")
Back to Journals » Journal of Inflammation Research » Volume 15
Inhibition of the cGAS-STING Pathway Attenuates Lung Ischemia/Reperfusion Injury via Regulating Endoplasmic Reticulum Stress in Alveolar Epithelial Type II Cells of Rats
Authors Huang R, Shi Q, Zhang S, Lin H, Han C , Qian X, Huang Y, Ren X, Sun J, Feng N, Xia C , Shi M
Received 11 March 2022
Accepted for publication 26 August 2022
Published 5 September 2022 Volume 2022:15 Pages 5103—5119
DOI https://doi.org/10.2147/JIR.S365970
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam D Bachstetter
Renhui Huang,1,2,* Qi Shi,1,* Shutian Zhang,1,2 Hong Lin,1,2 Chengzhi Han,1,2 Xinyi Qian,1 Yijun Huang,1 Xiaorong Ren,1 Jiayuan Sun,3 Nana Feng,4 Chunmei Xia,1 Meng Shi5
1Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai, 200032, People’s Republic of China; 2Department of Clinical Medicine, Shanghai Medical College, Fudan University, Shanghai, 200032, People’s Republic of China; 3Department of Respiratory Endoscopy, Department of Respiratory and Critical Care Medicine, Shanghai Chest Hospital, Shanghai Jiao Tong University, Shanghai, 200030, People’s Republic of China; 4Department of Respiratory and Critical Medicine, Shanghai Eighth People’s Hospital Affiliated to Jiangsu University, Shanghai, People’s Republic of China; 5Department of Thoracic and Cardiovascular Surgery, Huashan Hospital, Affiliated with Fudan University, Shanghai, 200040, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Meng Shi, Department of Thoracic and Cardiovascular Surgery, Huashan Hospital, Affiliated with Fudan University, 12 Wulumuqi Middle Road, Shanghai, 200040, People’s Republic of China, Tel +86 15921527577, Email [email protected] Chunmei Xia, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai, 200032, People’s Republic of China, Tel +86 13917406439, Email [email protected]
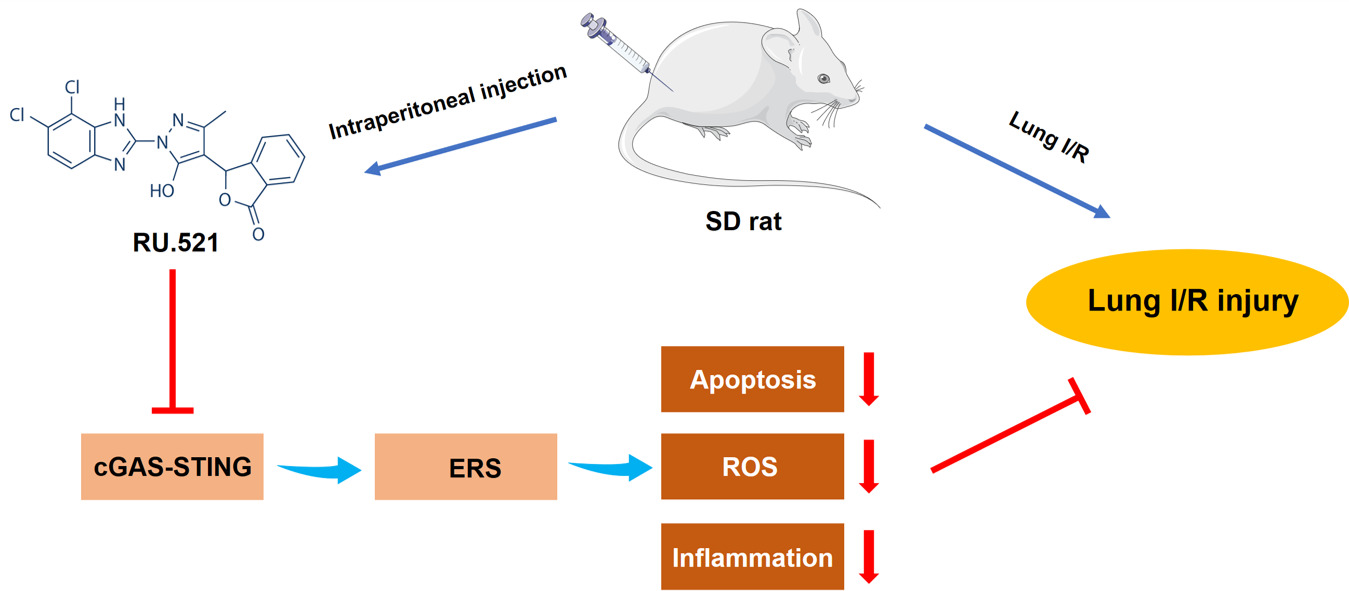
Purpose: Endoplasmic reticulum stress (ERS) plays an important role in the pathogenesis of lung ischemia/reperfusion (I/R) injury. Cyclic GMP-AMP synthase (cGAS) is a cytosol dsDNA sensor, coupling with downstream stimulator of interferon genes (STING) located in the ER, which involves innate immune responses. The aim of our present study was to investigate the effects of cGAS on lung I/R injury via regulating ERS.
Methods: We used Sprague-Dawley rats to make the lung I/R model by performing left hilum occlusion-reperfusion surgery. cGAS-specific inhibitor RU.521, STING agonist SR-717, and 4-phenylbutyric acid (4-PBA), the ERS inhibitor, were intraperitoneally administered in rats. Double immunofluorescent staining was applied to detect the colocalization of cGAS or BiP, an ERS protein, with alveolar epithelial type II cells (AECIIs) marker. We used transmission electron microscopy to examine the ultrastructure of ER and mitochondria. Apoptosis and oxidative stress in the lungs were assessed, respectively. The profiles of pulmonary edema and lung tissue injury were evaluated. And the pulmonary ventilation function was measured using a spirometer system.
Results: In lung I/R rats, the cGAS-STING pathway was upregulated, which implied they were activated. After cGAS-STING pathway was inhibited or activated in lung I/R rats, the ERS was alleviated after cGAS was inhibited, while when STING was activated after lung I/R, ERS was aggravated in the AECIIs, these results suggested that cGAS-STING pathway might trigger ERS responses. Furthermore, activation of cGAS-STING pathway induced increased apoptosis, inflammation, and oxidative stress via regulating ERS and therefore resulted in pulmonary edema and pathological injury in the lungs of I/R rats. Inhibition of cGAS-STING pathway attenuated ERS, therefore attenuated lung injury and promoted pulmonary ventilation function in I/R rats.
Conclusion: Inhibition of the cGAS-STING pathway attenuates lung ischemia/reperfusion injury via alleviating endoplasmic reticulum stress in alveolar epithelial type II cells of rats.
Keywords: cGAS-STING, endoplasmic reticulum stress, alveolar epithelial type II cells, lung ischemia/reperfusion injury
Graphical Abstract:
Introduction
Acute lung injury induced by ischemia/reperfusion (I/R) occurs in various clinical conditions, including lung transplantation, pulmonary thrombectomy, and cardiopulmonary bypass.1 Particularly in lung transplantation, lung I/R injury is the leading cause of primary graft dysfunction,2 which is responsible for nearly 50% of mortality within the first year after lung transplantation surgery and has no effective clinical therapy until now.3,4 Despite improved strategies intended to reduce lung I/R injury, both from the experimental and clinical aspects, such as inflammation inhibitors application, protection in ventilation and reperfusion, fibrinolytic treatment, and also, inhalation of therapeutic gases and substances,5–7 the fundamental improvement in prevention and treatment of lung I/R injury are still lacking, the underlying molecular mechanism remains unknown.
Recent studies showed that multiple mechanisms were involved during the process of lung I/R, including intracellular calcium overload, inflammation responses, oxidative stress, and apoptosis.8–10 Reactive oxygen species (ROS), damaging inflammation response and high level of alveolar epithelium apoptosis could lead to severe pathological injury of the alveoli and impairment of the alveolar-capillary barrier. AECII is a critical component of the alveolar epithelium, where they play a role in innate immunity, regulating transepithelial water and ion transport, and contributing to alveolar epithelial type I cell (AECI) trans-differentiation. AECIIs are also involved in the production and secretion of surfactant, which is essential for maintaining pulmonary ventilation function and homeostasis.11–13 A single AECII can create about 106 proteins per minute, with the endoplasmic reticulum (ER) processing about 30% of them.14 As a result, ER homeostasis is a need for AECII function, with any ER disruption resulting in downstream consequences.
The destruction of ER homeostasis induced by the lung I/R leads to an accumulation of unfolded or misfolded proteins, leading to prolonged ERS.15 Inflammation, apoptosis, and the development of oxidative stress are all exacerbated by severe and consecutive activation of ERS signaling in AECII, which is an important destructive mechanism of the alveolar-capillary barrier.16 Pulmonary edema and acute lung tissue pathological injury result from increased pulmonary vascular permeability and aberrant pulmonary inflammation, aggravating pulmonary ventilation dysfunction. Researches have shown that inhibition of ERS significantly reduces apoptotic cells while also inhibiting inflammation responses caused by the I/R process.17,18 As a result, preserving lung from I/R injury by preventing persistent ERS could be a viable option.
