")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 12
Inhibitors of the Autotaxin-Lysophosphatidic Acid Axis and Their Potential in the Treatment of Interstitial Lung Disease: Current Perspectives
Authors Zulfikar S, Mulholland S, Adamali H , Barratt SL
Received 11 March 2020
Accepted for publication 17 June 2020
Published 13 July 2020 Volume 2020:12 Pages 97—108
DOI https://doi.org/10.2147/CPAA.S228362
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Sabrina Zulfikar, Sarah Mulholland, Huzaifa Adamali, Shaney L Barratt
Bristol Interstitial Lung Disease Service, North Bristol NHS Trust, Bristol, UK
Correspondence: Shaney L Barratt
Consultant Respiratory Physician and Honorary Associate Researcher, North Bristol NHS Trust, Southmead Hospital, Southmead, Bristol BS10 5NB, UK
Email [email protected]
Abstract: Idiopathic pulmonary fibrosis is a progressive fibrosing interstitial lung disease for which there is no known cure. Currently available therapeutic options have been shown at best to slow the progression of the disease and thus there remains an urgent unmet need to identify new therapies. In this article, we will discuss the mechanisms of action, pre-clinical and clinical trial data surrounding inhibitors of the autotaxin-lysophosphatidic acid axis, which show promise as emerging novel therapies for fibrotic lung disease.
Keywords: autotaxin inhibitors, autotaxin-lysophosphatidic acid axis, idiopathic pulmonary fibrosis, interstitial lung disease, fibrotic lung disease
Introduction
Idiopathic Pulmonary Fibrosis (IPF) is an incurable, interstitial lung disease (ILD) characterised by chronic, progressive fibrosis and respiratory failure. Currently available therapies at best slow the inexorable decline in lung function1–3 and recent recommendations promoting high standards of clinical care in IPF, suggest a focus on best supportive care from the point of diagnosis.4 In this article, we will discuss the mechanisms of action, pre-clinical and clinical trial data surrounding inhibitors of the autotaxin-lysophosphatidic acid axis (ATX-LPA), novel drugs showing promise as emerging future therapeutic interventions in IPF.
Idiopathic Pulmonary Fibrosis (IPF)
Epidemiology
IPF is a chronic, progressive fibrotic lung disease of unknown origin, limited to the lung and occurring primarily in older adults. Morbidity and mortality associated with the disease is high, with a median survival of only 2–3 years from diagnosis.5,6 A systematic review of the global databases of IPF suggests that the prevalence of IPF is increasing worldwide, with incidence rates reported to be between 2.8 and 9.3 per 100,000 per year in North America and Europe7,8 with growing economic health care burden.9
Pathogenesis
Much has been learnt about the pathogenesis of this idiopathic fibrotic lung disease over the past decade. A comprehensive review of the factors thought to be important in the development of IPF is beyond the scope of this article and has been recently reviewed by others;10,11 thus, key concepts are outlined below.
Current paradigms suggest that the repetitive injury of an inherently dysfunctional alveolar epithelium is a crucial initiating factor in IPF, resulting in the activation of multiple pathways mediating the fibrotic cascade.12 Failure of the epithelium to regenerate disrupts the alveolar capillary membrane (ACM) barrier,13 promoting capillary leak of proteins, activation of the coagulation cascade14 and abnormal neovascularisation in an attempt at ongoing repair.15,16
Transforming growth factor β (TGF β) plays a central role in the mediation of the fibrotic process in IPF,17 promoting apoptosis, epithelial to mesenchymal transition, extracellular matrix deposition and recruitment and activation of fibroblasts with differentiation into myofibroblasts. Collections of fibroblasts/myofibroblasts (fibrotic foci) deposit extracellular matrix (ECM) in abundance, with progressive lung remodelling and disruption of the normal architecture, with evidence of temporal and spatial heterogeneity; a usual interstitial pneumonia (UIP) pattern of fibrosis that is the pathological hallmark of the disease.10,18
Anti-Fibrotic Medications
Two anti-fibrotic drugs are currently approved for the treatment of IPF; pirfenidone and nintedanib.1,2,19 Pirfenidone is a novel compound that was shown to exhibit both anti-inflammatory and anti-fibrotic properties in pre-clinical models.20,21 Pooled analyses of three large-scale Phase III multicentre trials (CAPACITY 1+2 and ASCEND) suggested that pirfenidone at 2403 mg/day (delivered in divided doses, three times daily) reduced the proportion of patients experiencing a forced vital capacity (FVC) of ≥10% or death by 43.8%.22 Nintedanib is a triple tyrosine kinase inhibitor of vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF).23 Parallel Phase III, multicentre randomised controlled trials (INPULSIS I and II) demonstrated a significant reduction in the rate of FVC decline over a 52-week period, in IPF patients receiving nintedanib compared to placebo. More recently, nintedanib has also been shown to slow FVC decline in a broader spectrum of progressive fibrosing ILDs other than IPF, in a large phase III clinical trial (INBUILD).24
Meta-analysis suggests that pirfenidone and nintedanib treatments demonstrate similar efficacy in terms of slowing the rate of FVC decline in IPF.25,26 So treatment decisions in this context are usually driven by tablet load or side effect profiles.27 Neither pirfenidone nor nintedanib has been able to demonstrate stabilisation or even improvement in lung function and there remains no cure for this devastating disease. The medications are often poorly tolerated leading to discontinuation in a significant proportion of patients.28 As such, best supportive care and symptom control are central to disease management. It is unclear whether antifibrotic medications improve symptoms such as breathlessness and cough or whether their beneficial effect on functional decline results in increased survival. As such, there is an ongoing need to develop more effective novel therapies.
The Autotaxin (ATX)-Lysophosphatidic Acid (LPA) Axis
Phospholipid growth factors (PLGFs) are a family of lipids with growth factor-like properties. Lysophosphatidic acid (LPA) is a member of the PLGF family that promotes a diverse range of physiological cellular functions by binding to specific G-protein-coupled receptors (LPAR 1–6), present within the plasma membrane (Figure 1). Downstream signalling cascades include those involved in cell proliferation, cell motility, cell invasion, apoptosis and the chronic inflammatory response.29 LPA is present in abundance in the circulation, with activated platelets being considered one of the primary sources,30 although it may be generated from numerous cells, including cancer cells,31 fibroblasts,32 adipocytes,33 alongside its presence in microvesicle exosomes released from activated inflammatory cells.30,34 There are two main pathways to produce LPA: firstly, LPA can be produced from phosphatidic acid (PA) by phospholipase A1 (PLA1) and phospholipase A2 (PLA2) family members. The majority of LPA in the circulation is produced via an alternative pathway, however, in which PLA1 and PLA2 first convert phospholipids to lysophospholipids, such as lysophosphatidylcholine (LPC),30 which can then be converted to LPA by autotaxin (ATX).
