")
Back to Journals » Journal of Pain Research » Volume 14
Inhibition of the Soluble Epoxide Hydrolase as an Analgesic Strategy: A Review of Preclinical Evidence
Authors Wang Y, Wagner KM, Morisseau C , Hammock BD
Received 13 October 2020
Accepted for publication 8 December 2020
Published 13 January 2021 Volume 2021:14 Pages 61—72
DOI https://doi.org/10.2147/JPR.S241893
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Michael Schatman
Yuxin Wang, Karen M Wagner, Christophe Morisseau, Bruce D Hammock
Department of Entomology and Nematology, UC Davis Comprehensive Cancer Center, University of California Davis, Davis, CA 95616, USA
Correspondence: Bruce D Hammock Email [email protected]
Abstract: Chronic pain is a complicated condition which causes substantial physical, emotional, and financial impacts on individuals and society. However, due to high cost, lack of efficacy and safety problems, current treatments are insufficient. There is a clear unmet medical need for safe, nonaddictive and effective therapies in the management of pain. Epoxy-fatty acids (EpFAs), which are natural signaling molecules, play key roles in mediation of both inflammatory and neuropathic pain sensation. However, their molecular mechanisms of action remain largely unknown. Soluble epoxide hydrolase (sEH) rapidly converts EpFAs into less bioactive fatty acid diols in vivo; therefore, inhibition of sEH is an emerging therapeutic target to enhance the beneficial effect of natural EpFAs. In this review, we will discuss sEH inhibition as an analgesic strategy for pain management and the underlying molecular mechanisms.
Keywords: epoxy fatty acids, chronic pain, molecular mechanisms
Introduction
Pain is a critical signal and a survival mechanism, but enhanced and persistent pain is an unpleasant sensation and emotional experience which has a profound impact on individuals and society.1,2 There are approximately 100 million Americans suffering from chronic pain, with an associated $560–635 billion yearly cost in direct medical expenses and lost productivity.3 In 2016, approximately 20% of US adults had chronic pain (approximately 50 million), and 8% of US adults (approximately 20 million) had high-impact chronic pain according to the Centers of Disease Control and Prevention (CDC).4 Opioids are major pharmaceutical treatments available to control pain; however, they have serious side effects, leading to increasing risks for abuse and overdose-related deaths.5 We need additional analgesic agents that can be integrated into multimodal pain control strategies.6 Therefore, pain management research has become one of the top priorities in the US and developing new therapeutic approaches for pain management that are both effective and safe is practically important for our society.
The natural purpose of pain is to protect body from damage or potentially damaging situation.7 However, chronic pain is not always related to tissue damage and does not always serve a protective function.8 Based on the biological and physiological processes involved, pain is classified as either nociceptive, neuropathic, or inflammatory.9 Nociceptive pain can be triggered by exposure to extreme heat, cold or noxious pressure.10 Neuropathic pain, which is caused by a lesion or disease of the somatosensory nervous system may occur spontaneously in the absence of stimuli or be evoked by sensory stimuli inducing hyperalgesia and allodynia.11 Inflammation is characterized by redness, heat, swelling, pain or hypersensitivity, and loss of function usually occurs subsequent to injury and involves the release of cytokines and immune cell infiltration.12 Since different forms of pain involve a variety of distinct biological processes, a better understanding of the molecular mechanisms underlying the pain is key for the development of more effective and safe therapies in the near future.
Eicosanoids are the metabolites of arachidonic acid (ARA) and related unsaturated fatty acids produced by three oxidative pathways, cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP450). They are important lipid signaling molecules involved notably in the regulation of inflammation and pain.13–15 While the COX and LOX pathways are well studied, the CYP450 is less understood and is at the center of this review. In this pathway, polyunsaturated fatty acids (PUFAs) including linoleic acid (LA), α-linolenic acid (ALA), dihomo-γ-linolenic acid (DGLA), arachidonic acid (ARA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and others are metabolized by CYP monooxygenases, especially CYP2C and CYP2J isoforms, to form a mixture of monohydroxy-fatty acids (hydroxyeicosatetraenoic acids (HETEs) from ARA) and epoxy-fatty acids (EpFAs; such as epoxy-eicosatrienoic acids (EETs) from ARA) with diverse biological actions, especially in inflammatory related diseases (Figure 1).13,16–18 In this review, we will discuss the roles of CYP450 and their metabolites in the pathology of chronic pain and the underlying mechanisms of action of these metabolites.
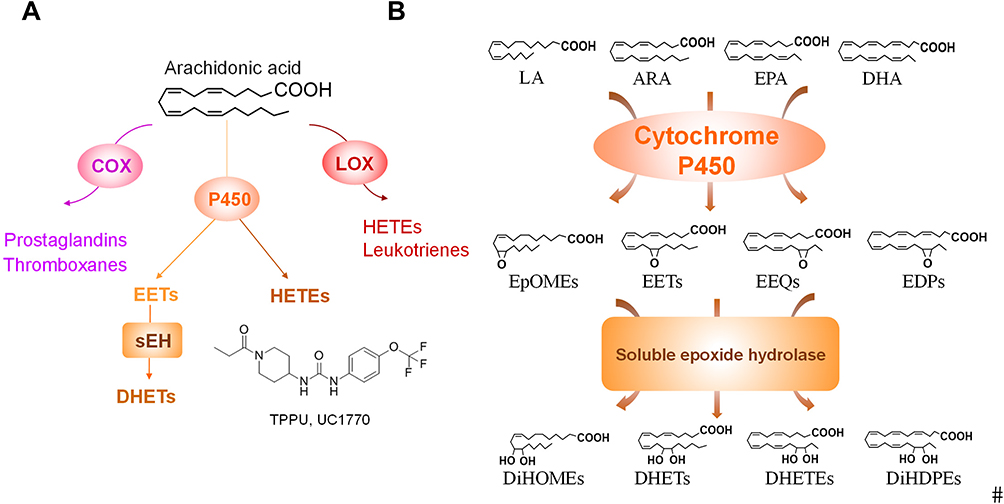 |
Figure 1 (A) Metabolism of ARA by COX, LOX, and CYP enzymes leads to formation of oxylipin metabolites. The structure of the sEHI TPPU is shown. (B) The CYP/sEH pathway that produces epoxy-fatty acids and corresponding diols. For simplicity, only one regioisomer of the epoxides and diols are shown here. Abbreviations: ARA, arachidonic acid; COX, cyclooxygenase; LOX, lipoxygenase; CYP, cytochromes P450; EETs, epoxyeicosatrienoic acid; HETEs, hydroxyeicosatetraenoic acids; DHETs, dihydroxy-eicosatrienoic acids; LA, linoleic acid; EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid; EpOMEs, epoxy-octadecenoic acid; EEQs, epoxy-eicosatetraenoic acid; EDPs, epoxy-docosapentaenoic acid; DiHOMEs, dihydroxy-octadecenoic acid; DiHETEs, dihydroxyicosa-tetraenoic acid; DiHDPEs, dihydroxy-docosapentaenoic acid. |
Soluble Epoxide Hydrolase and Pain
The Effect of Epoxy-fatty Acids in Pain Model