Pattern-recognition receptors (PRRs), such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), are sensors of cellular danger signals for innate immune systems.19 When cells are attacked and injured by pathogens or other cells, various kinds of cytosolic sensors are aggregated, ascribing to the sensation of PRRs. It is noted that cyclic GMP-AMP synthase (cGAS) is an endogenous sensor in innate immune signaling activated by cytosolic DNA, such as extranuclear chromatin and DNA released from impaired mitochondria.20 cGAS normally resides as an inactive protein in cytosol. Upon binding to cytosolic DNA, cGAS transforms its conformation to an active state and produces the second messenger cyclic GMP-AMP (cGAMP), using ATP and GTP, which could be subsequently detected by the stimulation of interferon genes (STING), a transmembrane protein located at the ER, responsible for sensing the cyclic-dinucleotide. Activation of STING by binding to cGAMP can subsequently trigger inflammatory responses and cell death in various ways via activating STING downstream signaling. Recent studies have demonstrated that change in STING activity is associated with ERS induction. Cur et al reported that STING exacerbates inflammatory response and apoptosis via activating the interferon regulatory factor 3 (IRF3) in an ERS-dependent manner.21 However, in this study, we found that ERS was also essential for the activation of STING and the induction of STING-dependent type I interferon program genes. Obviously, there is a reciprocal regulation between STING and ERS. Another research on heart inflammation and fibrosis has also demonstrated this type of reciprocal crosstalk.22 STING activation induced by angiotensin II was dependent upon ERS, while knockout of STING resulted in a decreased level of ERS following the aortic banding process in mice. Our present study was to investigate the effects of cGAS on lung ischemia/reperfusion injury via regulating ERS. We hypothesized that inhibition of cGAS-STING pathway improves pulmonary ventilation dysfunction induced by lung I/R via suppressing ERS in alveolar epithelial type II cells. cGAS might be a potential therapeutic target for protecting lung from I/R injury.
Materials and Methods
Regents and Antibodies
RU.521 (S6841) and SR-717 (S0853) were purchased from Selleckchem (Houston, Texas, USA); 4-PBA (S305161) was purchased from Aladdin (Shanghai, China). Hematoxylin and Eosin (HE) Staining Kit (C0105S), Dihydroethidium (DHE) (S0063), and Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End-Labeling (TUNEL) Apoptosis Assay Kit (C1089) were purchased from Beyotime (Shanghai, China). Antibodies: cGAS (ab252416), STING (ab13647), BiP (ab21685), Goat Anti-Rabbit IgG (ab150077), and Goat Anti-Mouse IgG (ab150115) were purchased from Abcam (Cambridge, UK); SP-C (SC-13979) was purchased from Santa Cruz Biotechnology (Dallas, USA); β-actin (3700) were purchased from Cell Signaling Technology (Boston, USA).
Experimental Design
Figure S1 shows the outline of the experimental design. Totally 40 rats were divided into five groups randomly (n = 8 for each group): (1) Sham group receiving the sham operation and 210 minutes ventilation without occlusion of left lung hilum, (2) Lung I/R modeling group receiving the lung ischemia/reperfusion (LI/R) surgery, (3) I/R + RU.521 group receiving the LI/R surgery one hour after intraperitoneal injection of RU.521 (5 mg/kg), (4) I/R + SR-717 group receiving the LI/R surgery one hour after intraperitoneal injection of SR-717 (3 mg/kg), and (5) I/R + SR-717 + 4-PBA group receiving the same intraperitoneal co-injection of SR-717 (3 mg/kg) and 4-PBA (an inhibitor of ERS, 40 mg/kg) one hour before modeling surgery. The lung I/R model of rats was established according to the previously reported method with a slight modification.23 Specifically, we performed the thoracotomy surgery and 90 minutes occlusion of left lung hilum for modeling ischemia phase, then following 120 minutes of reperfusion by losing the left lung hilum. The volume of vehicle was delivered for the Sham and the I/R group equivalently.
Lung Ischemia/Reperfusion Injury Model Induction
The male Sprague-Dawley rats ranging from 300 to 350 g were acquired from the Animal Laboratory Center of Fudan University. The rats were accommodated in a standard 24°C temperature-controlled and 12-hour light/dark cycle room, and they were supplied with food and water sufficiently. Greatest efforts have been made to minimize the usage number and the sufferings of animals. This study was approved by the Animal Care Committee of Fudan University, Shanghai, China. Animal care and all experimental procedures were conformed to the guidelines by the Institutional Ethics Committee.
Specifically, the rats were anesthetized using intraperitoneal injection of 20% urethane (1.5 g/kg). cGAS potent inhibitor RU.521 (5 mg/kg), which has high selectivity and nontoxicity,24 was dissolved with 0.5 mL of 10% dimethyl sulfoxide (DMSO) and injected intraperitoneally in the I/R + RU.521 group, while the same volume of 10% DMSO without RU.521 was administered in the I/R modeling group. The usage and dosage of RU.521 in our study was referenced to the previous researches.25 STING selective agonists SR-717 (3 mg/kg) and 4-PBA (40 mg/kg) and the ERS inhibitor were dissolved in 10% DMSO and 0.9% saline, respectively, then SR-717 was injected intraperitoneally in the I/R + SR-717 group singly and was administered in the I/R + SR-717 + 4-PBA group cooperated with 4-PBA injection.26,27 As the cGAS-specific inhibitor, RU.521 can bind to the catalytic domain of cGAS, thus inhibiting the activation of cGAS by dsDNA. SR-717 is a non-nucleotide cGAMP analogue, which was designed based on the structure of cGAMP and can activate STING directly. Throughout the experiment, the rectal temperature was measured and maintained at 37 ± 0.5°C using a heated operation table, and the inspiration oxygen fraction was kept at 100%.
Rats were performed tracheotomy and ventilated at the certain parameter: 8 mL/kg tidal volume (TV) and 80 breaths/minute respiratory rate with VentElite (Harvard, Massachusetts, US). For the rats in I/R modeling groups, the left thoracotomy surgery was performed subsequently, and the left hilum was occluded with a proper suture for 90 minutes. After 90 minutes of occlusion, we then released the suture, and reperfusion was performed by restoring blood flow and ventilation to the operated lung. The sham group rats were performed the sham operation, specifically, we surrounded through the left hilum with the same suture but without occlusion, and maintained bilateral ventilation for 210 minutes in total.
Pulmonary Ventilation Function Measurement
Rats were ventilated at the certain parameter throughout the lung ischemia/reperfusion surgery: 8 mL/kg TV and 80 breaths/minute respiratory rate with VentElite (Harvard, Massachusetts, US). After 120 minutes of reperfusion, the animals were then connected to the R&C system (EMMS, Alton, UK) to assess the pulmonary function. The TV and respiratory rates noted above were maintained at 8 mL/kg and 80 breaths/minute, respectively, and then, we measured the mean airway pressure, peak airway pressure, and dynamic compliance for 10 minutes (see Figure S2).
Pulmonary Edema Evaluation
The bilateral lung of separate groups (n = 6) was removed after the reperfusion phase, and the total lung weight was determined, thereafter cut the upper part of the left lung with a sharp blade and measured the wet weight immediately and dried it in an oven at 60°C for 48 hours when a stable dry weight was reached. The lung wet/dry (W/D) ratio and lung weight/body weight (LW/BW) ratio were calculated as an evaluation of pulmonary edema.28
Endoplasmic Reticulum Stress Evaluation
Endoplasmic reticulum stress was evaluated by detecting the content of BiP and observing the morphology of the ER. BiP, a chaperone, has been proposed to be a direct ER stress sensor, leading to UPR activation, in addition to its activity as a key ER chaperone.29 To determine the expression of BiP, we employed immunofluorescent staining and Western blot. In addition, we used transmission electron microscopy (TEM) to scan the ER’s structure.
Rats Perfusion Fixation
The anesthetic rat was placed on the shallow tray filled with crushed ice. The heart and the lungs of the rat were fully exposed with an incision from the abdominal to the thoracic wall. Then, a puncture was made at the apex of the heart with a perfusion needle, and an incision was made in the auricula dextra. Finally, 0.9% saline solution and 4% paraformaldehyde solution are pumped up in sequence. The lungs were harvested and put into 4% paraformaldehyde solution for post-fixation.
Hematoxylin and Eosin (HE) Staining and Lung Injury Scoring
The histopathological changes of lung tissue induced by I/R were visualized using HE staining. The left lungs of rats were harvested and fixed in 4% paraformaldehyde for 48 hours. The lung tissue was embedded in paraffin, and then, we sectioned it into 4-μm-thick slices and stained it with hematoxylin and eosin. Two independent pathologists were blinded to evaluate lung tissue pathological injury under microscopy (Olympus, Tokyo, Japan).
We used the lung injury scoring system reported previously to quantify the pathological injury of lung tissue.30 Specifically, the pathology indexes consisted of alveolar wall thickness, alveolar congestion, lung edema, hemorrhage, and infiltration of neutrophils into the airspace or vessel wall. The scoring was showed in detail as follows: lung hemorrhage (0 = no hemorrhage, 1 = mild hemorrhage, 2 = severe hemorrhage), lung interstitial edema (0 = no edema, 1 = mild edema, 2 = severe edema), alveolar wall thickness (0 = no alveolar wall thickness, 1 = mid-alveolar wall thickness, 2 = severe alveolar wall thickness, and 3 = severe alveolar wall thickness with >50% lung consolidation) and infiltration of inflammatory cells (0 = no inflammatory cell infiltration, 1 = mild infiltration of inflammatory cell infiltration, 2 = moderate inflammatory cell infiltration, 3 = severe inflammatory cell infiltration).