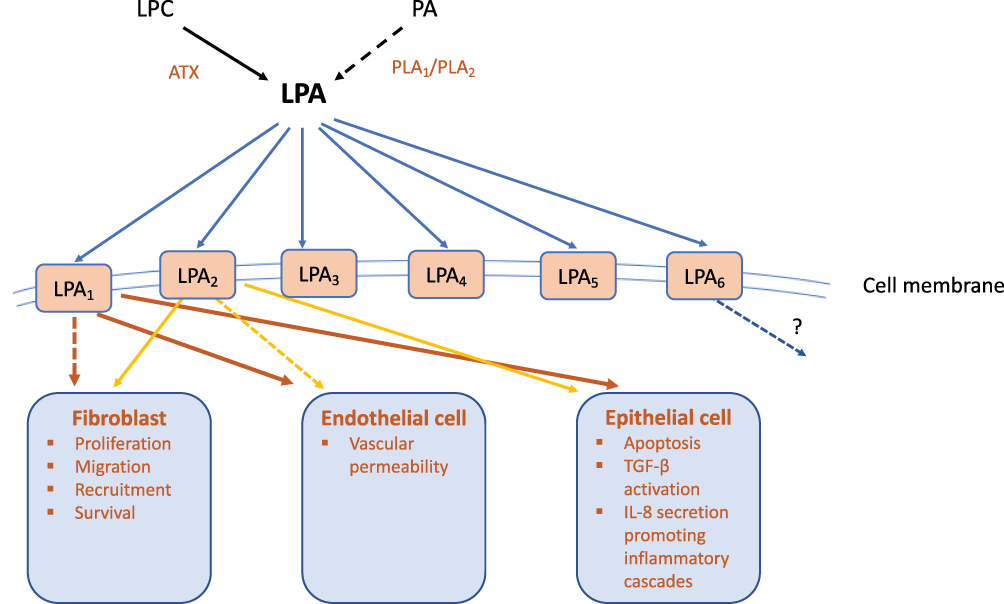 |
Figure 1 Schematic diagram of the Autotaxin-Lysophosphatidic acid (ATX-LPA) axis in fibrotic lung disease. The majority of LPA in the circulation is produced from hydrolysis of lysophosphatidylcholine (LPC), mediated by the secreted enzyme ATX. LPA is also produced from phosphatidic acid (PA) by its deacylation, catalysed by phospholipase A (PLA)-type enzymes. The biological roles of LPA are diverse and include developmental, physiological, and pathophysiological effects. This diversity is mediated through multiple downstream signalling pathways activated following the binding of LPA to membrane G protein-coupled receptors. One of the most important roles of the ATX-LPA axis following development, is in the mediation of wound healing and tissue remodelling through its pleiotropic effects on multiple cell types. The ATX-LPA axis has been implicated in the development of pulmonary fibrosis. Preclinical models have suggested that signalling through LPA1 and/or LPA2 receptors, LPA can disrupt the alveolar-capillary membrane by promoting epithelial cell apoptosis and increasing vascular permeability; a key initiating event in current paradigms of idiopathic pulmonary fibrosis (IPF). LPA stimulates fibroblast migration (predominantly through LPA2) and αvβ6-mediated TGF-β activation through epithelial cell activation – considered to be the archetypal fibrotic mediator. Through LPA1, LPA induces IL-8 secretion from epithelial cells, a powerful neutrophil chemoattractant and stimulator of the chronic inflammatory response. LPA2 has however been described as an inducer of both pro-inflammatory and anti-inflammatory responses. LPA6 receptor is abundantly expressed in the lung, but it’s possible role in pulmonary pathophysiology and fibrosis has yet to be explored. |
ATX (also known as ENPP2 – ectonucleotide pyrophosphatase-phosphodiesterase) is a secreted lysophospholipase D (lysoPLD) that derived its name based on its property as an “autocrine motility factor” when first isolated in melanoma cell culture.35 Five catalytically active isoforms are known to exist in humans, formed by alternative splicing of the ENPP2 gene.36 Although the exact role of each isoform is not fully understood, isoform β is most abundant, likely accounting for the majority of ATX/LPA pathophysiological effects.37
ATX is catalytically active in most biological fluids, including serum/plasma, bronchoalveolar lavage fluid (BALF), cerebrospinal fluid and urine.38 The major source of ATX in the circulation is considered to be adipose tissue, following the observation that conditional deletion of the ATX gene specifically in adipocytes, results in a reduction in plasma LPA levels of approximately 38%,39 whilst heterozygous ATX± gene deletion results in a 50% reduction.40,41
Role of ATX-LPA Axis in Embryological Development and Normal Lung Maintenance
The sequence and structure of ATX proteins are highly conserved in humans and mice, highlighting its physiological importance.42 In the developed lung ATX is constitutively expressed predominantly by bronchial epithelial cells and macrophages, whilst it can also be detected in BALF.43 Table 1 summarises the studies that have used transgenic mice to further understand the role of the ATX-LPA axis in normal development and lung homeostasis.