The EpFAs, especially EETs, which function primarily as both autocrine and paracrine signaling molecules, have well described beneficial effects on multiple cardiovascular diseases, the renal system, angiogenesis, inflammation, and cancer.19–21 Using an animal model of inflammatory pain, the total oxylipin concentrations were measured both in the spinal cord and brain of rats after base hydrolysis of the lipid esters. EpFAs, especially from ARA and DHA, but neither the parent fatty acid nor the corresponding diols, selectively modulate nociceptive pathophysiology, and spinal administration of epoxy-docosapentaenoic acids (EpDPE) reduces both mechanical and thermal pains associated with inflammation, supporting an important function of EpFAs in modulating nociceptive signaling.22 Additional studies showed that EpFAs are effective in both inflammatory and neuropathic pain models, suggesting them as potential novel therapeutics for pain management.23–25 However, in vivo, the EpFAs are rapidly metabolized by soluble epoxide hydrolase (sEH) to generate the corresponding and less-bioactive, even pro-inflammatory, dihydroxy-eicosatrienoic acids (DHETs) (Figure 1B).26–29 Therefore, sEH inhibitors (sEHIs) were developed to increase in vivo EpFAs levels, and thus reduce blood pressure, improve insulin sensitivity, and decrease inflammation.28,30–33 The sEH is considered an emerging therapeutic target for enhancing the beneficial function of EpFAs in numerous diseases, including cardiovascular diseases, inflammatory bowel diseases, hypertension, and metabolic disorders, which have inflammation as a common underlying cause.28,30–33
The Effect of sEHIs in Pain Model
In an inflammatory pain model induced with intraplantar injection of 10 μg lipopolysaccharides (LPS) in one hind paw of rats, using an sEHI 1-trifluoromethoxyphenyl-3-(1-acetylpiperidin-4-yl)-urea (TPAU) through intraplantar injection significantly blocked inflammatory pain in a dose-dependent manner.24 Moreover, an sEH metabolite 12,13-dihydroxy-9Z-octadecenoic acid (12,13-DiHOME) was increased in peripheral nervous tissue during acute zymosan- and complete Freund’s adjuvant (CFA)-induced inflammatory pain.34 In this CFA-induced inflammatory pain model, oral administration of 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl (TPPU) reduces 12.13-DiHOME concentrations and reduces zymosan- and CFA-induced thermal hyperalgesia in vivo.34 These results showed the analgesia effect of sEHIs in inflammatory pain. In addition, compared to the traditional nonsteroidal anti-inflammatory drug celecoxib, sEHIs are superior and have better efficacy in both diabetic neuropathy and inflammatory pain models.35
Wagner et al. demonstrated that compared with gabapentin, subcutaneously injection of the sEHI trans-4-[4-(3-trifluoromethoxyphenyl-1-ureido)-cyclohexyloxy]-benzoic acid (t-TUCB) elicited a similar degree of withdrawal threshold improvement without the same degree of spontaneous locomotion decline in mice with neuropathic pain.36 In diabetic Akita mice (Ins2Akita or Ins2C96Y), which progress naturally and are more similar to the human disease state than chemical ablation of beta islet cells. The results showed t-TUCB is an analgesic in diabetic neuropathy, and this effect is related to sexual dimorphism since the female mice are less susceptible to the diabetic phenotype.37 These results indicate the sEHI has analgesic effects with limited side effects in diabetic neuropathy pain.
Synergistic Effect of sEHI with Other Pharmaceutical Inhibitors in Pain
Besides potent effects from sEHI itself, recent research showed combinations of sEHI and other enzyme inhibitors might achieve greater analgesic efficacy therapeutically. For example, the combination treatment of nonsteroidal anti-inflammatory drugs (NSAIDs) and the sEHI 12-(3-adamantan-1-yl-ureido)dodecanoic acid n-butyl ester (AUDA-nBE) produced significantly beneficial effects for alleviating LPS-induced inflammatory pain in mice.38 The NSAIDs and sEHI combination therapy also reduced COX-2 protein expression and shifted oxylipin metabolomic profiles,38 suggesting that this therapy has efficacy in decreasing inflammation but also decreased side effects of NSAIDs in cardiovascular and gastrointestinal tract complications.38–40 A COX-2/sEH dual inhibitor, 4-(5-phenyl-3-{3-[3-(4-trifluoromethylphenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide (PTUPB) exhibited antiallodynic activity that was more effective than the same dose of either a COX-2 inhibitor (celecoxib) or a sEH inhibitor t-AUCB alone, as well as co-administration of both inhibitors in a nociceptive behavioral assay.41 In addition, fatty acid amide hydrolase (FAAH) is another enzyme catalyzing the hydrolysis of bioactive lipid mediators—fatty acid ethanolamides (FAEs). Previous results demonstrated combinations of a sEHI, TPPU, and FAAH inhibitor, URB937, showed high antihyperalgesia activity in two pain models: carrageenan-induced hyperalgesia in mice and streptozocin-induced allodynia in rats, revealing a possible functional crosstalk between FAEs and EpFAs in regulating pain responses.42 Moreover, phosphodiesterase-4 (PDE-4)-targeted therapies have shown promise for treating patients with a variety of autoimmune diseases.43 Co-inhibition of sEH and PDE-4, greatly increases the level of EpFAs and is thus more efficient at reducing acute pain perception.44 A novel PDE-4/sEH dual inhibitor N-(4-methoxy-2-(trifluoromethyl)benzyl)-1-propionylpiperidine-4-carboxamide (MPPA) at 3 mg/kg (oral administration) reduced LPS-induced inflammatory pain. MPPA also does not alter self-motivated exploration of rats with inflammatory pain or the withdrawal latency in control rats, suggesting that MPPA has good efficacy together with limited off-target effects.45
The Underlying Mechanisms of sEHI/EpFAs in Pain Management
Cyclic Adenosine Monophosphate (cAMP) Signaling Pathway in Pain
Cyclic adenosine 3′,5′-monophosphate (cAMP) was the first identified second messenger and plays a fundamental role in many cellular responses to hormones and neurotransmitters.46–48 The intracellular levels of cAMP are regulated by the balance between two enzymes: adenylyl cyclase (AC), which catalyzes cAMP formation from ATP,49 and cyclic nucleotide phosphodiesterase (PDE) that degrades intracellular cyclic nucleotides.50 PDE inhibitors have been shown as therapeutic approach to neuroprotection, repair, and cardiovascular system.51,52 Rolipram, a selective PDE-4 inhibitor and theophylline, a nonspecific PDE inhibitor exerted dose-dependent analgesic and anti-inflammatory effect against acetic acid-induced writhing in mice and carrageenan-induced paw edema in rats.53
Rolipram induced artificially elevated cAMP in healthy mice, while co-administration with the sEHI 1-trifluoromethoxyphenyl-3-(1-acetylpiperidin-4-yl) urea (TPAU) largely blunted pain-related behavior, which indicate the analgesic effect of sEH inhibitor and PDE-4 inhibitor.44 These results indicate the analgesic effect of sEHIs is dependent on cAMP. In another study, EETs or sEHI lead to antihyperalgesia and was correlated to upregulation of, steroidogenic acute regulatory protein (StARD1), a carrier protein which assists neuro-steroid production.54 In summary, these results further give mechanistic evidence showing the analgesic effect of sEHIs through cAMP signaling pathway (Figure 2).