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End-Labeling (TUNEL)
Cell apoptosis in lung was detected by using the TUNEL assay with an apoptosis detection kit (Beyotime, Shanghai, China). We fixed the left lung tissue in 4% paraformaldehyde for 24 hours, embedded in paraffin, and sectioned it into 4-μm-thick slices. The slices were photographed under a Fluorview FV300 laser scanning confocal microscope (Olympus, Tokyo, Japan). The TUNEL positive cells (nuclei stained red) were identified in five random and non-overlapping regions. The apoptosis index was calculated as the ratio of TUNEL positive cells to the total number of cells.31
In vivo ROS Detection
The in-situ production of ROS in the lung tissue was detected by using the dihydroethidium (DHE) fluorescent probe assay.32 Briefly, after the reperfusion phase, the fresh left lung tissue of rats was collected as mentioned above. The segments of lung tissue were sectioned to 5-μm-thick sections and then placed in a glass slide (DHE, Molecular Probes). The slices were treated with 1 mmol/L DHE in a dark and humidified chamber at 37°C for 30 minutes. Literally, DHE can be oxidized by superoxide to ethidium bromide, which binds to DNA and emits red fluorescence, thereby can be detected digitally captured by fluorescence microscope (Olympus, Tokyo, Japan).
Transmission Electron Microscopy Observation
Samples of fresh lung tissue (1 mm × 1 mm × 1 mm) were harvested carefully from rats, and then, they were fixed in 2.5% glutaraldehyde for 24 hours primarily. After that, we washed the lung samples with Sorenson’s Phosphate Buffer (pH = 7.4) and post-fixed them in 1% osmium tetroxide for 1 hour. We used propylene oxide to wash the tissues after post-fixation and dehydration procedures and then the tissues were embedded in the epoxy resin embedding media. Following these processes, we sectioned the tissue blocks into segments around 60 nanometers in thickness using the ultramicrotome (LKB Nova, Sweden). These sections were stained with uranyl acetate and lead citrate and then observed using the transmission electron microscope (HITACHI, Japan).
Immunofluorescent Staining Analysis
The lung sections were deparaffinized and subsequently washed in PBS and then used 0.25% Triton X-100 to permeabilize for 30 minutes followed by incubation with 10% fetal donkey or goat serum for 2 hours at room temperature to block non-specific protein. After blocking, the samples were incubated with primary antibody to cGAS, STING, BiP, and SP-C overnight at 4°C. Sections were incubated with corresponding secondary antibodies for 2 hours at room temperature, washed with PBS twice for 10 minutes, and incubated with DAPI for 5 minutes at room temperature. The fluorescence signal was detected under a Fluorview FV300 laser scanning confocal microscope (Olympus, Tokyo, Japan); Immunoreactivity was manifested in specific green, red or pink fluorescence. Totally, 3 slides of lung tissue sections were selected, and five separated files of each slide were selected randomly to be observed by 2 pathologists blinded to the experimental groups. We utilized ImageJ software to analyze the immunofluorescent images and the colocalization event of the segmented points from the dual channels was evaluated by using the ImageJ plugin Just Another Colocalization Plugin (JACoP). JACoP can calculate Pearson coefficients, which indicated the percentage of thresholded pixels in the red channel that was occupied by relevant thresholded pixels in the green channel. ANOVA analysis was carried out by using GraphPad Prism software to provide further information regarding the statistical analysis of colocalization.
Western Blot Analysis
Lung tissues from each rat were homogenized in a lysis buffer containing 5 mmol/L EDTA and 1 mmol/L PMSF as described previously.33 We extracted protein samples at the same amount from each rat to analyze protein expression by Western blot. Briefly, the protein samples (15μg per well) were subjected to SDS/PAGE in 10% gradient del (Beyotime, Shanghai, China) and then transferred to the PVDF membrane. Primary antibodies were incubated using the following dilution: cGAS (1:500), STING (1:500), BiP (1:1000), and β-actin (1:2000). After washing, the membranes were incubated with the secondary antibodies, horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1000) or goat anti-mouse IgG (1:1000). The ECL detection reagents (WBKLS0050; Millipore) were utilized to assess the amount of protein expression and the automatic chemiluminescence image analysis system (Tanon-5200; Tanon Science & Technology, Shanghai, China) was used to visualize and quantify the immunostaining band. The data were normalized by developing the β-actin as a loading control.
Statistical Analysis
We use SPSS 11.0 to analyze the data. The data were presented as mean ± standard error of the mean (SEM). The experimental groups were compared by variance analysis followed by One-way ANOVA with post hoc Bonferroni-Dunn using GraphPad Prism software. A P-value of less than 0.05 indicated the presence of a significant difference.
Results
cGAS-STING Signaling Was Activated and ERS Was Augmented After I/R in Rats
Figure 1A and B demonstrated that after I/R, cGAS in the I/R group significantly increased compared to the sham group (P<0.05), and simultaneously, the expression of STING was increased in lung tissue measured by immunofluorescent staining, compared to the sham group (Figure 1C and D, P<0.05). Besides cGAS-STING signaling activation, Figure 1E and F demonstrated that BiP expression in the I/R group was higher than that in the sham group (P<0.05). In the lung tissue of I/R rats, western blot analysis verified the increase in cGAS compared to the sham group (Figure 2F and I, P<0.05). Also, STING was shown to be higher in I/R rats’ lung tissue than in the sham group, according to Western blot analysis, while intraperitoneal treatment of RU.521, a cGAS inhibitor, significantly reduced STING content (Figure 2F and J, P<0.05). These results Suggested that RU.521 inhibited activation of the cGAS-STING pathway induced by I/R in rats. Western blot analysis verified the increased BiP expression in lung tissue of I/R rats compared to the sham group, which could be reversed by RU.521 (Figure 2F and K, P<0.05). Furthermore, the double immunofluorescent staining showed that the increased STING and BiP were predominantly expressed in the alveolar epithelial type II cells (AECIIs) (Figure 2C, H and Figure 2E, G, P<0.05), while the increased cGAS was hardly detectable in AECIIs (Figure 2A, B and D, P<0.05). These findings implied that I/R triggered cGAS-STING signaling activation and ERS augmentation in the lung.