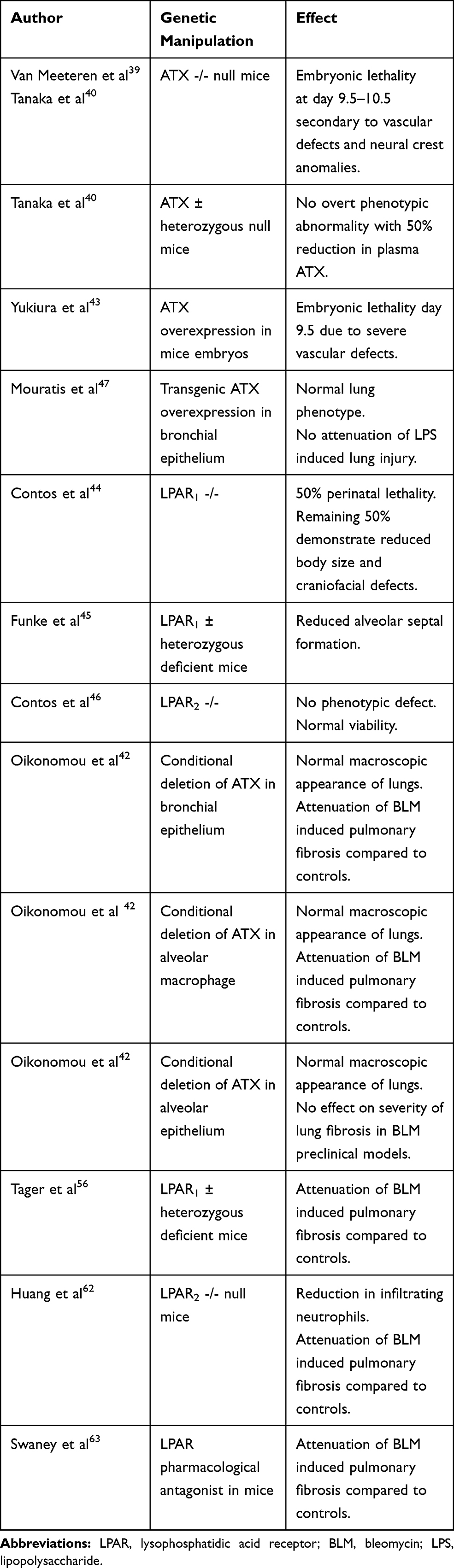 |
Table 1 Summary of preclinical studies manipulating the Autotaxin-Lysophosphatidic Acid (ATX-LPA) Axis |
ATX is absolutely critical for embryological development, since ATX null mice die at day 9.5, secondary to vascular and neural crest anomalies.40 Furthermore, ATX levels must be carefully regulated to ensure coordinated vascular formation, as ATX overexpression is also lethal due to severe vascular defects.44 Approximately 50% of LPAR1 null mice demonstrate perinatal lethality due to craniofacial defects and difficulties suckling,45 whilst in contrast, heterozygous LPAR1± deficient mice survive postnatally but demonstrate phenotypic lung abnormalities with impaired pulmonary alveolarisation.46 LPAR2 null mice have normal viability however and show no phenotypic abnormalities.47 LPA3 is essential for normal embryo implantation, as such, LPAR3 null mice demonstrate abnormalities in reproduction.48
Heterozygous ATX ± deficient mice or genetic abrogation of bronchial cell ATX expression does not affect the macroscopic appearances of the lungs of mice, suggesting small amounts of LPA within the lung, together with possible extrapulmonary sources of LPA30 are sufficient for normal lung development and maintenance.41,43,49 Furthermore, transgenic overexpression of ATX in the bronchial epithelium also results in a normal lung phenotype, suggesting that the mere presence of ATX and LPA is not adequate to induce lung damage in isolation.37,49
The ATX-LPA Axis in Pathological Circumstances
More generally, one of the most important roles of the ATX-LPA axis following development, is in the mediation of wound healing and tissue remodelling through its pleiotropic effects on multiple cell types, stimulating platelet aggregation and promotes the migration of immune cells, fibroblasts, endothelial cells into sites of tissue damage.30 The ATX-LPA axis has been implicated in a multitude of pathologies including the development of atherosclerosis, the regulation of glucose homeostasis and obesity-related insulin resistance and inflammatory arthritis; reviewed elsewhere.37
ATX-LPA signalling has long been associated with cancer initiation, progression and metastasis since the first isolation of ATX in melanoma cell culture by Stracke et al in 1992.35 The common pathway appears to relate to chronic inflammatory upregulation of ATX by cancer cells or the surrounding environment, with concomitant upregulation of LPA receptors and sustained downstream signalling promoting angiogenesis, alongside cancer cell growth and survival.42 It is beyond the scope of this review to fully describe the recent advances in understanding the role of the ATX-LPA axis in cancer, which has eloquently been summarised by others.42,50 Nevertheless, despite the apparent relevance of ATX and LPARs in cancer, no inhibitors have progressed to cancer-related clinical trials thus far.
Signalling through the ATX-LPA axis appears to be a common pathway signature of fibrosis; being implicated in the fibrosis of several organs including the liver, skin, kidney and lung.43,51-53 For example, plasma LPA levels and higher serum ATX activity correlate with histologic stage of fibrosis, which has thus been proposed as a novel biomarker in liver cirrhosis.54,55
Role of LPA-ATX Axis in Pulmonary Fibrosis
The ATX gene (Enpp2) was identified as a candidate gene controlling lung function, development and remodelling through genome-wide linkage analysis, coupled with expression profiling.56
Immunohistochemical analysis of the fibrotic lungs of mice challenged with bleomycin demonstrates increased ATX expression that is localised to the bronchiolar epithelial cells and macrophages, with additional weak alveolar cell staining and increased ATX levels in corresponding BALF samples.43 Similarly, ATX localises most prominently to the hyperplastic bronchial epithelium in human lung tissue of patients with IPF by immunohistochemistry, with additional staining of the alveolar epithelium, fibroblastic foci cells and alveolar macrophages.43 Levels of LPA and ATX are both elevated in the BALF and exhaled breath condensate of IPF patients.43,57,58 Conditional deletion of ATX in the bronchial epithelial cells or macrophages of mice, both attenuated bleomycin (BLM)-induced fibrosis as demonstrated through reduction in lung collagen and BAL cell counts compared to controls, whilst in contrast, specific-alveolar cell deletion of ATX had no significant protective effect.43 These findings corroborated the pulmonary sources of ATX whilst also establishing a pathogenic role for ATX.
Hydroxamic acids represent a novel class of ATX inhibitors. One of the most potent in vitro compounds (inhibitor 32) exhibited promising efficacy in a murine model of pulmonary fibrosis with reduced inflammatory cell influx (by approximately 50%), vascular permeability and collagen deposition in treated mice compared to BLM controls.59 Several other novel compounds including aromatic phosphonates and anti-ATX DNA aptamers (RB-014) have been developed demonstrating pharmacological inhibition of ATX and abrogation of BLM-induced pulmonary fibrosis in murine models.60,61 Conflicting results were obtained by Black et al, however, who reported that ATX inhibition with PAT-048 (Bristol Myers Squibb; WO2012024620) had no effect on BLM-induced pulmonary fibrosis;62 differences in experimental conditions or models may explain these contradictory findings. More recently, the therapeutic potential of targeting ATX has gained momentum, with the identification of a novel small molecule inhibitor (GLPG1690; Galapagos), that attenuated BLM-induced pulmonary fibrosis and has subsequently been entered into clinical trials (see later).63
In agreement with a pathogenic role for the ATX-LPA axis in the development of pulmonary fibrosis, genetic deletion mice of either LPAR158 or LPAR264 or pharmacological inhibition of LPA1 receptor65 protects against the development of bleomycin-induced pulmonary fibrosis. In these studies, reduced alveolar and bronchial epithelial TUNEL+ positive cells were observed in transgenic mice, suggesting that LPA may promote epithelial cell apoptosis through LPA1 and/or LPA2 as an initiating event in the development of fibrosis in this model and in keeping with current paradigms of IPF. Furthermore, LPAR1 deficient mice exhibited reduced vascular leak, suggesting a role for LPA in promoting alveolar-capillary membrane disruption or endothelial permeability upon injury.58 LPA also promotes re-organisation and proliferation of lung fibroblasts37 through LPA264 but not LPA1 receptors.58 Critical roles for LPA signalling through LPA1 have additionally been shown in skin and kidney pre-clinical models of fibrosis,51,52 considered to be an aberrant and excessive response to injury.