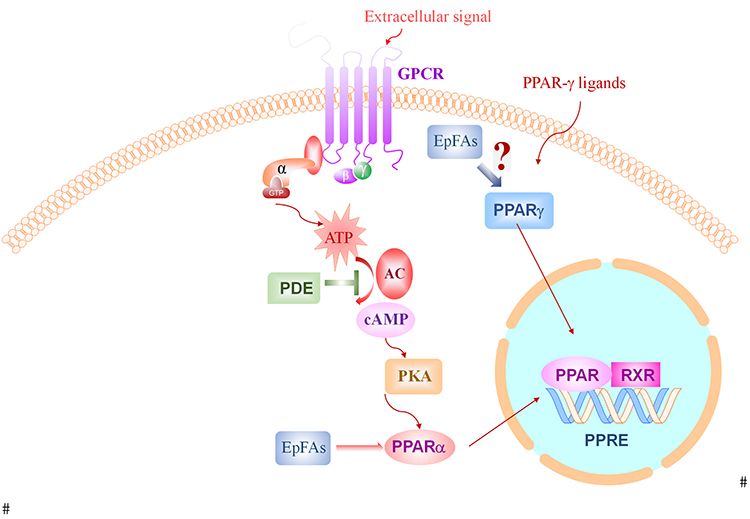 |
Figure 2 The effect of sEH inhibition and EpFAs on cAMP-PPAR signaling pathways. Abbreviations: GPCR, G-protein-coupled receptors; PPAR, peroxisome proliferator-activated receptor gamma; EpFAs, epoxy fatty acids; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; PDE, phosphodiesterase; RXR, retinoid X receptor; PPRE, peroxisome proliferator hormone response elements. |
PPARs Signaling in Pain
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors belonging to a nuclear hormone receptor superfamily, which contains three isoforms: PPARα, PPARβ/δ and PPARγ.55,56 The three PPARs share a high homology but differ in tissue distribution and ligand specificity.57 PPARs primary function as important fatty acid sensors which not only regulate lipid, carbohydrates, and amino acid metabolism, but also play key roles in various pathophysiology processes.55 Extensive research showed PPARs may also involve in the control of the nociceptive response and neuropathic pain.58,59 There is evidence showing cAMP is the major stimulator of PPAR activity and cAMP signaling pathway modulates PPAR function, possibly by transactivation,60,61 suggesting the link between cAMP and PPAR signaling pathways. Interestingly PPAR agonists are potent inducers of the sEH message and protein.
A relevant study on PPARs and pain investigated the activation of PPAR in the rat spinal cord following subcutaneous injection of CFA into a hind paw.62 The results showed only the PPARα isoform was activated using electrophoretic mobility-shift assay (EMSA) method. LoVerme et al. showed in mice that PPARα agonists suppress pain behaviors induced by tissue injury, nerve damage, or inflammation.63 In addition, PPARα–/– female mice are hypersensitive to the cold, mechanical allodynia, and heat hyperalgesia,64 suggesting genetic ablation of PPARα is involved in neuropathic and visceral nociception.64 Leukotriene B4, a potent agent that initiates, coordinates, and amplifies the inflammatory response, is an activating ligand for the transcription factor PPARα.65 These results suggest the critical role of PPARα in the control of nociception and inflammation. Thus, further studies of combined effects on sEHI and PPARα agonist in pain preclinical models are needed and development of this novel class of compounds could represent a useful new pharmacological approach for the pain relief.
PPARγ is another subtype of PPAR, which is present in several tissues and cell types.66 Intrathecally administered PPARγ agonists dose-dependently decreased mechanical and cold hypersensitivity in the rats,58 demonstrating the important role of PPARγ in the neuropathic pain. Further studies demonstrated that activation of PPARγ has beneficial effects of modification of astrocyte metabolism and mitochondrial function which are important in inflammation.67,68 In addition, the PPARγ agonist, rosiglitazone, attenuated CFA-induced inflammatory pain through induction of heme oxygenase (HO)-1, leading to the differentiation of pro-inflammatory M1 macrophages to anti-inflammatory M2 phenotype.69 In an angiotensin-II (AngII) induced cardiac hypertrophy model, sEH is upregulated by AngII. Rosiglitazone is a potent sEH inducer and the protective role of PPARγ activation in AngII-induced cardiac hypertrophy is partially through downregulating sEH.70,71 Thus, the beneficial actions of rosiglitazone should be enhanced and some of its side effects reduced by co-administration with sEHI since the combined administration of both pharmacological agents rosiglitazone and the sEH inhibitor t-AUCB led to synergistic improvement of vascular function and reduced fibrotic kidney damage.72
EETs, together with the sEHI 12-(3-adamantan-1-yl-ureido) dodecanoic acid (AUDA), increased PPARγ transcription activity in endothelial cells and 3T3-L1 preadipocytes and PPARγ antagonist GW9662 abolished the EET/AUDA-mediated anti-inflammatory effect, indicating PPARγ is an effector of EETs.73 Further study demonstrated that dual PPARγ/sEH inhibitor RB394 showed the ability to blunt diabetic complications such as hypertension, insulin resistance, hyperlipidemia, and kidney injury in metabolic syndrome modeled in obese spontaneously hypertensive (SHROB) rats and obese diabetic Zucker fatty/spontaneously hypertensive heart failure F1 hybrid (ZSF1) rats.74 Another study showed the dual inhibitor RB394 or combination of sEHI and PPARγ agonist significantly prevented renal fibrosis development by preventing renal inflammation and oxidative stress.75 Interestingly the sEHI will of course counter the sEHI induction and other possible deleterious side effects of high dose PPARγ agonists.76 Kim et al. demonstrated that sEHI t-TUCB could promote anti-inflammatory effects in ureteral obstruction in mice, the mechanism is mainly through increased levels of EETs and inhibition the PPARγ reduction.77 Altogether, these results indicate that PPARγ is an important effector in the anti-inflammatory effect of sEHI, while more evidence is still needed for the mechanism study of sEHI in the pain management through PPAR signaling pathways (Figure 2).
The Transient Receptor Potential (TRP) Superfamily in Pain
The superfamily of TRP channels play critical roles in the responses to the major classes of external stimuli, including light, sound, chemicals, temperature, and touch.78 Mutations in several TRP genes have been implicated in pain pathological states.79 Most TRPs are nonselective cation channels, only a few are highly Ca2+ selective, or permeable for highly hydrated Mg2+ ions.79
Transient receptor potential ankyrin 1 (TRPA1) is the most well-studied pain regulator among TRP superfamily, which is one of the Ca2+-permeable cation ion channels involved in the transduction of potentially harmful stimuli and in amplification of nociceptive transmission in their central terminals.80–83 Studies revealed TRPA1 is a potential target in pain relief. A TRPA1 selective antagonist significantly reduced mechanical hyperalgesia evaluated by the von Frey assay and completely inhibited the noxious cold hyperalgesia in CFA induced persistent inflammation in mice.80 Furthermore, TRPA1 also plays key roles in the inflammatory pain, neuropathic pain and migraine.84 Both genetic deletion of Trpa1 and pharmacological inhibition of TRPA1 abrogated pain-like behaviors in mice.85
In addition to TRPA1, transient receptor potential cation channel subfamily V member 1 (TRPV1) is another Ca2+ permeant nonselective member of TRP family which has been implicated in a variety of cellular and physiological processes, including noxious physical and chemical stimuli detection, making it one of the promising targets for pain-relieving drugs.86,87 TRPV1−/− mice showed no vanilloid-evoked pain behavior in the detection of painful heat and showed little thermal hypersensitivity in the inflammation, and TRPV1−/− mice showed an attenuated fever in response to LPS.88,89 These results conclude TRPV1 is essential in the inflammatory thermal hyperalgesia, nociception, and pain sensation. Finally, several TRPV1 agonists such as JNJ-39,439,335, NEO6860, and ABT-102 have been in clinical trials targeting pain relief.90–92 Altogether, these results suggest examination of TRPV1 as a mechanistic target for pain treatment.
sEH was shown to regulate pain via TRP channels. The sEH enzyme has been reported as co-localized with TRPV1 in the primary trigeminal ganglion neurons (TGNs).93 Pretreatment with 10 μM EETs antagonist 14.15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE) attenuated the calcitonin gene-related peptide (CGRP) release, which is a marker of neurogenic inflammation.93 The CGRP release is induced by TRPV1 agonist capsaicin or K+, but sEHI AUDA did not have this effect,93 suggesting that there are inadequate levels of EETs released to be stabilized by AUDA. These data suggest EETs may act as intracellular regulators of neuropeptide release, which may have important clinical implications for treatment of neurogenic inflammation. Further investigation into the therapeutic potential of sEHI through TRP channels is needed.