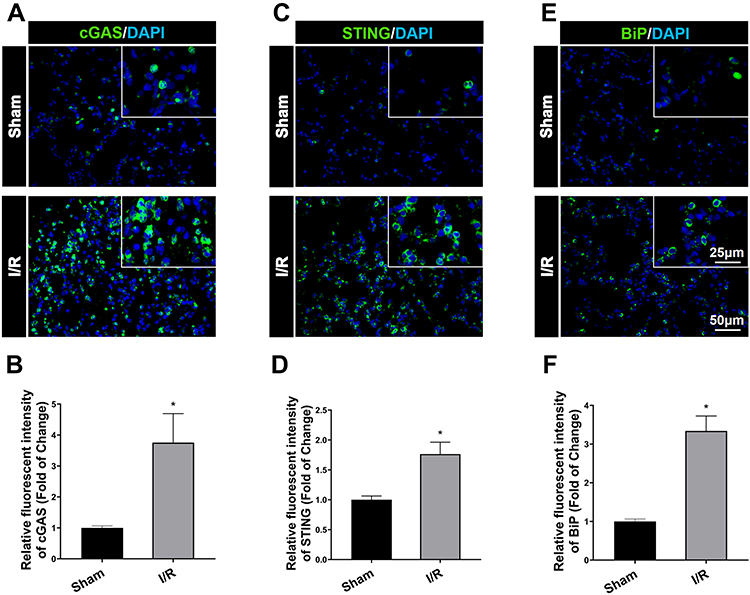 |
Figure 1 Immunofluorescent staining of cGAS, STING and BiP (ER stress marker protein) expression in lung tissue after IR in rats. (A) Immunofluorescent staining of cGAS expression in the lung tissue. (B) Immunofluorescent density analysis to quantify the expression of cGAS. (C) Immunofluorescent staining of STING. (D) Immunofluorescent density analysis to quantify the expression of STING. (E) Immunofluorescent staining of BiP. (F) Immunofluorescent density analysis to quantify the expression of BiP. Scale bar = 25μm or 50μm. Data are presented as mean ± SEM, n = 6 per group. *P < 0.05 vs sham group. |
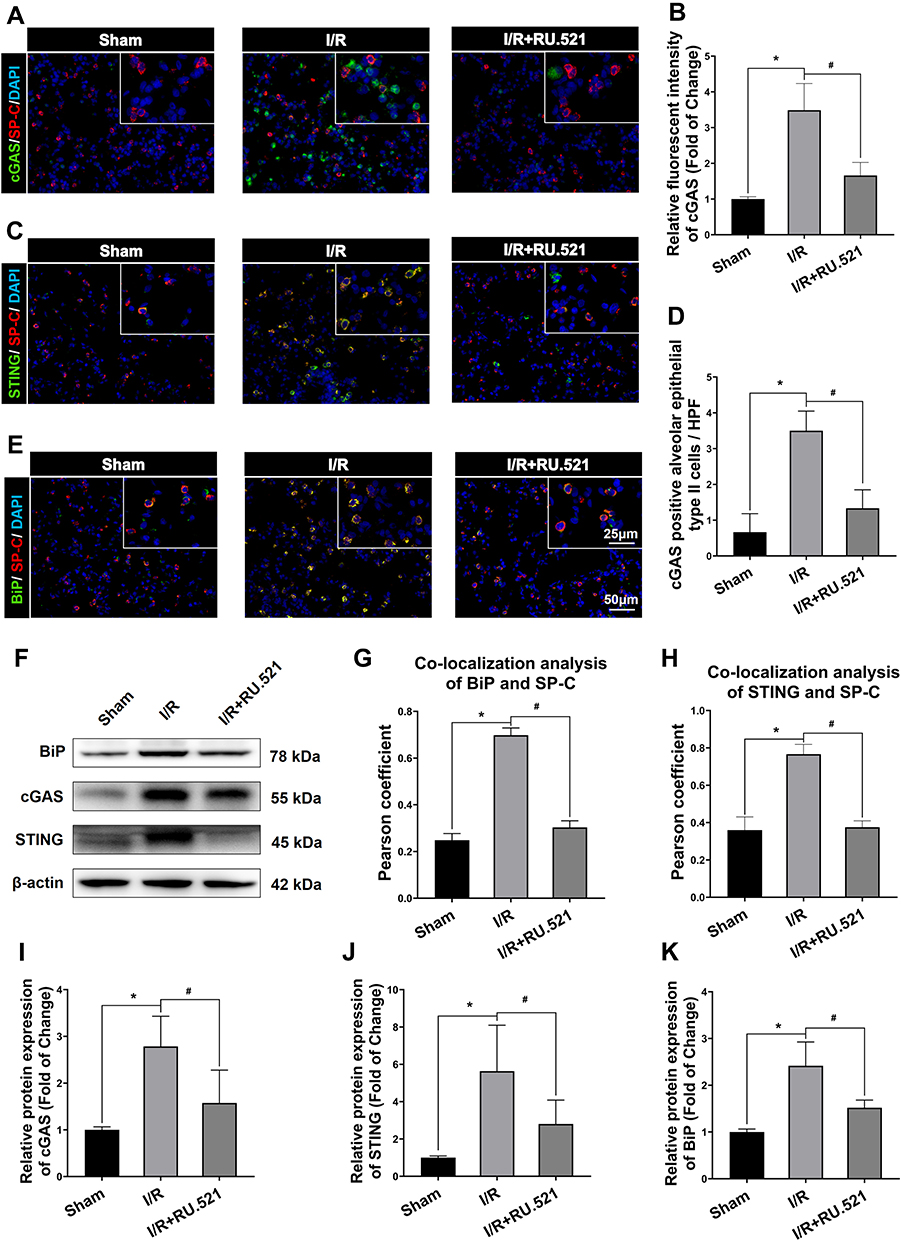 |
Figure 2 Western blot and colocalization of cGAS, STING and BiP with alveolar epithelial type II cells marker of rats. (A) Double immunofluorescent staining to colocalize cGAS with alveolar epithelial type II cells marker SP-C in lung tissue. (B) Immunofluorescent density analysis to quantify the expression of cGAS. (C) Double immunofluorescent staining to check localization of STING expression in alveolar epithelial type II cells. (D) Number of cGAS positive alveolar epithelial type II cells in high power field (HPF). (E) Colocalization of BiP with alveolar epithelial type II cells marker by using double immunofluorescent staining. (G) The quantified colocalization of BiP with alveolar epithelial type II cells marker were assessed using the Pearson coefficient. (H) The quantified colocalization of STING with alveolar epithelial type II cells marker were assessed using the Pearson coefficient. Scale bar = 25μm or 50μm. Data are presented as mean ± SEM, n = 6 per group. (F and I-K) Western blotting analysis to verify the expression of cGAS, STING and BiP, β-actin was used as the loading control. Data are presented as mean ± SEM, n = 3 per group. *P < 0.05 vs sham group, #P < 0.05 vs I/R group. |
cGAS-STING Signaling Activation Induced ERS in Alveolar Epithelial Type II Cells After I/R in Rats
Numerous physiological and pathological factors can cause ER environment disturbance, ERS is a condition which is accelerated by aggregation of unfolded/misfolded proteins.34 We interfered the cGAS-STING signaling by using the cGAS inhibitor RU.521 and the STING potent agonist SR-717, respectively, to investigate the causal relationship between cGAS-STING signaling and ERS. The ultrastructure of alveolar epithelial type II cells (AECIIs) was investigated using transmission electron microscopy (Figure 3). As Figure 3 shows, we found that I/R induced dramatical swelling of the ER and evident dilatation of the lumen, which could be reversed by RU.521. While to the contrary, the STING agonist, SR-717 further aggravated the ultra-pathological damage of the ER induced by I/R. Western blot analysis revealed the increased BiP expression in the lung tissue after I/R in rats (Figure 4C and D, P<0.05). As performed in Figure 4C and D, we found that RU.521 treatment deregulated BiP expression, while intraperitoneal injection of SR-717, a STING agonist, upregulated BiP expression, which indicated that cGAS-STING signaling activation might aggravate ERS responses. Furthermore, the double immunofluorescent staining and co-localization analysis showed that increased BiP was mainly present in AECII (Figure 4A and B, P<0.05). These results suggested that cGAS-STING signaling activation-induced ERS in the I/R rats.
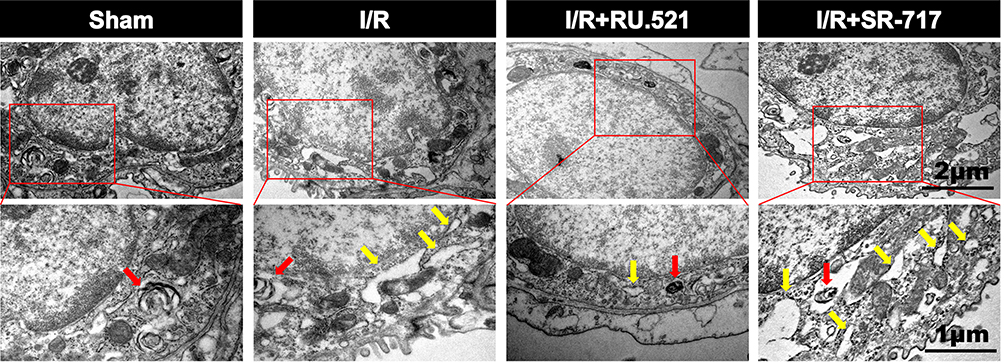 |
Figure 3 Electron micrographs showed the morphology of endoplasmic reticulum of alveolar epithelial type II cells in rats. Red arrow indicates the presence of lamellar bodies, which are characteristics of alveolar epithelial type II cells, yellow arrow notes swallowed and dilated endoplasmic reticulum. n = 3. Scale bar = 1μm or 2μm. |
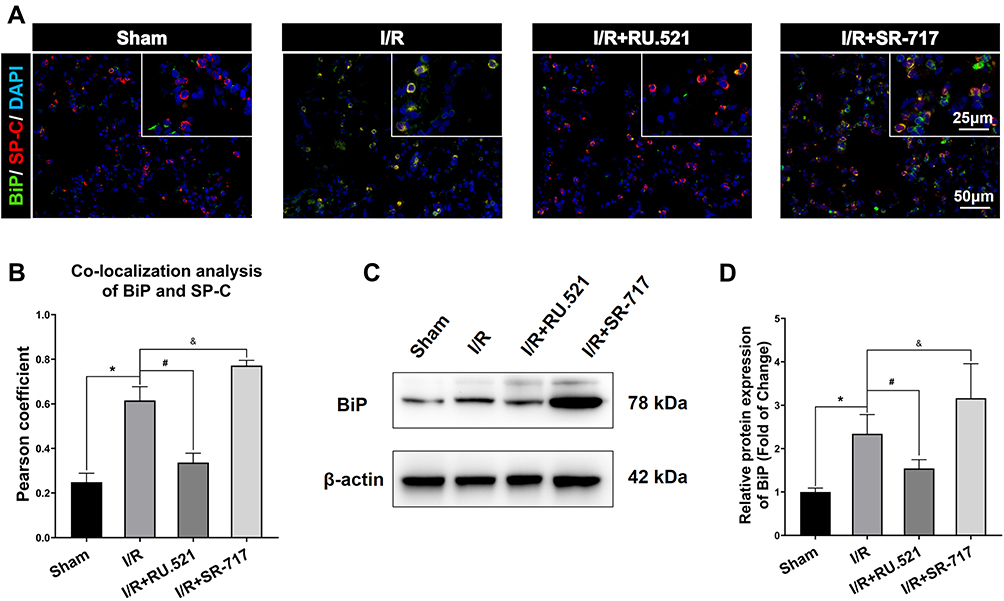 |
Figure 4 BiP expression in alveolar epithelial type II cells by using Western blot and double immunofluorescent staining respectively. (A) Double immunofluorescent staining to colocalize BiP with alveolar epithelial type II cells marker SP-C in lung tissue. Scale bar = 25μm or 50μm. (B) The quantified colocalization of BiP with alveolar epithelial type II cells marker was assessed using the Pearson coefficient. Data are presented as mean ± SEM, n = 6 per group. (C-D) Western blotting analysis to verify the expression of BiP. Data are presented as mean ± SEM, n = 3 per group. *P < 0.05 vs sham group, #P < 0.05 vs I/R group, &P < 0.05 vs I/R group. |
Activation of cGAS-STING Pathway Exacerbated Cell Apoptosis in Lung via Regulating ERS in I/R Rats
It was noted that a high level of alveolar epithelium apoptosis is critical in the pathogenesis of acute lung injury induced by I/R.35 To investigate the causality between ERS and cell apoptosis, we intervened the ERS by using the ERS inhibitor 4-PBA. The cell apoptosis in the lungs after I/R of rats was assessed using the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay. We discovered that the I/R group had significantly more apoptotic cells in lung tissue than the sham group, which could be reversed by RU.521, a cGAS inhibitor (Figure 5A and D, P<0.05). Furthermore, as compared to the I/R group, SR-717 administration exacerbated pulmonary cell apoptosis, whereas co-treatment with the ERS inhibitor 4-PBA reversed the effects. Furthermore, the double immunofluorescent staining revealed the increased apoptotic alveolar epithelial type II cells following I/R in rats (Figure 5B and E, P<0.05). These results suggested that cGAS-STING pathway activation aggravated cell apoptosis in lung via regulating ERS in the I/R rats.