It is acknowledged that inflammatory mediators play a role in the development and progression of pulmonary fibrosis, despite the failure of anti-inflammatory therapies. Through LPA1, LPA induces IL-8 secretion from epithelial cells, a powerful neutrophil chemoattractant and stimulator of the chronic inflammatory response37 and can thus theoretically affect the inflammatory component of pulmonary fibrosis. LPA2 has however been described as an inducer of both pro-inflammatory and anti-inflammatory responses.37 LPA6 receptor is also abundantly expressed in the lung, but the possible role in pulmonary pathophysiology and fibrosis has yet to be explored.37
TGF-β (Transforming growth factor-β) is considered an archetypal fibrotic factor, considered to mediate several pathologically important aspects of the fibrotic cascade, such as myofibroblast differentiation, extracellular matrix (ECM) deposition, epithelial to mesenchymal cell transition and downstream signalling following epithelial cell injury.66 Several reports suggest that the ATX-LPA axis may promote fibrosis through TGF-β activation; LPA signalling through LPA2 has been shown to induce αvβ6-mediated activation of TGF-β in lung epithelial cells,67 induces TGFβ expression in fibroblasts in vitro,68 and stimulate lung fibroblast to myofibroblast differentiation.69
In summary, the ATX-LPA axis has been shown to promote pleiotropic effects for multiple pulmonary cell types indicated in the pathogenesis of pulmonary fibrosis and provided proof of principle for the clinical development of pharmacological interventions in IPF.
Inhibitors of ATX-LPA Axis in Clinical Trial Development in IPF
A number of ATX and LPA signalling antagonists have now been entered into clinical trials and are summarised below (Table 2).
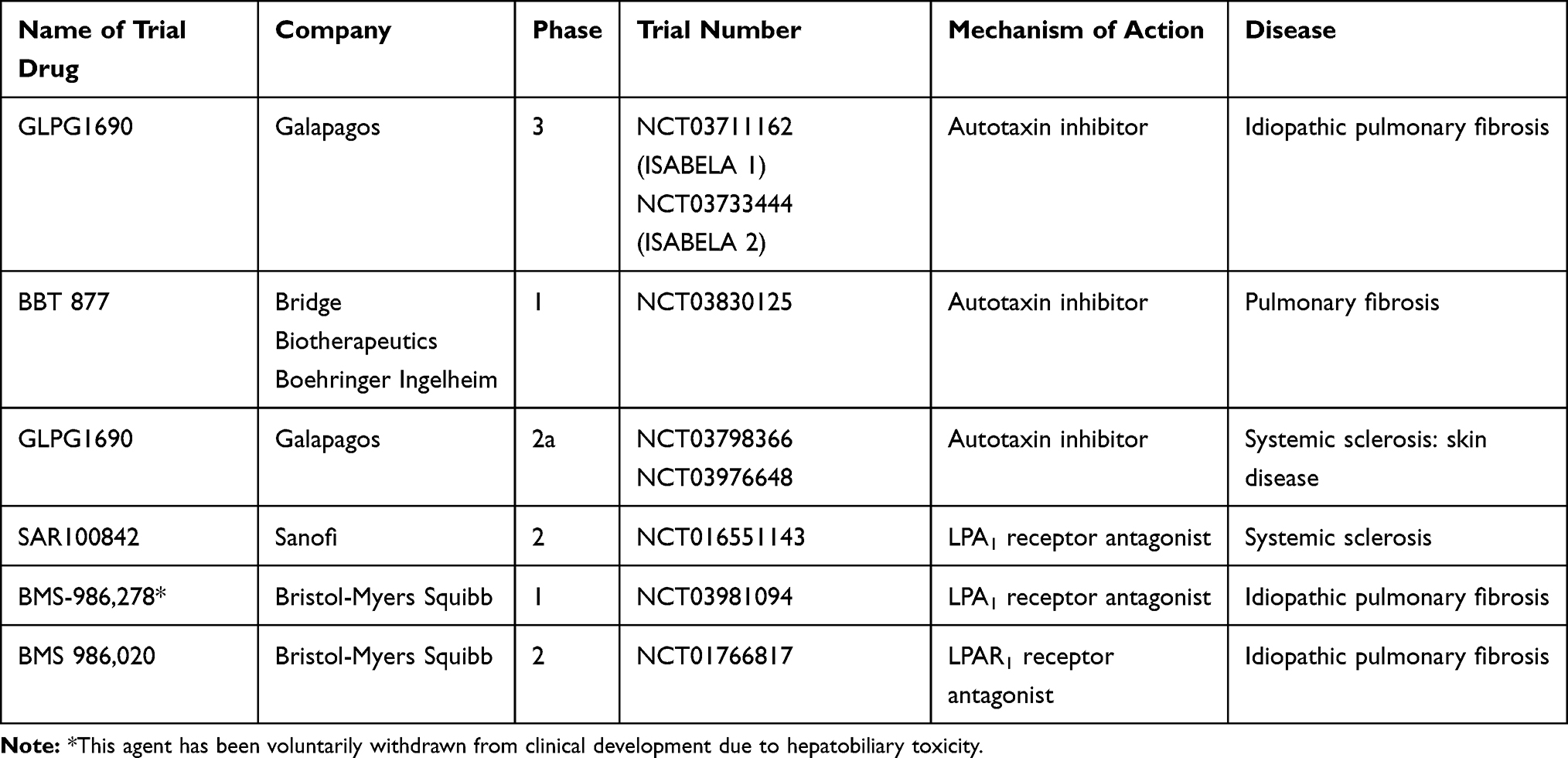 |
Table 2 Autotaxin (ATX) and Lysophosphatidic Acid (LPA) receptor antagonists in clinical development |
Galapagos: GLPG1690
Pre-Clinical Evaluation
High throughput screening (HTS) is a scientific method in which hundreds of experimental samples are simultaneously analysed under given conditions. Via this method, 2,3,6-trisubstituted imidazole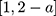 pyridine derivatives were identified as ATX inhibitors.63 After several structural modifications and improvements in pharmacokinetic properties and safety profile, a potent selective ATX inhibitor was identified; initially labelled as compound 11, it was subsequently named as GLPG1690.63
pyridine derivatives were identified as ATX inhibitors.63 After several structural modifications and improvements in pharmacokinetic properties and safety profile, a potent selective ATX inhibitor was identified; initially labelled as compound 11, it was subsequently named as GLPG1690.63
The ability of GLPG1690 to inhibit the in vivo conversion of LPC to LPA was initially demonstrated in rats, with a sustained reduction in plasma LPA levels following a single oral dose of GLPG1690.63 Subsequently, it was shown to reduce the histopathological appearances of BLM-induced pulmonary fibrosis in murine pre-clinical models, reducing ECM deposition by nearly 50% and reducing LPA concentrations in BALF by approximately 70% compared to BLM-controls, and with an efficacy comparable (at lower dose) or superior (at higher dose) to that of the reference compound pirfenidone.63
Preliminary work using explanted fibroblasts from IPF lung tissue suggested that this effect was in part mediated through inhibition of TGF-β induced expression of fibrotic mediators, including connective tissue growth factor (CTGF), interleukin-6 (IL-6) and endothelin-1 (ET-1); with an additive inhibitory effect of GLP1690 with nintedanib.70
Phase 1 Trials
Phase 1 clinical trials with GLPG1690 included a two-week randomised, double-blind, placebo-controlled trial assessing the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics of single or multiple doses of the compound, in healthy males (n=40)(NCT02179502). GLPG1690 was well tolerated up to a single dose of 1500 mg and up to 1000 mg twice daily for 14 days, with accompanied reduction in plasma LPA levels, occurring in a concentration-dependent manner. The most common treatment-emergent adverse effect (TEAE) was headache, followed by diarrhoea and nasopharyngitis, with no significant adverse effects on electrocardiogram (ECG) or vital signs. In one subject (1000 mg once daily cohort) aspartate aminotransferase (AST) levels rose to 2.25 times the upper limit of normal, but normalised after 7 days at follow up.71 A second randomized, open-label, crossover phase 1 trial compared the bioavailability of tablet and capsule formulations of GLPG1690 and evaluated the effect of food on the tablet formulation (NCT03143712).72 Tablet and capsule formulations had similar PK profiles, and no clinically significant food effect was observed when comparing tablets taken in fed and fasted states. These findings supported the ongoing clinical development of GLPG1690.