Endoplasmic Reticulum (ER) Stress Signaling Pathway in Pain
The endoplasmic reticulum (ER) is an organelle in which newly synthesized secretory and transmembrane proteins are assembled and folded into their correct tertiary structures.94 However, protein misfolding caused by various stimuli and gene mutations, leads to the disruption of ER function and activation of ER stress signaling pathway. Eukaryotic cells have developed an evolutionarily conserved adaptive mechanism called unfolded protein response (UPR), whose purpose is to clear unfolded proteins, promote proper folding by increased chaperones and reduced protein synthesis, and restore ER homeostasis.95 The UPR influences cellular metabolism through diverse mechanisms, including calcium and lipid transfer, which are key involvement in the pathogenesis of diseases, including pain, neurodegeneration, and cardiovascular disease.96–98 When unfolded proteins accumulate in the ER, the N-terminus in the lumen of the ER chaperone Grp78 releases transmembrane ER proteins involved in inducing the UPR to prevent their aggregation. However, when misfolded proteins accumulate, Grp78 releases, allowing aggregation of these transmembrane signaling proteins, launching and activating the UPR.99 The UPR is distinguished by the action of three signaling proteins named IRE1α (inositol-requiring protein-1α), PERK (protein kinase RNA (PKR)-like ER kinase), and ATF6 (activating transcription factor 6).95
The ER stress signaling pathway has been demonstrated as playing key roles in the pathogenesis of pain. The IRE1α–unspliced X-box-binding protein 1 (XBP1) axis operates as a crucial mediator of eicosanoid metabolism and prostaglandin synthesis in myeloid immune cells by promoting the expression of both COX-2 and microsomal prostaglandin E synthase-1 (mPGES-1), and genetic ablation or pharmacological inhibition of this pathway diminishes pain-related behaviors in mice.100 Other research showed that an IRE1α small interfering RNA (siRNA) improved the neurological morphology and reduced diabetic peripheral neuropathy (DPN) in rats.101 It also rescued ER stress-related apoptosis in the sciatic nerve,101 indicating IRE1α–XBP1 signaling may be helpful for the improvement of pain management. In addition, Lupachyk et al. examined the role of ER stress signaling in the development of peripheral neuropathy in streptozotocin (STZ)-induced diabetic rodents and found two structurally dissimilar chemical chaperones (trimethylamine oxide [TMAO] and 4-phenylbutyric acid [4-PBA]), which can counteract ER stress by promoting normal protein folding, significantly suppressed ER stress marker proteins whose upregulation was induced by STZ,102,103 reduced thermal and mechanical responses, and enhanced sensitivity to touch with diabetes.103 They also observed the neuropathic effects of CCAAT-enhancer-binding proteins (C/EBP) homologous protein (CHOP), one of the components of the ER stress-mediated apoptosis pathway in the DPN.104 The results showed genetic ablation of CHOP showed attenuation of motor and sensory nerve conduction velocity deficits, thermal hypoalgesia, and intraepidermal nerve fiber loss, while diabetes-induced mechanical hypoalgesia and tactile allodynia remained at similar levels in both CHOP−/− and wild-type mice,102 suggesting different aspects of ER stress and the UPR were targeted in diabetic neuropathy.
Numerous studies showed sEH is a physiological modulator of ER stress signaling involved in many disorders.105–107 Thus, the sEH enzyme is a nonchannel, non-neurotransmitter therapeutic and well characterized target for pain.108 Both pain and ER stress markers are elevated in peripheral nervous system of type I diabetic rats. Further results showed TPPU, a widely used potent sEH inhibitor, blocks pain-related behavior and suppresses markers of the ER stress signaling pathway (p-PERK, p-IRE1α, and cleaved-ATF6).108,109 In addition, TPPU reversed the tunicamycin (Tm) induced ER stress response and pain-related behaviors both alone and synergistically together with chemical chaperon 4-PBA.108 This observation suggests a beneficial drug interaction among chemical chaperones and sEHI. Another sEHI t-TUCB, attenuated neuropathic pain without the same degree of spontaneous locomotion that is observed with gabapentin.36 Altogether, these results indicate dosing with sEHI represents an analgesic strategy for pain relief through the ER stress signaling pathway (Figure 3).
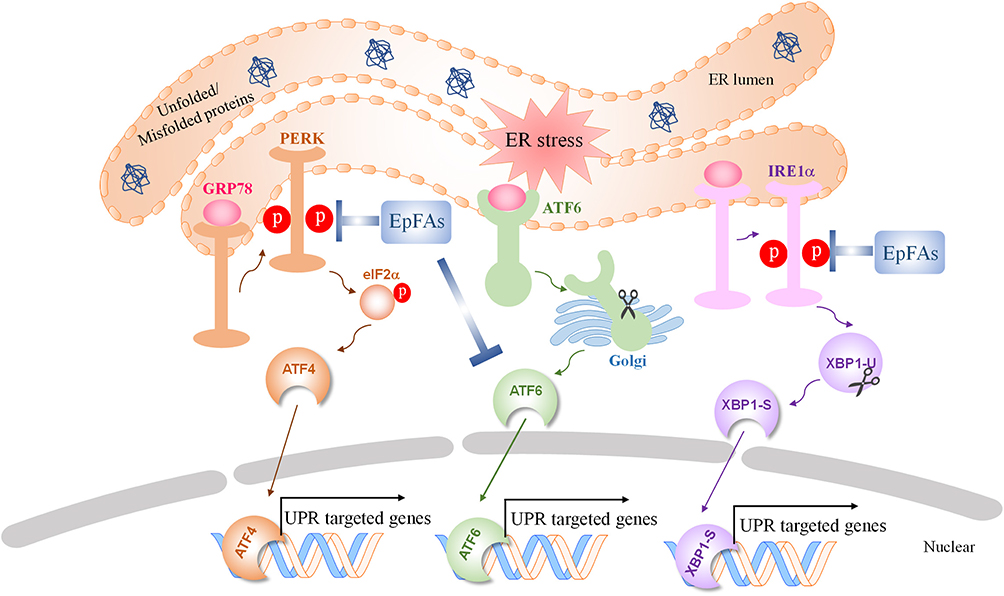 |
Figure 3 The effect of sEH inhibition and EpFAs on Endoplasmic Reticulum (ER) stress signaling pathways. Abbreviations: ER, endoplasmic reticulum; PERK, protein kinase R (PKR)-like endoplasmic reticulum kinase; GRP78, glucose-regulated protein; eIF2α, eukaryotic initiation factor 2α; ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; IRE1α, inositol-requiring enzyme 1α; XBP1-U, un-spliced X-box-binding protein 1; XBP1-S, spliced X-box-binding protein 1; EpFAs, epoxide fatty acids; UPR, unfolded protein response. |
Interaction and Complementarities of the Three Mechanistic Pathways
There is evidence showing an interaction among cAMP-PPAR, TRP channels and ER stress signaling pathways. These include ER stress regulated uncoupling protein 1 (UCP1) expression via PPARγ suppression in beige adipocytes,110 and UCP1 increased by both PPARγ stimulation and cAMP activation through their ability to stimulate the expression of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α).111 In Zn-induced lipolysis, Zn exposure evoked ER stress and dysregulation of Ca2+ homeostasis, and then activated cAMP/protein kinase A (PKA) pathway resulting in hepatic lipolysis,112 highlighting the importance of the ER stress–cAMP/PKA axis in Zn-induced lipolysis. However, there is no research about the interaction of these signaling pathways in pain research, illustrating additional studies are needed to explore the link between these signaling pathways in the pain biology and the putative role of EpFAs.
