au@Pt-NSs may be nontoxic toward normally dividing cells, which increases their potential as a novel therapeutic option for bladder cancer cells.
Au@Pt-NSs caused a dose-dependent accumulation of cells in the G1 phase of the cell cycle. This indicated that effectors, such as CDKs, cyclins, and CKIs, in G1 phase participate in the anti-proliferative action of Au@Pt-NSs. We demonstrated that p21WAF1, but not p27KIP1, is a key regulator of the Au@Pt-NSs-mediated anti-proliferative effect in bladder EJ cancer cells. However, p53, which is involved in ultraviolet (UV) irradiation-mediated ATM pathways, was not implicated in the mode of action of the Au@Pt-NSs. We expanded our investigation to MAPKs and AKT signaling, because these pathways have been suggested as key regulators in the progression of bladder cancer.7,36,37 Fernandez-Gallardo et al38 demonstrated that heterometallic NPs made with titanium and gold could inhibit the growth of renal cancers significantly, both in vitro and in vivo, by suppressing the activity of AKT. Using ovarian cancer cells and animal models, Arvizo et al39 reported that unmodified Au-NPs inhibited tumor growth and metastasis significantly by suppressing phosphorylation of ERK1/2 and reversing the epithelial–mesenchymal transition. In addition, Wang et al40 showed that NP realgar powder inhibited the proliferation of human histiocytic lymphoma U937 cells by inducing phosphorylation of JNK1/2. Thus, these previous studies indicated that the induction of MAPKs seems to be cell-type dependent. In this study, a time-course induction experiment showed that the phosphorylation of p38, but not ERK1/2 or JNK1/2, was increased significantly by Au@Pt-NSs. In addition, AKT phosphorylation was downregulated in the presence of Au@Pt-NSs. These results suggested that both p38 and AKT are the key mediators associated with the mechanism of action of Au@Pt-NSs in EJ bladder cancer cells.
Previous studies have demonstrated that tumor metastasis is a critical issue that accounts for the majority of cancer deaths.15,16 Therefore, targeting the migratory and invasive potential of tumor cells could be an effective strategy to manage bladder cancer. Accordingly, we investigated whether Au@Pt-NSs suppress the migration and invasion of bladder EJ cancer cells. Treatment with Au@Pt-NSs suppressed the metastatic potential of EJ cells significantly in a dose-dependent manner. However, normal HUCs were unchanged by treatment with Au@Pt-NSs. Based on the evidence from previous studies15,41 and by the notion that Au@Pt-NSs displayed inhibitory activity against the migratory and invasive potential of EJ cancer cells, we investigated the gelatinase activities of MMP-2 and MMP-9 in EJ cells treated with Au@Pt-NSs using zymography. Treatment with Au@Pt-NSs inhibited the enzymatic activities of MMP-2 and MMP-9 dose dependently, suggesting that Au@Pt-NSs suppress the metastatic potential of EJ tumors by interfering with MMP activity. Previous studies by others demonstrated that the expression of MMP-9 is correlated markedly with the advanced stages of bladder tumors;15,16 therefore, we examined the regulation of MMP-9 expression. Using an EMSA assay, we verified that transcription factors such as NF-κB and Sp-1, but not AP-1, participated in the inhibitory mechanism of Au@Pt-NSs against the migration and invasion of EJ cancer cells. The molecular signaling pathways identified in the study are shown in Figure 9.