Phase 2a (FLORA) Trial
The FLORA trial was a phase 2a trial randomised, multi-centre, placebo-controlled study to assess the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics of GLPG1690 in patients with IPF (NCT02738801).73 In this study, a total of 23 IPF patients (FVC≥50% and DLCO≥30%), were randomised 3:1 to once-daily GLPG1690 600 mg or placebo for 12 weeks. Exclusion criteria included treatment with pirfenidone, nintedanib or prednisolone >15mg/day, within 4 weeks of screening. Primary endpoints were safety and tolerability, with secondary endpoints that included FVC, changes in disease biomarkers, functional respiratory imaging and health-related quality of life (HRQOL) (assessed by St George’s Respiratory Questionnaire, SRGQ). Twenty participants completed the study (one in each group discontinued due to AEs; one on GLPG1690 withdrew consent).
The PK profile of GLPG1690 was equivalent to that seen in the phase 1 trials in healthy participants and resulted in a sustained reduction in plasma LPA levels, demonstrating target engagement. TEAE was mostly mild to moderate, occurring at a similar rate in both groups (n=4 placebo, n=11 GLPG1690 non-significant), the most common ones being infections and infestations (10 events) with no obvious difference from the placebo group. There was one report of cholangiocarcinoma in the GLPG1690 group; symptoms were noted on day 1 after intake of GLPG1690, but the symptoms had actually started during the month prior to randomisation. Two further serious adverse events were reported in the placebo group, one developing urinary tract infection, acute kidney injury and lower respiratory tract infection and the other showing second-degree atrioventricular block on ECG. All three subjects were discontinued from the study. There were no reports of death or exacerbation of IPF in either group. No significant change was noted in the SGRQ for either group, suggesting there was no deterioration in symptoms in the 12-week period.
Whilst the study was not powered to detect differences between treatment and placebo groups, preliminary efficacy evaluation revealed encouraging results with a mean change from baseline in forced vital capacity (FVC) of +25 mL for GLPG1690 (week 12) and –70 mL for placebo; the latter in keeping with that observed in placebo IPF groups in pooled analyses of pirfenidone and nintedanib in phase III clinical trials at 3 months.22,74 This study gave the confidence to further assess the novel ATX inhibitor in a larger population of IPF patients over a longer duration.
Phase 3 (ISABELA 1 and 2)
GLPG1690 is currently being evaluated in phase III clinical trials. ISABELA 1 and 2 (NCT03711162 and NCT03733444) are two identically designed, international, randomised, double-blind, parallel-group, placebo-controlled multicentre studies appraising the efficacy and safety of two doses of GLPG1690, in addition to standard of care (SOC), for a minimum of 52 weeks, in patients with IPF.75 Initiated in November 2018, the completion date is planned for late 2021. It has been powered such that approximately 750 participants with IPF will be enrolled in each study, randomised 1:1:1 to receive either 200 mg once daily of GLPG1690, 600 mg once daily of GLPG1690 or placebo in addition to standard of care (SOC) (defined as pirfenidone or nintedanib or neither).
The primary endpoint of the study is the rate of decline of FVC over 52 weeks, with planned secondary endpoints of disease progression (10% or more absolute decline in FVC) or all-cause mortality, time to first respiratory-related hospitalisation and change in HRQOL (SGRQ). The results of this trial will be eagerly awaited as the single dosing schedule of GLPG1690 may have advantages over current anti-fibrotic regimes.
BBT-877
BBT-877 is a selective potent ATX inhibitor that is being developed in collaboration with Bridge Biotherapeutics and Boehringer Ingelheim. The molecule is currently in phase 1 studies having shown promise in pre-clinical BLM-induced lung fibrosis animal models, reducing body weight loss, lung weight and Ashcroft score, as well as collagen content, compared to the vehicle-treated group.76 Only mild adverse events were noted in 80 healthy volunteers receiving oral treatment over two weeks, with reductions in plasma LPA concentrations, confirming target engagement.76 The phase 1 trial results are expected to be finalised shortly (NCT03830125) and entrance into Phase II trials within the next 12 months.
BMS-986,020/BMS-986,278
The safety and efficacy of BMS-986,020 (Bristol-Myers Squibb), an LPA1 receptor antagonist, has recently been assessed in a Phase II trial for the treatment of IPF (NCT01766817).77 This randomised, double-blind, placebo-controlled trial was conducted over a period of 26 weeks, with a primary end point of rate of change in FVC from baseline. Of 143 IPF patients randomised, 108 completed the 26-week dosing schedule. Whilst patients treated with BMS-986,020 experienced a statistically significantly slower rate of decline in FVC compared to placebo (−0.042 L vs 0.134 L, respectively), dose-related derangements in liver enzyme tests were observed and the trial was terminated early due to three cases of cholecystitis thought to be directly related to the trial drug, with voluntary withdrawal from further clinical development. Similar safety concerns were not observed in phase 1 trials of healthy volunteers and pre-clinical research comparing BMS-986,020 with a structurally distinct LPAR1 antagonist BMS-986,234 suggests that this may be an off-target effect specific to BMS-986,020, related to inhibition of hepatobiliary transporters and bile composition rather than a class-effect of LPA antagonists.78 BMS-986,278 is a next-generation LPAR1 receptor antagonist that does not show hepatobiliary toxicity in pre-clinical studies in rat, monkey and multiple in vitro systems79 and may be a future candidate for clinical trials.