Preclinical Research and Clinical Trials of sEHIs
Currently, several preclinical studies are evaluating the effects of sEHIs in the pain management in animals. TPPU, an potent sEHI, has multimodal analgesics effects in a rat chronic pain model without changing the motor control and functioning in control animals.113 There are also several reports evaluating the sEHIs in the pain management in veterinary medicine. In a chronic laminitic horses, sEH activity in the digital laminae is significantly higher (P= 0.01) than in healthy horses,114 and treatment with the sEHI t-TUCB for 10 days significantly reduced the forelimb lifts and the pain scores compared with baseline (P= 0.04).114 A follow-up study showed that no adverse effects were detected on clinical and laboratory examinations during and after t-TUCB administration. No new episodes of laminitis have been noted up to the 120 days following treatment.115 Consistent with these studies, in another randomized controlled trial which used LPS-induced inflammatory joint pain in adult mares, treatment of 1 mg/kg t-TUCB lowered the pain, lameness and tactile allodynia, further demonstrating the analgesia effect of t-TUCB.116 Additionally, administration of t-TUCB orally for five days significantly reduced pain at a dose of 5 mg/kg in aged dogs with natural arthritis.117 Together, sEHI have already shown efficacy for inflammatory and neuropathic pain in rodents, with no apparent adverse or addictive effects, as well as relieving natural-onset pathological pain in horses and dogs (Table 1). Since horses and dogs are sensitive to side effects of NSAIDs and COXIBs, the well-established synergism of sEH inhibitors with these drugs and their reduction of side effects offers an attractive drug combination in veterinary medicine.
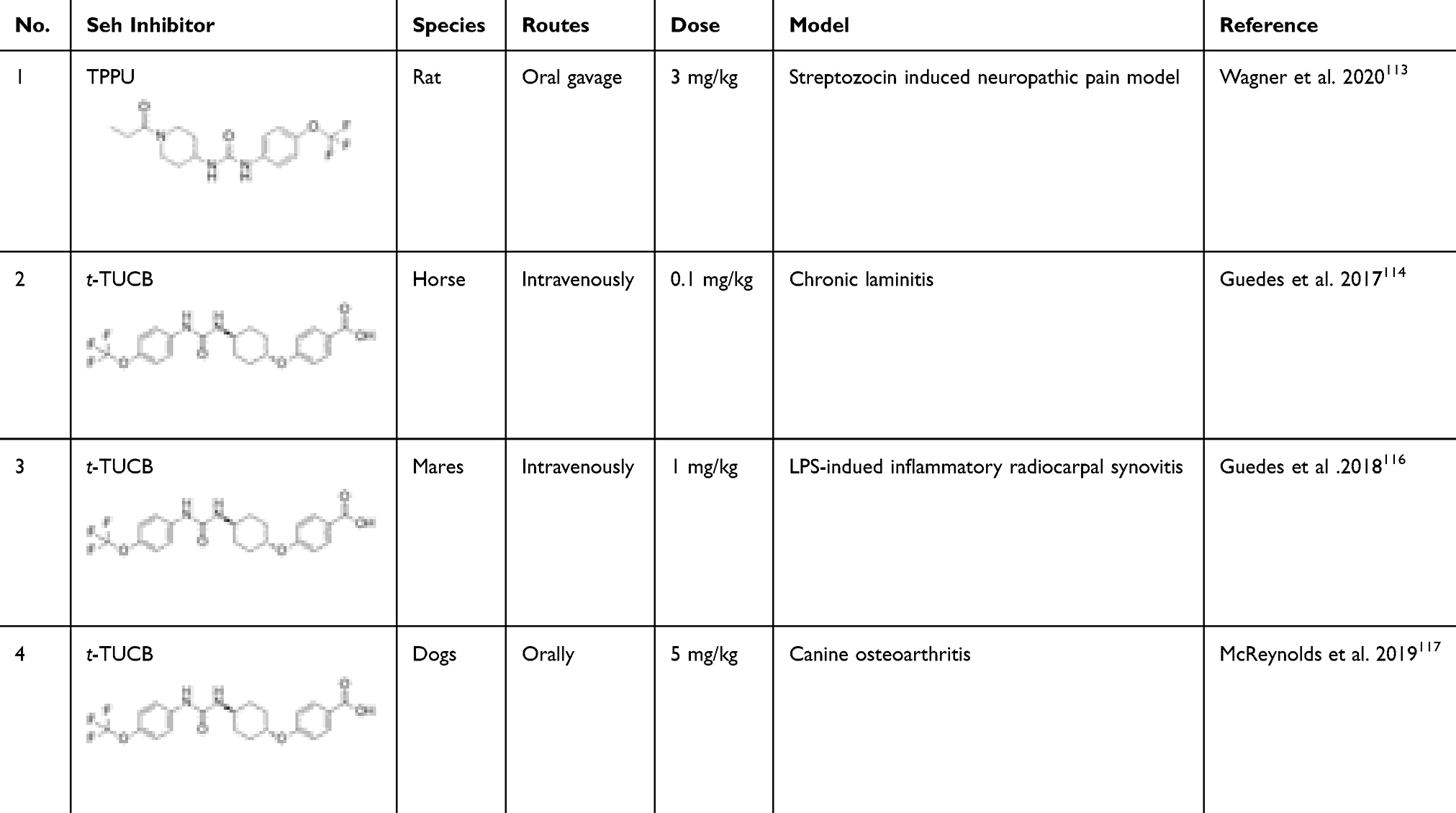 |
Table 1 Preclinical Studies of Soluble Epoxide Hydrolase Inhibitors in Pain Model |
In human, 1-(1-acetypiperidin-4-yl)-3-adamantanylurea (APAU), a potent and selective sEHI, has been in clinical development targeting hypertension and type 2 diabetes, and was well tolerated, no dose-related adverse events were observed during either study in healthy subjects.118 The sEHI GSK2256294 (chemical name: 1R,3S)-N-[[4-cyano-2-(trifluoromethyl)phenyl]methyl]-3-[[4-methyl-6-(methylamino)-1,3,5-triazin-2-yl]amino]-cyclohexanecarboxamide was well-tolerated and demonstrated sustained inhibition of sEH activity on COPD human patients.119 Recently a new class of oral non-narcotic analgesics based on inhibition of the sEH, EC5026 (chemical name: (S)-1‐[3‐fluoro‐4‐(trifluoromethoxy)phenyl]‐3‐{1‐(2‐methylbutanoyl] piperidin‐4‐yl}urea), has finished the Phase 1a clinical trial, showing no adverse effect on healthy volunteers.120 EC5026 is developed to treat neuropathic pain and should enter a Phase 2 clinical trial soon.
Conclusion
Currently, a large population in the US suffer with chronic pain, due to lack of efficacy, high expense, and safety problems, making chronic pain a serious health problem. It is important to identify novel therapeutic targets for chronic pain, to develop effective and safe methods for chronic pain treatment. Substantial studies have shown the EpFAs play essential roles in the pathology of inflammation and chronic pain, and our review further discusses the underlying molecular mechanisms of EpFAs/sEHI actions as an analgesic strategy for pain management. Recently several clinical trials of sEH inhibitors aiming at different diseases including chronic pain, hypertension, and COPD, emphasizing the importance of sEH as a promising therapeutic target. While more mechanisms need to be explored, inhibition of sEH to stabilize the beneficial effect of EpFAs is a potent and safe approach for pain management.
Acknowledgments
This study was supported, in part, by a grant from the National Institute of Environmental Health Sciences (NIEHS) Grant R35ES030443, and NIEHS Superfund Research Program P42 ES004699.
Disclosure
Karen M Wagner reports personal fees from EicOsis LLC, outside the submitted work. Christophe Morisseau reports grants from NIEHS, outside the submitted work. The University of California holds patents on the use of sEH inhibitors to treat inflammation, inflammatory pain, and neuropathic pain. Bruce D Hammock is a cofounder and Karen M Wagner is an employee of EicOsis L.L.C., a startup company advancing sEH inhibitors into the clinic. The authors report no other potential conflicts of interest in this work.