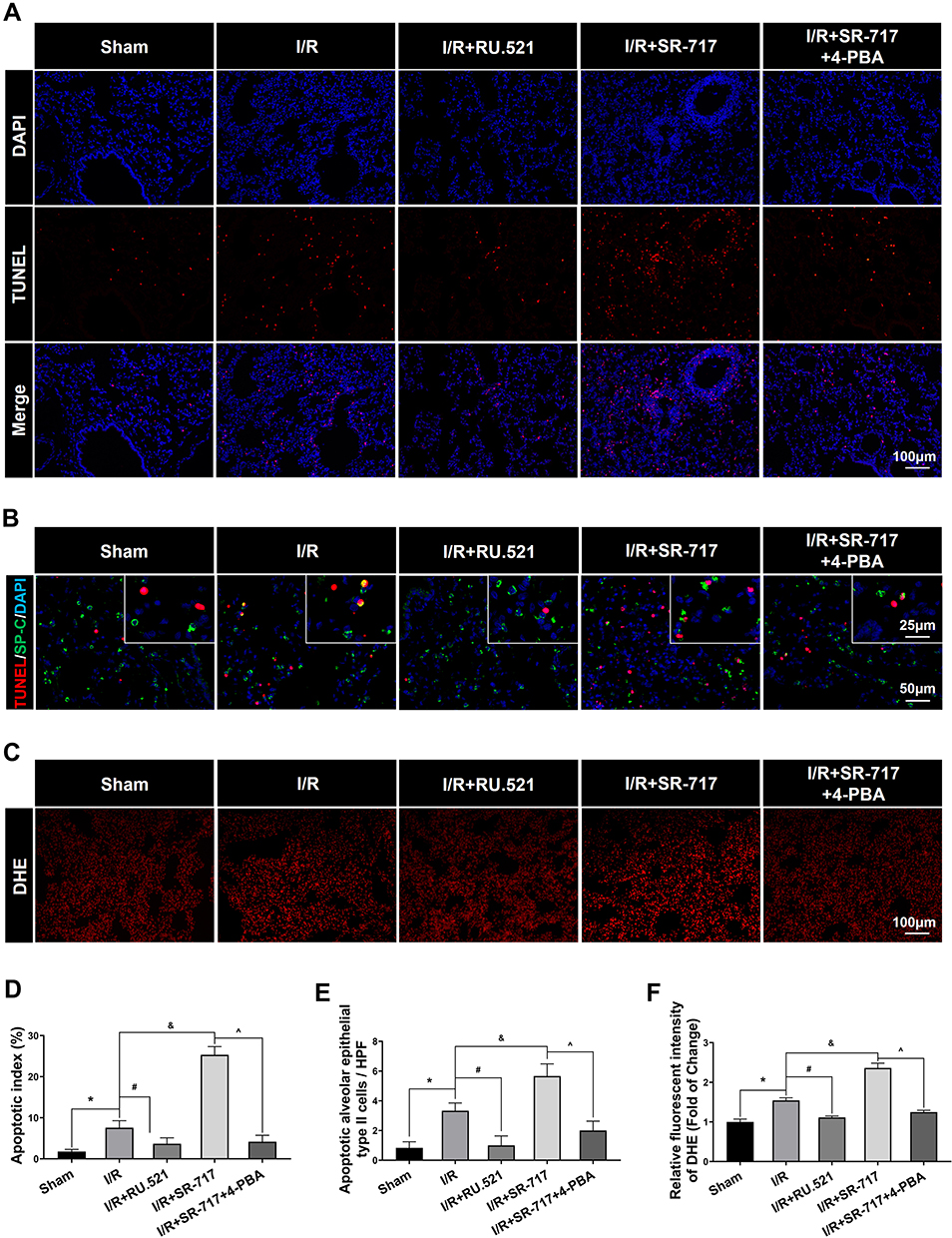 |
Figure 5 Cell apoptosis and oxidative stress detection in lung of rats. (A) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was used to detect apoptosis in lung. Scale bar = 100μm. (B) Double immunofluorescent staining to colocalize TUNEL with alveolar epithelial type II cells marker SP-C. Scale bar = 25μm or 50μm. (C) The representative figures of the DHE probe detecting superoxide anion in lung tissue. Scale bar = 100μm. (D) Quantified analysis of cell apoptosis in lung by using apoptosis index. (E) The quantification of apoptotic alveolar epithelial type II cells in high power field (HPF). (F) Immunofluorescent density evaluation of DHE staining. Data are presented as mean ± SEM, n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs I/R group, &P < 0.05 vs I/R group, ^P < 0.05 vs I/R + SR-717 group. |
ERS Triggered the Inflammatory Response and Oxidative Stress in Lung Tissue After I/R in Rats
Excessive ERS was found to exacerbate LPS-induced acute lung injury by initiating the inflammatory response and increasing oxidative stress.36 Up to date, however, little was known about the role of ER hormesis in I/R-induced inflammatory response and high level of oxidative stress. We found that SR-717 administration aggravated the I/R induced infiltration of inflammatory cells, while 4-PBA with SR-717 co-treatment alleviated infiltration of inflammatory cells in the lung tissue after I/R in rats (Figure 6A–C, P < 0.05). Furthermore, DHE superoxide anion probe detection revealed that SR-717 administration aggravated the I/R induced oxidative stress indicated by ROS production, while 4-PBA with SR-717 co-treatment evidently reduced the ROS production in the lung tissue after I/R in rats (Figure 5C and F, P < 0.05).
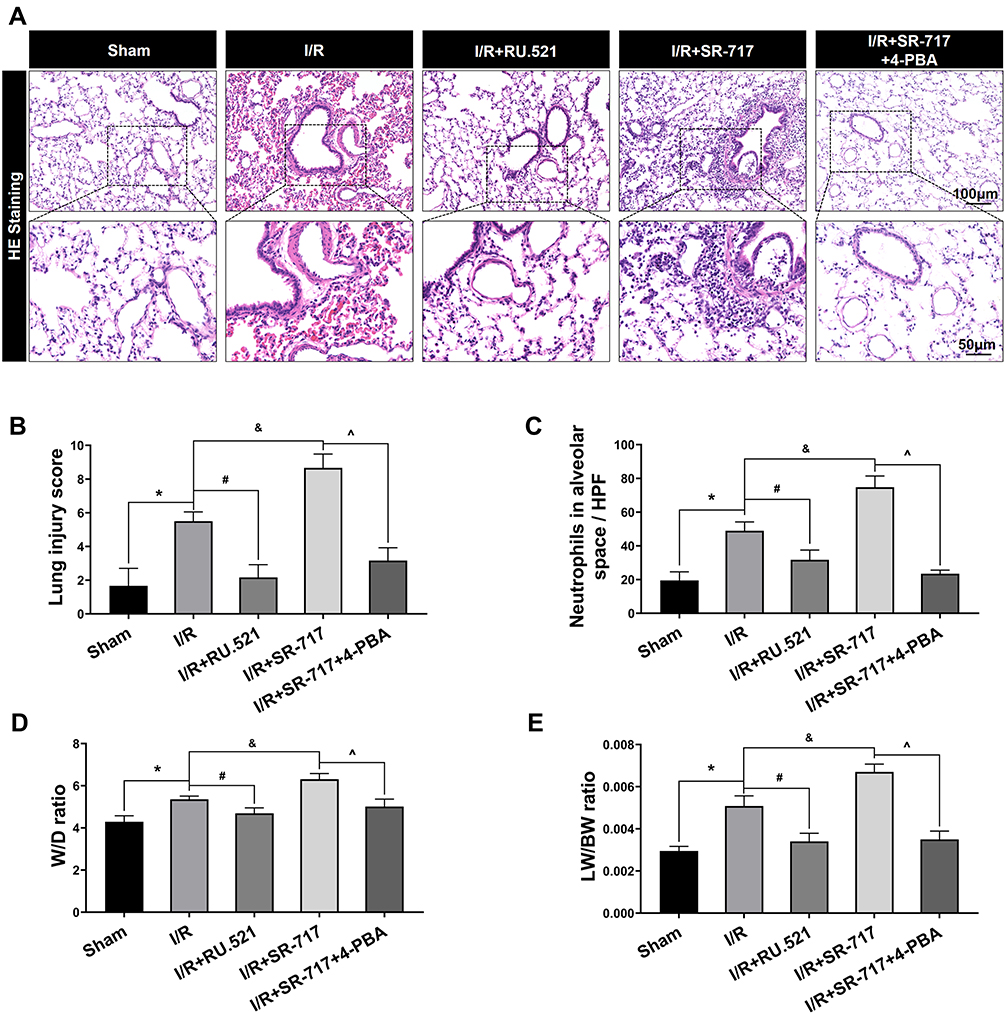 |
Figure 6 Hematoxylin and eosin staining of lung tissue and evaluation of pulmonary edema in rats. (A) The representative figures were used to evaluate the pathological injury of lung tissue. Scale bar = 50μm or 100μm. (B) The profiles of lung pathological injury were assessed with the lung injury scoring system. (C) The quantification of neutrophils infiltration in alveoli or interstitial space. (D and E) The W/D ratio and LW/BW ratio were used to assess pulmonary edema. Data are presented as mean ± SEM, n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs I/R group, &P < 0.05 vs I/R group, ^P < 0.05 vs I/R + SR-717 group. |
Inhibition of cGAS-STING Pathway Improved I/R Induced Pulmonary Ventilation Dysfunction by Attenuating Pulmonary Edema and Pathological Injury of Lung Tissue via Suppressing ERS
It was noted that excessive cell apoptosis, damaging ROS and aberrant inflammatory response aggravates injury of the alveoli and destruction of the alveolar-capillary barrier, which results in pulmonary edema and acute pathological injury of lung tissue, thus leading to I/R induced pulmonary ventilation dysfunction. The morphological changes showed that the left lung of rats following I/R had significant edema and hemorrhage (Figure S3). Furthermore, we used W/D ratio, LW/BW ratio, and hematoxylin and eosin staining, respectively, to evaluate the pulmonary edema and lung tissue pathological changes after I/R in rats. We found that the I/R group had severe pulmonary edema indicated by higher W/D ratio and LW/BW ratio, while RU.521 treatment significantly attenuated pulmonary edema caused by lung I/R (Figure 6D and E, P<0.05). As Figure 6A–C showed, we found that the I/R group had significantly damaged alveoli structure with interstitial edema and remarkably inflammatory cell infiltration, while RU.521 treatment significantly alleviated these pathological changes. Likewise, 4-PBA inhibited I/R-induced lung tissue pathological injury which was aggravated by SR-717 administration. Finally, the mean dynamic compliance, mean airway pressure, and peak airway pressure were used to quantify the pulmonary ventilation function of each group after I/R surgery. We found that 90 minutes left hilum clamp and 120 minutes reperfusion resulted in severe pulmonary ventilation dysfunction. By contrast, RU.521 treatment remarkably attenuated the pulmonary ventilation function after I/R, as indicated by the significantly lower mean/peak airway pressure and higher mean dynamic compliance. It was found that SR-717 administration aggravated the increased mean/peak airway pressure and the decreased mean dynamic compliance, while 4-PBA with SR-717 co-treatment improved I/R induced pulmonary ventilation dysfunction in rats (Figure 7A–C, P < 0.05).
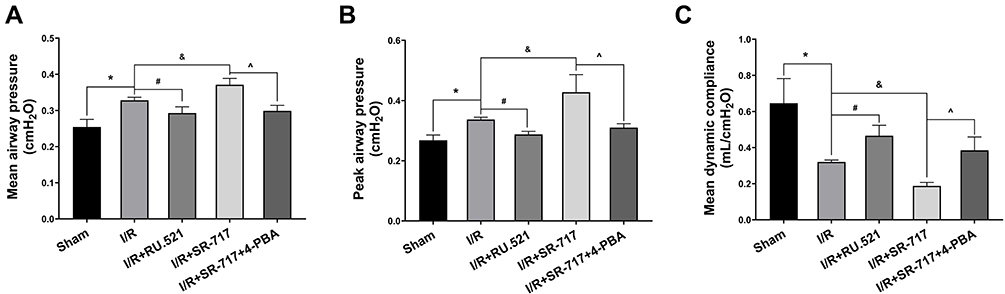 |
Figure 7 Measurement of pulmonary ventilation function in rats. (A–C) The mean airway pressure, peak airway pressure and mean dynamic compliance were used to evaluate pulmonary ventilation function. Data are presented as mean ± SEM, n = 6 per group. *P < 0.05 vs sham group, #P < 0.05 vs I/R group, &P < 0.05 vs I/R group, ^P < 0.05 vs I/R + SR-717 group. |
Discussion
Lung I/R injury is the fundamental cause of pulmonary dysfunction following lung transplantation surgery, which could lead to primary graft dysfunction that accounts for a high rate of mortality, even with intensive supportive therapy.37 Further researches have confirmed that over-activation of pulmonary inflammation and excessive apoptosis are important mechanisms of pulmonary dysfunction induced by the lung I/R process, but their initial molecular mediators are not fully understood.38