Inhibitors of ATX-LPA Axis in Clinical Trial Development in Systemic Sclerosis
Systemic sclerosis (SSc) is an autoimmune condition of the connective tissue and vasculature, characterised by inflammation, fibrosis and vasculopathy of multiple organs including the skin and lungs.80
The ATX-LPA axis has also been implicated in the skin fibrosis that accompanies SSc. In human skin, tissue injury increases LPA production81 and elevated circulating levels of LPA have been isolated from patients with SSc compared to controls, in the absence of any significant differences in ATX activity.82
Increased ATX staining has been detected in human lung samples of patients with fibrotic non-specific interstitial pneumonia (fNSIP); one of the commonest histopathological subtypes of fibrosis seen in SSc.43 In contrast, minimal staining was identified in samples of cellular NSIP which is classically corticosteroid responsive, possibly indicating that ATX up-regulation is associated with more fibrotic forms of interstitial lung disease than more inflammatory subtypes.49 In keeping with this hypothesis, in pre-clinical models of lipopolysaccharide (LPS)-induced acute lung injury (ALI), although BALF levels of ATX increased following instillation of LPS, neither genetic nor pharmacological targeting of ATX had beneficial effects in reducing ALI severity, suggesting differential involvement of the ATX-LPA axis in acute versus chronic pulmonary inflammation.49
ATX expression (both mRNA and protein) is elevated in the dermis of BLM-injected mice compared to controls.83 Furthermore, LPA signalling, specifically through LPA1, is required for dermal fibrosis, with both genetic deletion and pharmacological inhibition of ATX expression (ATX PAT-048) leading to attenuation of skin fibrosis through inhibition of an ATX-LPA-IL-6 amplification loop.51,83 This same group showed similar results in cultures of dermal fibroblasts from patients with SSc.83 Together, these results suggest that directed pharmacological inhibition of LPAR or the ATX-LPA-IL6 amplification loop may represent an innovative pathway to target fibrosis associated with SSc.82 Indeed, phase III clinical trials of Tocilizumab, a monoclonal antibody against the IL6-receptor have shown promising results both in terms of improving skin scores and stabilisation of lung fibrosis in SSc.84
Sanofi SAR100842, an oral antagonist of the LPA1 receptor, has entered Phase II trials for the treatment of SSc (NCT 01651143).85 The safety of SAR100842 was assessed in a double-blind, randomised, eight-week placebo-controlled study with a 16-week open-label extension study. Fifteen patients received the active trial drug (n=17 placebo) and the most frequent adverse events reported were headache, diarrhoea, nausea, and falling, all mild to moderate. The safety profile was acceptable during the open-label extension and gene signature analysis suggested target engagement. Whilst, the ATX inhibitor GLPG1690 (NCT03798366 and NCT03976648) has entered into phase 2a trials, evaluating the efficacy, safety and tolerability of the orally administered agent in patients with systemic sclerosis, with a primary endpoint of change in skin fibrosis scores. Further studies are eagerly awaited.
Conclusions
IPF remains a devastating progressive fibrotic lung disease for which there is no cure. Existing antifibrotic medications have been shown to slow disease progression but can be poorly tolerated and do not treat symptoms of the disease, and as such, there is an unmet need to develop more effective therapies. We are at an exciting time where several novel agents that interfere with LPA signalling could become viable therapeutic modalities for IPF in the future. In the context of sustained ATX inhibition, the production of small amounts of LPA by alternative pathways, such as the PLA1 and PLA2 enzymes, is likely to provide sufficient LPA for physiological homeostasis, without driving pathology. Whilst these novel therapies appear to be well tolerated thus far, it is imperative that ongoing trials continue to assess safety, particularly given the pleiotropic effects of LPA signalling, the important role LPA has in repair following tissue injury and the resultant potential for unintended off-target consequences.
Acknowledgments
SZ and SM provided an initial draft of the review, which was then verified by SB and HA. SB is the guarantor of the content of the manuscript.
Disclosure
Both SLB and HA have received financial support from Boehringer Ingelheim to attend medical conferences and have undertaken consultancy work for Boehringer Ingelheim. SM reports personal fees from Boehringer Ingelheim, non-financial support from Chiesi, non-financial support from Roche, outside the submitted work. The authors do not have any other conflicts of interest.
References
1. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
2. King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092. doi:10.1056/NEJMoa1402582
3. Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi:10.1056/NEJMoa1402584
4. NICE clinical guideline CG 163: idiopathic pulmonary fibrosis in adults: diagnosis and management. 2013. Available from: www.nice.org.uk.
5. Strongman H, Kausar I, Maher TM. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv Ther. 2018;35(5):724–736. doi:10.1007/s12325-018-0693-1
6. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi:10.1164/rccm.2009-040GL
7. Navaratnam V, Fleming KM, West J, et al. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax. 2011;66(6):462–467. doi:10.1136/thx.2010.148031
8. Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med. 2014;2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
9. Diamantopoulos A, Wright E, Vlahopoulou K, Cornic L, Schoof N, Maher TM. The burden of illness of idiopathic pulmonary fibrosis: a comprehensive evidence review. Pharmacoeconomics. 2018;36(7):779–807. doi:10.1007/s40273-018-0631-8
10. Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res. 2018;19(1):32. doi:10.1186/s12931-018-0730-2
11. Lederer DJ, Martinez FJ, Longo DL. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378(19):1811–1823. doi:10.1056/NEJMra1705751
12. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9(1):157–179. doi:10.1146/annurev-pathol-012513-104706
13. Basset F, Ferrans VJ, Soler P, Takemura T, Fukuda Y, Crystal RG. Intraluminal fibrosis in interstitial lung disorders. Am J Pathol. 1986;122(3):443–461.