References
1. Mills SEE, Nicolson KP, Smith BH. Chronic pain: a review of its epidemiology and associated factors in population-based studies. Br J Anaesth. 2019;123(2):e273–e283. doi:10.1016/j.bja.2019.03.023
2. Lumley MA, Cohen JL, Borszcz GS, et al. Pain and emotion: a biopsychosocial review of recent research. J Clin Psychol. 2011;67(9):942–968. doi:10.1002/jclp.20816
3. Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain. 2012;13(8):715–724. doi:10.1016/j.jpain.2012.03.009
4. Dahlhamer J, Lucas J, Zelaya C, et al. Prevalence of chronic pain and high-impact chronic pain among adults - United States, 2016. MMWR Morb Mortal Wkly Rep. 2018;67(36):1001–1006. doi:10.15585/mmwr.mm6736a2
5. Avril T, Chevet E. Stress signaling in pain control. Science. 2019;365(6450):224–225. doi:10.1126/science.aay2721
6. Kerns RD, Krebs EE, Atkins D. Making integrated multimodal pain care a reality: a path forward. J Gen Intern Med. 2018;33(Suppl 1):1–3. doi:10.1007/s11606-018-4361-6
7. Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57(1):1–164. doi:10.1016/s0301-0082(98)00048-3
8. Finnerup NB, Ropper AH. Nonnarcotic methods of pain management. N Engl J Med. 2019;380(25):2440–2448. doi:10.1056/NEJMra1807061
9. Orr PM, Shank BC, Black AC. The role of pain classification systems in pain management. Crit Care Nurs Clin North Am. 2017;29(4):407–418. doi:10.1016/j.cnc.2017.08.002
10. Dubin AE, Patapoutian A. Nociceptors: the sensors of the pain pathway. J Clin Invest. 2010;120(11):3760–3772. doi:10.1172/JCI42843
11. Treede RD, Rief W, Barke A, et al. Chronic pain as a symptom or a disease: the IASP classification of chronic pain for the international classification of diseases (ICD-11). Pain. 2019;160(1):19–27. doi:10.1097/j.pain.0000000000001384
12. Vasko MR. Inflammatory pain. In: Binder MD, Hirokawa N, Windhorst U, editors. Encyclopedia of Neuroscience. Springer Berlin Heidelberg; 2009:1952–1955.
13. Wang Y, Wang W, Sanidad KZ, Shih PA, Zhao X, Zhang G. Eicosanoid signaling in carcinogenesis of colorectal cancer. Cancer Metastasis Rev. 2018;37(2–3):257–267. doi:10.1007/s10555-018-9739-8
14. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15(8):511–523. doi:10.1038/nri3859
15. Petho G, Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev. 2012;92(4):1699–1775. doi:10.1152/physrev.00048.2010
16. Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276(39):36059–36062. doi:10.1074/jbc.R100030200
17. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi:10.1126/science.294.5548.1871
18. Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. 1999;274(34):23679–23682. doi:10.1074/jbc.274.34.23679
19. Sudhahar V, Shaw S, Imig JD. Epoxyeicosatrienoic acid analogs and vascular function. Curr Med Chem. 2010;17(12):1181–1190. doi:10.2174/092986710790827843
20. Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC. EET signaling in cancer. Cancer Metastasis Rev. 2011;30(3–4):525–540. doi:10.1007/s10555-011-9315-y
21. Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292(3):C996–1012. doi:10.1152/ajpcell.00402.2006
22. Morisseau C, Inceoglu B, Schmelzer K, et al. Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res. 2010;51(12):3481–3490. doi:10.1194/jlr.M006007
23. Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs). Prostaglandins Other Lipid Mediat. 2007;82(1–4):42–49. doi:10.1016/j.prostaglandins.2006.05.004
24. Wagner K, Inceoglu B, Gill SS, Hammock BD. Epoxygenated fatty acids and soluble epoxide hydrolase inhibition: novel mediators of pain reduction. J Agric Food Chem. 2011;59(7):2816–2824. doi:10.1021/jf102559q
25. Wagner K, Inceoglu B, Hammock BD. Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostaglandins Other Lipid Mediat. 2011;96(1–4):76–83. doi:10.1016/j.prostaglandins.2011.08.001
26. Zhang G, Kodani S, Hammock BD. Stabilized epoxygenated fatty acids regulate inflammation, pain, angiogenesis and cancer. Prog Lipid Res. 2014;53:108–123. doi:10.1016/j.plipres.2013.11.003
27. Hendrickson BA, Gokhale R, Cho JH. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin Microbiol Rev. 2002;15(1):79–94. doi:10.1128/cmr.15.1.79-94.2002
28. Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8(10):794–805. doi:10.1038/nrd2875
29. Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53(1):37–58. doi:10.1146/annurev-pharmtox-011112-140244
30. Zhang W, Li H, Dong H, Liao J, Hammock BD, Yang GY. Soluble epoxide hydrolase deficiency inhibits dextran sulfate sodium-induced colitis and carcinogenesis in mice. Anticancer Res. 2013;33(12):5261–5271.
31. Wang W, Yang J, Zhang J, et al. Lipidomic profiling reveals soluble epoxide hydrolase as a therapeutic target of obesity-induced colonic inflammation. Proc Natl Acad Sci U S A. 2018;115(20):5283–5288. doi:10.1073/pnas.1721711115
32. Lopez-Vicario C, Alcaraz-Quiles J, Garcia-Alonso V, et al. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: role for omega-3 epoxides. Proc Natl Acad Sci U S A. 2015;112(2):536–541. doi:10.1073/pnas.1422590112
33. Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39(2 Pt 2):690–694. doi:10.1161/hy0202.103788
34. Zimmer B, Angioni C, Osthues T, et al. The oxidized linoleic acid metabolite 12,13-DiHOME mediates thermal hyperalgesia during inflammatory pain. Biochim Biophys Acta. 2018;1863(7):669–678. doi:10.1016/j.bbalip.2018.03.012
35. Wagner K, Inceoglu B, Dong H, et al. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur J Pharmacol. 2013;700(1–3):93–101. doi:10.1016/j.ejphar.2012.12.015
36. Wagner K, Yang J, Inceoglu B, Hammock BD. Soluble epoxide hydrolase inhibition is antinociceptive in a mouse model of diabetic neuropathy. J Pain. 2014;15(9):907–914. doi:10.1016/j.jpain.2014.05.008
37. Wagner K, Gilda J, Yang J, et al. Soluble epoxide hydrolase inhibition alleviates neuropathy in Akita (Ins2 Akita) mice. Behav Brain Res. 2017;326:69–76. doi:10.1016/j.bbr.2017.02.048
38. Schmelzer KR, Inceoglu B, Kubala L, et al. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103(37):13646–13651. doi:10.1073/pnas.0605908103
39. Graham DJ. COX-2 inhibitors, other NSAIDs, and cardiovascular risk: the seduction of common sense. JAMA. 2006;296(13):1653–1656. doi:10.1001/jama.296.13.jed60058
40. Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2010;24(2):121–132. doi:10.1016/j.bpg.2009.11.005
41. Hwang SH, Wagner KM, Morisseau C, et al. Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J Med Chem. 2011;54(8):3037–3050. doi:10.1021/jm2001376
42. Sasso O, Wagner K, Morisseau C, Inceoglu B, Hammock BD, Piomelli D. Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacol Res. 2015;97:7–15. doi:10.1016/j.phrs.2015.04.001
43. Kumar N, Goldminz AM, Kim N, Gottlieb AB. Phosphodiesterase 4-targeted treatments for autoimmune diseases. BMC Med. 2013;11(1):96. doi:10.1186/1741-7015-11-96
44. Inceoglu B, Wagner K, Schebb NH, et al. Analgesia mediated by soluble epoxide hydrolase inhibitors is dependent on cAMP. Proc Natl Acad Sci U S A. 2011;108(12):5093–5097. doi:10.1073/pnas.1101073108
45. Blocher R, Wagner KM, Gopireddy RR, et al. Orally available soluble epoxide hydrolase/phosphodiesterase 4 dual inhibitor treats inflammatory pain. J Med Chem. 2018;61(8):3541–3550. doi:10.1021/acs.jmedchem.7b01804