Our present findings showed that the reactive cGAS-STING pathway leads to lung I/R injury by aggravating ERS in alveolar epithelial type II cells. Pulmonary edema and pathological lung tissue injury are the two main pathophysiological components of lung I/R injury. We discovered that RU.521, a specific inhibitor of cGAS, reduced cell apoptosis, production of reactive oxygen species (ROS), and infiltration of inflammatory cells in a rat lung I/R model; thereafter attenuated pulmonary edema and alleviated lung tissue pathological injury; and thus improved pulmonary ventilation function.
cGAS is a novel pattern recognition receptor (PRR) that functions as a double-stranded (dsDNA) sensor, which could be activated by cytosolic DNA and produces cGAMP, which binds to STING and activates its downstream pathway, thereby triggering the inflammatory response and modulating cell death.39 Cytosolic DNA is a potent trigger of cGAS activation. The foundational researches of cGAS indicate its requirement in response to DNA virus infection, and the exogenous microbial DNA remains the primary activator of cGAS.20 However, in addition to infection-related DNA entering the cytosol from viral or bacterial sources, cGAS has now been found to interact with various endogenous self-DNA according to recent studies. These types of endogenous DNA include cytosolic DNA origin from nuclei and mitochondria, chromatin in the nucleus, and DNA in the cytosolic micronuclei.40–42 DNA is restricted to nuclei and mitochondria under normal circumstances, but the high level of oxidative stress defined by overproduction of ROS could enhance the mitochondrial permeability transition, allowing self-DNA to leak from mitochondria into the cytosol.43 Increased amounts of intracellular DNA serve as a DAMP, which could be detected by PRRs involving cGAS. In our present study, we found that the production of ROS was upregulated in lung after I/R in rats, which was consistent with precious study.9 And the transmission electron microscopy analysis revealed that mitochondria were morphologically normal in pneumonocytes including alveolar epithelial type II and type I cells for rats in sham group. However, I/R induced evident deformation of the mitochondrial cristae and destruction of the mitochondrial membrane integrity (Figure S4). As a result, mtDNA might leak from the impaired mitochondria induced by lung I/R and trigger cGAS activation.
The main function of the cGAS-STING signaling activation is to initiate antiviral immunity and active adaptive immune system through inducing type I interferon pathway and upregulating the expression of type I interferon.19 Besides its primary function of triggering an inflammatory response, recent works showed that cGAS-STING signaling plays an important role in cell death modulation. cGAS-STING signaling activation promotes the formation of a complex between BAX (Bcl2-associated X gene) and IRF3, which triggers apoptosis through the mitochondrial pathway.44 Besides, type I interferon induced by cGAS-STING can stimulate expression of TNF-related apoptosis-inducing ligand (TRAIL), which act via death receptor 5 to induce apoptosis.45 Growing evidence has highlighted that apoptosis, inflammation, and ROS play a critical and detrimental role in the development of lung I/R injury, but the regulation of cGAS in cell apoptosis and pulmonary inflammation induced by lung I/R is largely unexplored. The linkage between cGAS-STING pathway activation and lung injury caused by the I/R process remains elusive.
Zhao et al reported that deletion of cGAS resulted in decreased hepatocytes autophagy induced by hypoxia and thus leading to increased apoptotic cell death during liver I/R, which revealed a novel role for cGAS in the protection of the liver from I/R injury.46 On the contrary, in a report on cerebral I/R injury, Liao et al described that activation of the cGAS-STING pathway served as a detrimental role via promoting the formation of a pro-inflammatory microenvironment, while deletion of cGAS attenuated I/R-induced neuroinflammation and brain injury.47 In our present study, we found that the expression of both cGAS and STING were upregulated dramatically in rat following I/R. Inhibition of cGAS significantly reduced cell apoptosis and pulmonary inflammation caused by the lung I/R, thereafter alleviated pulmonary edema and lung tissue pathological injury and attenuated pulmonary ventilation function. The important role of cGAS in the setting of lung I/R injury was indicated by our findings. We showed that the cGAS-STING pathway activation exacerbated I/R-induced lung injury via mediating apoptosis, oxidative stress and the inflammatory response. The cGAS inhibitor, RU.521 has been proven to be nontoxic in animals by previous studies,48 so it may serve as basic experimental evidence for the potential protective effect of RU.521 in clinical practice. Therefore, blocking cGAS might be an effective approach to inhibit excessive cell apoptosis, ROS production and aberrant inflammatory response, thus preventing the lung from I/R injury. However, the underlying mechanisms need to be further investigated.