14. Lin C, Borensztajn K, Spek CA. Targeting coagulation factor receptors - protease-activated receptors in idiopathic pulmonary fibrosis. J Thromb Haemost. 2017;15(4):597–607. doi:10.1111/jth.13623
15. Barratt SL, Creamer A, Hayton C, Chaudhuri N. Idiopathic Pulmonary Fibrosis (IPF): an overview. J Clin Med. 2018;7(8):201. doi:10.3390/jcm7080201
16. Cosgrove GP, Brown KK, Schiemann WP, et al. Pigment epithelium–derived factor in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2004;170(3):242–251. doi:10.1164/rccm.200308-1151OC
17. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776–1784. doi:10.1172/JCI200320530
18. King TE, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
19. Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. doi:10.1056/NEJMoa1103690
20. Kaneko M, Inoue H, Nakazawa R, et al. Pirfenidone induces intercellular adhesion molecule-1 (ICAM-1) down-regulation on cultured human synovial fibroblasts. Clin Exp Immunol. 1998;113(1):72–76. doi:10.1046/j.1365-2249.1998.00618.x
21. Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590(1–3):400–408. doi:10.1016/j.ejphar.2008.06.046
22. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47(1):243–253. doi:10.1183/13993003.00026-2015
23. Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1434–1445. doi:10.1183/09031936.00174914
24. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–1727. doi:10.1056/NEJMoa1908681
25. Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE. Drug treatment of idiopathic pulmonary fibrosis: systematic review and network meta-analysis. Chest. 2016;149(3):756–766. doi:10.1016/j.chest.2015.11.013
26. Loveman E, Copley VR, Scott DA, Colquitt JL, Clegg AJ, O’Reilly KM. Comparing new treatments for idiopathic pulmonary fibrosis – a network meta-analysis. BMC Pulm Med. 2015;15(1):37. doi:10.1186/s12890-015-0034-y
27. Hayton C, Chaudhuri N. Managing idiopathic pulmonary fibrosis: which drug for which patient? Drugs Aging. 2017;34(9):647–653. doi:10.1007/s40266-017-0488-0
28. Barratt SL, Mulholland S, Al Jbour K, et al. South–West of England’s experience of the safety and tolerability pirfenidone and nintedanib for the treatment of Idiopathic Pulmonary Fibrosis (IPF). Front Pharmacol. 2018;9:1480. doi:10.3389/fphar.2018.01480
29. Matralis AN, Afantitis A, Aidinis V. Development and therapeutic potential of autotaxin small molecule inhibitors: from bench to advanced clinical trials. Med Res Rev. 2019;39(3):976–1013. doi:10.1002/med.21551
30. Aoki J. Mechanisms of lysophosphatidic acid production. Semin Cell Dev Biol. 2004;15(5):477–489. doi:10.1016/j.semcdb.2004.05.001
31. Baker DL, Morrison P, Miller B, et al. Plasma lysophosphatidic acid concentration and ovarian cancer. JAMA. 2002;287(23):3081–3082. doi:10.1001/jama.287.23.3081
32. Fukami K, Takenawa T. Phosphatidic acid that accumulates in platelet-derived growth factor-stimulated Balb/c 3T3 cells is a potential mitogenic signal. J Biol Chem. 1992;267(16):10988–10993.
33. Valet P, Pagès C, Jeanneton O, et al. Alpha2-adrenergic receptor-mediated release of lysophosphatidic acid by adipocytes. A paracrine signal for preadipocyte growth. J Clin Invest. 1998;101(7):1431–1438. doi:10.1172/JCI806
34. Fourcade O, Simon MF, Viodé C, et al. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell. 1995;80(6):919–927. doi:10.1016/0092-8674(95)90295-3
35. Stracke ML, Krutzsch HC, Unsworth EJ, et al. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J Biol Chem. 1992;267(4):2524–2529.
36. Giganti A, Rodriguez M, Fould B, et al. Murine and human autotaxin alpha, beta, and gamma isoforms: gene organization, tissue distribution, and biochemical characterization. J Biol Chem. 2008;283(12):7776–7789. doi:10.1074/jbc.M708705200
37. Ninou I, Magkrioti C, Aidinis V. Autotaxin in pathophysiology and pulmonary fibrosis. Front Med. 2018;5:180. doi:10.3389/fmed.2018.00180
38. Barbayianni E, Kaffe E, Aidinis V, Kokotos G. Autotaxin, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog Lipid Res. 2015;58:76–96. doi:10.1016/j.plipres.2015.02.001
39. Dusaulcy R, Rancoule C, Grès S, et al. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J Lipid Res. 2011;52(6):1247–1255. doi:10.1194/jlr.M014985
40. van Meeteren LA, Ruurs P, Stortelers C, et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol. 2006;26(13):5015–5022. doi:10.1128/MCB.02419-05
41. Tanaka M, Okudaira S, Kishi Y, et al. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem. 2006;281(35):25822–25830. doi:10.1074/jbc.M605142200
42. Benesch MGK, MacIntyre ITK, McMullen TPW, Brindley DN. Coming of age for autotaxin and lysophosphatidate signaling: clinical applications for preventing, detecting and targeting tumor-promoting inflammation. Cancers (Basel). 2018;10(3):73. doi:10.3390/cancers10030073
43. Oikonomou N, Mouratis MA, Tzouvelekis A, et al. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2012;47(5):566–574. doi:10.1165/rcmb.2012-0004OC
44. Yukiura H, Kano K, Kise R, Inoue A, Aoki J, Yue J. Autotaxin overexpression causes embryonic lethality and vascular defects. PLoS One. 2015;10(5):e0126734. doi:10.1371/journal.pone.0126734
45. Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci U S A. 2000;97(24):13384–13389. doi:10.1073/pnas.97.24.13384
46. Funke M, Knudsen L, Lagares D, et al. Lysophosphatidic acid signaling through the lysophosphatidic acid-1 receptor is required for alveolarization. Am J Respir Cell Mol Biol. 2016;55(1):105–116. doi:10.1165/rcmb.2015-0152OC
47. Contos JJ, Ishii I, Fukushima N, et al. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2). Mol Cell Biol. 2002;22(19):6921–6929. doi:10.1128/MCB.22.19.6921-6929.2002
48. Ye X, Hama K, Contos JJA, et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature. 2005;435(7038):104–108. doi:10.1038/nature03505
49. Mouratis MA, Magkrioti C, Oikonomou N, et al. Autotaxin and endotoxin-induced acute lung injury. PLoS One. 2015;10(7):e0133619. doi:10.1371/journal.pone.0133619
50. Brindley DN, Lin FT, Tigyi GJ. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim Biophys Acta. 2013;1831(1):74–85. doi:10.1016/j.bbalip.2012.08.015
51. Castelino FV, Seiders J, Bain G, et al. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 2011;63(5):1405–1415. doi:10.1002/art.30262
52. Pradère JP, Klein J, Grès S, et al. LPA1 receptor activation promotes renal interstitial fibrosis. J Am Soc Nephrol. 2007;18(12):3110–3118. doi:10.1681/ASN.2007020196