46. Sutherland EW, Rall TW. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem. 1958;232(2):1077–1091.
47. Steuer Costa W, Yu SC, Liewald JF, Gottschalk A. Fast cAMP modulation of neurotransmission via neuropeptide signals and vesicle loading. Curr Biol. 2017;27(4):495–507. doi:10.1016/j.cub.2016.12.055
48. Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci U S A. 1994;91(18):8517–8521. doi:10.1073/pnas.91.18.8517
49. Raker VK, Becker C, Steinbrink K. The cAMP pathway as therapeutic target in autoimmune and inflammatory diseases. Front Immunol. 2016;7:123. doi:10.3389/fimmu.2016.00123
50. Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi:10.1146/annurev.biochem.76.060305.150444
51. Knott EP, Assi M, Rao SN, Ghosh M, Pearse DD. Phosphodiesterase inhibitors as a therapeutic approach to neuroprotection and repair. Int J Mol Sci. 2017;18(4). doi:10.3390/ijms18040696
52. Feneck R. Phosphodiesterase inhibitors and the cardiovascular system. Contin Educ Anaesth Crit Care Pain. 2008;8(2):76. doi:10.1093/bjaceaccp/mkn010
53. Kumar A, Jain NK, Kulkarni SK. Analgesic and anti-inflammatory effects of phosphodiesterase inhibitors. Indian J Exp Biol. 2000;38(1):26–30.
54. Inceoglu B, Jinks SL, Ulu A, et al. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A. 2008;105(48):18901–18906. doi:10.1073/pnas.0809765105
55. Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10(4):355–361. doi:10.1038/nm1025
56. Ahmadian M, Suh JM, Hah N, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557–566. doi:10.1038/nm.3159
57. Youssef J, Badr M. Peroxisome proliferator-activated receptors and cancer: challenges and opportunities. Br J Pharmacol. 2011;164(1):68–82. doi:10.1111/j.1476-5381.2011.01383.x
58. Churi SB, Abdel-Aleem OS, Tumber KK, Scuderi-Porter H, Taylor BK. Intrathecal rosiglitazone acts at peroxisome proliferator-activated receptor-gamma to rapidly inhibit neuropathic pain in rats. J Pain. 2008;9(7):639–649. doi:10.1016/j.jpain.2008.02.002
59. Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB(1), TRPV1 and PPARgamma receptors and neurotrophic factors. Pain. 2008;139(3):541–550. doi:10.1016/j.pain.2008.06.003
60. Lazennec G, Canaple L, Saugy D, Wahli W. Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol Endocrinol. 2000;14(12):1962–1975. doi:10.1210/mend.14.12.0575
61. Lim H, Dey SK. A novel pathway of prostacyclin signaling-hanging out with nuclear receptors. Endocrinology. 2002;143(9):3207–3210. doi:10.1210/en.2002-220159
62. Benani A, Heurtaux T, Netter P, Minn A. Activation of peroxisome proliferator-activated receptor alpha in rat spinal cord after peripheral noxious stimulation. Neurosci Lett. 2004;369(1):59–63. doi:10.1016/j.neulet.2004.07.056
63. LoVerme J, Russo R, La Rana G, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319(3):1051–1061. doi:10.1124/jpet.106.111385
64. Ruiz-Medina J, Flores JA, Tasset I, Tunez I, Valverde O, Fernandez-Espejo E. Alteration of neuropathic and visceral pain in female C57BL/6J mice lacking the PPAR-alpha gene. Psychopharmacology (Berl). 2012;222(3):477–488. doi:10.1007/s00213-012-2662-8
65. Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996;384(6604):39–43. doi:10.1038/384039a0
66. Corona JC, Duchen MR. PPARgamma as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic Biol Med. 2016;100:153–163. doi:10.1016/j.freeradbiomed.2016.06.023
67. Dello Russo C, Gavrilyuk V, Weinberg G, et al. Peroxisome proliferator-activated receptor gamma thiazolidinedione agonists increase glucose metabolism in astrocytes. J Biol Chem. 2003;278(8):5828–5836. doi:10.1074/jbc.M208132200
68. Lopez-Armada MJ, Riveiro-Naveira RR, Vaamonde-Garcia C, Valcarcel-Ares MN. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13(2):106–118. doi:10.1016/j.mito.2013.01.003
69. Hasegawa-Moriyama M, Kurimoto T, Nakama M, et al. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates inflammatory pain through the induction of heme oxygenase-1 in macrophages. Pain. 2013;154(8):1402–1412. doi:10.1016/j.pain.2013.04.039
70. De Taeye BM, Morisseau C, Coyle J, et al. Expression and regulation of soluble epoxide hydrolase in adipose tissue. Obesity (Silver Spring). 2010;18(3):489–498. doi:10.1038/oby.2009.227
71. Pang W, Li N, Ai D, Niu XL, Guan YF, Zhu Y. Activation of peroxisome proliferator-activated receptor-gamma downregulates soluble epoxide hydrolase in cardiomyocytes. Clin Exp Pharmacol Physiol. 2011;38(6):358–364. doi:10.1111/j.1440-1681.2011.05492.x
72. Hiesinger K, Wagner KM, Hammock BD, Proschak E, Hwang SH. Development of multitarget agents possessing soluble epoxide hydrolase inhibitory activity. Prostaglandins Other Lipid Mediat. 2019;140:31–39. doi:10.1016/j.prostaglandins.2018.12.003
73. Liu Y, Zhang Y, Schmelzer K, et al. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102(46):16747–16752. doi:10.1073/pnas.0508081102
74. Hye Khan MA, Kolb L, Skibba M, et al. A novel dual PPAR-gamma agonist/sEH inhibitor treats diabetic complications in a rat model of type 2 diabetes. Diabetologia. 2018;61(10):2235–2246. doi:10.1007/s00125-018-4685-0
75. Khan AH, Stavniichuk A, Yeboah MM, et al. A dual soluble epoxide hydrolase inhibitor/PPAR-γ agonist prevents renal fibrosis in mouse unilateral ureteral obstruction model. FASEB J. 2019;33(1_supplement):
76. Ota K, Hammock B. Cytosolic and microsomal epoxide hydrolases: differential properties in mammalian liver. Science. 1980;207(4438):1479–1481. doi:10.1126/science.7361100
77. Kim J, Yoon SP, Toews ML, et al. Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am J Physiol Renal Physiol. 2015;308(2):F131–9. doi:10.1152/ajprenal.00531.2014
78. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76(1):387–417. doi:10.1146/annurev.biochem.75.103004.142819
79. Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011;12(3):218. doi:10.1186/gb-2011-12-3-218
80. da Costa DS, Meotti FC, Andrade EL, Leal PC, Motta EM, Calixto JB. The involvement of the transient receptor potential A1 (TRPA1) in the maintenance of mechanical and cold hyperalgesia in persistent inflammation. Pain. 2010;148(3):431–437. doi:10.1016/j.pain.2009.12.002
81. Story GM, Peier AM, Reeve AJ, et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112(6):819–829. doi:10.1016/s0092-8674(03)00158-2
82. Jordt SE, Bautista DM, Chuang HH, et al. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427(6971):260–265. doi:10.1038/nature02282
83. Koivisto A, Jalava N, Bratty R, Pertovaara A. TRPA1 antagonists for pain relief. Pharmaceuticals. 2018;11(4):117. doi:10.3390/ph11040117
84. Nassini R, Materazzi S, Benemei S, Geppetti P. The TRPA1 channel in inflammatory and neuropathic pain and migraine. Rev Physiol Biochem Pharmacol. 2014;167:1–43. doi:10.1007/112_2014_18