Various studies have demonstrated that ERS plays a primary role in the development of I/R injury and inhibition of ERS has become a common intervention strategy for I/R injury in the heart, lung, liver, and other important organs.49–51 But its upstream target remains incompletely understood. ERS can be seen as a compensatory response responsible for maintaining cell homeostasis via correcting endoplasmic reticulum protein folding errors and promoting autophagy-associated degeneration of unfolded or misfolded proteins during the I/R process. Several researches on in vitro pharmacologic manipulation illustrated a reciprocal regulation between ERS and STING activation.52,53 ERS could induce STING activity, while activation of STING increased ERS and ERS-dependent autophagy. The linkage between ERS and the cGAS-STING signaling activation remains unknown in the setting of lung I/R injury. According to our present study, we found that lung I/R significantly activated the cGAS-STING signaling and pulmonary ERS. The expression of STING and BiP was downregulated when cGAS was inhibited, whereas activation of STING with the STING specific agonist SR-717 resulted in substantially increased ERS and subsequent lung injury. Taking these findings together, we suggested that cGAS-STING pathway might trigger the ERS responses. Severe and persistent ERS is considered to be detrimental due to its capacity to facilitate cell death and trigger an aberrant inflammatory response by activating the pathogenic cascades of various chronic disorders. Profound ERS results in cell death and serious inflammation via triggering further ROS damage, promoting overload of intracellular calcium, aggravating mitochondrial metabolic disorders, and depletion of ATP.54–56 Moreover, subsequent cascades may lead to a series of catastrophic reactions which ultimately augment the pro-apoptosis pathway activation and inflammatory activity.57
Alveolar epithelial type II cells (AECIIs), distinguished by the presence of lamellar bodies which can store and secrete pulmonary surfactant, are functionally important in regulating alveolar fluid levels and adjusting pulmonary ventilation function and contribute to the immune response as well.13 We found that RU.521 evidently downregulated the expression of STING and BiP induced by I/R in AECIIs. However, our current research revealed an intriguing and perplexing finding: increased I/R-induced cGAS was scarcely expressed in AECIIs, which was unexpected. A possible explanation might be because cGAS secondary message cGAMP can enter bystander cells to trigger downstream signaling pathways as well. According to various studies, it can spread over gap junctions to neighboring cells or enter distant cells by incorporating into viral capsids.58,59 Besides, some cGAMP can be released into the microenvironment upon conditions including cell damage or necrotic cell death, and this cGAMP can enter other cells via channel-dependent mechanisms.60 The regulation of cGAS-STING pathway is complex, and cGAMP generated by other pneumonocytes could diffuse into AECIIs and be detected by STING, thereby inducing ERS of AECIIs. However, the exact process by which cGAMP enters AECIIs is uncertain, and more research is needed.
In summary, we conclude that cGAS-STING pathway activated by lung ischemia/reperfusion promotes cell apoptosis, oxidative stress and inflammation via inducing ERS in alveolar epithelial type II cells. Inhibition of cGAS attenuates pulmonary ERS and improves pulmonary ventilation function of rats after lung I/R.
There are some limitations in our present study. First, cGAS and STING might localize in alveolar epithelial type II cells or other pneumonocyte types in lung; therefore, further experiments are required to define cell-specific effects of cGAS on lung I/R injury. Second, the interaction among different pneumonocyte types in the regulation of lung I/R requires further investigation.
Conclusion
To conclude, inhibition of the cGAS-STING pathway attenuates lung ischemia/reperfusion injury via regulating ERS in alveolar epithelial type II cells of rats (Figure 8). cGAS might be a potential target for treating lung ischemia/reperfusion injury.
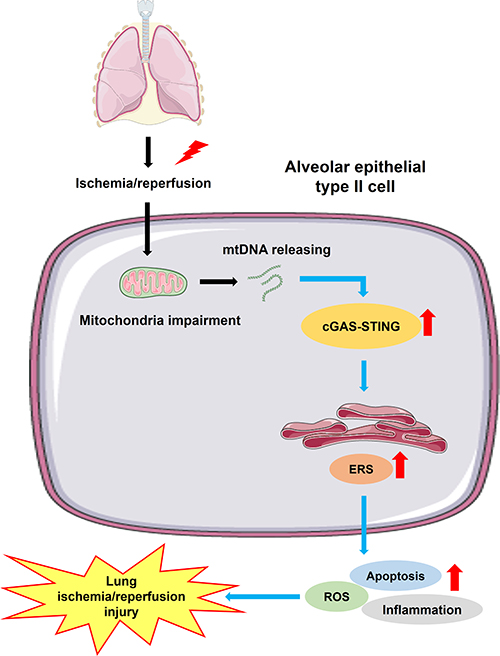 |
Figure 8 Schematic diagrams illustrating the effects of the cGAS-STING pathway in lung ischemia/reperfusion injury. The process of lung ischemia/reperfusion aggravates the impairment of mitochondria in alveolar epithelial type II cells, augmenting the releasing of mtDNA from mitochondria to cytosol. Subsequently, the downstream cGAS-STING signaling is activated, leading to enhanced endoplasmic reticulum stress (ERS). Severe and persistent ERS finally leads to excessive cell apoptosis, aberrant inflammation and a high level of oxidative stress, which result in lung ischemia/reperfusion injury. |
Abbreviations
cGAS, cyclic GMP-AMP synthase; STING, stimulation of interferon genes; ERS, endoplasmic reticulum stress; SP-C, surfactant protein-C; 4-PBA, 4-phenylbutyric acid; DMSO, dimethyl sulfoxide; mtDNA, mitochondrial DNA; I/R, ischemia/reperfusion; W/D, lung wet/dry ratio; LW/BW, lung weight/body weight ratio; HE, hematoxylin and eosin; TUNEL, terminal deoxynucleotidyl transferase-mediated nick end-labeling; DHE, dihydroethidium; TEM, transmission electron microscopy; ROS, reactive oxygen species; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (no. 30800500, 81900007), the Shanghai Science and Technology Committee (18140903400), the Shanghai Municipal Health Commission (20204Y0082), the Project of the Xuhui District Science Committee of Shanghai (SHXH201838) and Fudan Undergraduate Research Opportunities Program (FDUROP, 21077). We sincerely thank Dr. Hongyang Gao’s technical support in electron micrographs taken by transmission electron microscopy (School of Basic Medical Sciences, Fudan University, Shanghai). We thank Miss. Yun Lin (Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai) for her excellent technical support in rat’s pulmonary ventilation function measurement and Western blot analysis.
Author Contributions
All the authors have made an important contribution to the present work, whether that is in the study design, conception, execution, acquisition of data, analysis and interpretation, or in all these areas: taking part in drafting, critically reviewing or revising the article. All the authors have given final approval of the version to be published and agreed on the journal to which the article has been submitted as well as agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Iske J, Hinze CA, Salman J, Haverich A, Tullius SG, Ius F. The potential of ex vivo lung perfusion on improving organ quality and ameliorating ischemia reperfusion injury. Am J Transplant. 2021;21(12):3831–3839. doi:10.1111/ajt.16784
2. Lama VN. Update in lung transplantation 2008. Am J Respir Crit Care Med. 2009;179(9):759–764. doi:10.1164/rccm.200902-0177UP
3. Wang X, O’Brien ME, Yu J, et al. Prolonged cold ischemia induces necroptotic cell death in ischemia-reperfusion injury and contributes to primary graft dysfunction after lung transplantation. Am J Respir Cell Mol Biol. 2019;61(2):244–256. doi:10.1165/rcmb.2018-0207OC
4. Daud SA, Yusen RD, Meyers BF, et al. Impact of immediate primary lung allograft dysfunction on bronchiolitis obliterans syndrome. Am J Respir Crit Care Med. 2007;175(5):507–513. doi:10.1164/rccm.200608-1079OC
5. Jungraithmayr W. Novel strategies for endothelial preservation in lung transplant ischemia-reperfusion injury. Front Physiol. 2020;11:581420. doi:10.3389/fphys.2020.581420
6. Mrazkova H, Lischke R, Hodyc D, Herget J. The protective effect of hypercapnia on ischemia-reperfusion injury in lungs. Respir Physiol Neurobiol. 2015;205:42–46. doi:10.1016/j.resp.2014.10.002
7. Wu JX, Zhu HW, Chen X, et al. Inducible nitric oxide synthase inhibition reverses pulmonary arterial dysfunction in lung transplantation. Inflamm Res. 2014;63(8):609–618. doi:10.1007/s00011-014-0733-5
8. Zhang JP, Zhang WJ, Yang M, Fang H. Propofol attenuates lung ischemia/reperfusion injury though the involvement of the MALAT1/microRNA-144/GSK3β axis. Mol Med. 2021;27(1):77. doi:10.1186/s10020-021-00332-0
9. Ferrari RS, Andrade CF. Oxidative stress and lung ischemia-reperfusion injury. Oxid Med Cell Longev. 2015;2015:590987. doi:10.1155/2015/590987
10. Li J, Chen Q, He X, et al. Dexmedetomidine attenuates lung apoptosis induced by renal ischemia-reperfusion injury through α2AR/PI3K/Akt pathway. J Transl Med. 2018;16(1):78. doi:10.1186/s12967-018-1455-1
11. Weibel ER. On the tricks alveolar epithelial cells play to make a good lung. Am J Respir Crit Care Med. 2015;191(5):504–513. doi:10.1164/rccm.201409-1663OE
12. Jansing NL, McClendon J, Henson PM, et al. Unbiased quantitation of alveolar type II to alveolar type I cell transdifferentiation during repair after lung injury in mice. Am J Respir Cell Mol Biol. 2017;57(5):519–526. doi:10.1165/rcmb.2017-0037MA
13. Wu Y, Lv J, Feng D, et al. Restoration of alveolar type II cell function contributes to simvastatin-induced attenuation of lung ischemia-reperfusion injury. Int J Mol Med. 2012;30(6):1294–1306. doi:10.3892/ijmm.2012.1161
14. Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92(2):537–576. doi:10.1152/physrev.00027.2011
15. Men X, Han S, Gao J, et al. Taurine protects against lung damage following limb ischemia reperfusion in the rat by attenuating endoplasmic reticulum stress-induced apoptosis. Acta Orthop. 2010;81(2):263–267. doi:10.3109/17453671003587085
16. Korfei M, Ruppert C, Mahavadi P, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846. doi:10.1164/rccm.200802-313OC
17. Sun X, Liu H, Sun Z, et al. Acupuncture protects against cerebral ischemia-reperfusion injury via suppressing endoplasmic reticulum stress-mediated autophagy and apoptosis. Mol Med. 2020;26(1):105. doi:10.1186/s10020-020-00236-5
18. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol. 2020;15:493–518. doi:10.1146/annurev-pathmechdis-012419-032847
19. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21(9):501–521. doi:10.1038/s41580-020-0244-x
20. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791. doi:10.1126/science.1232458
21. Cui Y, Zhao D, Sreevatsan S, et al. Mycobacterium bovis induces endoplasmic reticulum stress mediated-apoptosis by activating IRF3 in a murine macrophage cell line. Front Cell Infect Microbiol. 2016;6:182.