53. Kaffe E, Katsifa A, Xylourgidis N, et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology. 2017;65(4):1369–1383. doi:10.1002/hep.28973
54. Nakagawa H, Ikeda H, Nakamura K, et al. Autotaxin as a novel serum marker of liver fibrosis. Clin Chim Acta. 2011;412(13–14):1201–1206. doi:10.1016/j.cca.2011.03.014
55. Nakagawa Y, Masuda M, Shiihara H, et al. Successful pulmonary thromboendarterectomy for chronic thromboembolic pulmonary hypertension associated with anticardiolipin antibodies: report of a case. Surg Today. 1992;22(6):548–552. doi:10.1007/BF00308902
56. Ganguly K, Stoeger T, Wesselkamper SC, et al. Candidate genes controlling pulmonary function in mice: transcript profiling and predicted protein structure. Physiol Genomics. 2007;31(3):410–421. doi:10.1152/physiolgenomics.00260.2006
57. Montesi SB, Mathai SK, Brenner LN, et al. Docosatetraenoyl LPA is elevated in exhaled breath condensate in idiopathic pulmonary fibrosis. BMC Pulm Med. 2014;14:5. doi:10.1186/1471-2466-14-5
58. Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14(1):45–54. doi:10.1038/nm1685
59. Nikolaou A, Ninou I, Kokotou MG, et al. Hydroxamic acids constitute a novel class of autotaxin inhibitors that exhibit in vivo efficacy in a pulmonary fibrosis model. J Med Chem. 2018;61(8):3697–3711. doi:10.1021/acs.jmedchem.8b00232
60. Jiang G, Madan D, Prestwich GD. Aromatic phosphonates inhibit the lysophospholipase D activity of autotaxin. Bioorg Med Chem Lett. 2011;21(17):5098–5101. doi:10.1016/j.bmcl.2011.03.068
61. Kato K, Ikeda H, Miyakawa S, et al. Structural basis for specific inhibition of autotaxin by a DNA aptamer. Nat Struct Mol Biol. 2016;23(5):395–401. doi:10.1038/nsmb.3200
62. Black KE, Berdyshev E, Bain G, et al. Autotaxin activity increases locally following lung injury, but is not required for pulmonary lysophosphatidic acid production or fibrosis. FASEB J. 2016;30(6):2435–2450. doi:10.1096/fj.201500197R
63. Desroy N, Housseman C, Bock X, et al. Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)thiazole-5-carbonitrile (GLPG1690), a first-in-class autotaxin inhibitor undergoing clinical evaluation for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2017;60(9):3580–3590. doi:10.1021/acs.jmedchem.7b00032
64. Huang LS, Fu P, Patel P, et al. Lysophosphatidic acid receptor-2 deficiency confers protection against bleomycin-induced lung injury and fibrosis in mice. Am J Respir Cell Mol Biol. 2013;49(6):912–922. doi:10.1165/rcmb.2013-0070OC
65. Swaney JS, Chapman C, Correa LD, et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br J Pharmacol. 2010;160(7):1699–1713. doi:10.1111/j.1476-5381.2010.00828.x
66. Munger JS, Huang X, Kawakatsu H, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–328. doi:10.1016/S0092-8674(00)80545-0
67. Xu MY, Porte J, Knox AJ, et al. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am J Pathol. 2009;174(4):1264–1279. doi:10.2353/ajpath.2009.080160
68. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208(7):1339–1350. doi:10.1084/jem.20110551
69. Yin Z, Watsky MA. Chloride channel activity in human lung fibroblasts and myofibroblasts. Am J Physiol Lung Cell Mol Physiol. 2005;288(6):L1110–L1116. doi:10.1152/ajplung.00344.2004
70. Coornaert B, Duys I, VdS J, Van der Aar EBH. Autotaxin inhibitor GLPG1690 affects TGF Β-induced production of the pro-fibrotic mediators CTGF, IL-6 and ET-1 in fibroblasts. Am J Respir Crit Care Med. 2020;201. A2404.
71. Van der Aar EM, Farad L, Desrivot J, et al. Favorable Human Safety, Pharmacokinetics and Pharmacodynamics of the Autotaxin Inhibitor GLPG1690, a Potential New Treatment in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2016;193:A2701.
72. van der Aar E, Desrivot J, Dupont S, et al. Safety, pharmacokinetics, and pharmacodynamics of the autotaxin inhibitor GLPG1690 in healthy subjects: phase 1 randomized trials. J Clin Pharmacol. 2019;59(10):1366–1378. doi:10.1002/jcph.1424
73. Maher TM, van der Aar EM, Van de Steen O, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir Med. 2018;6(8):627–635. doi:10.1016/S2213-2600(18)30181-4
74. Richeldi L, Cottin V, Du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS® trials. Respir Med. 2016;113:74–79. doi:10.1016/j.rmed.2016.02.001
75. Maher TM, Kreuter M, Lederer DJ, et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir Res. 2019;6(1):e000422. doi:10.1136/bmjresp-2019-000422
76. Lee G, Kang SU, Ryou JH, JJ L, YH L. BBT-877, a potent autotaxin inhibitor in clinical development to treat idiopathic pulmonary fibrosis. Eur Respir J. 2019;54(Suppl.63):PA1293.
77. Palmer SM, Snyder L, Todd JL, et al. Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest. 2018;154(5):1061–1069. doi:10.1016/j.chest.2018.08.1058
78. Rosen G, Sivaraman L, Cheng P, et al. LPA1 antagonists BMS-986020 and BMS-986234 for idiopathic pulmonary fibrosis: preclinical evaluation of hepatobiliary homeostasis. Eur Respir J. 2017;50:PA1038. doi:10.1183/13993003.00711-2017
79. Gill MW, Lakshmi S, Cheng PTW, et al. BMS-986278, an LPA1 receptor antagonist for idiopathic pulmonary fibrosis: preclinical assessments of potential hepatobiliary toxicity. Am J Respir Crit Care Med. 2019;199:A5882.
80. Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390(10103):1685–1699. doi:10.1016/S0140-6736(17)30933-9
81. Mazereeuw-Hautier J, Gres S, Fanguin M, et al. Production of lysophosphatidic acid in blister fluid: involvement of a lysophospholipase D activity. J Invest Dermatol. 2005;125(3):421–427. doi:10.1111/j.0022-202X.2005.23855.x
82. Tokumura A, Carbone LD, Yoshioka Y, et al. Elevated serum levels of arachidonoyl-lysophosphatidic acid and sphingosine 1-phosphate in systemic sclerosis. Int J Med Sci. 2009;6(4):168–176. doi:10.7150/ijms.6.168
83. Castelino FV, Bain G, Pace VA, et al. An autotaxin/lysophosphatidic acid/interleukin-6 amplification loop drives scleroderma fibrosis. Arthritis Rheumatol. 2016;68(12):2964–2974. doi:10.1002/art.39797
84. Khanna D, Denton CP, Lin CJF, et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann Rheum Dis. 2018;77(2):212–220. doi:10.1136/annrheumdis-2017-211682
85. Allanore Y, Distler O, Jagerschmidt A, et al. Lysophosphatidic acid receptor 1 antagonist SAR100842 for patients with diffuse cutaneous systemic sclerosis: a double-blind, randomized, eight-week placebo-controlled study followed by a sixteen-week open-label extension study. Arthritis Rheumatol. 2018;70(10):1634–1643. doi:10.1002/art.40547
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.