85. Trevisan G, Benemei S, Materazzi S, et al. TRPA1 mediates trigeminal neuropathic pain in mice downstream of monocytes/macrophages and oxidative stress. Brain. 2016;139(Pt5):1361–1377. doi:10.1093/brain/aww038
86. Jara-Oseguera A, Simon SA, Rosenbaum T. TRPV1: on the road to pain relief. Curr Mol Pharmacol. 2008;1(3):255–269. doi:10.2174/1874467210801030255
87. Premkumar LS, Sikand P. TRPV1: a target for next generation analgesics. Curr Neuropharmacol. 2008;6(2):151–163. doi:10.2174/157015908784533888
88. Caterina MJ, Leffler A, Malmberg AB, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288(5464):306–313. doi:10.1126/science.288.5464.306
89. Iida T, Shimizu I, Nealen ML, Campbell A, Caterina M. Attenuated fever response in mice lacking TRPV1. Neurosci Lett. 2005;378(1):28–33. doi:10.1016/j.neulet.2004.12.007
90. Manitpisitkul P, Brandt M, Flores CM, et al. TRPV1 antagonist JNJ-39439335 (mavatrep) demonstrates proof of pharmacology in healthy men: a first-in-human, double-blind, placebo-controlled, randomized, sequential group study. Pain Rep. 2016;1(4):e576. doi:10.1097/PR9.0000000000000576
91. Arsenault P, Chiche D, Brown W, et al. NEO6860, modality-selective TRPV1 antagonist: a randomized, controlled, proof-of-concept trial in patients with osteoarthritis knee pain. Pain Rep. 2018;3(6):e696. doi:10.1097/PR9.0000000000000696
92. Garami A, Shimansky YP, Rumbus Z, et al. Hyperthermia induced by transient receptor potential vanilloid-1 (TRPV1) antagonists in human clinical trials: insights from mathematical modeling and meta-analysis. Pharmacol Ther. 2020;208:107474. doi:10.1016/j.pharmthera.2020.107474
93. Iliff JJ, Fairbanks SL, Balkowiec A, Alkayed NJ. Epoxyeicosatrienoic acids are endogenous regulators of vasoactive neuropeptide release from trigeminal ganglion neurons. J Neurochem. 2010;115(6):1530–1542. doi:10.1111/j.1471-4159.2010.07059.x
94. Kadowaki H, Nishitoh H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes (Basel). 2013;4(3):306–333. doi:10.3390/genes4030306
95. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–3470. doi:10.1016/j.bbamcr.2013.06.028
96. Bravo R, Parra V, Gatica D, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–290. doi:10.1016/B978-0-12-407704-1.00005-1
97. Zhang E, Yi MH, Shin N, et al. Endoplasmic reticulum stress impairment in the spinal dorsal horn of a neuropathic pain model. Sci Rep. 2015;5(1):11555. doi:10.1038/srep11555
98. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10(1):173–194. doi:10.1146/annurev-pathol-012513-104649
99. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115(10):2656–2664. doi:10.1172/JCI26373
100. Chopra S, Giovanelli P, Alvarado-Vazquez PA, et al. IRE1alpha-XBP1 signaling in leukocytes controls prostaglandin biosynthesis and pain. Science. 2019;365(6450):eaau6499. doi:10.1126/science.aau6499
101. Yao W, Yang X, Zhu J, Gao B, Shi H, Xu L. IRE1alpha siRNA relieves endoplasmic reticulum stress-induced apoptosis and alleviates diabetic peripheral neuropathy in vivo and in vitro. Sci Rep. 2018;8(1):2579. doi:10.1038/s41598-018-20950-9
102. Cameron NE. Role of endoplasmic reticulum stress in diabetic neuropathy. Diabetes. 2013;62(3):696–697. doi:10.2337/db12-1469
103. Lupachyk S, Watcho P, Stavniichuk R, Shevalye H, Obrosova IG. Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes. 2013;62(3):944–952. doi:10.2337/db12-0716
104. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. doi:10.1038/sj.cdd.4401373
105. Bettaieb A, Nagata N, AbouBechara D, et al. Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue. J Biol Chem. 2013;288(20):14189–14199. doi:10.1074/jbc.M113.458414
106. Jiang XS, Xiang XY, Chen XM, et al. Inhibition of soluble epoxide hydrolase attenuates renal tubular mitochondrial dysfunction and ER stress by restoring autophagic flux in diabetic nephropathy. Cell Death Dis. 2020;11(5):385. doi:10.1038/s41419-020-2594-x
107. Inceoglu B, Bettaieb A, Haj FG, Gomes AV, Hammock BD. Modulation of mitochondrial dysfunction and endoplasmic reticulum stress are key mechanisms for the wide-ranging actions of epoxy fatty acids and soluble epoxide hydrolase inhibitors. Prostaglandins Other Lipid Mediat. 2017;133:68–78. doi:10.1016/j.prostaglandins.2017.08.003
108. Inceoglu B, Bettaieb A, Trindade da Silva CA, Lee KSS, Haj FG, Hammock BD. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc Natl Acad Sci U S A. 2015;112(29):9082–9087. doi:10.1073/pnas.1510137112
109. Rose TE, Morisseau C, Liu JY, et al. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J Med Chem. 2010;53(19):7067–7075. doi:10.1021/jm100691c
110. Yuliana A, Daijo A, Jheng HF, et al. Endoplasmic reticulum stress impaired uncoupling protein 1 expression via the suppression of peroxisome proliferator-activated receptor gamma binding activity in mice beige adipocytes. Int J Mol Sci. 2019;20(2):274. doi:10.3390/ijms20020274
111. Chen HY, Liu Q, Salter AM, Lomax MA. Synergism between cAMP and PPARgamma signalling in the initiation of UCP1 gene expression in HIB1B brown adipocytes. PPAR Res. 2013;2013:476049. doi:10.1155/2013/476049
112. Song YF, Hogstrand C, Wei CC, Wu K, Pan YX, Luo Z. Endoplasmic reticulum (ER) stress and cAMP/PKA pathway mediated Zn-induced hepatic lipolysis. Environ Pollut. 2017;228:256–264. doi:10.1016/j.envpol.2017.05.046
113. Wagner KM, Atone J, Hammock BD. Soluble epoxide hydrolase inhibitor mediated analgesia lacks tolerance in rat models. Brain Res. 2020;1728:146573. doi:10.1016/j.brainres.2019.146573
114. Guedes A, Galuppo L, Hood D, Hwang SH, Morisseau C, Hammock BD. Soluble epoxide hydrolase activity and pharmacologic inhibition in horses with chronic severe laminitis. Equine Vet J. 2017;49(3):345–351. doi:10.1111/evj.12603
115. Guedes AG, Morisseau C, Sole A, et al. Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis. Vet Anaesth Analg. 2013;40(4):440–448. doi:10.1111/vaa.12030
116. Guedes AGP, Aristizabal F, Sole A, et al. Pharmacokinetics and antinociceptive effects of the soluble epoxide hydrolase inhibitor t-TUCB in horses with experimentally induced radiocarpal synovitis. J Vet Pharmacol Ther. 2018;41(2):230–238. doi:10.1111/jvp.12463
117. McReynolds CB, Hwang SH, Yang J, et al. Pharmaceutical effects of inhibiting the soluble epoxide hydrolase in canine osteoarthritis. Front Pharmacol. 2019;10:533. doi:10.3389/fphar.2019.00533
118. Chen D, Whitcomb R, MacIntyre E, et al. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J Clin Pharmacol. 2012;52(3):319–328. doi:10.1177/0091270010397049
119. Lazaar AL, Yang L, Boardley RL, et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2016;81(5):971–979. doi:10.1111/bcp.12855
120. NINDS EHHINIoNDaS. Safety, tolerability, and pharmacokinetics of oral EC5026 in healthy subjects. Available from: https://clinicaltrials.gov/ct2/show/NCT04228302?term=EC5026&draw=2&rank=1.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.