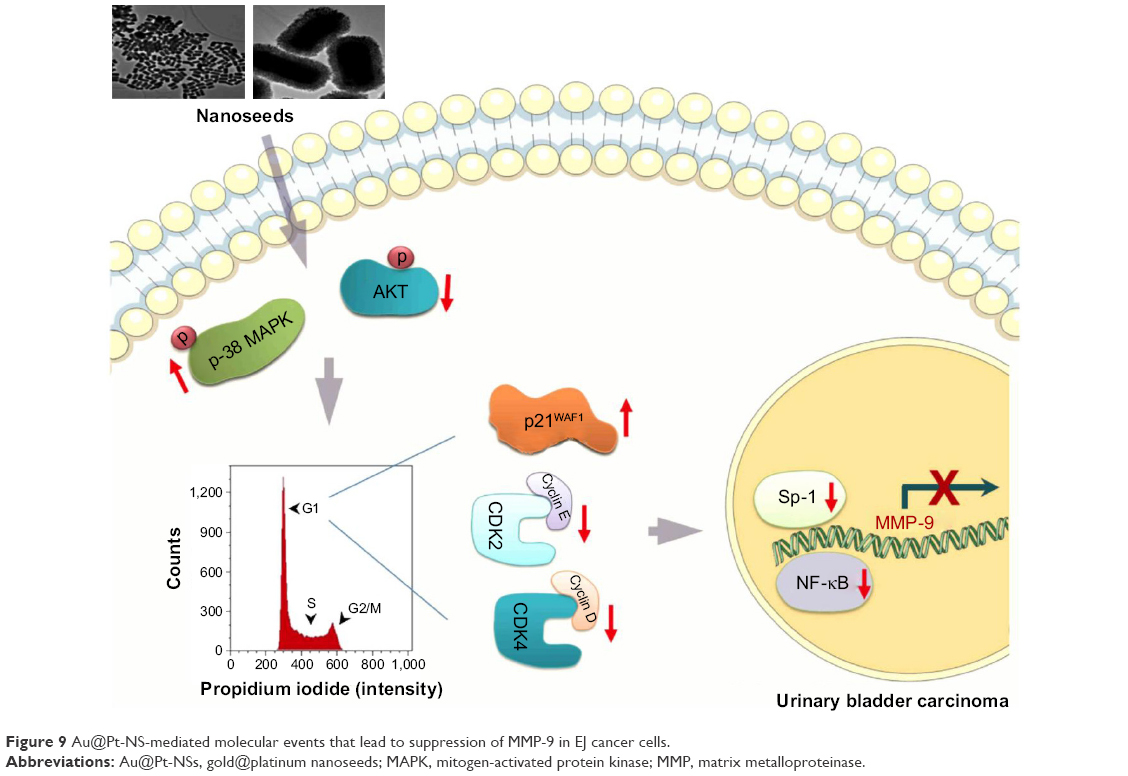 | Figure 9 Au@Pt-NS-mediated molecular events that lead to suppression of MMP-9 in EJ cancer cells. |
Conclusion
We demonstrated that Au@Pt-NSs markedly inhibited the proliferation of EJ bladder cancer cells by inducing accumulation in the G1 cell cycle phase. The accumulation was caused, at least in part, by the downregulation of CDK2, CDK4, cyclin D1, and cyclin E, which was mediated by the upregulation of p21WAF1. In addition, we demonstrated, for the first time, that Au@Pt-NSs exerts its inhibitory activity by inducing the phosphorylation of p38 and inhibiting the phosphorylation of AKT. Au@Pt-NSs also inhibited the migration and invasion of EJ cells by suppressing MMP-9 activity via reduced binding of NF-κB and Sp-1 transcription factors. Based on the results, we believe that Au@Pt-NSs, without side effects, might be a potential candidate for the prevention and treatment of bladder tumors. Further research is needed to investigate the efficacy of Au@Pt-NSs by using an animal model.