22. Zhang Y, Chen W, Wang Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomed Pharmacother. 2020;125:110022. doi:10.1016/j.biopha.2020.110022
23. Ueda S, Chen-Yoshikawa TF, Tanaka S, et al. Protective effect of necrosulfonamide on rat pulmonary ischemia-reperfusion injury via inhibition of necroptosis. J Thorac Cardiovasc Surg. 2022;163(2):e113–e122. doi:10.1016/j.jtcvs.2021.01.037
24. Vincent J, Adura C, Gao P, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. 2017;8(1):750. doi:10.1038/s41467-017-00833-9
25. Xu Q, Xiong H, Zhu W, Liu Y, Du Y. Small molecule inhibition of cyclic GMP-AMP synthase ameliorates sepsis-induced cardiac dysfunction in mice. Life Sci. 2020;260:118315. doi:10.1016/j.lfs.2020.118315
26. Chin EN, Yu C, Vartabedian VF, et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science. 2020;369(6506):993–999. doi:10.1126/science.abb4255
27. Chen X, Wang Y, Xie X, et al. Heme oxygenase-1 reduces sepsis-induced endoplasmic reticulum stress and acute lung injury. Mediators Inflamm. 2018;2018:9413876.
28. Lan CC, Peng CK, Tang SE, et al. Inhibition of Na-K-Cl cotransporter isoform 1 reduces lung injury induced by ischemia-reperfusion. J Thorac Cardiovasc Surg. 2017;153(1):206–215. doi:10.1016/j.jtcvs.2016.09.068
29. Kopp MC, Larburu N, Durairaj V, Adams CJ, Ali MMU. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat Struct Mol Biol. 2019;26(11):1053–1062. doi:10.1038/s41594-019-0324-9
30. Sun H, Zhao X, Tai Q, Xu G, Ju Y, Gao W. Endothelial colony-forming cells reduced the lung injury induced by cardiopulmonary bypass in rats. Stem Cell Res Ther. 2020;11(1):246. doi:10.1186/s13287-020-01722-7
31. Zhang Y, Zhang X, Rabbani ZN, Jackson IL, Vujaskovic Z. Oxidative stress mediates radiation lung injury by inducing apoptosis. Int J Radiat Oncol Biol Phys. 2012;83(2):740–748. doi:10.1016/j.ijrobp.2011.08.005
32. Wang J, Huang J, Wang L, et al. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-κB signaling pathway. J Thorac Dis. 2017;9(11):4398–4412. doi:10.21037/jtd.2017.09.135
33. Chu SJ, Tang SE, Pao HP, Wu SY, Liao WI. Protease-activated receptor-1 antagonist protects against lung ischemia/reperfusion injury. Front Pharmacol. 2021;30(12):752507. doi:10.3389/fphar.2021.752507
34. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi:10.1146/annurev-pathol-012513-104649
35. Yang C, Yang W, He Z, et al. Kaempferol alleviates oxidative stress and apoptosis through mitochondria-dependent pathway during lung ischemia-reperfusion injury. Front Pharmacol. 2021;12:624402. doi:10.3389/fphar.2021.624402
36. Huang CY, Deng JS, Huang WC, Jiang WP, Huang GJ. Attenuation of lipopolysaccharide-induced acute lung injury by hispolon in mice, through regulating the TLR4/PI3K/Akt/mTOR and Keap1/Nrf2/HO-1 pathways, and suppressing oxidative stress-mediated er stress-induced apoptosis and autophagy. Nutrients. 2020;12(6):1742. doi:10.3390/nu12061742
37. de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia-reperfusion-induced lung injury. Am J Respir Crit Care Med. 2003;167(4):490–511. doi:10.1164/rccm.200207-670SO
38. Talaie T, DiChiacchio L, Prasad NK, et al. Ischemia-reperfusion injury in the transplanted lung: a literature review. Transplant Direct. 2021;7(2):e652. doi:10.1097/TXD.0000000000001104
39. Zhang X, Bai XC, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. 2020;53(1):43–53. doi:10.1016/j.immuni.2020.05.013
40. Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, Jr GM. Interleukin-1β induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol Cell. 2019;74(4):801–815.e6. doi:10.1016/j.molcel.2019.02.038
41. Maekawa H, Inoue T, Ouchi H, et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019;29(5):1261–1273.e6. doi:10.1016/j.celrep.2019.09.050
42. Li C, Liu W, Wang F, et al. DNA damage-triggered activation of cGAS-STING pathway induces apoptosis in human keratinocyte HaCaT cells. Mol Immunol. 2021;131:180–190. doi:10.1016/j.molimm.2020.12.037
43. Zhao M, Wang Y, Li L, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. 2021;11(4):1845–1863. doi:10.7150/thno.50905
44. Chattopadhyay S, Marques JT, Yamashita M, et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010;29(10):1762–1773. doi:10.1038/emboj.2010.50
45. Zhu Q, Man SM, Karki R, Malireddi RKS, Kanneganti TD. Detrimental type I interferon signaling dominates protective AIM2 inflammasome responses during Francisella novicida infection. Cell Rep. 2018;22(12):3168–3174. doi:10.1016/j.celrep.2018.02.096
46. Lei Z, Deng M, Yi Z, et al. cGAS-mediated autophagy protects the liver from ischemia-reperfusion injury independently of STING. Am J Physiol Gastrointest Liver Physiol. 2018;314(6):G655–G667. doi:10.1152/ajpgi.00326.2017
47. Liao Y, Cheng J, Kong X, et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics. 2020;10(21):9644–9662. doi:10.7150/thno.47651
48. Gamdzyk M, Doycheva DM, Araujo C, et al. cGAS/STING pathway activation contributes to delayed neurodegeneration in neonatal hypoxia-ischemia rat model: possible involvement of LINE-1. Mol Neurobiol. 2020;57(6):2600–2619. doi:10.1007/s12035-020-01904-7
49. Feng D, Wang B, Wang L, et al. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J Pineal Res. 2017;62:3. doi:10.1111/jpi.12395
50. Liu H, Wang L, Weng X, et al. Inhibition of Brd4 alleviates renal ischemia/reperfusion injury-induced apoptosis and endoplasmic reticulum stress by blocking FoxO4-mediated oxidative stress. Redox Biol. 2019;24:101195. doi:10.1016/j.redox.2019.101195
51. Yang YF, Wang H, Song N, et al. Dexmedetomidine attenuates ischemia/reperfusion-induced myocardial inflammation and apoptosis through inhibiting endoplasmic reticulum stress signaling. J Inflamm Res. 2021;31(14):1217–1233.
52. Petrasek J, Iracheta-Vellve A, Csak T, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci USA. 2013;110(41):16544–16549. doi:10.1073/pnas.1308331110
53. Moretti J, Roy S, Bozec D, et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. 2017;171(4):809–823.e13. doi:10.1016/j.cell.2017.09.034
54. Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249. doi:10.1016/j.biopha.2019.109249
55. Namgaladze D, Khodzhaeva V, Brüne B. ER-mitochondria communication in cells of the innate immune system. Cells. 2019;8(9):1088. doi:10.3390/cells8091088
56. Sprenkle NT, Sims SG, Sánchez CL, Meares GP. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener. 2017;12(1):42. doi:10.1186/s13024-017-0183-y
57. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. doi:10.1038/nrm3270
58. Ablasser A, Schmid-Burgk JL, Hemmerling I, et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503(7477):530–534. doi:10.1038/nature12640
59. Gentili M, Kowal J, Tkach M, et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015;349(6253):1232–1236. doi:10.1126/science.aab3628
60. Luteijn RD, Zaver SA, Gowen BG, et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature. 2019;573(7774):434–438. doi:10.1038/s41586-019-1553-0
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.