Acknowledgments
This study was supported by the National Research Foundation (NRF) of Korea grant funded by the Korea government (Ministry of Science, The Information and Communications Technology and Future Planning [MSIP]; No 2017R1A2B4009384) and also by the International Science and Business Belt Program through MSIP (2017K000490).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Taylor JA III, Kuchel GA. Bladder cancer in the elderly: clinical outcomes, basic mechanisms, and future research direction. Nat Clin Pract Urol. 2009;6(3):135–144. | ||
Chromecki TF, Bensalah K, Remzi M, et al. Prognostic factors for upper urinary tract urothelial carcinoma. Nat Rev Urol. 2011;8(8):440–447. | ||
Dangle PP, Zaharieva B, Jia H, Pohar KS. Ras-MAPK pathway as a therapeutic target in cancer – emphasis on bladder cancer. Recent Pat Anticancer Drug Discov. 2009;4(2):125–136. | ||
Gerhardt D, Bertola G, Dietrich F, et al. Boldine induces cell cycle arrest and apoptosis in T24 human bladder cancer cell line via regulation of ERK, AKT, and GSK-3beta. Urol Oncol. 2014;32(1):36.e1–9. | ||
Zheng X, Ou Y, Shu M, et al. Cholera toxin, a typical protein kinase A activator, induces G1 phase growth arrest in human bladder transitional cell carcinoma cells via inhibiting the c-Raf/MEK/ERK signaling pathway. Mol Med Rep. 2014;9(5):1773–1779. | ||
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. | ||
Lee MH, Yang HY. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cell Mol Life Sci. 2001;58(12–13):1907–1922. | ||
Li A, Blow JJ. The origin of CDK regulation. Nat Cell Biol. 2001;3(8):E182–E184. | ||
Zetterberg A, Larsson O, Wiman KG. What is the restriction point? Curr Opin Cell Biol. 1995;7(6):835–842. | ||
Canavese M, Santo L, Raje N. Cyclin dependent kinases in cancer: potential for therapeutic intervention. Cancer Biol Ther. 2012;13(7):451–457. | ||
Paternot S, Bockstaele L, Bisteau X, Kooken H, Coulonval K, Roger PP. Rb inactivation in cell cycle and cancer: the puzzle of highly regulated activating phosphorylation of CDK4 versus constitutively active CDK-activating kinase. Cell Cycle. 2010;9(4):689–699. | ||
Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75(4):805–816. | ||
Harper JW, Elledge SJ, Keyomarsi K, et al. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995;6(4):387–400. | ||
Bianco FJ Jr, Gervasi DC, Tiguert R, et al. Matrix metalloproteinase-9 expression in bladder washes from bladder cancer patients predicts pathological stage and grade. Clin Cancer Res. 1998;4(12):3011–3016. | ||
Davies B, Waxman J, Wasan H, et al. Levels of matrix metalloproteases in bladder cancer correlate with tumor grade and invasion. Cancer Res. 1993;53(22):5365–5369. | ||
Zhou YT, He W, Wamer WG, et al. Enzyme-mimetic effects of gold@platinum nanorods on the antioxidant activity of ascorbic acid. Nanoscale. 2013;5(4):1583–1591. | ||
Fan J, Yin JJ, Ning B, et al. Direct evidence for catalase and peroxidase activities of ferritin-platinum nanoparticles. Biomaterials. 2011;32(6):1611–1618. | ||
Lee J, Yoon S, Lo M, Wu H, SY L, Moon B. Intrinsic polyphenol oxidase-like activity of gold@platinum nanoparticles. RSC Adv. 2015;5:63757–63764. | ||
Kim J, Takahashi M, Shimizu T, et al. Effects of a potent antioxidant, platinum nanoparticle, on the lifespan of Caenorhabditis elegans. Mech Ageing Dev. 2008;129(6):322–331. | ||
Watanabe A, Kajita M, Kim J, et al. In vitro free radical scavenging activity of platinum nanoparticles. Nanotechnology. 2009;20(45):455105. | ||
Lee J, Son J, Yoo K, Lo M, Moon B. Characterization of the antioxidant activity of gold@platinum nanoparticles. RSC Adv. 2014;4:19824–19830. | ||
Mateo D, Morales P, Avalos A, Haza AI. Oxidative stress contributes to gold nanoparticle-induced cytotoxicity in human tumor cells. Toxicol Mech Methods. 2014;24(3):161–172. | ||
Bhamidipati M, Fabris L. Multiparametric assessment of gold nanoparticle cytotoxicity in cancerous and healthy cells: the role of size, shape, and surface chemistry. Bioconjug Chem. 2017;28(2):449–460. | ||
Cho SK, Emoto K, Su LJ, Yang X, Flaig TW, Park W. Functionalized gold nanorods for thermal ablation treatment of bladder cancer. J Biomed Nanotechnol. 2014;10(7):1267–1276. | ||
Hsieh DS, Wang H, Tan SW, et al. The treatment of bladder cancer in a mouse model by epigallocatechin-3-gallate-gold nanoparticles. Biomaterials. 2011;32(30):7633–7640. | ||
Gehrke H, Pelka J, Hartinger CG, et al. Platinum nanoparticles and their cellular uptake and DNA platination at non-cytotoxic concentrations. Arch Toxicol. 2011;85(7):799–812. | ||
Pelka J, Gehrke H, Esselen M, et al. Cellular uptake of platinum nanoparticles in human colon carcinoma cells and their impact on cellular redox systems and DNA integrity. Chem Res Toxicol. 2009;22(4):649–659. | ||
Porcel E, Liehn S, Remita H, et al. Platinum nanoparticles: a promising material for future cancer therapy? Nanotechnology. 2010;21(8):85103. | ||
Yue Y, Wagner S, Medina-Kauwe L, et al. WE-FG-BRA-11: theranostic platinum nanoparticle for radiation sensitization in breast cancer radiotherapy. Med Phys. 2016;43(6):3826. | ||
Shmarakov I, Mukha I, Vityuk N, et al. Antitumor activity of alloy and core-shell-type bimetallic AgAu nanoparticles. Nanoscale Res Lett. 2017;12(1):333. | ||
Wenzel M, Bertrand B, Eymin MJ, et al. Multinuclear cytotoxic metallodrugs: physicochemical characterization and biological properties of novel heteronuclear gold-titanium complexes. Inorg Chem. 2011;50(19):9472–9480. | ||
Dano K, Romer J, Nielsen BS, et al. Cancer invasion and tissue remodeling – cooperation of protease systems and cell types. APMIS. 1999;107(1):120–127. | ||
Saha S, Xiong X, Chakraborty PK, et al. Gold nanoparticle reprograms pancreatic tumor microenvironment and inhibits tumor growth. ACS Nano. 2016;10(12):10636–10651. | ||
Qu Y, Lu X. Aqueous synthesis of gold nanoparticles and their cytotoxicity in human dermal fibroblasts-fetal. Biomed Mater. 2009;4(2):025007. | ||
Lv D, Wu H, Xing R, et al. HnRNP-L mediates bladder cancer progression by inhibiting apoptotic signaling and enhancing MAPK signaling pathways. Oncotarget. 2017;8(8):13586–13599. | ||
Qiu L, Zhou C, Sun Y, et al. Paclitaxel and ceramide synergistically induce cell death with transient activation of EGFR and ERK pathway in pancreatic cancer cells. Oncol Rep. 2006;16(4):907–913. | ||
Fernandez-Gallardo J, Elie BT, Sadhukha T, et al. Heterometallic titanium-gold complexes inhibit renal cancer cells in vitro and in vivo. Chem Sci. 2015;6(9):5269–5283. | ||
Arvizo RR, Saha S, Wang E, Robertson JD, Bhattacharya R, Mukherjee P. Inhibition of tumor growth and metastasis by a self-therapeutic nanoparticle. Proc Natl Acad Sci U S A. 2013;110(17):6700–6705. | ||
Wang XB, Gao HY, Hou BL, Huang J, Xi RG, Wu LJ. Nanoparticle realgar powders induce apoptosis in U937 cells through caspase MAPK and mitochondrial pathways. Arch Pharm Res. 2007;30(5):653–658. | ||
Lee EJ, Lee SJ, Kim S, et al. Interleukin-5 enhances the migration and invasion of bladder cancer cells via ERK1/2-mediated MMP-9/NF-kappaB/AP-1 pathway: involvement of the p21WAF1 expression. Cell Signal. 2013;25(10):2025–2038. |
Supplementary materials
Supplementary methods
Real-time quantitative polymerase chain reaction (qPCR)
Real-time qPCR was performed using Rotor Gene 6000 real-time PCR machine (Corbett Life Science, Mortlake, NSW, Australia). PCR was carried out using SYBR Premix EX taq (Takara Bio Inc., Otsu, Japan) in micro-reaction tubes (Corbett Life Science) according to the manufacturer’s protocol. Sequences of primers used are summarized in Table S1. Fluorescence intensity of the PCR was analyzed by Rotor Gene Q real-time analysis software 6.0 (Corbett Life Science). GAPDH was utilized in the reaction as an internal control. Conditions of the real-time qPCR were as follows: one cycle, 20 s, 96°C (initial denaturation); 40 cycles, 20 s, 96°C (denaturation), 15 s, 60°C (annealing), and 15 s, 72°C (extension). The heating rate of the reaction was 1°C/45 s. All samples were run in triplicate.
 | Figure S1 Relative mRNA expression of cell cycle regulators in heterometallic Au@Pt-NS-treated EJ cells. |
 | Figure S2 Relative mRNA expression of cell cycle regulators in heterometallic Au@Pt-NS-treated HUCs. |
 | Table S1 Sequences of primers used in the real-time qPCR |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.